

Abstract
The accumulation and removal of transposable elements (TEs) is a major driver of genome size evolution in eukaryotes. In plants, long terminal repeat (LTR) retrotransposons (LTR-RTs) represent the majority of TEs and form most of the nuclear DNA in large genomes. Unequal recombination (UR) between LTRs leads to removal of intervening sequence and formation of solo-LTRs. UR is a major mechanism of LTR-RT removal in many angiosperms, but our understanding of LTR-RT-associated recombination within the large, LTR-RT-rich genomes of conifers is quite limited. We employ a novel read-based methodology to estimate the relative rates of LTR-RT-associated UR within the genomes of four conifer and seven angiosperm species. We found the lowest rates of UR in the largest genomes studied, conifers and the angiosperm maize. Recombination may also resolve as gene conversion, which does not remove sequence, so we analyzed LTR-RT-associated gene conversion events (GCEs) in Norway spruce and six angiosperms. Opposite the trend for UR, we found the highest rates of GCEs in Norway spruce and maize. Unlike previous work in angiosperms, we found no evidence that rates of UR correlate with retroelement structural features in the conifers, suggesting that another process is suppressing UR in these species. Recent results from diverse eukaryotes indicate that heterochromatin affects the resolution of recombination, by favoring gene conversion over crossing-over, similar to our observation of opposed rates of UR and GCEs. Control of LTR-RT proliferation via formation of heterochromatin would be a likely step toward large genomes in eukaryotes carrying high LTR-RT content.
Keywords: gymnosperm, Picea, Pinus, angiosperm, retroelement, gene conversion, recombination suppression, genome size
Introduction
Transposable elements (TEs) are a major component of many eukaryotic genomes and long terminal repeat (LTR) retrotransposons (LTR-RTs) constitute the largest part of the DNA repetitive fraction in many plants (Feschotte et al. 2002). Because of their ability to quickly replicate and attain a very high copy number, LTR-RTs are often responsible for striking genome size variation, even between closely related species. The shrinkage of genomes via removal of LTR-RTs can also occur quickly as demonstrated in rice (Vitte et al. 2007), maize (SanMiguel et al. 1998), cotton (Hawkins et al. 2009), and Medicago truncatula (Wang and Liu 2008). There are two recombinant mechanisms that can remove LTR-RTs from host genomes: unequal recombination (UR), also called intrastrand homologous recombination, and illegitimate recombination (IR) (Devos et al. 2002; Ma et al 2004). UR occurs between LTRs of the same or different LTR-RTs and produces solo-LTRs in one step (Vicient et al. 1999), whereas IR, which unlike UR is not homology-driven, only gradually eliminates tracts of LTR-RT sequences and leaves incomplete elements in the genome (Devos et al. 2002; Ma et al 2004). So far, all angiosperm genomes studied show significant frequencies of solo-LTRs (e.g., SanMiguel et al. 1996; Vicient et al. 1999; Dubcovsky et al. 2001; Devos et al. 2002; Fu and Dooner 2002; Vitte and Panaud 2003), thus UR is a common process in angiosperms that can counteract genome expansion via LTR-RTs. The emerging scenario in conifers is quite different: LTR-RTs seem to accumulate slowly and consistently over tens of millions of years (Nystedt et al. 2013; Zuccolo et al. 2015), and our evidence to date suggests that the above mechanisms for LTR-RT removal have been largely inefficient (Nystedt et al. 2013). These findings could largely explain the huge sizes characterizing many conifer genomes.
Gene conversion events (GCEs) represent another homology-driven form of recombination and, when occurring between LTRs of an LTR-RT, are another possible outcome of intraelement recombination (Chen et al. 2007; Shi et al. 2010). In gene conversion, a recombination event transfers DNA information from a donor sequence to an acceptor sequence, modifying the acceptor sequence without significant sequence removal (contra UR). Gene conversion may occur between allelic haplotypes, but GCEs that occur between LTRs of a single LTR-RT are considered ectopic or interlocus events because they involve nonallelic sequences, similarly to the UR events that establish solo-LTRs. Although there are very few genome-wide studies on GCEs involving plant LTR-RTs, GCEs involving gene duplicates have been assessed in multiple angiosperms (Mondragon-Palomino and Gaut 2005; Wang and Paterson 2011; Guo et al. 2014). About 13% of duplicated genes in rice and sorghum experienced gene conversion after separation of these lineages (Wang et al. 2009). Physical proximity between paralogous genes facilitates gene conversion in these species (Wang and Paterson 2011), and notably, GCEs are more common in gene-rich regions, where the density of LTR-RTs is much lower than the whole-genome average (Wang and Paterson 2011). As would be expected for a homology-driven process, the intensity of gene conversion is also strongly associated with the sequence divergence of the loci involved, with higher divergence leading to fewer GCEs (Dooner and Martinez-Ferez 1997; Li et al. 2006; Chen et al. 2007).
A detailed examination of the frequency of GCEs between intraelement LTRs can also provide a more complete view of the genomic context of recombinative events involving LTR-RTs. Host genomes employ epigenetic mechanisms to suppress retroelement transcription and proliferation (Bucher et al. 2012), and areas that are particularly rich in retroelements can condense to interstitial heterochromatin (Lippman et al. 2004). Regions of heterochromatin, including those found at centromeres and telomeres, have long been thought to suppress homologous recombination. Recent studies contradict this assumption by indicating that it is not homology-driven repair that is suppressed within heterochromatin but rather resolution via crossing-over (Talbert and Henikoff 2010). In maize centromeres, crossing-over is entirely suppressed but GCEs are widespread (Shi et al. 2010), and in Drosophila, GCEs are common within centromeres and are also free of interference affecting crossing-over (Miller et al. 2016), perhaps due to features of double-stranded break (DSB) repair specific to heterochromatin (Chiolo et al. 2011; Peterson 2011). Thus, the fraction of genomic LTR-RTs occurring within heterochromatin could covary with relative rates of GCEs versus UR at LTR-RTs. Further evidence for the predominant genomic context of LTR-RTs in a species could be gained by determining whether structural features of LTR-RTs are associated with UR, as has been observed in some angiosperms (Vitte and Panaud 2003; Du et al. 2012; El Baidouri and Panaud 2013). Such associations could indicate that homology and other “local” features of the genome can affect rates of crossing-over, while the lack of such associations could indicate that the rate of crossing-over is more strongly affected by the “regional” context such as heterochromatin.
Which brings us again to the large, LTR-RT-rich genomes of conifers. To date, observations in conifers have been limited to just LTR-RT-associated UR affecting just three LTR-RT groups in a single species, Norway spruce (Picea abies) (Nystedt et al. 2013). Similarly, to our knowledge, there have been few studies addressing the intensity and features of GCEs between LTR-RT elements, and none involved multiple species (Kejnovsky et al. 2007; Shi et al. 2010; Sharma et al. 2013; Trombetta et al. 2016). Here, we analyze 23 different LTR-RT groups in P. abies and analyze 9 LTR-RT groups in three other conifers: the closely related species white spruce (P. glauca) and two species belonging to the genus Pinus that separated from Picea about 140 million years ago (Buschiazzo et al. 2012): loblolly pine (Pinus taeda) and sugar pine (Pinus lambertiana). We apply the same methodology to LTR-RT groups in seven angiosperm genomes: the herb Arabidopsis thaliana, the trees Amborella trichopoda and Populus trichocarpa, the woody vine Vitis vinifera, and the monocots/grasses Brachypodium distachyon, Oryza sativa (rice), and Zea mays (maize). The strategy we developed targeted tens of thousands of LTR-RT and solo-LTR copies at once. We also conducted a detailed analysis of rates of GCEs based on detailed investigation of hundreds of LTR-RT elements identified in angiosperms and in P. abies.
We show that the lowest rates of UR in the 11 species studied occur in the largest genomes: all 4 conifers as well as the angiosperm maize. We also show in our detailed analysis of GCEs that the highest rates of GCEs in the six species studied occur in the largest genomes, P. abies and maize. There is some variability in solo-LTR frequency between different LTR-RT groups in conifers, but we show in Norway spruce that this variation does not significantly correlate with any of the most evident structural features of the LTR-RT groups. Taken together, our results indicate a deep general difference in the genomic context of LTR-RTs in large, LTR-RT-rich plant genomes, and in light of other recent results, suggest that such differences may apply to eukaryotes with large genomes more generally.
