

Abstract
Asparagine-linked glycosylation is a common posttranslational protein modification regulating the structure, stability and function of many proteins. The N-linked glycosylation machinery involves enzymes responsible for the assembly of the lipid-linked oligosaccharide (LLO), which is then transferred to the asparagine residues on the polypeptides by the enzyme oligosaccharyltransferase (OST). A major goal in the study of protein glycosylation is to establish quantitative methods for the analysis of site-specific extent of glycosylation. We developed a sensitive approach to examine glycosylation site occupancy in Saccharomyces cerevisiae by coupling stable isotope labeling (SILAC) approach to parallel reaction monitoring (PRM) mass spectrometry (MS). We combined the method with genetic tools and validated the approach with the identification of novel glycosylation sites dependent on the Ost3p and Ost6p regulatory subunits of OST. Based on the observations that alternations in LLO substrate structure and OST subunits activity differentially alter the systemic output of OST, we conclude that sequon recognition is a direct property of the catalytic subunit Stt3p, auxiliary subunits such as Ost3p and Ost6p extend the OST substrate range by modulating interfering pathways such as protein folding. In addition, our proteomics approach revealed a novel regulatory network that connects isoprenoid lipid biosynthesis and LLO substrate assembly.
Glycosylation is a fundamental part of life whose impact and intricacy increase with the complexity of the organism (1). Glycans affect protein folding, stability and degradation, they mediate interactions of all cells and regulate essential biological, chemical and physical processes (2). Asparagine linked N-glycosylation is an essential modification of proteins conserved among eukaryotes, archaea and some bacteria (3). In eukaryotes, the reaction is catalyzed by oligosaccharyltransferase (OST)1 in the lumen of the endoplasmic reticulum (ER) (4). On arrival of protein substrate to the lumen of the ER, the preassembled oligosaccharide is transferred en bloc from the lipid carrier dolichol pyrophosphate (Dol-PP) to asparagines in selected glycosylation sequons (N-X-S/T; X≠P) (4, 5). After the oligosaccharide has been transferred, the N-linked glycan structure influences protein folding and helps ER quality control (ERQC) machinery to distinguish among folding intermediates, in which only properly folded proteins can reach their final destinations through the secretory pathway (6). If a polypeptide is not able to reach its native conformation, the misfolded glycoprotein will be recycled in the ER-associated degradation (ERAD) pathway (7). Hypoglycosylation of proteins will lead to the accumulation of unfolded proteins in the ER (ER stress) and to the activation of an unfolded protein response (UPR) pathway, in which transcriptional induction of UPR target genes allows the cell to adjust the capacity of protein folding in the ER (8, 9).
N-linked glycosylation starts with the assembly of the Glc3Man9GlcNAc2 lipid-linked oligosaccharide (LLO) on a Dol-PP carrier on the ER membrane (10, 11). The LLO is synthesized in a defined, stepwise manner by a series of enzymes belonging to the ALG (Asparagine-Linked Glycosylation) family (12). The process starts on the cytoplasmic side of the ER membrane, where Man5GlcNAc2 heptasaccharide is assembled on the Dol-PP by sequential action of the Alg7/Alg13/Alg14 and the Alg1/Alg2/Alg11 enzyme complexes (13, 14). These glycosyltransferases use nucleotide-activated sugars as donors. The LLO is then flipped onto the luminal side of the ER by a process requiring Rft1p (15). The LLO is further elongated by the action of Alg3p, Alg9p, Alg12p, and again Alg9p that add four additional mannosyl residues. The LLO is completed by the action of three glucosyltransferases Alg6, Alg8 and Alg10. Unlike the cytoplasmic glycosyltransferases, lumenal ALG enzymes use Dol-P-bound sugars as donors (16) and have a high LLO substrate specificity that ensures an assembly of an optimal OST LLO substrate that exposes the terminal α-1,2-linked glucose (17). Because of sequential nature of LLO biosynthesis, deficiencies in ALG enzymes result in accumulation of LLO intermediates. Truncated LLO structures will still be transferred by the OST though with a lower efficiency, resulting in the appearance of hypoglycosylated protein substrates (12).
In higher eukaryotes, OST is a multiprotein complex consisting of different subunits (Stt3p, Ost1p, Ost5p, Wbp1p, Ost2p, Swp1p, Ost3p/Ost6p, and Ost4p in S. cerevisiae. Some protozoan, archaeal, and bacterial species have a single protein OST, homologous to Stt3p, the catalytic protein subunit of the eukaryotic OST (4). Based on phylogenetic analysis, yeast STT3 is proposed to be a B type STT3 that is not associated with the translocon (18). As consensus sequons are required to be flexible to be accessible to the OST catalytic site, there is a competition between folding and glycosylation (19). Consequently, OST has evolved ways of competing with protein folding. In yeast, the presence of either Ost3p or Ost6p results in two alternative OST complexes (20). Both subunits have thioredoxin-like ER lumenal domains that transiently capture stretches of OST protein substrates in the ER through noncovalent or mixed disulfide bond (21–23). The proposed role of these subunits is to increase the efficiency at glycosylation sites by guiding the catalytic site of OST to nearby sequons and inhibiting local polypeptide folding (23, 24). The roles of the additional subunits in the eukaryotic OST are mostly unknown but may be necessary for directing the glycosylation toward protein substrates and glycans.
Mass spectrometry (MS) has become the method of choice for the identification and quantification of N-linked glycoproteins (25–33). MS assays rely on the elution time and precursor mass of the modified peptides, as well as the mass-to-charge ratio and relative intensity of specific fragment ions that indicate the sequence position of modified asparagine residues. Several MS techniques for measuring N-linked glycosylation site occupancy in yeast have been described using different modes of operation for targeted glycopeptide analysis, such as selected reaction monitoring (SRM), sequential window acquisition of targeted fragment ions (SWAT), and data-independent techniques, such as sequential window acquisition of all theoretical fragment ion spectra (SWATH) (32–34). These techniques have used isolation of the glycoprotein rich cell wall fraction for sample simplification. However, cell wall fractionation can introduce analytical bias as proteins that are inefficiently glycosylated in the ER tend to fold incorrectly and are prevented from trafficking out of the ER by the ERQC mechanism (6, 7). In this paper, we describe a complementary method for the identification and relative quantification of N-linked glycosylation occupancy and protein abundance of yeast membrane and lumenal proteins using SILAC (stable isotope labeling by amino acids in cell culture) strategy combined with a PRM (parallel reaction monitoring) based MS method. The PRM technique is currently the most sensitive mode of targeted proteomics analyses for quantitative measurements in biological samples and is now being exploited for quantitative analysis of N-linked glycosylation (35, 36).
EXPERIMENTAL PROCEDURES
Experimental Design and Statistical Rationale
The study aimed to define N-linked glycosylation occupancy of yeast proteins in distinct yeast mutants defective in the N-linked glycosylation pathway. SILAC coupled to PRM analysis was used to analyze quantitative changes in N-linked glycosylation occupancy. Each sample for MS analysis was generated by mixing two individual SILAC-labeled cell populations. Ribosomal proteins Rpl5 and Rsp1 were used as internal control to evaluate variability during mixing of light and heavy cells. All quantitative PRM experiments were performed in biological triplicates resulting in a total of 21 samples. Reproducibility of results was assessed by calculating coefficient of variation for each set of biological triplicates in Excel where > 83% (n = 62) of the data analyzed had CV value of < 20%. Statistical tests used to analyze significant differences indicated in the respective figure legends were performed using t test in Microsoft Excel.
Yeast Strains and Growth Conditions
All yeast strains and plasmids used in this study are listed in supplemental Table S1 and S2. Standard yeast genetic techniques were used (37, 38). All yeast strains were grown in an orbital shaker at 30 °C and 180 rpm to exponential phase (OD600 nm 1.0).
For MS assay, cells were grown in appropriate synthetic drop-out (S.D.) medium (0.67% (w/v) yeast nitrogen base, 2% (w/v) glucose with appropriate amino acid supplements) containing either 20 mg/L of light or heavy isotopes of arginine (13C6) and lysine (13C6-15N2) (Cambridge Isotope Laboratories). Cells were collected, frozen in liquid nitrogen and stored at −80 °C.
For sterol inhibition assay, mid-log phase cultures of the wild-type yeast cells in light S.D. medium were exposed to miconazole (0.3 μg/ml, dissolved in DMSO; Janseen Geel) whereas the controls received an equivalent amount of DMSO. After 2.5 h, cells were washed once with ice cold 50 mm sodium acetate, 10 mm EDTA buffer (pH 4.5) before they were collected, frozen in liquid nitrogen and stored at −80 °C.
Sample Preparation for Mass Spectrometry
Membrane proteins were prepared as described (39). In brief, yeast cells were lysed at 4 °C using glass beads, and the microsomal fraction was pelleted by centrifugation (16000 × g; 20 min), resuspended in 0.1 m Na2CO3, 1 mm EDTA, pH 11.3 buffer and pelleted again. Samples were resuspended in SDS buffer (2% SDS (w/v), 50 mm DTT, 0.1 m Tris HCl pH 7.6) and processed using the filter assisted sample preparation protocol (40). Membrane proteins were digested first with endopeptidase LysC (20 μg/ml; Wako Pure Chemical, Richmond, VA) at room temperature for 16 h followed by digestion with trypsin (20 μg/ml; Promega) at 37 °C for 4 h. For data-dependent acquisition (DDA)-based experiments in the discovery phase of PRM assay development, sample was enriched for glycopeptides using SPE ZIC-HILIC (SeQuant) column and collected in a single fraction as described (41). For PRM experiments protein digestion was directly followed by digestion of N-glycans with endo-β-N-acetylglucosaminidase H (EndoH; 500 U; New England Biolabs) in sodium citrate buffer (50 mm, pH 5.5) at 37 °C with agitation for 40 h. Peptides were desalted using Sep Pak C18 classic (Waters) and dried using speed vacuum. Desalted peptides were resuspended in ACN/H2O (3:97 (v/v)) with formic acid (FA; 0.1% (v/v)) and analyzed by LC-ESI-MS/MS.
