

Abstract
The subgenomic compositions of the octoploid (2n = 8× = 56) strawberry (Fragaria) species, including the economically important cultivated species Fragaria x ananassa, have been a topic of long-standing interest. Phylogenomic approaches utilizing next-generation sequencing technologies offer a new window into species relationships and the subgenomic compositions of polyploids. We have conducted a large-scale phylogenetic analysis of Fragaria (strawberry) species using the Fluidigm Access Array system and 454 sequencing platform. About 24 single-copy or low-copy nuclear genes distributed across the genome were amplified and sequenced from 96 genomic DNA samples representing 16 Fragaria species from diploid (2×) to decaploid (10×), including the most extensive sampling of octoploid taxa yet reported. Individual gene trees were constructed by different tree-building methods. Mosaic genomic structures of diploid Fragaria species consisting of sequences at different phylogenetic positions were observed. Our findings support the presence in octoploid species of genetic signatures from at least five diploid ancestors (F. vesca, F. iinumae, F. bucharica, F. viridis, and at least one additional allele contributor of unknown identity), and questions the extent to which distinct subgenomes are preserved over evolutionary time in the allopolyploid Fragaria species. In addition, our data support divergence between the two wild octoploid species, F. virginiana and F. chiloensis.
Keywords: polyploidy, evolution, amplicon sequencing, diploid progenitor
Introduction
Strawberry (Fragaria spp.) is among the many economically important fruit crops of the Rosaceae family (Hummer and Hancock 2009). According to Food and Agriculture Organization (FAO) of the United Nations, world production of strawberries reached 4.5 million tons (∼10 billion pounds) in 2012 (FAO STAT http://faostat.fao.org/site/567/DesktopDefault.aspx? PageID=567#ancor). Within the genus Fragaria, approximately 22 species have been identified (Folta and Davis 2006; Staudt 2008; Hummer and Hancock 2009). These species exist in five even-ploidy levels, ranging from diploid to decaploid. The modern cultivated strawberry, F. × ananassa, was derived from chance hybridization between representatives of its two progenitor octoploid species, F. chiloensis and F. virginiana in the mid-1700s (Hummer and Hancock 2009). As demonstrated by the recent employment of a reference genome from ancestral diploid F. vesca (Sargent et al. 2011; Shulaev et al. 2011) in the design and successful implementation of the first strawberry SNP array (Bassil and Davis et al. 2015), which has been adopted by many breeders, it is evident that increased knowledge of phylogenetic relationships, polyploid ancestries, and octoploid genome structure can open opportunities for further increasing the economic value of strawberry through marker-assisted breeding and other forms of genetic improvement.
Early studies on the origins of polyploid Fragaria species were based entirely or primarily on the observation of meiotic chromosome pairing. Fedorova (1946) proposed the first octoploid genomic composition model of AAAABBCC, where A, B, and C genome types might have been derived from tetraploid (AAAA) F. orientalis, diploid (BB) F. nipponica, and diploid (CC) F. vesca, respectively. A subsequent model by Senanayake and Bringhurst (1967) proposed the genomic formula AAA′A′BBBB, in which the A subgenome might have originated from F. vesca or F. viridis. On the basis of the accumulating observations of bivalent pairing (Byrne and Jelenkovic 1976), and genetic evidence from allozyme diversity and inheritance studies (Arulsekar et al. 1981), Bringhurst (1990) proposed a fully diploidized genome composition model: AAA′A′BBB′B′. This latter model implied the existence of two highly divergent subgenome types (A and A′ vs. B and B′), within which less divergent subgenome types (A vs. A′ and B vs. B′) were nested. Under this model, as many as four diploid sources may have each contributed two sets of chromosomes to the octoploid Fragaria × ananassa genome (Bringhurst 1990).
The first molecular analysis of phylogenetic relationships among Fragaria species was reported by Potter et al. (2000) using DNA sequence data from nuclear rDNA-ITS loci and the chloroplast trnL gene from 14 diploid and polyploid species, notably not including the unavailable diploids F. mandshurica and F. iinumae. Both ITS and trnL data supported the hypothesis that, among the studied diploids, F. vesca and F. bucharica (accessions formerly identified as F. nubicola: Folta and Davis 2006; Staudt 2006) displayed the closest relationship to the studied octoploids. However, rDNA sequences are problematic in polyploids because of their low levels of informative variants as mediated by concerted evolution (Wendel et al. 1995), which may thus preclude identification of more than one diploid ancestor of an allopolyploid on the basis of ITS data (Bailey et al. 2003). Later, a mitochondrial DNA sequence analysis identified a shared marker between F. iinumae and Fragaria octoploids, suggesting that F. iinumae may be the source of the octoploids’ mitochondrial genome (Mahoney et al. 2010). A recent study (Njuguna et al. 2013) using characters extracted from whole chloroplast genome sequences resolved F. vesca as the likely chloroplast genome donor to the octoploid species and to the decaploid species F. iturupensis. Although organelle genome ancestries were successfully traced, data from organelle genomes cannot provide the full picture of reticulate species phylogenies due to the typically uni-parental modes of organelle inheritance (Small et al. 2004), as confirmed in Fragaria for chloroplast (Davis et al. 2010) and mitochondrial (Mahoney et al. 2010) genomes.
To overcome these barriers to reticulate phylogenetic reconstruction, low-copy nuclear genes (LCNGs), which are normally considered as genes present in no more than four copies, or ideally as a single copy, per genome (Duarte et al. 2010) have been extensively used (Zimmer and Wen 2013; Tonnabel et al. 2014). LCNGs, when they are shared by different species, are more likely to be orthologous than are higher copy nuclear genes, most copies of which are necessarily related as paralogs. Rousseau-Gueutin et al. (2009) studied the sequences from two Fragaria LCNGs: GBSS1-2 and DHAR. Their results led to two alternatives, octoploid genomic composition hypotheses: Y1Y1Y1Y1ZZZZ, or Y1′Y1′Y1″Y1″ZZZZ, where Y1, Y1′, and Y1″ correspond to a genome or genomes related to F. vesca and/or F. mandshurica, whereas Z represents a genome related to F. iinumae. The phylogenetic tree inferred from the LCNG ADH2 (DiMeglio et al. 2014) was consistent with those of Rousseau-Gueutin et al. (2009) in revealing allele contribution to the octoploids by F. vesca and/or F. mandshurica, and also by F. iinumae. The study of Lundberg et al. (2011) was based on the data from an intragenic region between the genes RGA1 (Resistance Gene Analogue 1) and Subt (Subtilase). Their analysis suggested a possible contributory role of F. viridis to the octoploid lineage by way of the hexaploid intermediate, F. moschata.
Despite the progress reviewed earlier, only a small number of genomic loci were studied, taxon sampling was shallow, and discrepancies among the conclusions of previous studies require further clarification through broader sampling of phylogenetic informative loci and taxa. The development of next-generation sequencing technologies has provided promising solutions to generate sequencing data from multiple loci per plant sample. In the study of (Tennessen et al. 2014), thousands of genome-wide markers were obtained by target capture sequencing to provide an illuminating phylogenomic perspective. However, their taxon sampling was still very limited. An alternative technology is microfluidic PCR, where thousands of PCR amplifications are processed simultaneously in droplets before being pooled for barcoding and multiplexed sequencing (McCormack et al. 2013). Compared with other technologies, such as restriction digest-based methods (McCormack et al. 2013) and targeted sequence capture (Tennessen et al. 2014), microfluidic PCR can produce longer reads for LCNG-based amplicons from more samples.
In the present study, a bioinformatics pipeline was developed to identify multiple LCNGs, which were then used to investigate the phylogeny of Fragaria on a genome-wide scale, with emphasis on deep sampling of the octoploid taxa. Amplicon sequencing data were generated with the Fluidigm Access Array system in conjunction with the 454 sequencing platform. This microfluid PCR approach has been successfully applied in a previous phylogenetic study involving diploids and tetraploids (Richardson et al. 2012); but the present study constitutes its first use for higher ploidy levels involving a diversity of species. By employing the most extensive taxon sampling of Fragaria species to date, this study aimed to systematically survey the phylogenetic relationships of Fragaria species and to contribute increased insight into the diploid ancestries and the contemporary subgenomic compositions of the octoploid species.
Materials and Methods
Plant Materials and DNA Isolation
The studied Fragaria samples included 33 diploids representing eight species, one representative each of three tetraploid species, two representatives of hexaploid F. moschata, one representative each of two decaploids, one Fragaria sample of unknown ploidy, and 45 octoploids, including 14 representatives of F. virginiana, 12 of F. chiloensis, and 19 F. x ananassa cultivars (table 1). Six different species from the genera Potentilla, Drymocallis, Comarum, and Dasiphora, were represented as outgroups (table 1). Fragaria accessions within species were selected based on their collection sites to represent broad geographic distribution. Additionally, combined samples were constructed by mixing genome DNA from two or four different diploid species in specified ratios. Among them, a 2-way mix (sample ID: 2 equal mix) was made from DNA of F. vesca subsp. bracteata BC30 and F. iinumae FRA377 in a 1:1 ratio. Two replicates of a 4-way mix were made from DNA of BC30, FRA377, F. nilgerrensis FRA1358, and F. viridis FRA333 in a 1:1:1:1 ratio and were named as 4-equal-mix-a and 4-equal-mix-b. Another 4-way mix (sample ID: Unequal mix) was made from DNA of BC30, FRA377, FRA1358, and FRA333 at the ratio of 3: 1: 1: 1. These mixtures served as synthetic tetraploids and octoploids with known allelic constitutions, providing opportunity to test whether all alleles known to be present in a synthetic polyploid could in fact be detected. Genomic DNA was extracted from young, partially expanded leaves using a CTAB mini-prep protocol patterned after (Torres et al. 1993).