Materials and Methods
Species Sampled
We selected four conifer species and seven angiosperms species for study. The conifers (P. abies, P. glauca, P. taeda, and P. lambertiana) were the only gymnosperms with sufficient high-quality genomic sequence available at the start of the study. The angiosperms include both monocots and dicots and feature a range of genome sizes. Arabidopsis thaliana, B. distachyon, O. sativa, V. vinifera, and Z. mays have each been subject to earlier LTR-RT-related study relevant to facilitating comparisons and evaluating the pipeline described herein. Amborella trichopoda is the basal extant angiosperm, while P. trichocarpa has a high-quality genome and complete LTR-RT elements had been previously identified (Natali et al. 2015). Although the conifers examined include two congeneric pairs, the species are separated by considerable divergence time estimates that vary from the early Miocene for P. abies and P. glauca (∼14–20 Mya, Nystedt et al. 2013), around the origin of the genus Oryza (Zou et al. 2013), to the early Cretaceous for P. taeda and P. lambertiana (∼110–140 Mya, Saladin et al. 2017), roughly at the separation of the Amborella lineage from all other angiosperms (Amborella Genome Project 2013).
Identifying LTR-RT Groups in P. abies
LTR-RT groups were identified on the basis of phylogenetic analyses. Reverse transcriptase (RT) domains 100 amino acids long (supplementary table S4, Supplementary Material online) were used as queries in tBlastN searches of 100,000 P. abies 454 random sheared reads (ftp://congenie.org/Data/ConGenIE/) (Sundell et al. 2015). All significant hits (E-value < 1e–5) longer than 80 residues were retrieved, totalling 670 and 1410 paralogous sequences for each of the Ty1-copia and Ty3-gypsy superfamilies, respectively. Sequences were aligned separately for each superfamily using the software MUSCLE (Edgar 2004). The alignments (supporting Data sets S2 and S3, Supplementary Material online) were then used to build Neighbor-Joining phylogenetic trees using the software MEGA6 (Tamura et al. 2013). Overall we identified 7 Ty1-copia and 16 Ty3-gypsy groups supported by high bootstrap values (supplementary fig. S2, Supplementary Material online). We calculated the evolutionary divergence between identified LTR-RT groups using the Poisson-corrected number of amino acid substitutions per site (), averaged over all pairwise comparisons between groups as implemented in MEGA6 (Tamura et al. 2013). As expected, the evolutionary divergence between groups is greater than that within groups for all groups tested (supplementary table S8, Supplementary Material online). A representative reverse transcriptase sequence for each of the 23 groups was used to search the P. abies assembly scaffolds longer than 50 kbp using tBlastN (Camacho et al. 2009). Regions surrounding the best positive matches were inspected using dot-plot analyses (Sonnhammer and Durbin 1995) to identify regions containing complete LTR-RT elements. At least five complete LTR-RT elements for each group were identified and retrieved (supplementary table S5, Supplementary Material online).
Representative sequences for these and all other complete LTR-RT elements identified in studied species are provided in supporting Data set S1, Supplementary Material online.
Identifying Elements in Picea glauca, P. taeda, and P. lambertiana
A subset of the 23 LTR-RT groups identified in P. abies including four Ty3-gypsy and five Ty1-copia groups was further investigated in P. glauca. Included in this subset were the seven most abundant groups identified in P. abies as well as two Ty3-gypsy groups that were medium-abundant in P. abies. Complete LTR-RTs representing paralogous groups were identified by searching the P. glauca genome assembly sequence (Birol et al. 2013) using the LTR sequence of P. abies LTR-RT elements as query in similarity searches followed by dot plot analysis (Sonnhammer and Durbin 1995).
We manually searched 111 fully sequenced P. taeda BACs (Genbank accession numbers: AC241263–AC241362, GU477256, GU477266, HQ141589) (Kovach et al. 2010) for the presence of LTR-RTs using dot plot analysis (Sonnhammer and Durbin 1995). One hundred and twelve complete LTR-RT elements were identified, LTRs were aligned and the alignments were used to build Neighbor-Joining trees for phylogenetic analysis, similarly to what was done for P. abies above. Note that LTRs were used to build the trees for P. taeda, while RT sequences were used in P. abies; LTRs were used here because the number of elements considered was small enough to allow for manual curation. Complete elements were arranged into 16 groups on the basis of LTR sequence similarity, and the 9 most abundant groups were chosen for further investigation.
LTRs of representative elements of the nine LTR-RT groups selected in P. taeda were used to search 964,817 P. lambertiana contigs longer than 15 kb (http://dendrome.ucdavis.edu/ftp/Genome_Data/genome/pinerefseq/Pila/v1.0/pila.v1.0.scafSeq.gz; last accessed September 10, 2016). Representative elements for each of the nine groups in P. lambertiana were identified by dot plot analysis.
Identifying Elements in Angiosperm Genomes
For P. trichocarpa, full length LTR-RTs were from Natali et al. (2015). Full length LTR-RTs were downloaded from Repbase (Jurka et al. 2005) for A. thaliana, A. trichopoda, B. distachyon, O. sativa, V. vinifera, and Z. mays. These LTR-RTs were used to evaluate their abundance in the respective host genome using RepeatMasker (Smit et al. 2015) to search the corresponding genome assemblies. From three to five complete copies from each of the most abundant LTR-RTs group identified were retrieved for use in further analyses.
Estimating the Ratio of Solo-LTRs to Complete LTR-RT Elements
For each of the targeted LTR-RT group identified in the different species analyzed, we used the following strategy to infer the ratio of complete LTR-RTs to solo-LTRs, with the numbering of each step corresponds to that illustrated in figure 1:
Fig. 1.
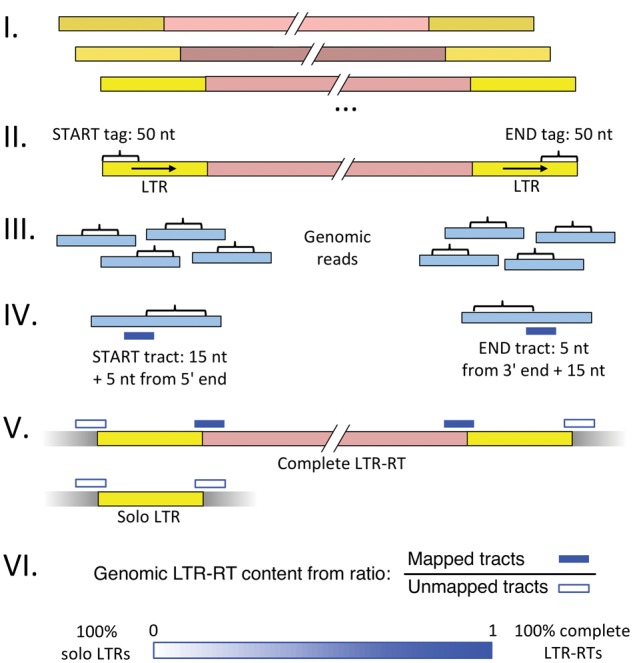
—Method to estimate ratio of solo-long terminal repeats (LTRs) to complete LTR-retrotransposons (RTs) within a species. (I) Retrieve or assemble 3 to 10 paralogs for each LTR-RT group. (II) Extract 50-nt START and END tags from LTRs of paralogs. (III) Find genomic reads matching START and END tags with RepeatMasker (Smit et al. 2015), allowing for mismatches. (IV) For each matching read, extract a 20-nt tract containing 5 nt from the tag and 15 nt flanking sequence. Tracts are taken from the 5′ or 3′ ends of START or END tag matches, respectively. (V) Map each tract to the LTR-RT paralogs collected in (I) using BWA ALN (Li and Durbin 2009), allowing for mismatches. Count the numbers of mapped (M) and unmapped (U) tracts. Genomic reads covering complete LTR-RTs yield tracts that are mapped and unmapped in equal numbers, while genomic reads covering solo LTRs produce only unmapped tracts. (VI) The relative genomic content of solo LTRs to complete LTR-RTs is inferred from the ratio of mapped to unmapped tracts. See “Methods” section for further details and pipeline validation results.
