

Abstract
The delivery of cancer chemotherapy to treat brain tumors remains a challenge, in part, because of the inherent biological barrier, the blood–brain barrier. While its presence and role as a protector of the normal brain parenchyma has been acknowledged for decades, it is only recently that the important transporter components, expressed in the tightly knit capillary endothelial cells, have been deciphered. These transporters are ATP-binding cassette (ABC) transporters and, so far, the major clinically important ones that functionally contribute to the blood–brain barrier are ABCG2 and ABCB1. A further limitation to cancer therapy of brain tumors or brain metastases is the blood–tumor barrier, where tumors erect a barrier of transporters that further impede drug entry. The expression and regulation of these two transporters at these barriers, as well as tumor derived alteration in expression and/or mutation, are likely obstacles to effective therapy.
Keywords: ABC transporter, CNS, blood–brain barrier, blood–tumor barrier, glioma, medulloblastoma, chemotherapy
1. ATP-Binding Cassette (ABC) Transporters in the Central Nervous System (CNS)
1.1. Overview of Blood–Brain Barrier (BBB)
The central nervous system (CNS) is separated from the blood by a vasculature comprised of highly specialized endothelial cells (ECs) that form part of the blood–brain barrier (BBB) (Figure 1). The BBB provides nutrients to the CNS by selective uptake of small molecules but also protects the CNS from deleterious endogenous and xenobiotic molecules. Importantly, transporters protect the CNS, by actively extruding xenobiotics at the BBB into the blood to create a “pharmacological sanctuary” for the brain. However, this sanctuary for the brain creates an obstacle to the effective delivery of drugs necessary for treating pathological conditions of the CNS from neurological diseases to cancer.
Figure 1.

Diagram of the components in the blood–brain barrier (BBB). ABCB1 (purple) and ABCG2 (green) are expressed on the luminal side of endothelial cells. They prevent xenobiotics (blue and pink) from entering the brain and frequently share common substrates. ABC, ATP-binding cassette.
The BBB was functionally identified in the early 1900s when an intravenous injection of various dyes failed to stain the CNS, whereas the dyes injected intrathecally into the subarachnoid space stained the brain [1]. In 1934, Stern proposed that the BBB not only protected the brain from toxic compounds, but also to maintaining homeostasis of the brain (as cited in [1]).
About 5% of the brain is composed of vascular cells and these cells provide an almost seamless protective barrier, the BBB [2,3]. Specialized ECs line the cerebral blood vessels, and properties of these cells, such as the expression of ATP-binding cassette (ABC) transporters, facilitate extrusion of undesirable molecules providing one level of barrier protection. A second level of protection is provided by a property of the ECs. Unlike ECs in other peripheral vessels, the cerebral vessel ECs form intercellular tight junctions (TJ) that are continuous. In combination, transporters and TJs provide a dual barrier to limit compounds from accessing the brain parenchyma. The integrity and maintenance of the BBB is further supported by astrocytes, neurons, microglial cells, pericytes, and the extracellular matrix (Figure 1) [4].
1.1.1. Development and Regulation of the BBB
The BBB develops by a multistep process requiring interplay among metabolic and signaling pathways emanating from a variety of cell types. Angiogenic stimuli from neural progenitor cells produce new vessels, sprouting from pre-existing vessels, equipped with properties of the BBB. The barrier function and integrity mature to provide maximum effect and must be maintained through extra- and intra-cellular signaling. Interestingly, some of these processes are disrupted in CNS tumors, as described later.
In brain ECs, the Wnt-β-catenin pathway promotes angiogenesis during embryogenesis [5]. Indeed, the transcriptional profile of brain ECs is enriched in the Wnt-β-catenin signaling pathway compared to peripheral ECs [3]. In the canonical Wnt pathway, secreted Wnt ligands bind to the Frizzled receptor on ECs plasma membrane. Binding of Wnt to the Frizzled receptor blocks phosphorylation of β-catenin by glycogen synthase kinase 3 (GSK3), allowing β-catenin to accumulate and translocate to the nucleus where it binds to TCF/LEF (T-cell factor/lymphoid enhancer factor) transcription factors to modulate expression of target genes [6]. The Wnt-β-catenin pathway also enhances BBB function by upregulating the expression of BBB-specific genes such as Glut-1 (Slc2a1), a nutrient uptake carrier. In addition, claudin-3 expression is increased by β-catenin. The Wnt-β-catenin pathway is implicated in the regulation of TJ formation during BBB maturation, because loss of β-catenin at postnatal days 4, 7, and 14, corresponding to days of BBB maturation, results in decreased claudin 3, an essential component of the BBB TJ [7]. This reduction in claudin 3 level associates with a decrease in BBB integrity as measured by evans blue staining [7]. Taken together, the Wnt-β-catenin pathway promotes angiogenesis and TJ formation at the BBB.