Table 1.
List of Plant Samples Included in This Study
| Taxon | Ploidy Level | Collection Site | Local Name | NCGR PI |
|---|---|---|---|---|
| Fragaria bucharica | 2× | Tajikistan | FRA1910.001 | 651569 |
| F. iinumae | 2× | Japan | FRA377.001 | 551751 |
| F. species | 2× | Japan | J1 | |
| F. iinumae | 2× | Japan | J4A(FRA1849.000) | 637963 |
| F. iinumae | 2× | Japan | J17(1855.000) | 637969 |
| F. mandschurica | 2× | Unknown | FME | |
| F. mandschurica | 2× | Mongolia | GS99-2D (FRA1947.001) | 657855 |
| F. mandschurica | 2× | Mongolia | GS99-C | |
| F. nilgerrensis | 2× | Yunnan, China | FRA1358.001 | 616672 |
| F. bucharica | 2× | Pakistan | FRA520.001 | 551851 |
| F. vesca | 2× | California, USA | HP6A | |
| F. species | 2× | Unknown | TMD_227D | |
| F. vesca | 2× | California, USA | DN3C | |
| F. vesca | 2× | California, USA | H1B | |
| F. vesca | 2× | Oregon, USA | S192-3 | |
| F. vesca | 2× | California, USA | U2A | |
| F. vesca | 2× | California, USA | TMD2(FRA1990.001) | 660765 |
| F. species | 2× | BC, Canada | BC5(FRA1988.001) | 660763 |
| F. vesca subsp. bracteata | 2× | BC, Canada | BC30(FRA1989.001) | 660764 |
| F. vesca subsp.vesca | 2× | Finland | FRA438.001 | 551792 |
| F. vesca subsp. vesca | 2× | Europe | FRA480 | 551827 |
| F. vesca subsp. vesca | 2× | Siberia | NOV 1C | |
| F. vesca subsp.vesca | 2× | Hawaii, USA | H4(FRA197.001) | 551572 |
| F. vesca subsp.californica | 2× | California, USA | FRA371.001 | 551749 |
| F. vesca subsp.americana | 2× | New Hampshire, USA | Pawt(FRA1948.001) | 657856 |
| F. vesca subsp.americana | 2× | New Hampshire, USA | WC6 | |
| F. species | 2× | Oregon, USA | FRA2001.002 | 658453 |
| F. vesca × F.viridis | 2× | Unknown | FRA364.002 | 551744 |
| F. viridis | 2× | Germany | FRA333.001 | 551741 |
| F. viridis | 2× | Unknown | GS91 | |
| F. viridis | 2× | Siberia | NOV 3A | |
| F. nipponica | 2× | Japan | J26(FRA1863.000 | 637976 |
| F. chinensis | 2× | Hebei, China | FRA202.001 | 551576 |
| F. corymbosa | 4× | Jilin, China | FRA1612.001 | 602942 |
| F. orientalis | 4× | Primorye, Russia | FRA1803.001 | 637934 |
| F. orientalis | 4× | Primorye, Russia | FRA1809.001 | 637940 |
| F. moschata | 6× | Europe | FRA157.001 | 551550 |
| F. moschata | 6× | Germany | FRA376.00# | 551741 |
| F. virginiana | 8× | Alaska, USA | PL1 | |
| F. virginiana | 8× | Colorado, USA | TMD227F | |
| F. virginiana | 8× | Alaska, USA | FM1 | |
| F. virginiana subsp. Grayana | 8× | Mississippi, USA | FRA1414.001 | 612569 |
| F. virginiana subsp. Glauca | 8× | BC, Canada | BC12 | |
| F. virginiana subsp. Glauca | 8× | BC, Canada | FRA1992.001 | 660767 |
| F. virginiana subsp. Glauca | 8× | Montana, USA | FRA1697.001 | 612495 |
| F. virginiana subsp. virginiana | 8× | Ont., Canada | FRA1699.001 | 612497 |
| F. virginiana subsp. virginiana | 8× | New Hampshire, USA | FRA1994.001 | 660769 |
| F. virginiana subsp. virginiana | 8× | New Hampshire, USA | FRA1995.001 | 660770 |
| F. virginiana subsp. virginiana | 8× | Maryland, USA | FRA67.001 | 452436 |
| F. virginiana subsp. virginiana | 8× | Unknown | BC Pink | |
| F. virginiana subsp.platypetala | 8× | California, USA | FRA58.002 | 551471 |
| F. virginiana subsp.platypetala | 8× | Oregon, USA | FRA1960.001 | 657868 |
| F. chiloensis subsp. lucida | 8× | Oregon, USA | FRA1691.001 | 612489 |
| F. chiloensis subsp.lucida | 8× | California, USA | FRA366.001 | 551734 |
| F. chiloensis subsp.lucida | 8× | BC, Canada | FRA34.002 | 551445 |
| F. chiloensis subsp.pacifica | 8× | California, USA | FRA357.002 | 551728 |
| F. chiloensis subsp.pacifica | 8× | Alaska, USA | FRA368.002 | 551735 |
| F. chiloensis subsp.pacifica | 8× | California, USA | FRA1692.001 | 612490 |
| F. chiloensis subsp.patagonica | 8× | Chile | FRA1088.002 | 612316 |
| F. chiloensis subsp.patagonica | 8× | Chile | FRA1092.002 | 612317 |
| F. chiloensis supsp.patagonica | 8× | Chile | FRA1100.002 | 602568 |
| F. chiloensis subsp.patagonica | 8× | Chile | FRA796.001 | 552091 |
| F. chiloensis subsp.chiloensis | 8× | Chile | FRA1108.002 | 602570 |
| F. chiloensis subsp.chiloensis | 8× | Chile | FRA743.001 | 552038 |
| F. × ananassa | 8× | California, USA | Albion | |
| F. × ananassa | 8× | Oregon, USA | Bountiful | 551855 |
| F. × ananassa | 8× | UK | EMR21 | |
| F. × ananassa | 8× | California, USA | Ca65.65-601 | |
| F. × ananassa | 8× | Maryland, USA | Earliglow | 551394 |
| F. × ananassa | 8× | France | Darselect | |
| F. × ananassa | 8× | Unknown | Cavendish | 616560 |
| F. × ananassa | 8× | Florida, USA | Florida_Belle | 551396 |
| F. × ananassa | 8× | Japan | Hogyoku | 616622 |
| F. × ananassa | 8× | New York, USA | Holiday | 551653 |
| F. × ananassa | 8× | New York, USA | Jewel | 551927 |
| F. × ananassa | 8× | Netherlands | Korona | |
| F. × ananassa | 8× | Maryland, USA | Lateglow | 551830 |
| F. × ananassa | 8× | California, USA | Seascape | 660779 |
| F. × ananassa | 8× | BC, Canada | Totem | 551501 |
| F. × ananassa | 8× | Florida, USA | Sweet_Charlie | |
| F. × ananassa | 8× | Oregon, USA | Valley_Red | |
| F. × ananassa | 8× | Maryland, USA | Tribute | 551953 |
| F. × ananassa | 8× | Unknown | Del_Norte | |
| F. cascadensis | 10× | Oregon, USA | FRA110.001 | |
| F. iturupensis | 10× | Sakhalin, Russia | FRA1841.013 | |
| F. species | ? × | Alaska, USA | F192 | |
| Drymocallis species | ? × | Colorado, USA | TMD223 | |
| P. nepalensis | ? × | Unknown | A436-993 | |
| P. recta | ? × | Unknown | Ben | |
| Dasiphora fruticosa | ? × | Unknown | PF | |
| Comarum palustris | ? × | Unknown | P.palustris | |
| Drymocallis glandulosa | 2× | Oregon, USA | S192D |
Gene Identification Pipeline
A bioinformatics pipeline was developed to search for candidate LCNGs and to design primers (fig. 1). The first step was to eliminate putative pseudogenes. Using the reference sequence version 1.1 of F. vesca “Hawaii 4” (FvH4) in FASTA and GFF3 formats as downloaded from the GDR database (https://www.rosaceae.org/organism/Fragaria/vesca; last accessed October 20, 2017), BLAST analyses of transcript sequences of all 31,213 predicted genes against a local cDNA database of sequences downloaded from NCBI were performed. This local database included cDNA sequences from Triticum, Fabaceae, Brassicales, Zea, Rosaceae, Oryza, Salicaceae, and Vitaceae. At the end of this step, any gene sequence longer than 900 bp and with 50% of transcript length aligned by a known cDNA sequence in the BLAST database was retained as a valid candidate gene. Then the full-length sequence and annotation of every candidate gene was retrieved from a local MySQL database for the following analyses.
Fig. 1.