I. We retrieved from 3 to 15 complete LTR-RT paralogs from the host genome for each group as described above. For each complete element from (I), we extracted the first 50 nt of the 5′ LTR and the last 50 nt of the 3′ LTR. We refer to these LTR-RT-derived sequences as tags, in particular START tags for those originating from the 5′ of the element and END tags for those originating from the 3′ end of the element. If no divergence has occurred between LTRs of an inserted element and thus the LTRs remain identical in sequence, the START and END tags would each match both LTRs perfectly.
II. Tags were mapped onto Illumina reads derived from the host genome using RepeatMasker (Smit et al. 2015).
III. Reads from (III) were filtered, retaining all the matches which met the following conditions: for START tags, the longest unmatched regions were 3 and 5 nucleotides at the 5′ and 3′ ends, respectively; for END tags, the longest unmatched regions were 5 and 3 nucleotides at the 5′ and 3′ ends, respectively. For each matching read passing filtering, we extracted a 20 nt region we call a tract. For START tags, the START tract included 5 nt from the 5′ end of the LTR together with the upstream 15 nt; for END tags the END tract included 5 nt from the 3′ end of the LTR together with the downstream 15 nt. Constructed in this way, a START tract will include interior sequence from a complete LTR-RT when the START tag from which it is derived matches the 3′ LTR of the complete LTR-RT, while for an END tract, this is true when it matches the 5′ LTR of a complete LTR-RT.
VI. Tracts were then mapped using BWA ALN (Li and Durbin 2009) onto the complete LTR-RT paralog sequences used in (I), with the settings k = 2, n = 4, l = 12.
V. The numbers of mapped (M) and unmapped (U) tracts were determined from BWA output and used to infer relative genomic content of complete LTR-RT elements and solo-LTRs.
Genomic reads covering a complete LTR-RT should, on average, produce the same amount of mapped and unmapped tracts, whereas genomic reads covering a solo-LTR should produce only unmapped tracts. The amount of mapped versus unmapped tags in a genome mostly containing complete LTR-RTs should be approximately equal, resulting in an M/U ratio of approximately 1. On the other hand, the presence of solo-LTRs in the genome should produce a notable reduction of this ratio from 1. There may be a bias toward unmapped reads, depending on the degree of divergence among genomic LTR-RTs; this can be controlled by ensuring START and END tags are derived from a variety of LTR-RT paralogs. We have endeavoured to be comprehensive for the groups studied, nevertheless a general caution for all genomic analyses of repetitive elements also applies here: because related elements within the same genome can show quite remarkable divergence, the results should be considered to be characteristic of the specific LTR-RT groups studied. Note also that some LTR-RT paralogs retrieved from assemblies contained N-gaps (supporting Data set S1, Supplementary Material online); in all cases these gaps are not present at LTR borders, thus they do not affect this analysis.
The ratios of solo-LTRs (S) per complete LTR-RT (C), as well as the reciprocal ratio of complete LTR-RTs per solo-LTR, can be quantified using the relations:
The pipeline was run for each species analyzed, using a whole-genome shotgun Illumina reads data set assumed to represent an unbiased sample of each genome (see supplementary table S1, Supplementary Material online for ENA accession numbers). For most read sets, a subset of reads were used; additionally, for paired-end data sets, only the first read of each pair was used. The amounts of read sequence used from each read set and relative genomic coverage provided by each reads data set are also detailed in supplementary table S1, Supplementary Material online.
Pipeline Validation
The reliability of the above pipeline was tested in P. abies and P. taeda using alternative approaches and other data sources. In P. abies, we randomly selected 4,348 sequences (175 Mbp in total, provided in supporting Data set S4, Supplementary Material online) from a large collection of fosmid pool scaffolds and estimated the M/U ratio for the Ty3-gypsy group Alisei. Each fosmid pool contained ∼40 Mbp of fosmid sequence, representing ∼0.2% of the total genome of P. abies, and is more representative of the true content of repetitive sequences in the genome than is the whole-genome shotgun assembly (Nystedt et al. 2013). The assembled fosmid sequences were manually searched for the presence of Alisei LTRs using dot plot analyses (Sonnhammer and Durbin 1995). We identified 171 complete elements and 18 solo-LTRs, giving an M/U ratio (0.90) consistent with the one estimated by the pipeline (0.89).
In P. taeda, representative LTRs from each LTR-RT group were also used to manually search the previously mentioned 111 fully sequenced BACs (totalling ∼11 Mbp) (Kovach et al. 2010) using dot plot analysis (Sonnhammer and Durbin 1995). Positive matches were checked to see if they belonged to a complete LTR-RT or to a solo-LTR. In total, 243 sites were identified: 187 complete LTR-RTs and 56 solo-LTRs. These figures translated to an M/U ratio of 0.77 that is somewhat less than the pipeline estimate of 0.88.
The underestimation of the M/U ratio for P. taeda, in contrast to the close agreement for P. abies, could simply be a stochastic effect of a lesser amount of high-quality sequences available for P. taeda versus P. abies (11 Mbp vs. 175 Mbp). Our restriction of the search in P. abies to a single LTR-RT group (Alisei) might have compensated for this to some degree, as indicated by the similar numbers of complete elements recovered, but this also could have allowed for greater tolerance for divergence when recovering solo-LTRs and thus greater relative numbers of solo-LTRs within the P. taeda BACs (see below), where this restriction was not applied. Nevertheless, for both species validation data provide further support for a strong under-representation of solo-LTRs.
We also specifically tested the accuracy of pipeline step (III) which maps tags onto Illumina reads using RepeatMasker. In particular, we evaluated the average similarity of the positive matches as well as the fraction of positive matches having a similarity value smaller than 80%. The latter fraction could include artefactual matches to very divergent elements or unrelated elements. The overall similarity is above 90% for all species with the exception of A. trichopoda at 87.84% (supplementary table S6, Supplementary Material online). These values are well above the lowest similarity value (80%) proposed by Wicker et al. (2007) for defining a LTR-RT family. Furthermore, the fraction of matches having similarity lower than 80% is quite limited, usually under 2% of the total, with the highest value reaching 2.38%, again in A. trichopoda (supplementary table S6, Supplementary Material online).
We evaluated the potential for tracts to be erroneously classified as “unmapped” during pipeline step (V) by collecting all unmapped tracts and clustering them using CD-HIT (Fu et al. 2012). Our reasoning is that unmapped tracts should reflect the random distribution of sequences adjacent to LTR-RT insertions and therefore should mostly differ from each other. Any large cluster of highly similar unmapped sequences would be suggestive of artefactual errors. We screened all of our unmapped tracts for such instances and no suspicious cases were identified (results not shown).
We also evaluated the potential for biases in mapping percentages during pipeline step (V) introduced by the generation of START and END tags from different ends of representative retroelements. If cases of element truncation are common, a clear difference in the mapped/unmapped (M/U) ratios should be apparent when calculated using tracts derived from START and END tags separately. In the overall majority of the cases for both angiosperms and gymnosperms, these ratios are in very good agreement and we observed no systematic bias involving tags from either origin or in gymnosperms versus angiosperms (supplementary table S7, Supplementary Material online).
We also considered the possible confounding effect of differences in relative genomic coverage provided by reads data sets among the studied species, as this negatively covaries with genome size (supplementary table S1, Supplementary Material online), an important factor in our conceptual models. We attempted to separate these effects by evaluating linear models in which M/U ratio was dependent on both relative coverage and genome size. A fully specified model showed neither coverage, genome size, nor their interaction to be individually significant (P > 0.24 for genome size, P > 0.75 for coverage and interaction) though the full model was (F3, 112 = 17.45, P < 1 × 10−8). Dropping the interaction term did not significantly weaken the model (likelihood ratio test, P = 0.89), and a model lacking the interaction term showed genome size to be a significant predictor of M/U (P < 1 × 10−5) while relative coverage was not (P > 0.74). Although sample size is limited, we interpret these results to indicate that genome size is a predictor of M/U and relative coverage is not.
Finally, we compared our estimated solo-LTR to complete LTR-RT ratios with the literature, which included five of the seven angiosperm species considered in this study (supplementary table S9, Supplementary Material online). Our results are in good agreement with those calculated in Z. mays by SanMiguel et al. (1996) and El Baidouri and Panaud (2013), with those calculate in O. sativa by Ma et al. (2004) and El Baidouri and Panaud (2013) and with those assessed in B. distachyon by El Baidouri and Panaud (2013). The most apparent discrepancy was seen for V. vinifera, for which we report a slight excess of solo-LTRs (ratio 1.28) while El Baidouri and Panaud (2013) report a slight deficit (ratio 0.84). It is however important to consider that data available in literature were obtained using a wide array of different strategies as well as varying definitions of solo-LTRs. Because of this, the direct comparisons of data from such different sources are not straightforward.