Data-dependent Acquisition and Targeted Proteomics Analysis
Mass spectrometric analysis
Samples were subjected to a Q Exactive HF mass spectrometer (Thermo Scientific) coupled to a nano EasyLC 1000 (Thermo Fisher Scientific) system and to an Acquity UPLC M-class System (Waters). Samples were loaded onto a self-made column (75 μm × 150 mm) packed with reverse-phase C18 material (ReproSil-Pur 120 C18-AQ, 1.9 μm, Dr. Maisch GmbH) when the EasyLC 1000 was coupled. ACQUITY UPLC M-Class column (75 μm × 150 mm) packed with reverse-phase C18 material (Waters HSS T3 100 C18, 1.8 μm) was used when the Acquity UPLC M-class system was coupled. Peptides were separated at a flow rate of 300 nL/min using a linear gradient of 1% to 35% solvent B (0.1% formic acid in acetonitrile) over 90 min, followed by an increase to 98% B over 2 min and held at 98% B for 5 min before returning to initial conditions of 1% B.
In DDA-MS, the mass spectrometer was set to acquire full-scan MS spectra (300–1700 m/z) at a resolution of 60,000 after accumulation to an automated gain control (AGC) target value of 3e6 and a maximum injection time of 15 ms. Charge state screening was enabled and unassigned charge states and singly charged precursors were excluded. Ions were isolated using a quadrupole mass filter with a 1.2 m/z isolation window. A maximum injection time of 45 ms was set. HCD fragmentation was performed at a normalized collision energy (NCE) of 28%. Selected ions were dynamically excluded for 20 s.
For PRM measurements, the Q Exactive HF performed MS1 scans (400–1200 m/z) followed by 12 MS/MS acquisitions in PRM mode. The full scan event was collected at a resolution of 60,000 (at m/z 400) and a AGC value of 3e6 and a maximum injection time of 120 ms. The PRM scan events used an Orbitrap resolution of 30,000 or 60,000, maximum fill time of 55 ms or 110 ms respectively, with an isolation width of 2 m/z and an AGC value of 2e5. HCD fragmentation was performed at a normalized collision energy (NCE) of 28% and MS/MS scans were acquired with a starting mass of m/z 120. Scan windows were set to 10 min for each peptide in the final PRM method to ensure the measurement of 6–10 points per LC peak per transition. All samples were analyzed using two PRM methods based on scheduled inclusion lists containing the 175 target precursor ions, including Biognosys iRT standard peptides, at an Orbitrap resolution of 30 000 and two PRM methods based on scheduled inclusion lists containing the 128 target precursor ions, including Biognosys iRT standard peptides (Biognosys AG, Zurich), at an Orbitrap resolution of 60,000 (supplemental Table S3).
Protein Identification and Spectral Library Building
MS and MS/MS spectra generated from enriched and nonenriched glycopeptides extracts were converted to Mascot generic format (MGF) using Proteome Discoverer, v1.4 (Thermo Fisher Scientific, Bremen, Germany) using the automated rule based converter control (42). The MGFs were searched with Mascot Server v.2.5.1.3 (www.matrixscience.com) using the following parameters: a precursor ion mass tolerance of 15 ppm, product ion mass tolerance of 0.05 Dalton, enzyme specificity to trypsin and LysC and up to two missed cleavages were allowed, as variable modifications methionine oxidation and asparagine N-linked HexNAc (CID/HCD) glycosylation, and as fixed modification carbamidomethylation of cysteine, for the glycoenriched samples. Searches were made against the Saccharomyces cerevisiae reference proteome database (containing 6649 entries, downloaded 24/04/2012), concatenated to a reversed decoyed FASTA database and 261 common mass spectrometry protein contaminants. A Mascot score >20 and an expectation value <0.05 were considered to identify the correctly assigned peptides. The Mascot search results (dat. files) were imported into Scaffold software (v4.0) and Scaffold PTM software (v2.1.3) to assign the localization score for HexNAc peptides where sites with Ascore > 20 corresponding to the localization probability of > 99% were considered to identify correctly assigned glycosylation sites (43, 44). Majority of software used to visualize and examine MS/MS data, including Scaffold PTM software, do not discriminate among asparagines located in consensus glycosylation sequons, thus the localization score reported for some glycopeptides is 0. Such sites were accepted given that they only contain one asparagine as possible glycosylation site (corresponding to known glycosylation motif Asn-X-Ser/Thr) (supplemental Table S4).
The Mascot search results (dat. files) were imported into the Skyline software (v2.6.0) (45) and spectral libraries were built using the BiblioSpec algorithm (46). Additionally, published spectral libraries (47) were used to populate the final PRM assays. For glycopeptides in which ion libraries could not be obtained at the time, fragment ions were manually selected based on MS2 spectra obtained directly from the mascot search results. We have used the normalized retention time (iRT) concept described before to calculate iRT values of the target peptides obtained in the DDA-MS (48). The retention time of the target peptides was normalized and transformed into iRT values using the coordinates of the spiked iRT-C18 reference peptides from Biognosys AG, Zurich.
Data Processing and Analysis
Skyline software (v2.6.0) with standard settings was used for data processing (45). The abundance of peptides was analyzed by summing the integrated areas of at least four fragment ions per peptide. Peptide peaks were analyzed manually, and correct identification was assigned on the basis of the following criteria: (1) retention time matching to spectral library within 5% of the gradient length, (2) coelution of light and heavy peptides, (3) dot product between light peptide precursor ion isotope distribution intensities and theoretical > 0.9, (4) dot product between library spectrum intensities and light peptides > 0.8 (in some cases lower dot product was accepted if the other criteria were fulfilled), and (5) matching peak shape for precursor and product ions from light and heavy peptides. The reported light to heavy intensity ratios (L/H) were normalized for proportionate mixing of heavy labeled with light labeled cells, by dividing the L/H intensity ratio of all measured peptides by the median of L/H intensity reported for four peptides belonging to two control proteins, Rps1a and Rpl5. L/H ratio for glycopeptides modified with HexNAc was used to calculate the relative site occupancy for the given peptide/glycosylation site compared with the wild-type reference strain. The relative site occupancy was normalized for expression differences between heavy labeled wild-type reference strain (H) and the mutant light strains (L) by dividing the L/H intensity ratio for the occupied glycopeptide by the median of L/H intensity ratios reported for all peptides that do not containing a NxT/S sequon from the same protein. Protein level quantification was calculated as the median of L/H intensity ratios reported for all peptides without glycosylation sequon belonging to the same protein, normalized for proportionate mixing of heavy labeled with light labeled cells as described above.
PRM MS Quantification of Ergosterol Pathway Protein Abundance
SWATH-MS data published earlier (47) was used as ion library source to build a PRM assay for targeted analysis of ergosterol pathway proteins. Ion library information for peptides belonging to 20 from 26 proteins known to be involved in mevalonate, ergosterol and dolichol pathway were extracted and targeted PRM analysis was performed on Q Exactive HF mass spectrometer as described before. Scheduled inclusion list contained 100 target precursor ions, including Biognosys iRT standard peptides (supplemental Table S5).
The abundance of peptides was analyzed using Skyline software with standard settings, in which peptide peaks were analyzed manually as described above. The reported L/H intensity ratios were normalized for proportionate mixing of heavy labeled with light labeled cells, by dividing the L/H intensity ratio of all measured peptides by the median of L/H intensity reported for peptides belonging to two control proteins, Rps1a and Rpl5. Protein level quantification was calculated as the median of L/H intensity ratios reported for all peptides belonging to the same protein. In the case of sterol inhibition assay, resulting protein abundance was further normalized to the protein abundance of the control samples treated with DMSO.
Data Deposit
The mass spectrometry proteomics data, from tier 3 measurements, were handled using the local laboratory information management system (LIMS) (49) and all relevant data have been deposited to the ProteomeXchange Consortium via the PRIDE (http://www.ebi.ac.uk/pride) partner repository with the data set identifier PXD006435 (Username: reviewer63730@ebi.ac.uk; Password: h7qCP1Jk).