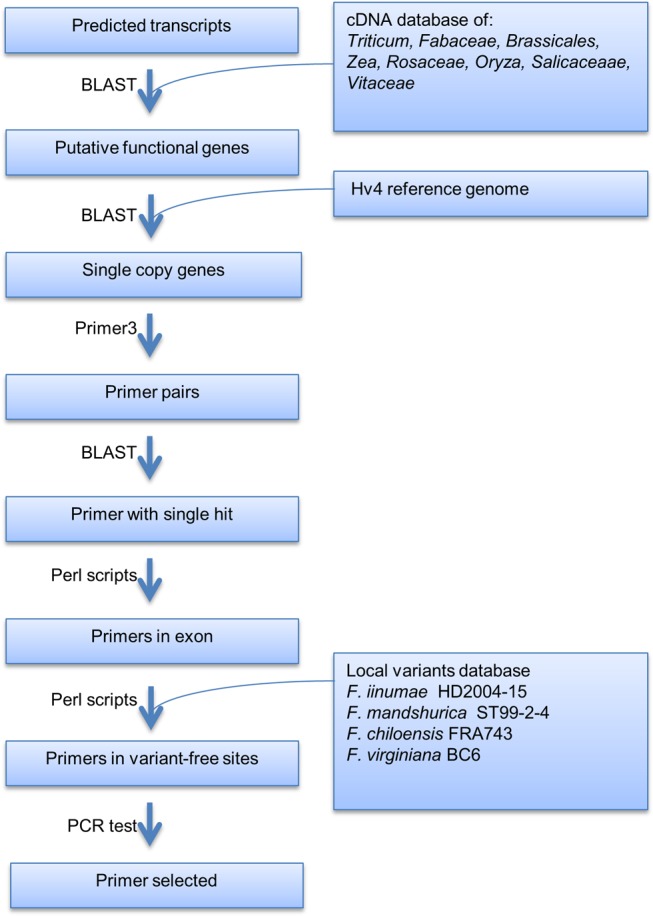
—Bioinformatic pipeline to identify target loci and design primers.
To identify LCNGs, potential single copy genes were detected by performing BLAST analyses of full-length sequences of candidate genes against the FvH4 v1.1 reference genome. The criteria were set as the following: the number of hits was <4, the e-value of the best hit was lower than 1e-15, and if a second-best hit existed, the second e-value was >5 times the first e-value, and the bit score of the first hit was >6 times that of the second-best hit score.
To identify potential variants within primer sites, where such variants could affect primer annealing to the template DNA and reduce the successful rate of PCR, Illumina sequencing data from a group of taxa [F. iinumae HD2004-15 (NCGR PI 637963), F. mandshurica GS99-2-4 (PI 657855), F. chiloensis FRA743 (PI 552038), and F. virginiana BC6 (PI 660767)] (data obtained from Bassil and Davis et al. 2015) were used. Sequencing protocols, read mapping, and variant detection were as described in (Bassil and Davis et al. 2015). Variant information was stored in a MySQL database for subsequent analyses.
For each gene that had passed the previous filters, 10 primer pairs were designed using Primer3 v2.3.4 (Untergasser et al. 2012), PCR product size was set as between 900 and 1,200 bp. The exact coordinates and numbers of hits on the reference genome of every primer sequence were determined by performing local BLAST against the FvH4 v1.1 reference genome. Primers with single hits were screened with the following parameters and requirements: the number of hits with e-value <0.5 were ≤3, the e-value of the best hit was less than 1e-15. If present, the e-value of the next best hit was >5 times the first e-value, and the bit score was less than one-sixth of the first bit score. By searching against the local database of variants, primers with any single variant in the primer site were removed. Finally, 40 target genes were selected for subsequent PCR test with the aim of achieving an even distribution among the seven pseudochromosomes of the FvH4 v1.1 assembly, and arbitrary decisions were made if multiple loci met the above criteria.
Target Amplification and Sequencing
Candidate primer pairs and all DNA templates were first evaluated by performing at least one individual PCR to validate the PCR product size and PCR profile. PCR amplifications were performed in 8 μl reactions using 1 μl 10× Buffer solution, 5% DMSO, 62.5 μM each dNTP, 0.5 unit Faststart Roche polymerase, 0.5 μl loading reagent, 200 ng template DNA, and 4 μM each primer. DMSO and loading reagent were provided by Fluidigm. The PCR protocol was based on the Access Array protocol (Fluidigm Corporation, South San Francisco, CA) with the following modifications: the first 94 °C incubation was 4 min; annealing temperature is 58 °C; time for 72 °C extension was 1.5 min; the first 3-step cycle was repeated 13 times. Products were visualized on 1% agarose TBE gels stained with ethidium bromide.
Based upon their reliability in PCR evaluations, 24 primer pairs (one for each target gene) and 96 DNA templates were eventually chosen for testing on the Access Array IFC, which was performed by MOGene (MOGene, LC, St. Louis, MO), for a total of 2,304 gene site × accession combinations. When these primers were synthesized, a universal forward (CS1-ACACTGACGACATGGTTCTACA) or reverse (CS2-TACGGTAGCAGAGACTTGGTCT) tag was added to the end of each forward or reverse primer, respectively, according to Fluidigm Access Array barcode library construction (www.fluidigm.com; last accessed October 20, 2017). Information about target genes and primer sequences is provided in table 2. During the PCR on the Access Array IFC, unique barcodes and 454 sequence adapter A (CGTATCGCCTCCCTCGCG CCATCAG) and B (CTATGCGCCTTGCCAGCCCGCTCAG) were added to the PCR products to identify each individual sample. PCR products were then collected and distributed on two 454 pico titer plate (PTP) regions identified by adapter A and B. Sequencing that was initiated with these adapters represented two ends of each amplicon.
Table 2.
List of Target Gene Primer Pairs
| Gene Name | Linkage GROUP | Loci Starta | Loci Endb | PCR Product Size | Forward Data set | Reverse Data set | Right Primer | Left Primer |
|---|---|---|---|---|---|---|---|---|
| G14746 | LG1 | 8647737 | 8648873 | 1,137 | R5 | R2 | AAGAGGAACATTGTGGTGGC | GGTGTCCTGCAAAACCAACT |
| G14770 | LG1 | 8746622 | 8747585 | 963 | R2 | R5 | TTGAGCACCACATCAAGCTC | GGCGGAGGAAAGATGATACA |
| G31441 | LG1 | 13856068 | 13856967 | 900 | R5 | R2 | GGAGGCGATATCAGGATTCA | CTGGAGCTGGTGACATGCTA |
| G20570 | LG1 | 20140186 | 20141100 | 914 | R2 | R5 | AGCAAATGACTCCCACATCC | GATTGGTACTCCGGCAAAGA |
| G31901 | LG2 | 4507467 | 4508621 | 1,155 | R2 | R5 | GCATGAAGGATGAAGCCATT | AATCGGATGATTCAGCTTGG |
| G08197 | LG2 | 12307791 | 12308726 | 936 | R2 | R5 | ATGCTGCTCTTGATTTGCCT | GAGGGAACCGATGTACGAGA |
| G08827 | LG2 | 20397775 | 20398801 | 1,027 | R5 | R2 | GCCCATATCCAAGAAAAGCA | ATGGCGTCTTTATCGGTCAC |
| G03299 | LG3 | 12234001 | 12234914 | 914 | R2 | R5 | ATGCCATTCGATCCATGACT | GCTCAGTTAGCAAACTTAAATGGA |
| G07945 | LG3 | 22910969 | 22912053 | 1,085 | R2 | R5 | AACATACTGGGGAGCTGTGG | CCAGCAATTTCCTTCACCAT |
| G20659 | LG3 | 30313242 | 30314344 | 1,102 | R5 | R2 | TCATGCTGCTTTGGTTCAAG | GATTCTGTCCGGATTGGAGA |
| G09999 | LG4 | 13686356 | 13687321 | 966 | R2 | R5 | CTTCTCAGTCCGGCAGAAAC | CTGAAATCATTGCCACATCG |
| G06709 | LG4 | 18602603 | 18603800 | 1,197 | R2 | R5 | TCCTCCTCAAGTCCCATCAC | CGCTTCCCATCTCTGACTTC |
| G03631 | LG4 | 24620053 | 24621159 | 1,107 | R2 | R5 | CCAACAAGCACACTCTCCAA | CCGTCAACATCACAAACGTC |
| G32075 | LG5 | 2660085 | 2661153 | 1,068 | R5 | R2 | TCTCAACCCCAACACAATGA | CCGAACCCACCACTAAGAAA |
| G08977 | LG5 | 9313007 | 9313977 | 971 | R2 | R5 | ATCATCATCTTCTGGGGCAG | GCAATCGAGGAGGTCAACAT |
| G31464 | LG5 | 19914899 | 19915899 | 1,001 | R2 | R5 | CTGGGTCGTCAAGCTTCTTC | CACGAACATCCACCACAGTC |
| G16711 | LG6 | 993574 | 994689 | 1,115 | R5 | R2 | GCTGCACAATGAGCCTGTTA | AACGGAGCCCTTGTCCTTAT |
| G00282 | LG6 | 3630541 | 3631733 | 1,193 | R5 | R2 | CAACCACAAAAATGAGCCCT | ACAAGCTCAAGCTCGGAGAG |
| G17793 | LG6 | 21004619 | 21005615 | 997 | R5 | R2 | AAGGACTTGCCTGTGCAGTT | TTGGAAAAACTTGCATGCTG |
| G25734 | LG6 | 25276790 | 25277942 | 1,153 | R5 | R2 | TCCTGGGATACCTGTGAAGC | GGTCACAACACTGGTCGATG |
| G23870 | LG6 | 35148747 | 35149771 | 1,025 | R5 | R2 | TGGTGTGGCATTGCACTATT | CACTTTGGAGGCTTGCTAGG |
| G26957 | LG7 | 5722825 | 5723880 | 1,056 | R5 | R2 | GATTGGAGGGCGTGAGATAA | CCTTGTTGACGCGAATTTTT |
| G23405 | LG7 | 13532248 | 13533315 | 1,068 | R5 | R2 | ATTGGGGATGACTTGAACCA | CTCTTTGGGCATGGTGCTAT |
| G12770 | LG7 | 20093279 | 20094389 | 1,111 | R2 | R5 | AACCCAAGATTAACAGGGGC | ACCAGACCAAAGATTGCTGG |
a,bCoordinate on the FvH4 v1.1 reference genome.