During revision, we learned of a similar method employing LTR-RT-derived tags described by Macas et al. (2015) when examining genome size variation in the legume tribe Fabeae. Although methodological details differ and our sampling of representative LTR-RTs, tag sites, and pipeline validation are more extensive, we would expect that both methods would produce broadly similar results. We would expect our method to be more stable when applied to taxa such as conifers, in which TEs can be quite old and diverged, where a Blast-based method might produce an unreasonably large number of element groups; we have not subjected this to test.
Intraelement LTR Gene Conversion
GCEs between LTRs of complete elements were detected using the software GENECONV (Sawyer 1999). We identified a total of 137 complete elements from angiosperm genomes and 353 complete elements from the P. abies 1.0 genome assembly (Nystedt et al. 2013) and fosmid pool assemblies (295 elements from the genome assembly and 58 elements from fosmids) using the same method as that described above to identify complete LTR-RT elements in P. abies. Each LTR sequence was extracted from the full-length copy element using BEDTools (Quinlan and Hall 2010) and the two LTRs of each element were compared locally against each other using BLAST+ 2.2.29 (Camacho et al. 2009) with the following settings: blastn–task blastn–dust no–e-value 1e–05. Alignments from the BLASTN results were parsed using custom Perl scripts and utilized to search for gene conversion segments using GENECONV (Sawyer 1999). Through permutation analyses of sequence alignments, GENECONV determines the probability that regions of the alignment showing a high level of nucleotide similarity derive from GCEs rather than stochastic variation of nucleotide substitutions. Recent GCEs appear as stretches of identical nucleotides in alignments of homologous sequences; converted segments derived from older GCEs tend to accumulate substitutions between the donor and acceptor sequences, thus appearing as shorter identical stretches interrupted by single-nucleotide substitutions or larger indels in the alignments.
The following GENECONV settings were used: /w123/lp/f/eb/g0 [or/g1 or/g2]-include_monosites. These settings allowed to search for gene conversion segments in alignments with two sequences only and to consider run of missing data sites or indel sites as single “polymorphisms.” Each aligned sequence was run through GENECONV three times with three different values for the gscale (g) option: 0, 1, and 2. The gscale value determines the mismatch penalties associated with conversion segments. A gscale value of 0 allows no mismatches in the segments, gscale 1 applies the lowest mismatch penalties and often results in more segments being detected, and gscale 2 applies more strict mismatch penalties and tends to identify a number of segments intermediate between the results of gscale 0 and 1 (Sawyer 1999). Segments discovered using different gscale values usually overlapped, although segments observed with gscale 0 tend to be shorter and to represent younger GCEs, while segments identified using gscale 1 tend to be the longest and could represent older segments that have accumulated more mismatches.
Results
Using Representative LTR-RTs and Short Reads to Estimate the Ratio of Solo-LTRs to Complete LTR-RTs
We developed the method shown in figure 1 to infer the rate of UR by estimating the ratio of solo-LTRs to complete LTR-RTs (S-to-C ratio). Our method uses representative full-length LTR-RT sequences and short-read sequence data and determines the numbers of tracts spanning the 5′ and 3′ ends of the LTR that could be mapped (M) and could not be mapped (U) to the representative complete LTR-RT elements. The rationale of this approach is that genomic reads covering a complete LTR-RT should, on average, produce the same amount of mapped and unmapped tracts, whereas genomic reads covering a solo-LTR should produce only unmapped tracts. If the host genome contains only complete LTR-RT elements, then the amount of mapped versus unmapped tags should be approximately equal, resulting in an M/U ratio of ∼1; due to stochastic error the ratio may occasionally slightly exceed 1. On the other hand, any notable reduction of this ratio from 1 indicates the presence of solo-LTRs in the genome (fig. 1). The ratio of solo-LTRs to complete LTR-RT elements (S-to-C) can be readily calculated as . We have extensively evaluated the consistency of the pipeline, including comparisons with results obtained via our own manual curation, evaluation of several possible biases affecting whether tracts are mapped or unmapped, establishing that relative coverage of reads data sets does not bias M/U ratios, and comparisons with previous estimates from the literature. Further details are available in “Pipeline validation” section, and in supplementary tables, Supplementary Material online indicated there.
We analyzed LTR-RT groups belonging to the Ty1-copia and Ty3-gypsy superfamilies in four conifer species and seven angiosperm species; sources of short-read sequence data and estimates of LTR-RT content and genome size for each studied species are provided in supplementary table S1, Supplementary Material online. See “Materials and Methods” section for complete details of group identification and selection in the study species.
In the conifer P. abies, we identified 23 abundant LTR-RT groups (7 from the Ty1-copia superfamily and 16 from Ty3-gypsy) using phylogenetic analysis (supplementary fig. S2, Supplementary Material online) and applied our method to a sequence data set containing more than 39 million 100-bp Illumina reads, corresponding to a total of 3.9 Gbp or about 0.2× coverage of the whole genome (supplementary table S1, Supplementary Material online). For the related P. glauca, we examined the nine most abundant of the 23 P. abies LTR-RT groups (5 Ty1-copia and 4 Ty3-gypsy) in a data set of 43 million 100-bp Illumina reads (4.3 Gbp, 0.21× genomic coverage). We studied nine abundant LTR-RT groups in P. taeda (supplementary fig. S3, Supplementary Material online) using 39.4 million 128-bp Illumina reads (5.04 Gbp, 0.23× coverage), and analyzed these same nine LTR-RT groups in P. lambertiana using a data set of 39.4 million 128-bp Illumina reads (5.04 Gbp, 0.17× coverage) (supplementary table S1, Supplementary Material online). Representative sequences for all studied LTR-RT groups are provided in supporting Data set D1, Supplementary Material online.
Variation in Ratio of Solo-LTRs to Complete LTR-RTs among Species
In P. abies, we analyzed 146,028 tracts, 50,825 for Ty1-copia, and 95,203 for Ty3-gypsy (supplementary table S2A, Supplementary Material online), reflecting the relative abundances of these LTR-RT superfamilies in the genome (Nystedt et al. 2013). Assuming the read data set is an unbiased representation of the whole genome, these figures indicate several tens of thousands elements belonging to each of these groups in the complete P. abies genome. The overall M/U ratio is 0.85, corresponding to an S-to-C ratio of 0.18, roughly 1 solo-LTR for every 5.6 complete LTR-RTs (fig. 2, supplementary table S2A, Supplementary Material online). In the closely related species P. glauca, we analyzed 86,410 tracts (supplementary table S2B, Supplementary Material online). The overall M/U ratio was 0.81, with roughly one solo-LTR for every four complete LTR-RT elements (fig. 2, supplementary table S2B, Supplementary Material online). Although the underrepresentation of solo-LTRs versus complete LTR-RT is less pronounced in P. glauca than in P. abies, the M/U ratios for the LTR-RT groups tested were not significantly different between the two Picea species (P = 0.21, Wilcoxon test).
Fig. 2.—
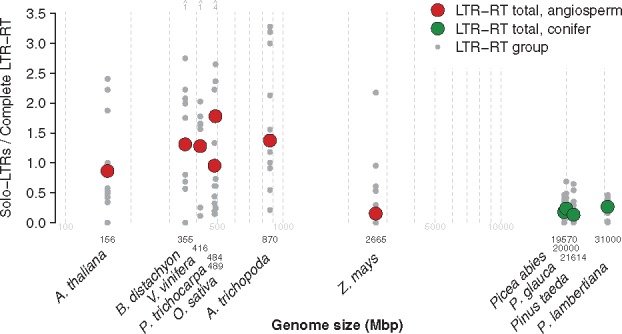
Ratios of solo-LTRs to complete LTR-RT elements, as a proxy for rates of unequal recombination, from seven angiosperm species and four conifer species versus genome size (log10 axis). For each species, ratios for separate LTR-RT groups are shown together with the total ratio of solo-LTRs to complete LTR-RT elements for all tracts. Shown above Brachypodium distachyon, Vitis vinifera, and Oryza sativa are the numbers of LTR-RT groups from each species with ratios that exceed the upper limit of the y-axis. See supplementary table S1, Supplementary Material online for genome size references and supplementary tables S2 and S3, Supplementary Material online for all LTR-RT group ratios.