RESULTS
SILAC Based PRM MS for Quantitative Profiling of N-glycoproteins in Yeast
To design targeted MS assays for site-specific quantification of N-linked glycosylation site occupancy in yeast, we focused on glycoproteins originating from microsomal fractions (39). A schematic overview of our assay development and workflow is presented in Fig. 1. In a discovery phase, glycopeptides originating from microsomal fractions were enriched after proteinase digestion (Fig. 1A). Glycoproteins originating from microsomal fractions were digested sequentially with LysC and trypsin proteinase. The use of both proteinases, compared with the use of trypsin proteinase alone, has been shown to result in higher yields, higher reproducibility and more accurate quantification (50). We enriched for glycopeptides by solid-phase extraction (SPE) using zwitterionic hydrophilic interaction chromatography (ZIC-HILIC) to ensure high coverage of hydrophilic N-linked glycopeptides as described previously (41). Glycans were released with endoglycosidase H that cleaves yeast oligomannose glycans leaving previously glycosylated asparagine residues tagged with a single GlcNAc residue and identified by DDA-MS. The use of Endo H offers an additional advantage over other analyses that have used PNGase F for deglycosylation, as measurements of tryptic peptides deglycosylated by PNGase F failed to robustly differentiate nonglycosylated from deglycosylated versions of the same peptide (34). We could identify 166 high confidence glycosylation sites corresponding to 105 glycoproteins using data collected from three glycoenriched samples (supplemental Table S4). Most of the glycoproteins were represented with either one or two glycopeptides, the exception were glycoproteins like Pdi1p, Ape3p, Fet3p, Plb1p, Ero1p, Plb2p, and Rax2p that were represented with four or more glycopeptides. Although some of these glycoproteins have more glycosylation sites, these peptides are often suboptimal to get identified in a regular proteomics experiment or might possess poor ionization properties, therefore it is not surprising that majority of glycoproteins were represented only with one or two glycopeptides. These results were the basis for a PRM-based approach, in which glycopeptides were monitored together with the peptides without glycosylation sequon belonging to the same glycoprotein.
Fig. 1.

Schematic representation of SILAC PRM MS assay development and workflow. A, During discovery phase yeast cells were grown in light medium, microsomal fractions were prepared using previously published method (39) and proteins digested with LysC and trypsin proteinases. Glycan containing peptides were enriched using SPE ZIC-HILIC method (41) before glycan release with Endo H endoglycosidase. Glycopeptides containing previously glycosylated asparagine residues tagged with a single GlcNAc residue were identified using DDA MS. B, Obtained data from the discovery phase was used for PRM assay build up in which yeast cells were grown in light medium and mixed 1:1 with the cells grown in heavy medium. Membrane derived peptides were prepared as described above, glycans were released with EndoH and targeted PRM analysis was performed.
To test whether we could target the peptides identified in the DDA-MS analysis but omitting the glycopeptide enrichment step, we mixed yeast wild-type cells grown in medium with heavy arginine and lysine isotopes with cells grown in medium with light arginine and lysine isotopes in an equal ratio and performed targeted PRM analysis (Fig. 1B). Samples enriched for microsomal fractions were digested with LysC and Trypsin proteinases and EndoH was used to generate single GlcNAc glycopeptides. Of 166 glycopeptides corresponding to 105 glycoproteins measured in the enriched samples we were able to detect 62 belonging to 43 glycoproteins in the unenriched samples from three biological replicates with high confidence (i.e. coelution of light and heavy peaks, good correlation of relative intensity of specific fragment ions with the reference library, reproducible retention times across different samples) (Table I).
Table I. List of all glycoproteins and the corresponding glycosylated peptides used in the SILAC PRM- MS analysis of N-linked glycosylation occupancy.
| UniProt ID | Protein | Glycosylation Site | Modified Peptide Sequence | Protein Description |
|---|---|---|---|---|
| P00729 | CPY | N124 | ILGIDPN[HexNAc]VTQYTGYLDVEDEDK | Carboxypeptidase Y |
| P00729 | CPY | N479 | VRN[HexNAc]WTASITDEVAGEVK | Carboxypeptidase Y |
| P12684 | HMG2 | N150 | IPTELVSEN[HexNAc]GTK | 3-hydroxy-3-methylglutaryl-coenzyme A reductase 2 |
| P17967 | PDI | N425 | LAPTYQELADTYAN[HexNAc]ATSDVLIAK | Protein disulfide-isomerase |
| P17967 | PDI | N117 | NSDVN[HexNAc]NSIDYEGPR | Protein disulfide-isomerase |
| P17967 | PDI | N82 | N[+203.1]ITLAQIDC[+57]TENQDLC[+57]M[+16]EHNIPGFPSLK | Protein disulfide-isomerase |
| P17967 | PDI | N174 | IDADFN[HexNAc]ATFYSMANK | Protein disulfide-isomerase |
| P17967 | PDI | N155 | QSQPAVAVVADLPAYLAN[HexNAc]ETFVTPVIVQSGK | Protein disulfide-isomerase |
| P22146 | GAS1 | N40 | FFYSNN[HexNAc]GSQFYIR | 1,3-beta-glucanosyltransferase |
| P23797 | GPI12 | N110 | VRELN[HexNAc]ESAALLLHNER | GlcANc-phosphatidylinositol de-N-acetylase |
| P27810 | KTR1 | N120 | N[HexNAc]VTSALVSGTTK | Alpha-1,2 mannosyltransferase |
| P27825 | CNE1 | N416 | N[HexNAc]VTEAQIIGNK | Calnexin homolog |
| P31382 | PMT2 | N403 | GLPSWSEN[HexNAc]ETDIEYLKPGTSYR | Dol-P-Man-protein mannosyltransferase 2 |
| P32353 | ERG3 | N40 | LLGLNSGFSN[HexNAc]STILQETLNSK | C-5 sterol desaturase |
| P32623 | CRH2 | N310 | N[HexNAc]GTSAYVYTSSSEFLAK | Probable glycosidase CRH2 |
| P32623 | CRH2 | N233/N237 | N[HexNAc]ETYN[HexNAc]ATTQK | Probable glycosidase CRH2 |
| P33302 | PDR5 | N734 | GPAYAN[HexNAc]ISSTESVCTVVGAVPGQDYVLGDDFIR | Pleiotropic ABC efflux transporter of multiple drugs |
| P33754 | SEC66 | N12 | FSNN[HexNAc]GTFFETEEPIVETK | Translocation protein |
| P33767 | WBP1 | N332 | LTLSPSGN[HexNAc]DSETQYYTTGEFILPDR | Oligosaccharyltransferase subunit WBP1 |
| P33767 | WBP1 | N60 | LEYLDIN[HexNAc]STSTTVDLYDK | Oligosaccharyltransferase subunit WBP1 |
| P36016 | LHS1 | N458 | LSN[HexNAc]ESELYDVFTR | Heat shock protein 70 homolog |
| P36051 | MCD4 | N90 | SLVMNN[HexNAc]ATYGISHTR | GPI ethanolamine phosphate transferase 1 |
| P36051 | MCD4 | N198 | HLDQLFHN[HexNAc]STLNSTLDYEIR | GPI ethanolamine phosphate transferase 1 |
| P36091 | DCW1 | N203 | YTGN[HexNAc]QTYVDWAEK | Mannan endo-1,6-alpha-mannosidase |
| P37302 | APE3 | N96 | LAN[HexNAc]YSTPDYGHPTR | Aminopeptidase Y |
| P37302 | APE3 | N150 | IISFN[HexNAc]LSDAETGK | Aminopeptidase Y |
| P37302 | APE3 | N162 | SFAN[HexNAc]TTAFALSPPVDGFVGK | Aminopeptidase Y |
| P37302 | APE3 | N85 | IKVDDLN[HexNAc]ATAWDLYR | Aminopeptidase Y |
| P38244 | PFF1 | N121 | SILFQQQDPFN[HexNAc]ESSR | Probable zinc metalloprotease YBR074W |
| P38248 | ECM33 | N304 | VQTVGGAIEVTGN[HexNAc]FSTLDLSSLK | Cell wall protein ECM33 |
| P38843 | CHS7 | N31 | THLILSN[HexNAc]STIIHDFDPLNLNVGVLPR | Chitin synthase export chaperone |
| P38875 | GPI16 | N184 | SYASDIGAPLFN[HexNAc]STEK | GPI transamidase component |
| P38993 | FET3 | N244 | N[HexNAc]VTDMLYITVAQR | Iron transport multicopper oxidase FET3 |
| P38993 | FET3 | N359 | NGVNYAFFNN[HexNAc]ITYTAPK | Iron transport multicopper oxidase FET3 |
| P39007 | STT3 | N539 | TTLVDNNTWN[HexNAc]NTHIAIVGK | Oligosaccharyltransferase subunit STT3 |
| P39105 | PLB1 | N215 | DAGFN[HexNAc]ISLADVWGR | Lysophospholipase 1 |
| P39105 | PLB1 | N489 | N[HexNAc]LTDLEYIPPLIVYIPNSR | Lysophospholipase 1 |
| P40345 | PDAT | N439 | SSSEDALNN[HexNAc]NTDTYGNFIR | Phospholipid:diacylglycerol acyltransferase |
| P40533 | TED1 | N266 | DNYWIEYETN[HexNAc]TTHPWR | Protein TED1 |
| P40557 | EPS1 | N299 | FPN[HexNAc]ITEGELEK | ER-retained PMA1-suppressing protein 1 |
| P40557 | EPS1 | N264 | VALVLPN[HexNAc]K | ER-retained PMA1-suppressing protein 1 |
| P41543 | OST1 | N217 | FSSN[HexNAc]ETLAIVYSHNAPLNQVVNLR | Oligosaccharyltransferase subunit OST1 |
| P43561 | FET5 | N364 | YAFFNN[HexNAc]ITYVTPK | Iron transport multicopper oxidase FET5 |
| P43561 | FET5 | N24 | LN[HexNAc]YTASWVTANPDGLHEK | Iron transport multicopper oxidase FET5 |
| P43611 | OSW7 | N297 | NLDDLN[HexNAc]TTVNEQLVFLDSK | Uncharacterized protein YFR039C |
| P46982 | MNN5 | N136 | LN[HexNAc]FSIPQR | Alpha-1,2-mannosyltransferase MNN5 |
| P46992 | YJR1 | N219 | N[HexNAc]SSSIGYYDLPAIWLLNDHIAR | Cell wall protein YJL171C |
| P52911 | EXG2 | N50 | FASYYAN[HexNAc]DTITVK | Glucan 1,3-beta-glucosidase 2 |
| P52911 | EXG2 | N157 | NLYIDN[HexNAc]ITFNDPYVSDGLQLK | Glucan 1,3-beta-glucosidase 2 |
| P53379 | MKC7 | N286 | STAYSLFAN[HexNAc]DSDSK | Aspartic proteinase MKC7 |
| P54003 | SUR7 | N47 | FYWVQGN[HexNAc]TTGIPNAGDETR | Protein SUR7 |
| Q03103 | ERO1 | N458 | YTIENIN[HexNAc]STK | Endoplasmic oxidoreductin-1 |