Sequence Quality Control
When the first sequencing run from adapter A produced a very low number of reads (data set R1), a repeated run was conducted to generate the data set R4. These two data sets were combined throughout the following analyses, and were thereafter named as R5. The data set from adapter B was named as R2. Raw data files in SFF (standard flowgram file) format generated from the 454 sequencing machine were demultiplexed into separate FASTQ files for each DNA sample using the sffinfo tool obtained from Roche, and were uploaded to the NCBI SRA (Bioproject Accession PRJNA314268). All 454 reads were trimmed and filtered using FlowClus (Gaspar 2014) with the following settings: a constant value of 0.5 was specified to call bases from a range of flow values, minimum sequence length was set to 200 bp, no more than two ambiguous bases were allowed in a read and a minimum of two mismatches to the primer sequence were allowed for a read before being trimmed, the length of the sliding window used to calculate average quality scores was 50 bp, and the minimum average quality score of sliding windows was 20. Sequences from each PCR surviving the above filters were grouped into clusters by FlowClus based on their identities, and the longest sequence was extracted from each cluster as the representative sequence. The number of sequences in a cluster was indicated in the header of the respective consensus sequence. Consensus sequences were input to UCHIME v4.2.4 (Edgar et al. 2011) to detect and remove PCR recombinants. For UCHIME parameters, the weight of “no” vote was set at 3, the minimum divergence between the query and the most abundant sequence was 0.2, the minimum number of different nucleotides in a segment was 2, and the minimum score was 0.18.
Phylogenetic Analysis
Because most reads sequenced from the two ends of each amplicon did not overlap, the phase of these reads could not be determined and they could not be coupled as read pairs. Thus, reads that passed quality control and with cluster size of three or higher were collected into 48 individual FASTA files, one for each combination of target gene and PTP data set [R5 or R2]. Thus, the two sequenced ends of each gene site were treated as separate loci and were used individually for phylogenetic reconstruction.
Sequences in each FASTA file were subjected to two rounds of alignment using MAFFT v7.221 (Katoh and Standley 2013). After the first round of alignment, poorly aligned positions were either fixed by eye or eliminated, and sequences were trimmed at the 3′ end to allow most of the sequences to be equal in length. After the final alignment, JModeltest (Darriba et al. 2012) was used to select for the best nucleotide substation model (supplementary table S1, Supplementary Material online). Multiple sequence alignment files were then converted into the MrBayes compatible NEXUS format using FastaConvert (Hall 2004). Bayesian analysis was performed using the settings of two independent runs with four chains, the default priors, sampling every 100 generations, and calculating the convergence diagnostic every 1,000 generations. The temperature for heating the chains was 0.2. Convergence of the runs was assessed by exploring the average standard deviation of split frequencies and the potential scale reduction factor (PSRF). The analysis was terminated when the average standard deviation of split frequency was <0.01, or when PSRF was close to 1.000, or after 15,000,000 generations (meaning they would not likely to reach convergence even if given more time). A burn-in of 25% (discarded the first 25% of samples) was used before summarizing the saved trees. The phylogenetic tree from each locus was viewed using Figuretree v1.4.0. (Rambaut 2009). Data matrixes from several loci (indicated in supplementary table S1, Supplementary Material online) that did not reach convergence in Bayesian analyses were then analyzed using Maximum likelihood (ML) through MEGA platform (Tamura et al. 2013) to reconstruct phylogenetic trees. For ML analyses, the HKY substitution model was used with gamma distributed rates among sites. 500 bootstrap replications were made. Gaps or missing data were partially deleted between pairwise sequence comparisons, and all other parameters were set as default. Each individual tree was rooted with the clade containing the most alleles from outgroup species (data matrices and trees are available at http://purl.org/phylo/treebase/phylows/study/TB2: S18992 or upon request).
Results
Data Quality
The numbers of reads returned after sequencing was 352,841 from the R5 data set and 372,688 from the R2 data set. After quality control, 120,192 sequences from the R5 data set and 282,944 sequences from the R2 data set remained for subsequent analyses. Given the large number of samples from diverse genetic backgrounds, a nonuniform level of read coverage for all 2,304 gene sites × accession combinations was anticipated. The distribution pattern of read depth among genes was similar between the R5 and R2 data sets. The two genes represented by the fewest total reads in the combined R5 and R2 data sets were genes G25734 and G06709, with 235 and 623, reads, respectively. All other genes were represented by at least 1,813 total reads, with the highest read total of 48,024 occurring in gene G00282. Genes G00282, G20570, G31441, and G03299 ranked as the four genes having the highest read depths within each of the R5 and R2 data sets (supplementary tables S2 and S3, Supplementary Material online). The R5 and R2 data sets displayed distinct distribution patterns of reads across plant samples. For example, the F. iinumae accession J4 had 9,802 sequences that passed quality control in the R2 data set but only had 1,383 sequences that passed in the R5 data set. Another interesting observation was that substantially lower numbers of reads were generated from gene site G00282 in both R5 and R2 data sets for F. vesca accessions than for other diploid species, such as F. viridis. The average numbers of gene site G00282 reads per F. vesca accession were 3.9 (eight accessions) in the R5 data set and 15.5 (11 accessions) in the R2 data set, whereas the average numbers of reads in the three F. viridis accessions were 798.7 and 1,725 for the R5 and R2 data sets, respectively.
A major concern was whether octoploid plants were represented by sufficient reads for each gene. For octoploid strawberries, including wild species and cultivars, the average read depth per gene × accession combination after data quality control was 41.6 in the R5 data set and 130.6 in the R2 data set. If a minimum of 64 reads were required to be able to sample all homoeologue alleles as adopted by (Rousseau-Gueutin et al. 2009), there were 188 and 450 gene × accession combinations that reached this threshold in the R5 and R2 data sets, respectively. Combining them together, 455 gene × accession combinations from 22 genes and 43 octoploid plant accessions had more than 64 quality filtered reads in at least one sequencing direction. Therefore, the read depths were sufficient to enable representative allele sampling for 22 genes in at least one sequencing direction.
Selection of a Subset of Phylogenetic Trees
Out of a total of 48 sequence data matrices, two matrices: G06709-R5 and G25734-R5 were eliminated from further consideration on the basis of low read depth. Thus, 46 phylogenetic trees could be reconstructed with the BI and/or the ML approach (supplementary figs. S1–S46, Supplementary Material online). These phylogenetic trees were not equally informative; instead they showed varied levels of resolution of the relationships among major taxonomic groups.
Since the allele composition of synthetic polyploid samples could be predicted based upon the alleles that were recovered from the individual contributing diploids, sequences from mixtures were expected to be easily distinguishable from each other and to cluster with sequences of their respective diploid contributors. The source of an allele would be uncertain if it resided in a polytomous clade containing more than one possible diploid contributor. The identification of sequences from two or more species in a mixture not only indicated the high possibility of sufficient data being obtained from different plant species but also suggested that such trees had a level of informativeness that was at least sufficient to resolve real differences among alleles despite any artifacts. The contributing diploids that could be recovered from the synthetic polyploid samples among all 46 phylogenetic trees were summarized in supplementary table S4, Supplementary Material online. Accordingly, a subset of 24 trees was selected for the subsequent analyses (table 3). Those trees recovered at least two different contributory species from among four synthetic polyploid samples, and resolved the phylogenetic relationships between at least two Fragaria species. An association between total read depth and tree informativeness was apparent (supplementary tables S2 and S3, Supplementary Material online). In the R2 data set, 12 out of 14 trees of intermediate total read depth (between 1,000 and 4,000) were deemed informative, in contrast to only one out of ten trees with read depths outside this range. In the R5 data set, 11 out of 18 trees of intermediate total read depth (between 3,000 and 16,000) were deemed informative, in contrast to only one out of six trees with read depths outside this range. Six of the eight highest read counts came from data sets that yielded rejected trees.
Table 3.
Summary of the Most Closely Related Diploid Species of Polyploid Species
| LG | Gene | Data set | Fragaria corymbosa | F. moschata | F. virginiana | F. chiloensis | F. × ananassa | F. cascadensis | F. iturupensis |
|---|---|---|---|---|---|---|---|---|---|
| 1 | G14746 | R2 | NA | F. vesca | F. vesca | F. vesca | F. vesca | NA | Unresolved |
| 1 | G14770 | R2 | NA | Unresolved | F. vesca | Unresolved | Unresolved | NA | F. iinumae |
| 1 | G31441 | R5 | F. viridis, F. chinensis | Unresolved | Unresolved | Unresolved | F. vesca | Unresolved | F. iinumae |
| 2 | G08197 | R2 | NA | F. vesca, F.mandshurica, F. viridis | F. vesca, F. viridis, | F. vesca | F. vesca | NA | F. vesca |
| 2 | G08197 | R5 | NA | F. vesca, F. viridis | F. vesca, F. viridis | F. viridis | F. viridis | NA | NA |
| 2 | G08827 | R5 | NA | F. viridis | Unresolved | Unresolved | Unresolved | NA | Unresolved |
| 2 | G31901 | R2 | NA | F. vesca | F. iinumae | F. iinumae | F. iinumae | NA | NA |
| 3 | G07945 | R5 | NA | F. viridis | Unresolved | Unresolved | Unresolved | NA | F. iinumae |
| 3 | G20659 | R2 | NA | Unresolved | F. vesca | F. vesca | F. vesca | NA | NA |
| 4 | G03631 | R5 | NA | NA | F. vesca | F. vesca | F. vesca | NA | NA |
| 4 | G03631 | R2 | NA | F. bucharica | F. iinumae | F. iinumae | F. iinumae | NA | F. iinumae |
| 4 | G09999 | R5 | NA | F. vesca, F. viridis | F. vesca | F. vesca, F. iinumae | F. vesca, F. iinumae | NA | F. iinumae |
| 5 | G08977 | R5 | Unresolved | F. viridis | Unresolved | Unresolved | Unresolved | Unresolved | NA |
| 5 | G31464 | R5 | NA | Unresolved | Unresolved | Unresolved | Unresolved | NA | NA |
| 5 | G32075 | R2 | NA | F. vesca, F. viridis | F. vesca, F. iinumae | F. vesca, F. iinumae | F. vesca, F. iinumae | NA | F. iinumae |
| 5 | G32075 | R5 | NA | NA | F. iinumae | F. vesca | F. vesca | NA | NA |
| 6 | G16711 | R5 | NA | Unresolved | F. bucharica | F. bucharica | F. bucharica | NA | NA |
| 6 | G16711 | R2 | NA | Unresolved | F. iinumae | F. iinumae | F. iinumae | NA | F. iinumae |
| 6 | G17793 | R2 | Unresolved | NA | F. iinumae | F. iinumae | F. iinumae | NA | F. iinumae |
| 6 | G23870 | R5 | NA | F. viridis | F. iinumae, F. viridis, | F. iinumae, F. viridis | F. iinumae, F. viridis | NA | NA |
| 6 | G23870 | R2 | NA | Unresolved | F. iinumae | F. iinumae | F. iinumae | NA | F. iinumae |
| 7 | G12770 | R2 | Unresolved | F. vesca, F. mandshurica | Unresolved | Unresolved | Unresolved | NA | F. iinumae |
| 7 | G26957 | R2 | NA | F. vesca | F. vesca, F. iinumae | F. vesca, F. iinumae | F. vesca, F. iinumae | F. vesca | F. vesca, F. iinumae |
| 7 | G26957 | R5 | NA | Unresolved | F. iinumae | F. iinumae | F. iinumae | NA | NA |
“Unresolved,” no such clade was found; NA, missing data.