In the conifer P. taeda, we analyzed 153,229 tracts, yielding an overall M/U ratio of 0.88, corresponding to 1 solo-LTR to ∼7.5 complete LTR-RTs (fig. 2, supplementary table S2C, Supplementary Material online). In its congener P. lambertiana, we analyzed 122,518 tracts (supplementary table S2D, Supplementary Material online). The overall M/U ratio was 0.79, translating to 1 solo-LTR to ∼3.7 complete LTR-RTs (fig. 2, supplementary table S2D, Supplementary Material online). The M/U ratios for the LTR-RT groups studied in the two Pinus species did not differ significantly (P = 0.67, Wilcoxon test).
Turning to the seven studied angiosperms, we identified LTR-RT groups and applied the same method; representative LTR-RT sequences are available in supporting Data set D1, Supplementary Material online. M/U ratios calculated for the most abundant LTR-RT groups taken as a whole are consistently lower than those calculated in conifers and S-to-C ratios are consistently higher, with the exception of Z. mays, which has the largest genome by far of the angiosperms studied (fig. 2, supplementary table S3, Supplementary Material online). The lowest M/U ratio among angiosperms was in O. sativa (0.39) and the ratios of the other analyzed species (excluding Z. mays) consistently indicate an excess of solo-LTRs (fig. 2, supplementary table S3, Supplementary Material online).
Previous studies in angiosperms have shown that the ratio of solo-LTRs to complete LTR-RTs is positively correlated with element features such as the LTR length (Du et al. 2012) and the ratio of LTR length to internal region length (El Baidouri and Panaud 2013), suggesting that, at least in angiosperms, structural features of LTR retrotransposon impact solo-LTR formation. We applied a similar analysis to the P. abies data set because it contained many more LTR-RT groups than the other three conifers. In contrast to the earlier results for angiosperms, neither of these structural features correlated with the M/U ratios of the groups (LTR length: Spearman’s rs = –0.24, P = 0.86; LTR length/internal region length: Spearman’s rs = –0.18, P = 0.8).
We extended this analysis to test two other element features, total LTR-RT abundance and LTR-RT GC content, and found no correlation between M/U ratio and either feature (element abundance: Spearman’s rs = 0.15, P = 0.76; LTR-RT GC content: Spearman’s rs = –0.05, P = 0.59).
Variation in Intraelement Gene Conversion Rate among Species
To identify GCEs, sequence alignments of intraelement LTRs were screened using GENECONV (Sawyer 1999), one of the most widely used programs in gene conversion studies (e.g., Drouin 2002; Xu et al. 2008; Casola et al. 2010; Casola et al. 2012). Because of the high substitution rate experienced by TEs including LTR-RTs, the initial complete identity between converted regions of LTRs tends to be quickly eroded (SanMiguel et al. 1998). To account for this, we combined results from several GENECONV runs at various levels of stringency for mismatches between LTR alignments (see “Methods” section). We found intraelement GCEs in 55% of P. abies LTR-RTs from fosmids (fig. 3). In the 1.0 genome assembly, we observed GCEs in 36% of LTR-RTs, affecting 40% of Ty3-gypsy elements, and 27% of Ty1-copia elements. The lower percentage of GCEs in the P. abies genome assembly is downward biased; the fraction of repetitive sequence within fosmid assemblies is more closely approximating that inferred to be in the P. abies genome in vivo than does the lower fraction of repetitive sequence observed in the genome assembly (Nystedt et al. 2013).
Fig. 3.—

Proportion of examined LTR-RTs with intraelement gene conversion events (GCEs) between LTRs versus genome size (log10 axis). Pooled results for all identified GCEs are shown, together with separate results for Gscale parameters in order of increasing stringency against mismatches for detection of GCEs between aligned sequences; see “Methods” section for further details. Species are colored as in figure 2.
In angiosperms, a lower fraction of LTR-RTs showed signs of gene conversion compared with P. abies fosmids, again with the exception of Z. mays, with an average of 23% of LTR-RTs across all studied angiosperms. This ranged from a single GCE observed in A. thaliana up to 40% elements with GCEs in Z. mays (fig. 3). Parallel differences in levels of gene conversion were also observed when comparing GENECONV analyses with varying stringency levels. Perfectly identical and presumably more recent gene conversion segments (Gscale = 0) were observed in 34% of P. abies fosmid LTR-RTs (fig. 3) and 16% of assembly LTR-RTs, while in angiosperms, conversion segments were identified in 5–30% of LTR-RTs, with A. thaliana and Z. mays again at the extremes of this frequency spectrum (fig. 3) and only Z. mays approaching the frequency observed in P. abies. GENECONV analyses with the lowest stringency threshold (Gscale = 1) resulted in slight increases of the proportion of converted elements, with the notable exception of the P. abies LTR-RTs from the genome assembly (fig. 3).
Despite the high fraction of observed GCEs in some species, conversion segments in all species were relatively short, and ranged between 222 and 428 bp except in rice (supplementary fig. S1, Supplementary Material online). As expected, higher-stringency GENECONV analyses detected much shorter stretches of perfectly identical conversion segments between LTRs, and revealed especially short segments in the two genomes with the oldest elements, A. trichopoda and P. abies.
The structure and sequence composition of converted and nonconverted LTR-RTs and their LTRs were further inspected to disentangle the possible role of these local features in promoting GCEs. Across most species, longer full-length elements, and especially longer LTRs were associated with gene conversion, with the exception of B. distachyon for both traits and Z. mays for LTR length (fig. 4A and B). In line with these findings, the alignments used to detect GCEs are much longer in converted versus nonconverted LTRs of most species (fig. 4C). Thus, there could be bias in the GENECONV analyses toward increasing the number of detected GCEs in longer elements, because longer alignments tend to contain more overall substitutions than shorter ones, which in turn increases the statistical power for detection. However, this association is absent in B. distachyon and Z. mays, which show similar alignment lengths in converted and nonconverted LTRs. A comparable trend was observed for the ratio of LTR length to internal length (fig. 4D). Overall, the length of LTR-RTs and LTRs appears to be a major determinant of the frequency of GCEs in P. abies and in most examined angiosperms.
Fig. 4.—

Characteristics of examined LTR-RTs inferred to contain (values on x-axis) or lack (values on y-axis) GCEs; the diagonal dashed lines represent equal values in both cases. Plotted values are within-species means ± standard error. Separate Picea abies values are shown for LTR-RTs in fosmid pool assemblies (filled circles) and the genome assembly (open circles); the latter contains a biased, lower proportion of repetitive sequences than the P. abies genome in vivo, see main text. Arabidopsis thaliana is excluded due to just one observed GCE. Species are colored as in figure 2.
The sequence identity was similar in converted LTRs compared with nonconverted LTRs, including for LTRs in Norway spruce and A. trichopoda which showed notably lower overall identity than in the other studied species (fig. 4E). This is counter to the trend typically observed between gene copies, for which paralogous genes with GCEs tend to share higher sequence similarity than nonconverted paralogs (Xu et al. 2008; Casola et al. 2010). Taken together with the length-related results earlier, this suggests the GCEs we observed within LTR-RTs may have been facilitated primarily by LTR length, rather than sequence similarity. As for the M/U ratio, we did not find a significant difference in GC-content between converted and nonconverted LTRs (fig. 4F).
One possible source of bias resulting from an interaction of GCEs and mapping success could be due to GCEs between internal regions of LTR-RTs and the flanking DNA of these elements (Vitte and Panaud 2003; Ma et al. 2005). If common, such events could skew the proportion of mapped reads onto full-length LTR-RT sequences compared with solo LTR-RTs. However, only 3/77 full-length elements were found to show evidence of internal-to-flanking DNA gene conversion in one study (Vitte and Panaud 2003), whereas a single example was described among 53 LTR-RTs analyzed in the orthologous Orp regions of maize, sorghum, and rice (Ma et al. 2005). The low frequency of gene conversion between internal LTR-RT sequences and their flanking regions observed in these studies suggests that this process is unlikely to have introduced a significant bias in our mapping data.
This comparison of the structure and composition of converted and nonconverted LTR-RTs and their LTRs indicates that while these factors may be important in determining when individual GCE events may occur, as has also been found by other studies already cited, these factors do not differ systematically among our studied species in a manner that could explain the differences we observe in GCE events in large plant genomes (fig. 3).