| Q03281 | HEH2 | N520 | SN[HexNAc]NTNYIYR | Inner nuclear membrane protein |
| Q03674 | PLB2 | N193 | SIVNPGGSN[HexNAc]LTYTIER | Lysophospholipase 2 |
| Q03674 | PLB2 | N217 | SDAGFN[HexNAc]ISLSDLWAR | Lysophospholipase 2 |
| Q03691 | ROT1 | N139 | YN[HexNAc]QTETFK | Protein ROT1 |
| Q06689 | YL413 | N429 | ILNSAVN[HexNAc]MTTITPEQLK | Cell membrane protein YLR413W |
| Q06689 | YL413 | N49 | IN[HexNAc]VTK | Cell membrane protein YLR413W |
| Q07830 | GPI13 | N411 | N[HexNAc]ISNTPPTSDPEK | GPI ethanolamine phosphate transferase 3 |
| Q12465 | RAX2 | N88 | EIGPETSSHGLVYYSN[HexNAc]NTYIQLEDASDDTR | Bud site selection protein |
| Q12465 | RAX2 | N677 | N[HexNAc]QTIQGDVHGITK | Bud site selection protein |
| Q12465 | RAX2 | N640 | N[HexNAc]SSLYADIYDNK | Bud site selection protein |
As it is known that changes in glycosylation can have an impact on glycoprotein steady-state levels, we determined the relative protein abundance as well (10, 51). To quantify protein abundance of each targeted glycoprotein in our analysis, we extended the target list by adding up to two peptides without glycosylation sequon for each glycoprotein analyzed. In the final set-up, the full analytical list consisted of 62 glycopeptides and 74 peptides without glycosylation sequon corresponding to 43 glycoproteins (supplemental Table S3). Furthermore, to account for any variance during mixing of light and heavy cells we also targeted peptides belonging to the ribosomal proteins Rpl5 and Rsp1, respectively, used as housekeeping proteins in previous studies (39). To ensure high-quality quantification of low abundance proteins and proteins containing peptides with poor MS properties, such proteins were analyzed using a PRM method with higher resolution (supplemental Table S3). The simultaneous accurate quantification of modified peptides as well as peptides that do not contain a glycosylation sequon enabled us to follow changes in the occupancy of the glycosylation sites analyzed as well as relative glycoprotein abundance.
Evaluation of the Functional Studies of the Glycosylation Machinery
To validate the newly developed SILAC PRM MS method, we investigated the roles of the Ost3 and Ost6 subunits in OST function in vivo. It has been described previously that these subunits with oxidoreductase activity facilitate glycosylation in a site-specific manner (24, 32, 34). As yeast OST contains either Ost3p or Ost6p, we generated strains in which either the OST3 or OST6 loci were deleted. Overexpression of plasmid-encoded OST3 or OST6 ensured normal levels of uniform OST (20). This allowed us to compare the phenotype of strains expressing either Ost3p- or Ost6p-containing OST complexes at equal levels. Equal amounts of wild-type reference cells grown in heavy medium and of cells with a deletion in one OST subunit grown in light medium were pooled. Cells were lysed, microsomal fractions collected, proteins prepared for MS, and light-to-heavy ratios for all peptides measured by PRM. Glycopeptide abundance relative to wild type was calculated. Values were normalized to the average of two control proteins, Rps1 and Rpl5 and to the average of peptides without glycosylation sequon belonging to the same glycoproteins (supplemental Table S6).
SILAC PRM MS analysis revealed that 84% of the glycosylation sites were hypoglycosylated (where the glycosylation occupancy ratio compared with wild-type reference strain was lower than 0.75) in the strain lacking Ost3 protein, whereas less severe hypoglycosylation was observed in the strain with OST6 being overexpressed with 39% of the glycosylation sites being hypoglycosylated (Fig. 2). Severe hypoglycosylation in Δost3 strain was therefore a combination of a reduced level of fully assembled OST and the lack of OST3 function. These results highlight the importance of using yeast strains expressing normal amounts of fully assembled OST, but lacking either Ost3p or Ost6p for the functional analysis of these subunits. The extent of hypoglycosylation is only mild in strains lacking Ost6p, in which only 2% of the glycosylation sites were hypoglycosylated in combination with overexpressed OST3. These results indicated that Ost3p-containing complex has a broader substrate specificity as compared with Ost6p-containing complex (Fig. 2). The data agree with previously published work demonstrating that Ost3p has a more evident role in N-linked glycosylation compared with Ost6p (21, 23, 24, 32). Ost3p affects polypeptide substrate specificity, as in a strain with Ost6p-containing OST only a hypoglycosylation on a subset of glycosylation sites (24 out of 62 sites analyzed, corresponding to 21 out of 43 glycoproteins) was observed (supplemental Table S6). We detected 17 novel Ost3p substrates (supplemental Table S6, in bold italic), in which the glycosylation of these protein substrates was dependent on the presence of the Ost3 subunit. Interestingly, when we examined Ost3p substrates with available structure or structural model, most of these proteins required oxidative folding (disulfide bond formation) (data not shown). These results are in accordance with previously proposed function of Ost3 protein to transiently form mixed disulfide bonds with OST polypeptide substrates (22, 24).
Fig. 2.

Glycosylation occupancy analysis at different glycosylation sites of various OST mutant strains and Δalg9 strain compared with wild-type cells. OST mutant cells, Δost3, Δost3 complemented with OST6, Δost6 and Δost6 complemented with OST3, and cells deleted in ALG9 loci were grown in light medium, mixed 1:1 with the wild-type reference strain grown in heavy medium and membrane derived peptides were prepared. Peptide abundance was measured by PRM MS. Intensity ratios of glycosylated light to heavy peptides were normalized for expression differences in mutant cells and wild-type cells. The resulting ratios represent the site occupancy for the mutant cells relative to the wild-type reference strain presented in a heatmap. Data is the mean of biological triplicates. Color is mapped from black (0%) to red (100%), gray is for data not being obtained. Data from supplemental Table S6.
An alternative way to affect OST activity in vivo is to prevent complete synthesis of the lipid-linked oligosaccharide substrate (12). Yeast Alg9p has a dual role in LLO biosynthesis. It is responsible for the addition of two α-1,2-linked Man residues, whereby the assembly of b-branch is a requirement for the assembly of c-branch (52). We generated a strain in which the ALG9 locus was deleted, resulting in the accumulation of Man6GlcNAc2 LLO and determined site-specific glycosylation efficiencies. As expected, in the alg mutant strain many sequons were not efficiently glycosylated and the level of hypoglycosylation was increased as compared with the strain lacking Ost3p complemented with OST6 (Fig. 2). Absence of Alg9p resulted in hypoglycosylation (i.e. glycosylation lower than 0.75) of 64% of the sites analyzed, whereas the lack of Ost3p complemented with OST6 resulted in hypoglycosylation of 39% of the sites analyzed (supplemental Table S6). Although more severe as compared with the loss of Ost3p complemented with OST6, the effect of suboptimal glycan donor appears to have a site-specific effect on the OST activity as more than one third of the sites in our analysis were not affected when the ALG9 loci was deleted.
To examine if the presence of specific amino acids in the local environment of the glycosylation sites influenced N-glycosylation efficiency, sequence analysis was performed on glycosylation sequons plus ten residues upstream and downstream from the glycosylation sites. Two sample logo analysis (53) was used to visualize differences between efficiently and poorly glycosylated sequences surrounding the glycosylation sites. Glycosylation efficiency of Ost6-containing OST (in Δost3 strain complemented with OST6) was not dependent on the presence of specific amino acids in the local surrounding of the glycosylation site (Fig. 3A). In contrast, poorly glycosylated sites in Δalg9 strain were enriched in N-X-S sequons, as compared with the more efficiently modified N-X-T sequons (Fig. 3B). Sequons that contain glutamic acid at +1 position are also less likely to be efficiently glycosylated. Furthermore, sites that are surrounded by basic arginine and lysine residues and polar amino acids appear to be disfavored when OST is presented with a suboptimal glycan donor. When we examined the localization of efficiently glycosylated sites in respect to their structural environment, for the substrates with available structure or structural model, we discovered that the glycosylation efficiency was not dependent on the type of secondary structures glycosylation sites were placed on in Δost3 strains nor Δalg9 strain (data not shown).
Fig. 3.