Note.—Phylogenetic trees can be found in the supplementary figures, Supplementary Material online. The most closely related diploids species of polyploids were determined by the smallest clade including the polyploid species and a single diploid Fragaria species.
Prior to the phylogeny interpretation, the identities of two Fragaria accessions FRA2001 and BC5 were found to require reconsideration based on the placement of their alleles in trees. FRA2001 had been originally identified as F. vesca subsp. bracteata. In this study, FRA2001 contained alleles distributed in multiple clades being sister to different diploid species in 11 trees (supplementary table S4, Supplementary Material online), thus indicating that it is an allopolyploid. Its polyploidy was then confirmed by flow cytometry analysis (data not shown). The plant BC5 had been initially identified as F. vesca subsp. vesca, but in addition to sequences that clustered with those of F. vesca, BC5 sequences also clustered with those of F. viridis in eight trees (supplementary table S4, Supplementary Material online). Combined with flow cytometry analysis confirming that BC5 was a diploid (data not shown), the phylogenetic placement of its sequences suggested that the plant labeled as BC5 was in fact a diploid hybrid between F. vesca and F. viridis. Finally, accession FRA364, which had been identified prior to the study to be a hybrid between F. vesca and F. viridis, contributed alleles to multiple clades in several trees, thereby confirming its hybridity. Thus, although included in the tree constructions, the alleles from accessions FRA2001, BC5, and FRA364 were ignored in the context of tree interpretation, as were the sequences from the various synthetic polyploids. Thus, inferences of phylogenetic relationships between diploid and polyploid Fragaria species (summarized in table 3) were determined using only the sequences from properly identified, nonhybrid diploid accessions and those from polyploid accessions.
Summary of Phylogenetic Relationships between Polyploidy and Diploid Fragaria Species
Sequences from tetraploid F. corymbosa accession FRA1612 were represented by three or more copies in only 4 of the 24 informative trees (table 3). In the G31441-R5 tree (fig. 2A and supplementary fig. S1, Supplementary Material online), one small clade consisted exclusively of sequences from F. corymbosa and F. viridis, whereas another consisted exclusively of sequences from F. corymbosa and F. chinensis. In the G08977-R5 tree (supplementary fig. S26, Supplementary Material online), F. corymbosa sequences shared a clade with sequences from only two diploids, F. viridis and F. chinensis, as well as from hexaploid F. moschata.
Fig. 2.

—Phylogenetic relationships between diploid and polyploid (tetraploid, hexaploid, and decaploid) Fragaria species are revealed by representative clades. (A) Two clades from the tree of G31441-R5 revealed the diploid ancestors of F. corymbosa (4×) to be F. viridis (1) and F. chinensis (2); (B) four putative diploid ancestors of F. moschata (6×) were F. vesca (1) from the tree of G14746-R2, F. viridis (2) from the tree of G08197-R2, F. mandschurica (3) from the tree of G08197-R2, and F. bucharica (4) from the tree of G03631-R2; and (C) two clades from the tree of G26957-R2 revealed that F. vesca was the most closely related diploid species to both F. cascadensis and F. iturupensis in clade (1), and that F. iinumae was the other diploid ancestor of F. iturupensis in clade (2). The complete images of these phylogenetic trees were included in the Supplementary Material online.
Allohexaploid F. moschata was represented by two accessions: FRA157 and FRA376. Of the 21 trees that included sequences from one or both F. moschata accessions, 13 trees displayed sister relationships between specific F. moschata alleles with those of specific diploid species. Fragaria vesca alleles clustered with those of F. moschata FRA157 in five trees and FRA376 in six trees, including both FRA157 and FRA376 alleles in four trees. Clades that contained F. vesca as the only diploid species being sister to F. moschata were identified in eight trees (table 3 and fig. 2B1). Alleles of F. mandshurica clustered with F. moschata alleles in two trees, which also included F. vesca alleles, but did not cluster with alleles of FRA376. Fragaria viridis alleles clustered with F. moschata FRA376 alleles in seven trees, but with FRA157 alleles in only two trees. A set of eight trees (table 3) supported a sister relationship of F. moschata sequences to F. viridis sequences (fig. 2B2).
For the octoploid species, F. vesca and F. iinumae were found to be the most closely related diploid species (table 3). In addition, clades containing octoploid accessions but without either F. vesca or F. iinumae sequences were also found. For example, there were three trees wherein the most closely related diploid species to octoploid sequences was identified as F. viridis (table 3 and fig. 3A). In one tree, F. bucharica was placed as sister to octoploid species (fig. 3B). Moreover, in at least five trees, octoploid sequences were placed in clades distant from all these four diploid species: F. vesca, F. iinumae, F. viridis, and F. bucharica. For example, one of these trees (G32075-R2; fig. 4) resolved octoploid sequences into two distinct clades, all distant from the F. vesca, F. iinumae, and other diploid sequences in the tree.
Fig. 3.

—Representative clades revealed the phylogenetic relationships between octoploid and diploid Fragaria species. (A) Fragaria viridis was revealed as the most closely related diploid species to octoploids in clade 1 from the tree of G23870-R5, clade 2 from the tree of G08197-R2, and clade 3 from the tree of G08197-R5 and (B) one clade from the tree of G16711-R2 revealed the most closely related diploid species to octoploids was F. bucharica.
Fig. 4.

—Phylogenetic tree of G32075-R2 reveals three types of clades containing octoploid sequences.
A number of well-supported (Posterior probability value > 80%) clades were composed of sequences exclusively from accessions of F. chiloensis or F. virginiana. Clades specific to F. chiloensis were identified in five trees (table 3), and all of them received posterior probability support ≥95%. For example, out of 12 F. chiloensis accessions sampled in this study, Clade 7 in the tree of G14770-R2 (supplementary fig. S3, Supplementary Material online) contains sequences from 10 F. chiloensis accessions without any F. virginiana sequences. Clades specific to F. virginiana were found in two trees: G14770-R2 (supplementary fig. S3, Supplementary Material online), and G03631-R2 (supplementary fig. S4, Supplementary Material online) (table 3).
With respect to the two decaploid species, sequences of F. cascadensis were represented in three trees. Only the tree G26957-R2 (fig. 2C1 and supplementary fig. S5, Supplementary Material online) confirmed one of the most closely related diploid species was F. vesca. Fragaria iturupensis sequences were represented in 14 trees. It was placed as sister clades to F. vesca in two trees, and shared the same clade with F. iinumae in 11 trees (table 3 and fig. 2C2).
Discussion
Overview of the Study
The phylogenetic study of polyploid species has been a great challenge due to their reticulate relationships with species of lower ploidy levels and the presence of multiple alleles of the same gene within their genomes. This study reports a genome-scale investigation of diploid ancestry and octoploid subgenome composition in Fragaria by using large-scale data sets from multiple nuclear loci and thorough taxon sampling. In this study, 44 phylogenetic trees were constructed with the data from 24 target genes. Among them, 24 trees corresponding to 18 genes were considered as potentially informative. The plant material used included 8 out of the 12 wild diploid species, and all 4 subspecies of F. vesca, and 5 diploids were represented by two or more accessions. As by far the most extensive sampling of octoploid Fragaria taxa to date, our study included multiple accessions of each of seven subspecies of the ancestral octoploids F. chiloensis and F. virginiana, as well as 19 F. x ananassa cultivars. Fragaria taxa not represented in our study were limited to diploids (F. daltoniana, F. nubicola, F. pentaphylla) and tetraploids (F. moupinensis, F. tibetica, F. gracilis) which, in previous studies (Rousseau-Gueutin et al. 2009; DiMeglio et al. 2014), had been shown to belong to clades of Asian species distant from the Fragaria octoploids and containing no octoploid-derived alleles.