Largest Genomes Have Lowest Fractions of Solo-LTRs and Highest Rates of GCEs
Considering the solo-LTR to complete LTR-RT fractions and LTR-RT-associated GCE rates together (fig. 5), these rates are positively correlated in the species with small- to medium-sized genomes (A. thaliana to A. trichopoda; n = 6, Spearman’s rs = 0.841, P = 0.036) while the correlation reverses and weakens to nonsignificance when including the large-genome species Z. mays and P. abies (n = 8, Spearman’s rs = −0.216, P = 0.61). As UR and gene conversion are both homology-driven processes which differ in whether they do or do not resolve in crossing-over, this suggests the possibility that resolution via crossing-over around LTR-RTs occurs at much lower rates in large-genomed species.
Fig. 5.—
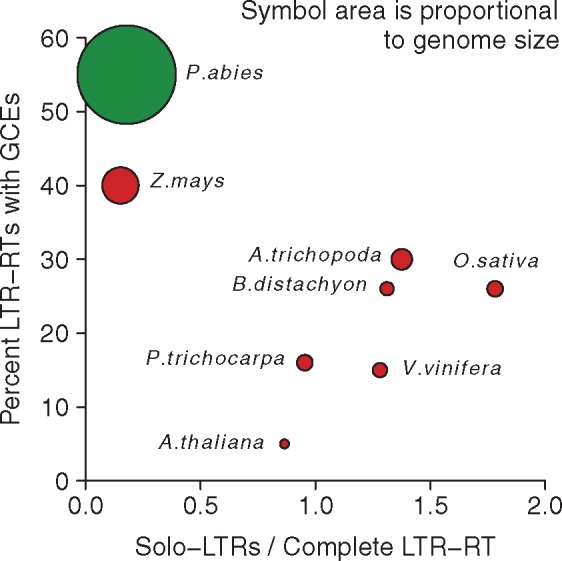
Proportion of examined LTR-RTs with intraelement GCEs between LTRs versus the total ratio of solo-LTRs to complete LTR-RT elements, as a proxy for rates of unequal recombination. Proportion of GCEs shown is for all identified GCEs (equivalent to solid dots in fig. 3). Species are colored as in figure 2 and symbol area is proportional to genome size of each species. The correlation among the six small- to medium-genome species is positive (Spearman’s ρ = 0.841, rs = 0.036) while including the two large-genome species reverses and weakens the correlation to nonsignificance (Spearman’s rs = −0.216, P = 0.61).
Discussion
Our results indicate that general, genome-wide differences in the resolution of LTR-RT-associated recombinative events covary with plant genome size. For GCEs, this is a positive and roughly linear relationship, with the highest rates in the largest genomes (fig. 3). For UR leading to solo-LTRs, our results suggest the occurrence of two distinct regimes: for small- to medium-sized genomes, rates of solo-LTR production are positively correlated and roughly linear, while rates are much lower in species with larger genomes, on the order of maize or larger (fig. 2). The occurrence of two distinct regimes is even more apparent when the rates are plotted together, for those species in which both were estimated (fig. 4).
The degree of solo-LTR under-representation in conifers shows some variability between different LTR-RT groups, but this variation does not significantly correlate with any of the most evident structural features of LTR-RTs in P. abies. This contrasts with previous results in angiosperms showing positive correlations between LTR-associated UR and LTR length-related features (Vitte and Panaud 2003; Du et al. 2012; El Baidouri and Panaud 2013), as well as our observed frequency of GCEs, which positively correlates with lengths of element features (fig. 4A). The highest levels of LTR-RT-associated GCEs were observed in the genomes of the two species where LTR-associated UR appears to be most strongly suppressed, which were also the two studies species with the largest genomes: P. abies and maize (fig. 5).
The contextual suppression of UR may be achieved via several potentially co-occurring processes, including reduction in homologous recombination via reduced numbers of DSBs, a preference for nonhomologous DNA repair pathways such as IR, and the favoring of alternative outcomes of homologous recombination that do not result in crossing-over, in particular gene conversion. Our results support the existence of this latter process in plants with large genomes. One mechanism that could underly this process would be the formation of heterochromatin in LTR-RT-rich regions via methylation. Evidence supporting a possible role of methylation in limiting and/or controlling the recombination processes has been collected in both animals and plants, albeit limited to particular cellular developmental stages such as meiosis. DNA methylation can restrain TEs from adopting chromatin features amenable to meiotic recombination in mice (Zamudio et al. 2015). In the germ line of honeybees, methylated genes show a reduced rate of crossing-over (CO) events (Wallberg et al. 2015). Similarly, DNA methylation and chromatin states were identified as key factors in explaining the striking variation of meiotic CO rate along A. thaliana chromosomes (Colomè-Tatchè et al. 2012; Mirouze et al. 2012). Yelina et al. (2015) demonstrated that DNA methylation has a pivotal role in establishing domains of meiotic recombination along chromosomes and it is sufficient to silence CO hot spots in Arabidopsis.
Genome size-associated differences in the regulation of LTR-RT-associated heterochromatin which thereby affects recombination seems the most plausible mechanism which could explain our results. Alternatively, there may exist significant differences in the regulation of the recombination process between seed plants with small- to medium-sized genomes and those with large, LTR-RT-rich genomes. In favor of a heterochromatin-based mechanism, we would predict that genome-wide methylation levels would covary with rates of LTR-RT-associated GCEs, not only in plants with large genomes but also in other taxa. Methylation is certainly elevated in the genomes of conifers, occurring at more than 83% of the total cytosines in P. abies (Ausin et al. 2016) and at more than 64.4% of the cytosines analyzed in Pinus pinea (Sáez-Laguna et al. 2014) and is consistently higher than that of other annual and perennial plants (Avramidou et al. 2015; Ausin et al. 2016).
Other factors may contribute to the observed variation in GCEs between species. For instance, the retroelements sampled from species with higher rates of GCEs may experience particularly high frequency of gene conversion compared with other LTR-RT families. Given that we selected several distantly related families from each species for our analyses, including elements from both Ty3-gypsy and Ty1-copia groups (supplementary figs. S2 and S3, Supplementary Material online), this is unlikely to have influenced our results significantly.
A recent study examining a limited number of LTR-RT families in four species of salamanders (Frahry et al. 2015) has provided similar evidence of a relationship between UR suppression and large genome size. Salamander genomes are huge, having sizes ranging from ∼14 Gbp up to ∼120 Gbp, this largest over six times that of Norway spruce. Low amounts of solo LTRs were detected and no single LTR-RT structural feature was identified as being a strong predictor of solo-LTR underrepresentation (Frahry et al. 2015). That eukaryotes as evolutionarily far apart as conifers and salamanders share these features regarding LTR-RT removal, with both also characterized by very large genome sizes, is suggestive of a more general mechanism related to the control of TE amplification and removal in large genomes. We predict that these salamander genomes also show an elevated rate of LTR-RT-associated GCEs.
Taken together, our results are also consistent with the hypothesis recently put forward by Fedoroff (2012) to explain the accumulation of large amounts of repetitive elements in eukaryote genomes despite the presence of mechanisms leading to their removal by UR or IR. She suggested that TEs can accumulate in huge quantities because of, not in spite of, the epigenetic mechanisms used to control their proliferation. These epigenetic mechanisms maintain heterochromatin where repeats are rich, suppressing the expression and transposition of TEs, and also simultaneously reducing recombinational events that could lead to TE removal. The largest genomes we studied—the four conifers plus maize—are also the genomes with the strongest evidence for suppression of sequence-removing UR. As the studies cited above indicate in maize and Drosophila, heterochromatin does not suppress all forms of recombination, rather just those that lead to crossing-over and hence UR and IR. While the epigenetic status and the chromatin state within and among the LTR-RT groups were not examined in the present study, our results do suggest an important interplay between LTR-RT content, recombination outcomes and heterochromatin, and are entirely consistent with Fedoroff’s hypothesis.