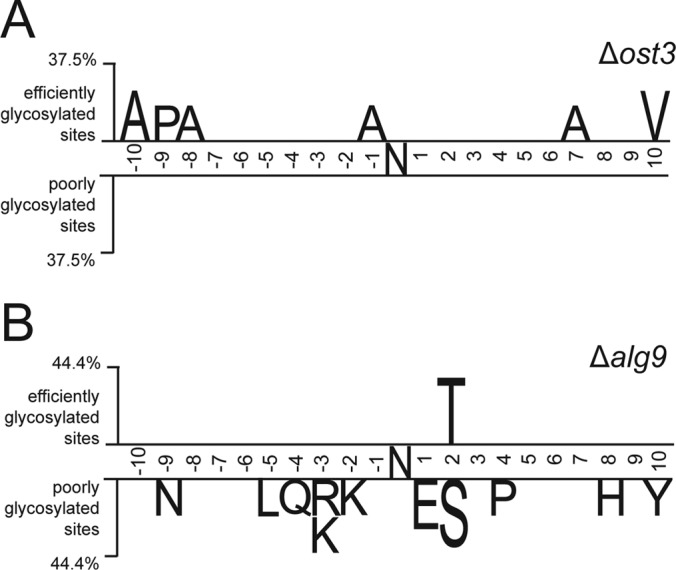
N-glycosylation efficiency is influenced by the primary protein sequence surrounding the glycosylation site. Two sample logo analysis (53) was used to calculate and visualize residues surrounding glycosylation sites that are significantly enriched in either efficiently or poorly glycosylated sites in Δost3 complemented with OST6 (A) and Δalg9 (B) strains. Sequence size analyzed contained ten amino acids upstream and downstream from the glycosylation site. Efficiently glycosylated sites are considered where glycosylation occupancy ratio compared with wild type is more than 0.75 and poorly glycosylated sites are considered where glycosylation occupancy ratio compared with wild type is less than 0.25.
Based on our results, we concluded that the SILAC PRM MS method was a reproducible, sensitive and useful approach for determining the efficiency of the N-linked glycosylation machinery in yeast.
Protein Structural Domains Influence Protein Specific Glycosylation Efficiency
Our novel analytical approach allowed us to analyze the N-glycosylation process of defined proteins in more detail. We followed the modifications of the ER resident protein disulfide isomerase, Pdi1p, that carries five N-linked glycans. Pdi1p is composed of four thioredoxin folds termed a, b, b', and a' that can fold independently (54). The a and a' domains contain two and one disulfide bonds, respectively, one per domain is directly involved in the catalytic activity of the isomerase. The five glycosylation sites are located in domain a (2 sites), b (2 sites), and a' (1 site), all of them in structured area of the protein: site 1 (N82, domain a) and site 2 (N117, domain a) are placed in structured loops, site 3 (N155, domain b), site 4 (N174, domain b) and site 5 (N425, domain a') are in α-helices (54) (Fig. 4).
Fig. 4.
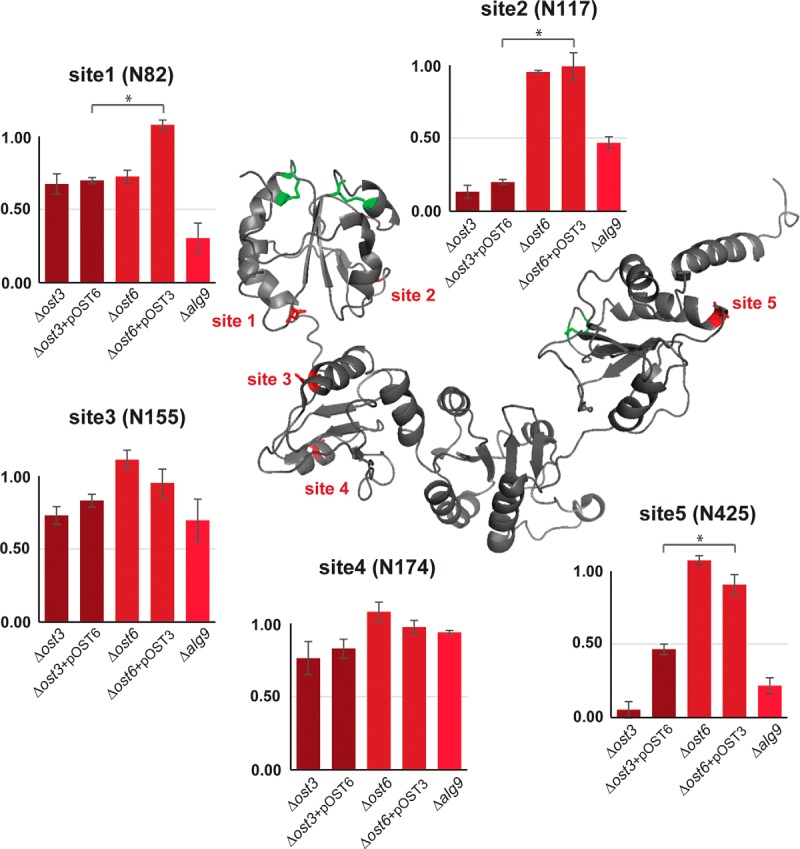
Site-specific glycosylation occupancy of Pdi1p is influenced by the localization of glycosylation sites in respect of distinct protein domains. Glycosylation occupancy ratio compared with wild type at five N-linked glycosylation sites (N82, N117, N155, N174 and N425) of Pdi1p in Δost3, Δost3 complemented with OST6, Δost6, Δost6 complemented with OST3 and Δalg9 cells. Values are a mean of biological triplicates. Error bars represent S.D. in three biological replicates.*p < 0.05. Pdi1p protein structure is modified from Tian et al. 2001 Cell. Data from Supplemental Table 6.
Glycosylation occupancy of any of the five sites was not severely affected in strains lacking Ost6p, except for site one (N82) being hypoglycosylated in Δost6 strain. This hypoglycosylation was rescued by overexpression of OST3. However, absence of the OST3 component had strong, but site-specific effects on Pdi1p glycosylation. In the Δost3+pOST6 strain, sites one (N82), two (N117) and five (N425) were hypoglycosylated in the absence of Ost3p (with 0.70, 0.20 and 0.47 glycosylation occupancy that was significantly lower compared with the strain lacking Ost6p, respectively), whereas site 3 (N155) and site 4 (N174) were much less affected. We noted that these three sites are in the a and a' domains of the protein that contain disulfide bonds (54). Sites three (N155) and four (N174), located in the b domain, were not hypoglycosylated (with 0.84 and 0.83 glycosylation occupancy, respectively) (Fig. 4). Pdi1p glycosylation occupancy analysis in Δalg9 strain revealed similar results as in Δost3+pOST6 strain, where the glycosylation of sites one (N82), two (N117) and five (N425) was more severely affected (with 0.31, 0.47 and 0.22 glycosylation occupancy, respectively) as compared with sites three (N155) and four (N174) (with 0.70 and 0.95 glycosylation occupancy, respectively) (Fig. 4).
Defects in LLO Biosynthesis Lead to Changes in Ergosterol Pathway
We determined the relative protein abundance of glycoproteins across different strains and measured by PRM MS peptides without glycosylation sequon belonging to 42 glycoproteins (supplemental Table S7). We noted a correlation between hypoglycosylation and unfolded protein response: Δost3 strains and Δalg9 strain displayed the most significant increase of six proteins known to be controlled by this regulatory network (Ero1p, Rot1p, Lhs1p, Pdi1p, Mcd4p and Eps1p). Most of these proteins act as molecular chaperons and are involved in either ERAD or UPR (9, 55). However, one particular protein, ERG3 C-5 sterol desaturase, involved in ergosterol biosynthesis, showed a 4-fold increase in abundance in Δalg9 strain only and not in strains with mutations affecting OST components (Fig. 5A and 5B; supplemental Table S7).
Fig. 5.

Protein abundance of several ergosterol pathway proteins is influenced by defects in dolichol pathway. A, Schematic representation of mevalonate, ergosterol and dolichol pathway (adapted from 67, 68). B, Deletion of ALG9 loci results in increased abundance of several proteins involved in UPR as well as Erg3p. Δalg9 cells were grown in light medium, mixed 1:1 with the wild-type reference strain grown in heavy medium and membrane derived peptides were prepared. Peptide abundance was measured by SILAC PRM. Intensity ratios of light to heavy peptides were normalized for expression differences in mutant cells and wild-type cells. The resulting ratios represent the protein abundance for the mutant cells relative to the wild-type reference strain. Error bars represent S.D. in three biological replicates. Data from supplemental Table S7. C, Deletion of ALG9 and ALG3 loci, as well as treatment of cells with sterol inhibitor miconazole results in increased protein abundance of multiple ERG proteins. Δalg9, Δalg3, cells treated with miconazole or DMSO were grown in light medium, mixed 1:1 with the wild-type reference strain grown in heavy medium and membrane derived peptides were prepared. Peptide abundance was measured by PRM MS with an inclusion list containing peptides belonging to mevalonate, ergosterol and dolichol pathway proteins. Data was normalized to the data obtained for control cells treated with DMSO. Error bars represent S.D. in three biological replicates. Data from supplemental Table S8.