As detailed below, we have presented evidence of mosaic genome compositions at the diploid and polyploid levels in Fragaria, thus questioning the appropriateness of octoploid subgenome composition models that assume the evolutionary preservation of intact, ancestrally derived subgenomes. In addition, we have added to evidence that as yet unknown diploid species have contributed alleles to the octoploid genomes. Thus, our results add justification to continued germplasm exploration and evaluation in Fragaria. By documenting genomic divergence between F. chiloensis and F. virginiana, our findings are relevant to efforts to reconstruct Fragaria x ananassa, and may help to explain reproductive barriers operating between these two octoploids and even within strawberry breeding programs. Finally, we have identified informative genetic loci as candidates for use in future phylogenetic studies within and beyond Fragaria.
Phylogenetic Relationships among Diploid Species
Regarding the relationships among diploid species and genomes as illuminated by the present study, F. vesca was often positioned as sister to one or more of the diploids F. mandshurica, F. bucharica, F. nilgerrensis, F. viridis, F. nipponica, and F. chinensis. In one tree G17793-R2 (supplementary fig. S6, Supplementary Material online), F. vesca and F. mandshurica formed a clade separate from all other diploid species, adding evidence that they are each other’s closest relatives. Fragaria vesca sequences constituted an exclusive clade in three trees (G08197-R5, G03631-R5, and G26957-R2) (supplementary figs. S7, S8, and S5, Supplementary Material online), representing a very strong signal of the monophyly of F. vesca. With respect to the phylogenic placement of other diploid species, our research provided extensive documentation of incongruence among phylogenies. Fragaria nilgerrensis clustered with F. iinumae in three trees: G14770-R2, G03631-R2, and G23870-R5 (supplementary figs. S3, S4, and S9, Supplementary Material online), and was sister to F. vesca in five trees: G14746-R2, G16711-R5, G17793-R2, G12770-R2, and G26957-R2 (supplementary figs. S5, S6, and S10–S12, Supplementary Material online). In the tree of G08827-R5 (supplementary fig. S13, Supplementary Material online), F. nilgerrensis branched off early on the tree and was placed as sister to all other Fragaria taxa. In several trees, F. viridis displayed a close relationship with different groups of diploid species on the basis that two or more alleles from F. viridis were found to be placed in distinct clades in each gene tree. For diploid species F. nipponica and F. chinensis, data from both species were available in seven genes. They were resolved as each other’s closest relative with the only exception of gene G09999-R5 (supplementary fig. S14, Supplementary Material online), where they were placed in different clades. In addition, F. nipponica and F. chinensis are both distributed in Southeast and East Asia, and they share a common pollen grain morphology (Staudt 2008). The similar phylogenetic positions of F. nipponica and F. chinensis suggested that they are very closely related and perhaps worthy of being considered for merger into a single species.
Incongruence among Phylogenetic Trees Assessed Using Diploid Species
Among the 24 selected trees, six pairs of trees were based on the respective R2 and R5 read sets from the same gene. Incongruent phylogenies between trees based on the forward and reverse reads of the same gene were found from three genes: G32075, G03631, and G08197. In the tree of G08197, phylogenetic conflict referred to the position of F. bucharica, which was placed as sister to F. vesca and F. mandschurica in the tree of G08197-R2 (supplementary fig. S16, Supplementary Material online), whereas it was placed at an early diverged branch being sister to all other Fragaria species in the tree of G08197-R5 (supplementary fig. S7, Supplementary Material online). Similarly, F. viridis was placed in the clade as sister to F. iinumae in the tree of G32075-R5 and G03631-R2 (supplementary figs. S4 and S15, Supplementary Material online), but it was placed in an early branched clade being sister to the remainder of Fragaria species in the tree of G32075-R2 (supplementary fig. S2, Supplementary Material online) and G03631-R5 (supplementary fig. S8, Supplementary Material online). Such conflicts in phylogenetic relationships of F. viridis and F. bucharica relative to other Fragaria species may be explained by the differing numbers of variations accumulated on two ends of each amplicon in F. viridis and F. bucharica. Since variations between species do not occur evenly along the gene or the chromosome, phylogenetic trees based on short sequences were susceptible to sampling error due to failure to recover an equal amount of phylogenetic signal from both ends of amplicons. Due to the missing data from different samples and to the large numbers of unresolved sequences, the extent of agreements among genes on the same versus different chromosomes could be assessed only to a limited degree, as illustrated by the placement of F. nilgerrensis in six trees. With respect to three gene trees on LG 1, F. nilgerrensis was resolved as sister lineage to F. vesca or F. iinumae in the trees of G14746-R2 and G14770-R2 (supplementary figs. S3 and S10, Supplementary Material online), respectively. But its position could not be resolved in the tree of G31441-R5 (supplementary fig. S1, Supplementary Material online). On LG 6, the tree of G17793-R2 (supplementary fig. S6, Supplementary Material online) placed F. nilgerrensis in the clade along with F. vesca, F. yezoensis, F. chinensis, and F. viridis, but it was placed in a distinct clade being sister to F. iinumae by the tree of G23870-R5 (supplementary fig. S9, Supplementary Material online) on LG 6.
Discrepancies among phylogenetic trees inferred for diploid Fragaria species have also been reported in previous investigations. Fragaria nilgerrensis, F. bucharica, and F. nipponica have each been placed in three different clades in terms of their clade memberships (Rousseau-Gueutin et al. 2009; DiMeglio et al. 2014; Njuguna et al. 2013; Tennessen et al. 2014), and the position of F. viridis was variously shown to be sister to F. vesca or to F. iinumae in previous studies (Rousseau-Gueutin et al. 2009; Tennessen et al. 2014). The conflicts among trees in this study, and between this study and those of previous studies might result from incomplete lineage sorting, hybridization, and/or introgression. Considering the young age of the Fragaria genus (Njuguna et al. 2013) and the nonoverlapped distribution area for some of these Fragaria species, hybridization and introgression may not be prevalently involved in the formation of new species. For example, F. viridis was found to include sequences being sister to F. iinumae, but F. viridis is distributed in Europe and central Asia (Staudt 1999), and it is geographically isolated from F. iinumae, which is found mainly in Japan and some adjacent locations. The lack of monophyletic Fragaria clades and the presence of polytomous relationships between Fragaria species at many gene sites suggest that incomplete lineage sorting might be a more plausible factor underlying the divergences of Fragaria species.
Phylogenetic Relationships between Polyploid and Diploid Species
In the phylogenetic analysis of allopolyploids (Smedmark et al. 2003), gene copies inherited from different diploid ancestors are expected to be represented in separate clades, and to be sister to the alleles of the respective extant diploids if present in the same tree. In the 24 trees considered informative in the present study, the positions of many alleles, both diploid- and polyploid-derived, were unresolved, thus posing a level of “noise” not seen in prior, gene-specific studies (Rousseau-Gueutin et al. 2009; DiMeglio et al. 2014). Nevertheless, clustering of polyploid- and diploid-derived alleles was evident in many trees. In the present study, data from one phylogenetic tree support that F. corymbosa is an allotetraploid resulted from the hybridization between F. chinensis and F. viridis. The contribution of four diploid species (F. viridis, F. bucharica, F. vesca, and F. mandshurica) to the genome of F. moschata received support from multiple phylogenetic trees in the present study. These results align with the previous study which has shown that tetraploid F. corymbosa was the descendant of F. chinensis, and that hexaploid F. moschata was a hybrid between F. viridis and/or F. bucharica ×F. vesca and/or F. mandshurica (Staudt 2008). Based on these findings, we proposed that F. moschata may possess a complex genome that was derived from three or more diploid ancestors.
The clustering of octoploid and diploid sequences variously involved diploids F. vesca, F. iinumae, F. viridis, and F. bucharica alleles (table 3), thus agreeing with prior studies implicating F. vesca (and/or F. mandshurica) (Fedorova 1946; Byrne and Jelenkovic 1976; Potter et al. 2000; Rousseau-Gueutin et al. 2009; Njuguna et al. 2013; DiMeglio et al. 2014; Tennessen et al. 2014) and F. iinumae (Rousseau-Gueutin et al. 2009; DiMeglio et al. 2014; Tennessen et al. 2014) as ancestral allele donors to the octoploids, whereas also drawing attention to F. viridis and F. bucharica as warranting further scrutiny. Five trees displayed two instances of octoploid–diploid clustering (table 3), of which two trees (G32075-R2 and G26957-R2) implicated both F. vesca and F. iinumae as allele donors, two (G08197-R2 and G08197-R5) implicated both F. vesca and F. viridis, and one (G23870-R5) implicated both F. iinumae and F. viridis. The involvement of F. viridis in the evolution of octoploid strawberries has received support from a previous phylogenetic study based on the nuclear low/single copy intragenic region between the two genes RGA1 (Resistance Gene Analogue 1) and Subt (Subtilase) (Lundberg et al. 2011).
When octoploids and the hexaploid F. moschata were sharing the same clade, only F. vesca was found to be the diploid species most closely related to both hexaploid and octoploid species. Supporting evidences come from four trees (G08197-R2, G08197-R5, G09999-R5, and G26957-R2, supplementary figs. S5, S7, S14, and S16, Supplementary Material online, respectively), each of them containing a clade that includes octoploid and hexaploid sequences and F. vesca as the only diploid member. Such findings suggest that at least some of the F. vesca-related sequences found in octoploid genomes may have been acquired from hexaploid F. moschata.
Two previous studies have proposed that F. vesca subsp. bracteata is the F. vesca subspecies most closely related to octoploids (Njuguna et al. 2013; Tennessen et al. 2014). Based on our data from 11 accessions of F. vesca, no consistent subspecies grouping pattern was identified. However, when only one F. vesca subspecies was resolved as the sole diploid Fragaria species being sister to octoploids in three phylogenetic trees, the diploid sister was F. vesca subsp. vesca, not subsp. bracteata. In the tree of G32075-R2 (supplementary fig. S2, Supplementary Material online) and G20659-R2 (supplementary fig. S17, Supplementary Material online), these F. vesca accessions most closely related to octoploids were FRA438A (F. vesca subsp. vesca), and H4 (F. vesca subsp. vesca). In the other tree G31441-R5 (supplementary fig. S1, Supplementary Material online), the F. vesca accession clustered with octoploids is NOV1-1 C (F. vesca subsp. vesca). Therefore, F. vesca subsp. vesca could be the potential subgenome donor to the octoploid species.