Our results across seed plants emphasize the importance of another prediction arising from Fedoroff’s (2012) hypothesis. When TE proliferation is more rapid than TE removal, runaway increases in genome size can occur if controls on TE activity develop after proliferation but before significant removal, with the relative balance determined by characteristics of the mechanisms employed to control TE activity. Further investigations of the relationship between epigenetic status, chromatin configuration, and the resolution of homology-dependent recombination in LTR-RT elements across many more taxonomic groups will be required to address the overall impact of TEs in genome size evolution across eukaryotes.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We are grateful to Alexander Suh for helpful discussions, and to the anonymous referees for helpful comments that greatly improved the paper.
Funding
This project was funded by Scuola Superiore Sant'Anna, Pisa, Italy (grant reference APOMIS11AZ). The Norway spruce genome project together with P. abies fosmids sequencing was originally supported by the Knut and Alice Wallenberg Foundation. Some computations were performed on resources provided by UPPNEX/SNIC through the Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) project b2010042.
Literature Cited
- Amborella Genome Project 2013. The Amborella genome and the evolution of flowering plants. Science 342:1241089. doi: 10.1126/science.1241089 [DOI] [PubMed] [Google Scholar]
- Ausin I, et al. 2016. DNA methylome of the 20-gigabase Norway spruce genome. Proc Natl Acad Sci U S A. 113(50):E8106–E8113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avramidou EV, Doulis AG, Aravanopoulos FA.. 2015. Determination of epigenetic inheritance, genetic inheritance, and estimation of genome DNA methylation in a full-sib family of Cupressus sempervirens L. Gene 562(2):180–187.http://dx.doi.org/10.1016/j.gene.2015.02.068 [DOI] [PubMed] [Google Scholar]
- Baucom RS, et al. 2009. Exceptional diversity, non-random distribution, and rapid evolution of retroelements in the B73 maize genome. PLoS Genet. 5(11):e1000732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MD, Leitch IJ.. 2012. Plant DNA C-values database (Release 6.0, Dec. 2012). Available from: http://data.kew.org/cvalues/. Last Accessed 20 April 2016.
- Birol I, et al. 2013. Assembling the 20 Gb white spruce (Picea glauca) genome from whole-genome shotgun sequencing data. Bioinformatics 29(12):1492–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucher E, Reinders J, Mirouze M.. 2012. Epigenetic control of transposon transcription and mobility in Arabidopsis. Curr Opin Plant Biol. 15(5):503–510.http://dx.doi.org/10.1016/j.pbi.2012.08.006 [DOI] [PubMed] [Google Scholar]
- Buschiazzo E, Ritland C, Bohlmann J, Ritland K.. 2012. Slow but not low: genomic comparisons reveals slower evolutionary rate and higher dN/dS in conifers compared to angiosperms. BMC Evol Biol. 12(1):8.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, et al. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casola C, Ganote CL, Hahn MW.. 2010. Nonallelic gene conversion in the genus Drosophila. Genetics 185(1):95–103.http://dx.doi.org/10.1534/genetics.110.115444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casola C, Conant GC, Hahn MW.. 2012. Very low rate of gene conversion in the yeast genome. Mol Biol Evol. 29(12):3817–3826.http://dx.doi.org/10.1093/molbev/mss192 [DOI] [PubMed] [Google Scholar]
- Chen J-M, Cooper DN, Chuzhanova N, Férec C, Patrinos GP.. 2007. Gene conversion: mechanisms, evolution and human disease. Nat Rev Genet. 8(10):762–775. [DOI] [PubMed] [Google Scholar]
- Chiolo I, et al. 2011. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 144(5):732–744.http://dx.doi.org/10.1016/j.cell.2011.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colomé-Tatché M, et al. 2012. Features of the Arabidopsis recombination landscape resulting from the combined loss of sequence variation and DNA methylation. Proc Natl Acad Sci U S A. 109(40):16240–16245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos KM, Brown JK, Bennetzen JL.. 2002. Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Res. 12(7):1075–1079.http://dx.doi.org/10.1101/gr.132102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooner HK, Martinez-Ferez IM.. 1997. Recombination occurs uniformly within the bronze gene, a meiotic recombination hotspot in the maize genome. Plant Cell 9(9):1633–1646.http://dx.doi.org/10.1105/tpc.9.9.1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin G. 2002. Characterization of the gene conversions between the multigene family members of the yeast genome. J Mol Evol. 55(1):14–23.http://dx.doi.org/10.1007/s00239-001-0085-y [DOI] [PubMed] [Google Scholar]
- Du J, et al. 2012. Evolutionary conservation, diversity and specificity of LTR-retrotransposons in flowering plants: insights from genome-wide analysis and multi-specific comparison. Plant J. 63:584–598. [DOI] [PubMed] [Google Scholar]
- Dubcovsky J, et al. 2001. Comparative sequence analysis of colinear barley and rice bacterial artificial chromosomes. Plant Physiol. 125(3):1342–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5):1792–1797.http://dx.doi.org/10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Baidouri M, Panaud O.. 2013. Comparative genomic paleontology across plant kingdom reveals the dynamics of TE-driven genome evolution. Genome Biol Evol. 5(5):954–965.http://dx.doi.org/10.1093/gbe/evt025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoroff NV. 2012. Presidential address. Transposable elements, epigenetics, and genome evolution. Science 338(6108):758–767.http://dx.doi.org/10.1126/science.338.6108.758 [DOI] [PubMed] [Google Scholar]
- Feschotte C, Jiang N, Wessler SR.. 2002. Plant transposable elements: where genetics meets genomics. Nat Rev Genet. 3(5):329–341.http://dx.doi.org/10.1038/nrg793 [DOI] [PubMed] [Google Scholar]
- Frahry MB, Sun C, Chong RA, Mueller RL.. 2015. Low levels of LTR retrotransposon deletion by ectopic recombination in the gigantic genomes of salamanders. J Mol Evol. 80(2):120–129.http://dx.doi.org/10.1007/s00239-014-9663-7 [DOI] [PubMed] [Google Scholar]
- Fu H, Dooner HK.. 2002. Intraspecific violation of genetic colinearity and its implications in maize. Proc Natl Acad Sci U S A. 99(14):9573–9578.http://dx.doi.org/10.1073/pnas.132259199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Niu B, Zhu Z, Wu S, Li W.. 2012. CD-HIT: accelerated for clustering the next generation sequencing data. Bioinformatics 28(23):3150–3152.http://dx.doi.org/10.1093/bioinformatics/bts565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, et al. 2014. Extensive and biased intergenomic nonreciprocal DNA exchanges shaped a nascent polyploid genome, Gossypium (cotton). Genetics 197(4):1153–1163.http://dx.doi.org/10.1534/genetics.114.166124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins HS, Proulx SR, Rapp RA, Wendel JF.. 2009. Rapid DNA loss as a counterbalance to genome expansion through retrotransposon proliferation in plants. Proc Natl Acad Sci U S A. 106(42):17811–17816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Brachypodium Initiative. 2010. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463:763–768.http://dx.doi.org/10.1038/nature08747 [DOI] [PubMed] [Google Scholar]
- International Rice Genome Sequencing Project. 2005. The map-based sequence of the rice genome. Nature 436(7052):793–800.http://dx.doi.org/10.1038/nature03895 [DOI] [PubMed] [Google Scholar]
- Jaillon O, et al. 2007. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449(7161):463–467. [DOI] [PubMed] [Google Scholar]
- Jurka J, et al. 2005. Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 110(1-4):462–467. [DOI] [PubMed] [Google Scholar]
- Kejnovsky E, et al. 2007. High intrachromosomal similarity of retrotransposon long terminal repeats: evidence for homogenization by gene conversion on plant sex chromosomes? Gene 390(1-2):92–97. [DOI] [PubMed] [Google Scholar]
- Kovach A, et al. 2010. The Pinus taeda genome is characterized by diverse and highly diverged repetitive sequences. BMC Genomics 11:420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 25(14):1754–1760.http://dx.doi.org/10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Jean M, Belzile F.. 2006. The impact of sequence divergence and DNA mismatch repair on homeologous recombination in Arabidopsis. Plant J. 45(6):908–916.http://dx.doi.org/10.1111/j.1365-313X.2006.02657.x [DOI] [PubMed] [Google Scholar]
- Lippman Z, et al. 2004. Role of transposable elements in heterochromatin and epigenetic control. Nature 430(6998):471–476.http://dx.doi.org/10.1038/nature02651 [DOI] [PubMed] [Google Scholar]
- Lu Y, Ran J-H, Guo D-M, Yang Z-Y, Wang X-Q.. 2014. Phylogeny and divergence times of gymnosperms inferred from single-copy nuclear genes. PLoS ONE 9(9):e107679.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Devos KM, Bennetzen JL.. 2004. Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 14(5):860–869.http://dx.doi.org/10.1101/gr.1466204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, SanMiguel P, Lai J, Messing J, Bennetzen JL.. 2005. DNA rearrangement in orthologous orp regions of the maize, rice and sorghum genomes. Genetics 170(3):1209–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macas J, et al. 2015. In-depth characterization of repetitive DNA in 23 plant genomes reveals sources of genome size varation in the legume tribe Fabeae. PLoS ONE 10(11):e0143424.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DE, et al. 2016. Whole-genome analysis of individual meiotic events in Drosophila melanogaster reveals that noncrossover gene conversions are insensitive to interference and the centromere effect. Genetics 203(1):159–171.http://dx.doi.org/10.1534/genetics.115.186486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirouze M, et al. 2012. Loss of DNA methylation affects the recombination landscape in Arabidopsis. Proc Natl Acad Sci U S A. 109(15):5880–5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondragon-Palomino M, Gaut BS.. 2005. Gene conversion and the evolution of three leucine-rich repeat gene families in Arabidopsis thaliana. Mol Biol Evol. 22(12):2444–2456.http://dx.doi.org/10.1093/molbev/msi241 [DOI] [PubMed] [Google Scholar]
- Natali L, Cossu RM, Mascagni F, Giordani T, Cavallini A.. 2015. A survey of Gypsy and Copia LTR-retrotransposon superfamilies and lineages and their distinct dynamics in the Populus trichocarpa (L.) genome. Tree Genet Genomes 11(5):107. [Google Scholar]
- Neale DB, et al. 2014. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 15(3):R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystedt B, et al. 2013. The Norway spruce genome sequence and conifer genome evolution. Nature 497(7451):579–584.http://dx.doi.org/10.1038/nature12211 [DOI] [PubMed] [Google Scholar]
- Paul R, et al. 2015. Repeat sequence characterization in sugar pine (Pinus lambertiana) and loblolly pine (Pinus taeda). Available from: http://pinegenome.org/pinerefseq/files/PAG2015_Paul_PineRefSeq.pdf, accessed 10 September 2016.