Because the initial steps in dolichol (the lipid involved in LLO biosynthesis) and ergosterol biosynthesis are connected, we investigated whether defects in LLO assembly affect other proteins involved in sterol biosynthesis (56). PRM assays were developed to analyze the abundance of enzymes required in the mevalonate, ergosterol or dolichol pathways (supplemental Table S5). Peptides belonging to 20 out of the 26 proteins were identified and quantified (Fig. 5A, black). We also included an alg3 mutant strain in the analysis. Yeast Alg3p is a α-1,3 mannosyltransferase responsible for the initiation of glycosylation on the LLO b-branch at the ER luminal side, which is then subsequently elongated by Alg9p (12). Protein abundance of Erg3p protein showed a 3 to 3.5-fold increase in both Δalg3 and Δalg9 strains, suggesting that the changes in protein levels of this protein are a result of general defects in LLO assembly (Fig. 5C). Moreover, the abundance of three other proteins (Erg11, Erg25, and Erg5, respectively), all acting in ergosterol biosynthetic pathway, showed an increased protein level ratio compared with wild-type cells in both Δalg3 and Δalg9 strains. Additionally, Erg1p was significantly increased in Δalg3 strain (Fig. 5C). Both defects in LLO assembly resulted in a 3- to 4-fold increase in protein amounts of Erg11p, Erg25p, and Erg3p and 1-to 2-fold increase of Erg5p. Earlier measurements using promoter fusions and mutations that alter sterol biosynthesis found upregulation of genes encoding for the same proteins in various genetic backgrounds (57–59). Furthermore, exposure of yeast cells to sterol inhibitors upregulated the expression of the same genes as well (60). To examine if the same phenotype was observed using our proteomic setup, we treated wild-type yeast cells with miconazole, an inhibitor of lanosterol 14-α-demethylase Erg11p (Fig. 5A) (61). Equal amounts of wild-type cells grown in heavy medium and of cells treated with miconazole or DMSO were pooled and processed as described above. The protein levels were normalized to the ones of the control cells treated with DMSO (supplemental Table S8). Like strains defective in LLO assembly, the same proteins displayed a significant increase in protein level ratio compared with cells, including proteins Erg11, Erg25, Erg3, and Erg5 (Fig. 5C).
DISCUSSION
N-linked glycosylation machinery, with OST as the central enzyme, modifies many proteins with glycans that can be fundamental in many ways for cell viability, yet the crucial mechanisms responsible for the regulation of N-linked glycosylation are not well understood. To increase the resolution of measurement of OST function in vivo, we developed a novel analytical tool to quantify relative N-glycosylation occupancy at many glycosylation sites in different membrane and luminal proteins. We have shown that its sensitivity and consistency make PRM MS especially suitable for the analysis of N-linked glycoproteins, sometimes present in medium to low amounts in cell extracts without the need of extensive fractionation. Coupled with SILAC our approach offers a powerful analytical tool to study the efficiency of N-linked glycosylation machinery in a sensitive, effective and reproducible way providing results that agreed with previously published data. Our method is a complementary analytical approach to the recently reported methods in which SWATH and SWAT MS were used to quantify relative glycosylation occupancy of a lower number of predefined glycosylation sites in glycoproteins extracted from yeast cell wall (32, 34, 62). Compared with other approaches, the analysis of yeast membrane and luminal glycoproteins eliminates the analytical bias present in the complementary approaches in which only mature, successfully secreted proteins from yeast cell wall extracts are analyzed (21). We are now able to investigate the glycosylation extent of glycoproteins involved in N-linked glycosylation, ERQC and UPR, in a resolved, quantifiable and reproducible way. Simultaneous analysis of protein abundance allows us to examine distinct influence of glycosylation on steady-state levels of individual substrate proteins or a global influence evident in ERQC and UPR activation.
The use of the SILAC method makes it possible to easily combine the PRM technology with yeast genetic techniques. Differentially labeled extracts from wild-type and mutant cells yield a precise relative quantification of peptide and glycopeptide levels, a clearly defined phenotype of the mutation(s) analyzed. In addition, the targeted analytical approach (PRM or SRM), combined with pulse or pulse-chase SILAC labeling can reveal information about (glyco)protein turnover (39) or the kinetics of glycoprotein processing.
In the present study, we have used this approach to investigate protein substrate specificities of OST isoforms containing either Ost3p or Ost6p. In agreement with other reports, both isoforms affected OST substrates specifically and displayed site-specific preferences for a subset of glycosylation sites. This effect was not caused by a general reduction in OST activity as distinct sites were affected in the absence of Ost3p or Ost6p. Furthermore, Ost3p was required for efficient glycosylation of more than a third of glycosylation sites analyzed here, compatible with a broader substrate array as compared with Ost6p isoform (21, 32, 34). We could detect novel Ost3p substrates and a majority of these protein substrates required oxidative folding. It has been demonstrated that both Ost3 and Ost6 subunits have thioredoxin-like domains that transiently interact with their protein substrates through mixed disulfide bonds (63). However, the importance of noncovalent interactions has been demonstrated as well, explaining why Ost3p influenced glycosylation at asparagines in substrate proteins that do not contain disulfide bonds (22–24, 64–66).
In the process of N-linked glycosylation the OST enzyme displays a preference for both the polypeptide substrate and LLO donor, in which suboptimal LLO structures are used, although with lower efficiency (12). We have examined the effect of the Man6GlcNAc2 suboptimal glycan donor generated in Δalg9 strain on the glycosylation efficiency of OST in vivo. Absence of Alg9p resulted in a more severe hypoglycosylation phenotype as compared with the loss of Ost3p complemented with OST6. Furthermore, the presence of a suboptimal glycan donor had a site-specific effect on the OST activity. This effect was not dependent on the protein secondary structure, e.g. the folding of the acceptor protein. However, glycosylation efficiency was sensitive to the primary sequence of the OST substrate: in Δalg9 strain, poorly glycosylated sites were enriched in N-X-S sequons as compared with the more efficiently modified N-X-T sequons. Crystal structure of PglB, a single subunit OST from Campylobacter jejuni, a homolog of the STT3 subunit of multisubunit OSTs revealed that the WWD domain, not directly involved in the catalysis, bound the N-X-T sequon more efficiently (19). We propose that the combination of a suboptimal LLO substrate and the low affinity sequon N-X-S is the cause for the reduced glycosylation of these sites. Similarly, our analysis revealed that sites that have a negative charge at −2 position and sites that are adjacent to basic and polar amino acids were disfavored when OST was presented with a suboptimal glycan donor. Therefore, the analysis of site-specific N-glycosylation in alg mutant strains revealed a peptide substrate preference of the OST. Importantly, this preference was not observed in ost3 or ost6 mutant strains. Glycosylation efficiency of Ost3/Ost6p-containing OSTs did not reveal a primary sequence specificity indicating that Ost3 and Ost6 subunits are not involved in defining the general polypeptide binding affinity of OST. Kinetic experiments with yeast OST have revealed that there is a single binding site for the polypeptide substrate whereas the LLO substrate is bound in a cooperative manner (5, 67, 68). A substrate activation model suggests that the binding of the LLO donor substrate to the regulatory LLO binding site is a prerequisite for the binding of both the polypeptide substrate and second LLO donor substrate to the catalytic site (67). Reducing LLO substrate affinity therefore disfavors glycosylation on low affinity peptide sequons.
The possibility to investigate the status of all glycosylation sites of Pdi1p, an ER-resident protein whose protein structure is known, allowed us to analyze the N-glycosylation process in more detail. N-linked glycosylation of sites located in two domains of Pdi1p that also contain disulfide bonds (a and a' respectively) were Ost3p dependent, in which the site closest to the nonactive disulfide bond (site two, N117) was the one most affected in the cells lacking Ost3p. We postulate that the formation of a mixed disulfide between Ost3p and Pdi1p slows down the folding and increases the glycosylation efficiency of this sequon. Protein folding is a critical competitor of N-linked glycosylation and indeed, the N117 site of Pdi1p is in a protein sequence with secondary structure not competitive with N-glycosylation. The same site of Pdi1p is also hypoglycosylated in Δalg9 strain, supporting our hypothesis that reducing OST activity affects sites in which folding is in competition with glycosylation. On the other hand, glycosylation of sites three (N155) and four (N174) were less affected as compared with other sites in ost3/6 or alg9 cells even though sites three and four are in α-helices. Folding of the b domain might be slower than the folding of the a and a' domains, leaving an OST with enough time to efficiently glycosylate sequons present in this region. Alternatively, additional OST components might prevent rapid folding of this domain.
Glycosylation and folding of proteins in the ER coevolved and suboptimal glycosylation can affect the folding of specific proteins. In a system in which not-properly folded proteins are degraded, this leads to an altered steady-state level of glycoproteins (2, 7, 69). In addition, the unfolded protein response (UPR) can be activated (8, 9). We observed reduced steady-state level on reduced glycosylation (Δalg9) for only a few glycoproteins tested (Yjr1p, Yl413p, Ape3p, Pdr5p) but we detected an upregulation of the know UPR proteins such as Rot1p, Ero1p, Pdi1p (Fig. 5). The largest increase in abundance was observed for Erg3p in the Δalg9 strain, which was not a result of UPR activation nor hypoglycosylation as the level of this protein was not altered in any other mutant strain, such as Δost3 or Δost6, tested. We showed that the deficiency in LLO assembly affected several other enzymes involved in the ergosterol but not mevalonate pathway. Genes encoding for the upregulated proteins were found previously to be upregulated on mutations that altered sterol biosynthesis or on exposure to sterol biosynthesis inhibitors. We could increase the steady-state levels when cells were treated with the sterol biosynthesis inhibitor miconazole and therefore postulated a novel regulatory mechanism that links the LLO biosynthetic pathway with ergosterol biosynthesis. In view of the topology of the different pathways involved, dolichyl pyrophosphate might act as a mediator in this regulation process.