In the tree of G32075-R2 (fig. 4 and supplementary fig. S2, Supplementary Material online), the clade “4,” which includes F. iinumae as the sole diploid species, was further diverged into several subclades. And the inner clades were more closely related to F. iinumae than others, suggesting various levels of divergence among alleles originated from F. iinumae, thus supporting the hypothesis that partial octoploid subgenomes may arise from the F. iinumae lineage, including F. iinumae itself and unknown ancestors probably close to F. iinumae as proposed by (Tennessen et al. 2014). Intriguingly, in addition to implicating F. vesca and F. iinumae as allele donors, tree G32075-R2 contains two other clades of octoploid alleles that are distant from both F. vesca and F. iinumae, as well as from F. viridis, F. bucharica, and all other diploids in the tree. Moreover, trees numbered G14770-R2, G20659-R2, and G31441-R5 contain clades of octoploid alleles without a clear diploid association. This finding is in line with the recent study of (Sargent et al. 2015) which investigated the identity of haploSNPs used for a F. ×ananassa mapping population and successfully identified two sets of discrete subgenomes derived from F. vesca and F. iinumae as well as subgenomic contributions from one or more unknown diploid ancestors. Thus, the octoploid genomes may harbor allele contributions from yet unknown diploid sources.
Model of Octoploid Subgenome Composition
The findings summarized above and considered in greater detail below have several implications regarding the modeling of octoploid subgenome composition. Importantly, because our data do not include information about the allele coupling relationships for genes on the same chromosome, we cannot draw conclusions about the existence, or lack thereof, of discrete, octoploid subgenomes inherited intact from diploid ancestors. However, we can assess the extent to which our data are consistent or inconsistent with the variously proposed models, as follows.
Aspects of Fedorova (1946) AAAABBCC model are contradicted by our findings and those of others. Specifically, in this model, the B genome designation was assigned to diploid F. nipponica (aka F. yezoensis). None of the molecular phylogenetic studies to date have placed F. nipponica and octoploid alleles in the same clade or sister to one another. Fragaria nipponica is among the group of Asian taxa previously designated as clade X by Rousseau-Gueutin et al. (2009), and clade B1 by DiMeglio et al. (2014), and as such falls outside the scope of further interest, except perhaps as an outgroup, in studies of octoploid subgenome composition. Like the Fedorova (1946) study, the other cytologically based models did not include meiotic analysis of hybrids involving F. iinumae, and made no mention of this important ancestral diploid. However, the Bringhurst (1990) models both invoke two major subgenome types, and hence predict two major phylogenetic clades, with one or both bifurcating into subclades. What they do not predict is the possibility of other, equally divergent allele clades pointing to the possibility of additional ancestral diploids not sister to either F. vesca or F. iinumae.
It is of both basic and practical interest to determine whether the genome of the octoploid cultivated strawberry is partitioned into discrete subgenomes, each having descended from a particular ancestral diploid. Discrete subgenome composition has been established for some other important polyploid crop species, such as bread wheat (AABBDD), where the A, B, and D subgenomes are evolutionarily derived from or related to ancestral diploids Triticum urartu (AA), Aegilops speltoides (BB), and Aegilops tauschii (DD) (Petersen et al. 2006). Other subgenomically characterized polyploid crop species include cotton (Reinisch et al. 1994), peanuts (Kochert et al. 1996; Seijo et al. 2007), and oilseed rape (Allender and King 2010).
Our findings of “orphan clades” of octoploid alleles lacking diploid cladistic partners conflicted not only with the A versus B (or Y1 vs. Z) subgenomic models (Fedorova 1946; Byrne and Jelenkovic 1976; Rousseau-Gueutin et al. 2009; Tennessen et al. 2014) but may cast doubt upon the maintenance of subgenomic integrity beyond that of the well supported AA subgenomic contribution from F. vesca (Fedorova 1946; Byrne and Jelenkovic 1976; Potter et al. 2000; Rousseau-Gueutin et al. 2009; Njuguna et al. 2013; DiMeglio et al. 2014; Tennessen et al. 2014). Our results do not support a universal formula that implies that all subgenomes are distinct from each other, and that all seven chromosomes within a subgenome have the same ancestral source. In contrast, extensive homogeneity within octoploid genomes was observed based on 12 trees that could not differentiate F. vesca and F. iinumae sequences. This observation is consistent with the identification of low polymorphism regions in the F. ×ananassa genome (Sargent et al. 2012), and by the polysomic chromosome pairing observed from segregation patterns of linkage groups in coupling and repulsion phases (Lerceteau-Kohler et al. 2003; Rousseau-Geutin et al. 2009). Being aware of such limited differences between subgenomes, future genome assembly projects could adopt more practical approaches to assemble subgenomes of octoploid strawberries. For example, it would become necessary to employ a high density of subgenome specific loci along the genome for anchoring purposes to accurately differentiate homoeologous chromosomes.
Other Findings of This Study
It has been recognized that there are significant morphological distinctions between F. chiloensis and F. virginiana. For example, F. chiloensis plants have thick, coriaceous leaves in dark green color, large achenes, and long spreading hairs, whereas F. virginiana plants have thin leaves from green to bluish green and smaller achenes (Staudt 1999). The separation of F. virginiana and F. chiloensis as distinct species has received support from cluster analysis of simple sequence repeat markers (Hokanson et al. 2006). Our results provided further support for the divergence between these two wild octoploid species. Well-supported clades comprised of sequences exclusively from F. chiloensis were observed in eight trees, and clades specific to F. virginiana were observed in two trees. However, the ancestral state of these loci could not be determined, and it is not clear whether the higher number of F. chiloensis specific clades than F. virginiana was caused by gain of derived characters in F. chiloensis or by loss of ancestral characters in F. virginiana. More plant samples from lower ploidy levels (tetraploids and hexaploids) and higher ploidy levels (decaploids) must be sequenced at these loci to resolve such questions.
Finally, it is of interest to evaluate the usefulness of the utilized gene sites in relation to future phylogenetic studies and other uses in Fragaria and perhaps other taxa. For six of the gene sites, both the forward and reverse read directions provided useful information. With technical modification to allow for correct phasing, the forward and reverse haplotypes could be properly merged, enhancing the robustness of the phylogenetic signal. Usefully, these six gene sites are distributed across six different chromosomes, leaving only chromosome I unrepresented. However, two gene sites on chromosome I (G31441-R5 and G14770-R2, supplementary figs. S1 and S3, Supplementary Material online) identified orphan clades in the octoploids, thus suggesting their future usefulness for studies of polyploidy in Fragaria.
Conclusions
In summary, we have presented evidence of mosaic genome compositions at the diploid and polyploid levels in Fragaria, and added to evidence that as yet unknown diploid species have contributed alleles to the octoploid genomes. Thus, our results add justification to continued germplasm exploration and evaluation in Fragaria. By documenting genomic divergence between F. chiloensis and F. virginiana, our findings prompt reconsideration of efforts to reconstruct Fragaria x ananassa, and may help to explain reproductive barriers operating between these two octoploids and even within strawberry breeding programs.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
Partial funding was provided by the New Hampshire Agricultural Experiment Station. This is Scientific Contribution Number 2758. This work is/was supported by the USDA National Institute of Food and Agriculture (Hatch) Projects NH00588 and NH00632. Support was also provided by United States Department of Agriculture, Cooperative State Research, Education, and Extension Service (USDA-CSREES) National Research Initiative (NRI) Plant Genome Program [grant number 2008-35300-04411], and by the University of New Hampshire (UNH) Department of Biological Sciences and the UNH Graduate School Summer Fellowship program. We thank Melanie Shields for helpful editorial assistance.