- Pereira V. 2004. Insertion bias and purifying selection of retrotransposons in the Arabidopsis thaliana genome. Genome Biol. 10:R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson CL. 2011. The ins and outs of heterochromatic DNA repair. Dev Cell 20(3):285–287.http://dx.doi.org/10.1016/j.devcel.2011.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM.. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26(6):841–842.http://dx.doi.org/10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sáez-Laguna E, et al. 2014. Epigenetic variability in the genetically uniform forest tree species Pinus pinea L. PloS ONE 9(8):e103145. doi: 10.1371/journal.pone.0103145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saladin B, et al. 2017. Fossils matter: improves estimates of divergence times in Pinus reveal older diversification. BMC Evol Biol. 17:95.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SanMiguel P, et al. 1996. Nested retrotransposons in the intergenic regions of the maize genome. Science 274(5288):765–768.http://dx.doi.org/10.1126/science.274.5288.765 [DOI] [PubMed] [Google Scholar]
- SanMiguel P, Gaut BS, Tikhonov A, Nakajima Y, Bennetzen JL.. 1998. The paleontology of intergene retrotransposons of maize. Nat Genet. 20(1):43–45. [DOI] [PubMed] [Google Scholar]
- Sawyer SA. 1999. GENECONV: a computer package for the statistical detection of gene conversion. Available from: http://www.math.wustl.edu/∼sawyer/geneconv/, accessed 10 September 2016.
- Schnable PS, et al. 2009. The B73 maize genome: complexity, diversity, and dynamics. Science 326(5956):1112–1115.http://dx.doi.org/10.1126/science.1178534 [DOI] [PubMed] [Google Scholar]
- Sharma A, Wolfgruber TK, Presting GG.. 2013. Tandem repeats derived from centromeric retrotransposons. BMC Genomics 14:142.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, et al. 2010. Widespread gene conversion in centromere cores. PLoS Biol. 8(3):e1000327.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer EL, Durbin R.. 1995. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene 167:1–10. [DOI] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P.. 2015. RepeatMasker Open-4.0. Available from: http://www.repeatmasker.org, accessed 10 September 2016.
- Stevens KA, et al. 2016. Sequence of the Sugar Pine megagenome. Genetics 204(4):1613–1626.http://dx.doi.org/10.1534/genetics.116.193227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundell D, et al. 2015. The plant genome integrative explorer resource: PlantGenIE.org. New Phytol. 208(4):1149–1156. doi: 10.1111/nph.13557 [DOI] [PubMed] [Google Scholar]
- Talbert PB, Henikoff S.. 2010. Centromeres convert but don’t cross. PLoS Biol. 8(3):e1000326.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson P, Filipski A, Kumar S.. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30(12):2725–2729. doi: 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombetta B, Fantini G, D'Atanasio E, Sellitto D, Cruciani F.. 2016. Evidence of extensive non-allelic gene conversion among LTR elements in the human genome. Sci Rep. 6:28710. doi: 10.1038/srep28710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuskan GA, et al. 2006. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313(5793):1596–1604.http://dx.doi.org/10.1126/science.1128691 [DOI] [PubMed] [Google Scholar]
- Vicient CM, et al. 1999. Retrotransposon BARE-1 and its role in genome evolution in the genus Hordeum. Plant Cell 11(9):1769–1784.http://dx.doi.org/10.1105/tpc.11.9.1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitte C, Panaud O.. 2003. Formation of solo-LTRs through unequal homologous recombination counterbalances amplifications of LTR retrotransposons in rice Oryza sativa L. Mol Biol Evol. 20(4):528–540.http://dx.doi.org/10.1093/molbev/msg055 [DOI] [PubMed] [Google Scholar]
- Vitte C, Panaud O, Quesneville H.. 2007. LTR retrotransposons in rice (Oryza sativa, L.): recent burst amplifications followed by rapid DNA loss. BMC Genomics 8(1):218..http://dx.doi.org/10.1186/1471-2164-8-218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallberg A, Glémin S, Webster MT.. 2015. Extreme recombination frequencies shape genome variation and evolution in the honeybee, Apis mellifera. PLoS Genet. 11(4):e1005189.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Liu J-S.. 2008. LTR retrotransposon landscape in Medicago truncatula: more rapid removal than in rice. BMC Genomics 9:382.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XY, Paterson AH.. 2011. Gene conversion in angiosperm genomes with an emphasis on genes duplicated by polyploidization. Genes (Basel) 2(1):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Tang H, Bowers JE, Paterson AH.. 2009. Comparative inference of illegitimate recombination between rice and sorghum duplicated genes produced by polyploidization. Genome Res. 19(6):1026–1032.http://dx.doi.org/10.1101/gr.087288.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegrzyn JL, et al. 2014. Unique features of the loblolly pine (Pinus taeda L.) megagenome revealed through sequence annotation. Genetics 196(3):891–909.http://dx.doi.org/10.1534/genetics.113.159996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, et al. 2007. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 8(12):973–982.http://dx.doi.org/10.1038/nrg2165 [DOI] [PubMed] [Google Scholar]
- Xu S, et al. 2008. Gene conversion in the rice genome. BMC Genomics 9:93.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelina NE, et al. 2015. DNA methylation epigenetically silences crossover hot spots and controls chromosomal domains of meiotic recombination in Arabidopsis. Gene Dev. 29(20):2183–2202.http://dx.doi.org/10.1101/gad.270876.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamudio N, et al. 2015. DNA methylation restrains transposons from adopting a chromatin signature permissive for meiotic recombination. Gene Dev. 29(12):1256–1270.http://dx.doi.org/10.1101/gad.257840.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou X-H, Yang Z, Doyle JJ, Ge S.. 2013. Multilocus estimation of divergence times and ancestral effective population sizes of Oryza species and implications for the rapid diversification of the genus. New Phytol. 198(4):1155–1164. [DOI] [PubMed] [Google Scholar]
- Zuccolo A, Scofield DG, De Paoli E, Morgante M.. 2015. The Ty1-copia LTR retroelement family PARTC is highly conserved in conifers over 200 MY of evolution. Gene 568(1):89–99.http://dx.doi.org/10.1016/j.gene.2015.05.028 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.