In summary, the SILAC PRM MS method provided adequate selectivity and sensitivity for the quantitative analysis of a large number of glycosylation sites on yeast N-glycoproteins. Compared with other analytical methods, our assay allowed detection of novel Ost3p and Ost6p glycoprotein substrates and made it possible to target specific substrates such as Pdi1p for a more detailed analysis. The targeted analytical approach, in combination with a time resolved SILAC approach, will be instrumental for a detailed description of the role of N-glycosylation in the in vivo protein maturation at a molecular level.
DATA AVAILABILITY
The mass spectrometry proteomics data, from tier 3 measurements, were handled using the local laboratory information management system (LIMS) (49) and all relevant data have been deposited to the ProteomeXchange Consortium via the PRIDE (http://www.ebi.ac.uk/pride) partner repository with the data set identifier PXD006435.
Supplementary Material
Acknowledgments
We thank the Functional Genomic Center Zurich for technical and analytical support. We further thank Prof. Dr. Bruno Alfred Gander for providing us with miconazole.
Footnotes
* This work was supported by Swiss National Science Foundation Grants 310030B_144083 and 310030_16236 to M.A.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- OST
- Oligosaccharyltransferase
- N
- Asparagine
- ER
- Endoplasmatic Reticulum
- GlcNAc
- N-acetyl-glucosamine
- LLO
- Lipid-linked oligosaccharide
- PRM
- Parallel reaction monitoring
- SILAC
- Stable isotope labelling with amino acids in cell culture.
REFERENCES
- 1. Zielinska D. F., Gnad F., Schropp K., Wiśniewski J. R., and Mann M. (2012) Mapping N-glycosylation sites across seven evolutionarily distant species reveals a divergent substrate proteome despite a common core machinery. Mol. Cell. 46, 542–548 [DOI] [PubMed] [Google Scholar]
- 2. Varki A. (1993) Biological roles of oligosaccharides: all of the theories are correct. Glycobiology 3, 97–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Szymanski C. M., and Wren B. W. (2005) Protein glycosylation in bacterial mucosal pathogens. Nat. Rev. Microbiol. 3, 225–237 [DOI] [PubMed] [Google Scholar]
- 4. Kelleher D. J., and Gilmore R. (2006) An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology 16, 47–62 [DOI] [PubMed] [Google Scholar]
- 5. Kelleher D. J., Banerjee S., Cura A. J., Samuelson J., and Gilmore R. (2007) Dolichol-linked oligosaccharide selection by the oligosaccharyltransferase in protist and fungal organisms. J. Cell Biol. 177, 29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aebi M., Bernasconi R., Clerc S., and Molinari M. (2010) N-glycan structures: recognition and processing in the ER. Trends Biochem. Sci. 35, 74–82 [DOI] [PubMed] [Google Scholar]
- 7. Zattas D., and Hochstrasser M. (2015) Ubiquitin-dependent Protein Degradation at the Yeast Endoplasmic Reticulum and Nuclear Envelope. Crit. Rev. Biochem. Mol. Biol. 50, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chapman R., Sidrauski C., and Walter P. (1998) Intracellular signaling from the endoplasmic reticulum to the nucleus. Annu. Rev. Cell Dev. Biol. 14, 459–485 [DOI] [PubMed] [Google Scholar]
- 9. Travers K. J., Patil C. K., Wodicka L., Lockhart D. J., Weissman J. S., and Walter P. (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101, 249–258 [DOI] [PubMed] [Google Scholar]
- 10. Helenius A., and Aebi M. (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049 [DOI] [PubMed] [Google Scholar]
- 11. Breitling J., and Aebi M. (2013) N -Linked Protein Glycosylation in the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 10.1101/cshperspect.a013359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Burda P., and Aebi M. (1999) The dolichol pathway of N-linked glycosylation. Biochim. Biophys. Acta - Gen. Subj. 1426, 239–257 [DOI] [PubMed] [Google Scholar]
- 13. Gao X. D., Nishikawa A., and Dean N. (2004) Physical interactions between the Alg1, Alg2, and Alg11 mannosyltransferases of the endoplasmic reticulum. Glycobiology 14, 559–570 [DOI] [PubMed] [Google Scholar]
- 14. Lu J., Takahashi T., Ohoka A., Nakajima K., Hashimoto R., Miura N., Tachikawa H., and Gao X. D. (2012) Alg14 organizes the formation of a multiglycosyltransferase complex involved in initiation of lipid-linked oligosaccharide biosynthesis. Glycobiology 22, 504–516 [DOI] [PubMed] [Google Scholar]
- 15. Helenius J., Ng D. T., Marolda C. L., Walter P., Valvano M. A., and Aebi M. (2002) Translocation of lipid-linked oligosaccharides across the ER membrane requires Rft1 protein. Nature 415, 447–450 [DOI] [PubMed] [Google Scholar]
- 16. Lairson L. L., Henrissat B., Davies G. J., and Withers S. G. (2008) Glycosyltransferases: Structures, Functions, and Mechanisms. Annu. Rev. Biochem. 77, 521–555 [DOI] [PubMed] [Google Scholar]
- 17. Burda P., and Aebi M. (1998) The ALG10 locus of Saccharomycescerevisiae encodes the alpha-1,2 glucosyltransferase of the endoplasmic reticulum: the terminal glucose of the lipid-linked oligosaccharide is required for efficient N-linked glycosylation. Glycobiology 8, 455–462 [DOI] [PubMed] [Google Scholar]
- 18. Ruiz-Canada C., Kelleher D. J., and Gilmore R. (2009) Cotranslational and Posttranslational N-Glycosylation of Polypeptides by Distinct Mammalian OST Isoforms. Cell 136, 272–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lizak C., Gerber S., Numao S., Aebi M., and Locher K. P. (2011) X-ray structure of a bacterial oligosaccharyltransferase. Nature 474, 350–355 [DOI] [PubMed] [Google Scholar]
- 20. Spirig U., Bodmer D., Wacker M., Burda P., and Aebi M. (2005) The 3.4-kDa Ost4 protein is required for the assembly of two distinct oligosaccharyltransferase complexes in yeast. Glycobiology 15, 1396–1406 [DOI] [PubMed] [Google Scholar]
- 21. Schulz B. L., and Aebi M. (2009) Analysis of glycosylation site occupancy reveals a role for Ost3p and Ost6p in site-specific N-glycosylation efficiency. Mol. Cell. Proteomics 8, 357–364 [DOI] [PubMed] [Google Scholar]
- 22. Mohd Yusuf S. N., Bailey U. M., Tan N. Y., Jamaluddin M. F., and Schulz B. L. (2013) Mixed disulfide formation in vitro between a glycoprotein substrate and yeast oligosaccharyltransferase subunits Ost3p and Ost6p. Biochem. Biophys. Res. Commun. 432, 438–443 [DOI] [PubMed] [Google Scholar]
- 23. Jamaluddin M. F. B., Bailey U.-M., and Schulz B. L. (2014) Oligosaccharyltransferase subunits bind polypeptide substrate to locally enhance N-glycosylation. Mol. Cell. Proteomics 10.1074/mcp.M114.041178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schulz B. L., Stirnimann C. U., Grimshaw J P., Brozzo M. S., Fritsch F., Mohorko E., Capitani G., Glockshuber R., Grütter M. G., and Aebi M. (2009) Oxidoreductase activity of oligosaccharyltransferase subunits Ost3p and Ost6p defines site-specific glycosylation efficiency. Proc. Natl. Acad. Sci. U.S.A. 106, 11061–11066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang H., Li X. J., Martin D. B., and Aebersold R. (2003) Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 21, 660–666 [DOI] [PubMed] [Google Scholar]
- 26. Lazar I. M., Lee W., and Lazar A. C. (2013) Glycoproteomics on the rise: established methods, advanced techniques, sophisticated biological applications. Electrophoresis 34, 113–125 [DOI] [PubMed] [Google Scholar]
- 27. Liu Y., Hüttenhain R., Surinova S., Gillet L. C., Mouritsen J., Brunner R., Navarro P., and Aebersold R. (2013) Quantitative measurements of N-linked glycoproteins in human plasma by SWATH-MS. Proteomics 13, 1247–1256 [DOI] [PubMed] [Google Scholar]
- 28. Parker B. L., Thaysen-Andersen M., Solis N., Scott N. E., Larsen M. R., Graham M. E., Packer N. H., and Cordwell S. J. (2013) Site-specific glycan-peptide analysis for determination of N-glycoproteome heterogeneity. J. Proteome Res. 12, 5791–5800 [DOI] [PubMed] [Google Scholar]
- 29. Song W., Mentink R. A., Henquet M. G., Cordewener J. H., van Dijk A. D., Bosch D., America A. H., and van der Krol A. R. (2013) N-glycan occupancy of Arabidopsis N-glycoproteins. J. Proteomics 93, 343–355 [DOI] [PubMed] [Google Scholar]
- 30. Pan S. (2014) Quantitative glycoproteomics for N-glycoproteome profiling. Methods Mol. Biol. 1156, 379–388 [DOI] [PubMed] [Google Scholar]
- 31. Xu Y., Bailey U. M., Punyadeera C., and Schulz B. L. (2014) Identification of salivary N-glycoproteins and measurement of glycosylation site occupancy by boronate glycoprotein enrichment and liquid chromatography/electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 28, 471–482 [DOI] [PubMed] [Google Scholar]
- 32. Yeo K. Y. B., Chrysanthopoulos P. K., Nouwens A. S., Marcellin E., and Schulz B. L. (2016) High-performance targeted mass spectrometry with precision data-independent acquisition reveals site-specific glycosylation macroheterogeneity. Anal. Biochem. 510, 106–113 [DOI] [PubMed] [Google Scholar]