Literature Cited
- Allender CJ, King GJ.. 2010. Origins of the amphiploid species Brassica napus L. investigated by chloroplast and nuclear molecular markers. BMC plant biology 10(1):54.http://dx.doi.org/10.1186/1471-2229-10-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arulsekar S, Bringhurst R, Voth V.. 1981. Inheritance of PGI and LAP isozymes in octoploid cultivated strawberries. J Am Soc Hortic Sci. 106:679–683. [Google Scholar]
- Bailey CD, Carr TG, Harris SA, Hughes CE.. 2003. Characterization of angiosperm nrDNA polymorphism, paralogy, and pseudogenes. Mol Phylogenet Evol. 29(3):435–455.http://dx.doi.org/10.1016/j.ympev.2003.08.021 [DOI] [PubMed] [Google Scholar]
- Bassil NV, Davis TM, Zhang H, Ficklin S, Mittmann M, Webster T, Mahoney L, Wood D, Alperin ES, Rosyara UR.. 2015. . Development and preliminary evaluation of a 90 K Axiom® SNP array for the allo-octoploid cultivated strawberry Fragaria× ananassa. BMC Genomics 16(1):155.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringhurst RS. 1990. Cytogenetics and evolution in American Fragaria. HortScience 25(8):879–881. [Google Scholar]
- Byrne D, Jelenkovic G.. 1976. Cytological diploidization in the cultivated octoploid strawberry Fragaria × ananassa. Can J Genet Cytol. 18(4):653–659. [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D.. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat methods 9(8):772.http://dx.doi.org/10.1038/nmeth.2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis TM, et al. 2010. Chloroplast DNA inheritance, ancestry, and sequencing in Fragaria. Acta Hort. 859:221–228. [Google Scholar]
- DiMeglio LM, Staudt G, Yu H, Davis TM, Peace C.. 2014. A phylogenetic analysis of the genus Fragaria (strawberry) using intron-containing sequence from the ADH-1 gene. PLoS One 9(7):e102237.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte JM, Wall PK, Edger PP, Landherr LL, Ma H, Pires JC, Leebens-Mack J.. 2010. Identification of shared single copy nuclear genes in Arabidopsis, Populus, Vitis and Oryza and their phylogenetic utility across various taxonomic levels. BMC Evol Biol. 10(1):61.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R.. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27(16):2194–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorova NJ. 1946. Crossability and phylogenetic relations in the main European species of Fragaria. Compil Natl Acad Sci USSR 52:545–547. [Google Scholar]
- Folta KM, Davis TM.. 2006. Strawberry genes and genomics. Crit Rev Plant Sci. 25(5):399–415.http://dx.doi.org/10.1080/07352680600824831 [Google Scholar]
- Gaspar JM, Thomas WK.. 2015. FlowClus: efficiently filtering and denoising pyrosequenced amplicons. BMC bioinformatics. 16(1):105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BG. 2004. Phylogenetic trees made easy: a how-to manual. Massachusetts: Sinauer Associates Sunderland. [Google Scholar]
- Hokanson K, Smith M, Connor A, Luby J, Hancock JF.. 2006. Relationships among subspecies of New World octoploid strawberry species, Fragaria virginiana and Fragaria chiloensis, based on simple sequence repeat marker analysis. Botany 84(12):1829–1841. [Google Scholar]
- Hummer KE, Hancock J. (2009). Strawberry genomics: botanical history, cultivation, traditional breeding, and new technologies Genetics and Genomics of Rosaceae. New York: Springer; p. 413–435. [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4): 772-780.http://dx.doi.org/10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochert G. et al. 1996. RFLP and cytogenetic evidence on the origin and evolution of allotetraploid domesticated peanut, Arachis hypogaea (Leguminosae). Am. J. Bot. pp.1282-1291. [Google Scholar]
- Lerceteau-Köhler E, Guerin G, Laigret F, Denoyes-Rothan B.. 2003. Characterization of mixed disomic and polysomic inheritance in the octoploid strawberry (Fragaria× ananassa) using AFLP mapping. Theor Appl Genet. 107(4): 619-628. [DOI] [PubMed] [Google Scholar]
- Lundberg M, Eriksson T, Zhang Q, Davis T. (2011). New insights into polyploid evolution in Fragaria (Rosaceae) based on the single/low copy nuclear intergenic region RGA1-Subtilase.
- Mahoney L, Quimby M, Shields M, Davis T.. 2010. Mitochondrial DNA transmission, ancestry, and sequences in Fragaria. Acta Hort. 859:301–308. [Google Scholar]
- McCormack JE, Hird SM, Zellmer AJ, Carstens BC, Brumfield RT.. 2013. Applications of next-generation sequencing to phylogeography and phylogenetics. Mol Phylogenet Evol. 66(2):526–538. [DOI] [PubMed] [Google Scholar]
- Njuguna W, Liston A, Cronn R, Ashman T-L, Bassil N.. 2013. Insights into phylogeny, sex function and age of Fragaria based on whole chloroplast genome sequencing. Mol Phylogenet Evol. 66(1):17–29. [DOI] [PubMed] [Google Scholar]
- Petersen G, Seberg O, Yde M, Berthelsen K, 2006. Phylogenetic relationships of Triticum and Aegilops and evidence for the origin of the A, B, and D genomes of common wheat (Triticum aestivum). Mol phylogenet Evol. 39(1):70-82. [DOI] [PubMed] [Google Scholar]
- Potter D, Luby JJ, Harrison RE.. 2000. Phylogenetic relationships among species of Fragaria (Rosaceae) inferred from non-coding nuclear and chloroplast DNA sequences. Syst Bot. 25(2):337–348.http://dx.doi.org/10.2307/2666646 [Google Scholar]
- Rambaut A, 2009. FigTree version 1.3. 1 [computer program]. Website http://tree. bio. ed. ac. uk/software/figtree/, last accessed October 21, 2017.
- Reinisch AJ, Dong JM, Brubaker CL, Stelly DM, Wendel JF, Paterson AH.. 1994. A detailed RFLP map of cotton, Gossypium hirsutum x Gossypium barbadense: chromosome organization and evolution in a disomic polyploid genome. Genetics 138(3): 829–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson BA, Page JT, Bajgain P, Sanderson SC, Udall JA.. 2012. Deep sequencing of amplicons reveals widespread intraspecific hybridization and multiple origins of polyploidy in big sagebrush (Artemisia tridentata; Asteraceae). Am J Bot. 99(12):1962–1975. [DOI] [PubMed] [Google Scholar]
- Rousseau-Gueutin M, Gaston A, Aïnouche A, Aïnouche ML, Olbricht K, Staudt G, Richard L, Denoyes-Rothan B.. 2009. Tracking the evolutionary history of polyploidy in Fragaria L.(strawberry): new insights from phylogenetic analyses of low-copy nuclear genes. Mol Phylogenet Evol. 51(3):515–530. [DOI] [PubMed] [Google Scholar]
- Sargent D, Yang Y, Šurbanovski Y, Bianco N, Buti L, Velasco M, Giongo RL, Davis T.. 2015. HaploSNP affinities and linkage map positions illuminate subgenome composition in the octoploid, cultivated strawberry (Fragaria × ananassa). Plant Sci. 242:140–150. [DOI] [PubMed] [Google Scholar]
- Sargent DJ, et al. 2011. Simple sequence repeat marker development and mapping targeted to previously unmapped regions of the strawberry genome sequence. Plant Genome 4(3):165–177.http://dx.doi.org/10.3835/plantgenome2011.05.0014 [Google Scholar]
- Sargent DJ, Passey T, Šurbanovski N, Girona EL, Kuchta P, Davik J, Harrison R, Passey A, Whitehouse A, Simpson D.. 2012. A microsatellite linkage map for the cultivated strawberry (Fragaria × ananassa) suggests extensive regions of homozygosity in the genome that may have resulted from breeding and selection. Theor Appl Genet. 124(7):1229–1240. [DOI] [PubMed] [Google Scholar]
- Seijo G, et al. 2007. Genomic relationships between the cultivated peanut (Arachis hypogaea, Leguminosae) and its close relatives revealed by double GISH. Am J Bot. 94(12):1963–1971.http://dx.doi.org/10.3732/ajb.94.12.1963 [DOI] [PubMed] [Google Scholar]
- Senanayake Y, Bringhurst R.. 1967. Origin of Fragaria polyploids. I. Cytological analysis. Am J Bot. 54(2):221–228.http://dx.doi.org/10.2307/2440801 [Google Scholar]
- Shulaev V, et al. 2011. The genome of woodland strawberry (Fragaria vesca). Nat Genet. 43(2):109–116.http://dx.doi.org/10.1038/ng.740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small RL, Cronn RC, Wendel JF.. 2004. LAS Johnson Review No. 2. Use of nuclear genes for phylogeny reconstruction in plants. Aust Syst Bot. 17(2):145–170.http://dx.doi.org/10.1071/SB03015 [Google Scholar]
- Smedmark JEE, Eriksson T, Evans RC, Campbell CS, Mason-Gamer R.. 2003. Ancient allopolyploid speciation in Geinae (Rosaceae): evidence from nuclear granule-bound starch synthase (GBSSI) gene sequences. Syst Biol. 52(3):374–385. [DOI] [PubMed] [Google Scholar]
- Staudt G. (1999). Systematics and geographic distribution of the American strawberry species Taxonomic studies in the genus Fragaria (Rosaceae: Potentilleae) (Vol. 81). University of California Press. [Google Scholar]
- Staudt G. 2006. Himalayan species of Fragaria (Rosaceae). Bot Jahrbücher 126(4):483–508. [Google Scholar]
- Staudt G. (2008). Strawberry biogeography, genetics and systematics. VI International Strawberry Symposium 842:71–84. [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S.. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30(12):2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen JA, Govindarajulu R, Ashman T-L, Liston A.. 2014. Evolutionary origins and dynamics of octoploid strawberry subgenomes revealed by dense targeted capture linkage maps. Genome Biol Evol. 6(12):3295–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonnabel J, Olivieri I, Mignot A, Rebelo A, Justy F, Santoni S, Caroli S, Sauné L, Bouchez O, Douzery EJ.. 2014. Developing nuclear DNA phylogenetic markers in the angiosperm genus Leucadendron (Proteaceae): a next-generation sequencing transcriptomic approach. Mol Phylogenet Evol. 70:37–46. [DOI] [PubMed] [Google Scholar]
- Torres A, Weeden N, Martin A.. 1993. Linkage among isozyme, RFLP and RAPD markers in Vicia faba. Theor Appl Genet. 85(8):937–945. [DOI] [PubMed] [Google Scholar]
- Untergasser A, et al. 2012. Primer3—new capabilities and interfaces. Nucleic Acids Res. 40(15): e115–e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel JF, Schnabel A, Seelanan T.. 1995. Bidirectional interlocus concerted evolution following allopolyploid speciation in cotton (Gossypium). Proc Natl Acad Sci U S A. 92(1):280–284.http://dx.doi.org/10.1073/pnas.92.1.280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer EA, Wen J.. 2013. Reprint of: using nuclear gene data for plant phylogenetics: progress and prospects. Mol Phylogenet Evol. 66(2):539–550.http://dx.doi.org/10.1016/j.ympev.2013.01.005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.