- 33. Zacchi L. F., and Schulz B. L. (2016) SWATH-MS glycoproteomics reveals consequences of defects in the glycosylation machinery. Mol. Cell. Proteomics 15, 2435–2447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu Y., Bailey U. M., and Schulz B. L. (2015) Automated measurement of site-specific N-glycosylation occupancy with SWATH-MS. Proteomics 15, 2177–2186 [DOI] [PubMed] [Google Scholar]
- 35. de Godoy L., Olsen J., de Souza G., Li G., Mortensen P., and Mann M. (2006) Status of complete proteome analysis by mass spectrometry: SILAC labeled yeast as a model system. - PubMed - NCBI. Genome Biol. 7, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Peterson A. C., Russell J. D., Bailey D. J., Westphall M. S., and Coon J. J. (2012) Parallel Reaction Monitoring for High Resolution and High Mass Accuracy Quantitative, Targeted Proteomics. Mol. Cell. Proteomics 11, 1475–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Güldener U., Heck S., Fielder T., Beinhauer J., and Hegemann J. H. (1996) A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res. 24, 2519–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Knop M., Siegers K., Pereira G., Zachariae W., Winsor B., Nasmyth K., and Schiebel E. (1999) Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast 15, 963–972 [DOI] [PubMed] [Google Scholar]
- 39. Mueller S., Wahlander A, Selevsek N., Otto C., Ngwa E. M., Poljak K., Frey A. D, Aebi M., and Gauss R. (2015) Protein degradation corrects for imbalanced subunit stoichiometry in OST complex assembly. Mol. Biol. Cell. 26, 2596–2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wiœniewski J. R., Zougman A., Nagaraj N., and Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 41. Neue K., Mormann M., Peter-Katalinic J., and Pohlentz G. (2011) Elucidation of glycoprotein structures by unspecific proteolysis and direct nanoESI mass spectrometric analysis of ZIC-HILIC-enriched glycopeptides. J. Proteome Res. 10, 2248–2260 [DOI] [PubMed] [Google Scholar]
- 42. Barkow-Oesterreicher S., Türker C., and Panse C. (2013) FCC – An automated rule-based processing tool for life science data. Source Code Biol. Med. 8, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Searle B. C. (2010) Scaffold: A bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics 10, 1265–1269 [DOI] [PubMed] [Google Scholar]
- 44. Vincent-Maloney N., Searle B. C., and Turner M. (2011) Probabilistically Assigning Sites of Protein Modification with Scaffold PTM. J. Biomol. Tech. 22, S36 [Google Scholar]
- 45. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., and MacCoss M. J. (2010) Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Frewen B., and MacCoss M. J. (2007) Using BiblioSpec for creating and searching tandem MS peptide libraries. Curr. Protoc. Bioinformatics Chapter 13, Unit 13.7 [DOI] [PubMed] [Google Scholar]
- 47. Selevsek N., Chang C.-Y., Gillet L. C., Navarro P., Bernhardt O. M., Reiter L., Cheng L.-Y., Vitek O., and Aebersold R. (2015) Reproducible and consistent quantification of the Saccharomyces cerevisiae proteome by SWATH-MS. Mol. Cell. Proteomics 10.1074/jbc.M114.604033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Escher C., Reiter L., MacLean B., Ossola R., Herzog F., Chilton J., MacCoss M. J., and Rinner O. (2012) Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics. 12, 1111–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Türker C., Akal F., and Schlapbach R. (2011) Life sciences data and application integration with B-fabric. J. Integr. Bioinform. 8, 159. [DOI] [PubMed] [Google Scholar]
- 50. McDonald W. H., Ohi R., Miyamoto D. T., Mitchison T. J., and Yates J. R. (2002) Comparison of three directly coupled HPLC MS/MS strategies for identification of proteins from complex mixtures: single-dimension LC-MS/MS, 2-phase MudPIT, and 3-phase MudPIT. Int. J. Mass Spectrom. 219, 245–251 [Google Scholar]
- 51. Molinari M. (2007) N-glycan structure dictates extension of protein folding or onset of disposal. Nat. Chem. Biol. 3, 313–320 [DOI] [PubMed] [Google Scholar]
- 52. Frank C. G., and Aebi M. (2005) ALG9 mannosyltransferase is involved in two different steps of lipid-linked oligosaccharide biosynthesis. Glycobiology 15, 1156–1163 [DOI] [PubMed] [Google Scholar]
- 53. Vacic V., Iakoucheva L. M., and Radivojac P. (2006) Two Sample Logo: A graphical representation of the differences between two sets of sequence alignments. Bioinformatics 22, 1536–1537 [DOI] [PubMed] [Google Scholar]
- 54. Tian G., Xiang S., Noiva R., Lennarz W. J., and Schindelin H. (2006) The crystal structure of yeast protein disulfide isomerase suggests cooperativity between its active sites. Cell 124, 61–73 [DOI] [PubMed] [Google Scholar]
- 55. Masato T., Yukio K., and Kenji K. (2008) Saccharomyces cerevisiae Rot1 Is an Essential Molecular Chaperone in the Endoplasmatic Reticulum. Mol. Biol. Cell. 19, 3514–3525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chojnacki T., and Dallner G. (1988) The biological role of dolichol. Biochem. J. 251, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Smith S. J., Crowley J. H., and Parks L. W. (1996) Transcriptional regulation by ergosterol in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 16, 5427–5432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kennedy MA., Barbuch R., and Bard M. (1999) Transcriptional regulation of the squalene synthase gene (ERG9) in the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 1445, 110–122 [DOI] [PubMed] [Google Scholar]
- 59. Henry K. W., Nickels J. T., and Edlind T. D. (2002) ROX1 and ERG regulation in Saccharomyces cerevisiae: Implications for antifungal susceptibility. Eukaryot. Cell 1, 1041–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bammert G. F., and Fostel J. M. (2000) Genome-wide expression patterns in Saccharomyces cerevisiae: Comparison of drug treatments and genetic alterations affecting biosynthesis of ergosterol genome-wide expression patterns in Saccharomyces cerevisiae: Comparison of drug treatments and genetics. Antimicrob. Agents Chemother. 44, 1255–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Joseph-Horne T., and Hollomon D. W. (1997) Molecular mechanisms of azole resistance in fungi. FEMS Microbiol. Lett. 149, 141–149 [DOI] [PubMed] [Google Scholar]
- 62. Zacchi L. F., and Schulz B. L. (2016) SWATH-MS glycoproteomics reveals consequences of defects in the glycosylation machinery. Mol. Cell. Proteomics 15, 2435–2447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mohorko E., Owen R. L., Malojcic G., Brozzo M. S., Aebi M., and Glockshuber R. (2014) Structural basis of substrate specificity of human oligosaccharyl transferase subunit N33/Tusc3 and its role in regulating protein N-glycosylation. Structure 22, 590–601 [DOI] [PubMed] [Google Scholar]
- 64. Fetrow J. S., Siew N., Di Gennaro J. A., Martinez-Yamout M., Dyson H. J., and Skolnick J. (2001) Genomic-scale comparison of sequence- and structure-based methods of function prediction: does structure provide additional insight? Protein Sci. 10, 1005–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schwarz M., Knauer R., and Lehle L. (2005) Yeast oligosaccharyltransferase consists of two functionally distinct sub-complexes, specified by either the Ost3p or Ost6p subunit. FEBS Lett. 579, 6564–6568 [DOI] [PubMed] [Google Scholar]
- 66. Jamaluddin M. F., Bailey U. M., Tan N. Y., Stark A. P., and Schulz B. L. (2011) Polypeptide binding specificities of saccharomyces cerevisiae oligosaccharyltransferase accessory proteins Ost3p and Ost6p. Protein Sci. 20, 849–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Karaoglu D., Kelleher D. J., and Gilmore R. (2001) Allosteric regulation provides a molecular mechanism for preferential utilization of the fully assembled dolichol-linked oligosaccharide by the yeast oligosaccharyltransferase. Biochemistry 40, 12193–12206 [DOI] [PubMed] [Google Scholar]
- 68. Kelleher D. J., Karaoglu D., Mandon E. C., and Gilmore R. (2003) Oligosaccharyltransferase isoforms that contain different catalytic STT3 subunits have distinct enzymatic properties. Mol. Cell. 12, 101–111 [DOI] [PubMed] [Google Scholar]
- 69. Larkin A., and Imperiali B. (2011) The expanding horizons of asparagine-linked glycosylation. Biochemistry 50, 4411–4426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Grabińska K., and Palamarczyk G. (2002) Dolichol biosynthesis in the yeast Saccharomyces cerevisiae: An insight into the regulatory role of farnesyl diphosphate synthase. FEMS Yeast Res. 2, 259–265 [DOI] [PubMed] [Google Scholar]
- 71. Liang R. M., Cao Y. B., Fan K. H., Xu Y., Gao P. H., Zhou Y., Dai B. D., Tan Y. H., Wang S. H., Tang H., Liu H. T., and Jiang Y. Y. (2009) 2-Amino-nonyl-6-methoxyl-tetralin muriate inhibits sterol C-14 reductase in the ergosterol biosynthetic pathway. Acta Pharmacol. Sin. 30, 1709–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data, from tier 3 measurements, were handled using the local laboratory information management system (LIMS) (49) and all relevant data have been deposited to the ProteomeXchange Consortium via the PRIDE (http://www.ebi.ac.uk/pride) partner repository with the data set identifier PXD006435.