The sonic hedgehog (SHH) pathway also regulates BBB development. Unlike the Wnt-β-catenin pathway, SHH signaling is not required for angiogenesis in the brain, but it regulates the TJ function through the expression of TJ proteins such as occludin [8]. SHH secreted from astrocytes binds and inactivates the SHH receptor patched 1 (PTCH1) on the cell surface of brain ECs. PTCH1 then releases the co-receptor smoothened (SMO) to activate a downstream signaling cascade leading to the translocation of the transcription factor GLI (glioma-associated oncogene)into the nucleus. Blocking SHH signal transduction, by cyclopamine, an inhibitor of SMO, produces an extravasation of exogenous and endogenous molecules secondary to breakdown of the BBB. As brain hemorrhage was not observed, these results suggest SHH signaling ensures the integrity of brain EC tight junctions.
The EC transcriptome of BBB differs from peripheral ECs. The EC transcriptome displays a regulatory signature: the nuclear receptor, retinoic x receptor (RXR)/pregnane x receptor (PXR) and the “LPS (lipopolysaccharide)/IL-1 (interleukin-1) mediated inhibition of RXR function” pathway when compared to the peripheral ECs. The nuclear receptor RXRα regulates expression of brain EC-enriched transcripts, including transporters Abcb1 and Abcc4 [3]. Notably, while the RXRα-mediated transcription is upregulated in brain ECs, peripheral ECs downregulate these pathways.
Pericytes and astrocytes contribute to BBB development and maintenance. Pericytes envelope the outer walls of capillaries and provide structural stability. Pericytes have more than a physical role and are required for proper BBB development during embryogenesis [9]. Pericytes affect vessel permeability by regulating TJ formation. Astrocytes, on the other hand, are absent during initial vascularization, but maintain BBB function and integrity once formed. Astrocytes, as mentioned above, secrete signaling molecules such as SHH that support TJ integrity.
Exactly how the BBB protects the developing brain has been an incremental process. Wislocki, in 1920, showed that the brain from a mid-gestation guinea embryo was refractory to dye staining, thus revealing, by intravenous trypan blue (a molecule < 1000 Da) the BBB during fetal development (as cited in [1]). Similar results were obtained in human fetuses, demonstrating the conservation of BBB function in mammals. Ultrastructural studies revealed that TJ are formed in rat and rabbit embryos suggesting that this component of the BBB forms early in development [1,10]. Interestingly, the expression of genes for uptake and efflux transporters, in ECs vary during development into adulthood, possibly contributing to the notion of “immaturity” of the fetal or post-natal BBB compared to the adult BBB. As expected, some uptake carriers that provide nutrients are higher in fetal brain ECs. These are likely important in increasing the brain concentration of certain amino acids, but suggest the BBB is “leaky”. An efflux transporter Abcb1a transcript increases in adult mice compared to post-natal pups, which is likely to selectivity restrict the molecules allowed into the brain.
1.1.2. Disruption of the BBB in Diseases
The BBB integrity and functionality must be maintained throughout life to protect CNS against a host of toxins and pathogens; however, some insults and diseases compromise the BBB. Oxidative stress is known to disrupt BBB and is commonly seen in neurological disorders such as multiple sclerosis and stroke [11,12]. Reactive oxygen species (ROS) generated during oxidative stress can also disrupt the BBB [13]. Interestingly, increased ROS accumulation is associated with aging, thus it is conceivable that the BBB integrity and functionality declines as people age (discussed further in Section 1.3). The CNS tumors such as glioblastoma and Wnt-subtype of medulloblastoma are also known to disrupt BBB function via different mechanisms that will be described later (Section 2 and Section 3).
1.2. ABC Transporters Provide Critical Barrier Function of BBB
Efflux transporters that belong to the ABC transporter superfamily provide critical barrier function at the brain ECs through the efflux of toxic compounds. In particular, two transporters, ABCB1 (also known as P-glycoprotein or multidrug resistance protein 1 (MDR1)) and ABCG2 (breast cancer resistance protein (BCRP)) are highly expressed in the brain ECs. Other ABC transporters such as ABCC2, ABCC4, and ABCC5 have been detected, but this review will primarily focus on ABCB1 and ABCG2, as they are also the major transporters involved in multidrug resistance (MDR) due to their ability to transport a wide range of chemotherapeutics. Transcriptomic and mass-spectroscopy analysis showed enrichment of both Abcb1 and Abcg2 mRNA and protein in the brain vessels further confirming the importance of these transporters in BBB function. Of interest, ABCC4 is highly expressed in the choroid plexus and plays a critical role in providing barrier function at the blood–cerebrospinal fluid (CSF) barrier. At the BBB, ABCC4 protein levels were >10-fold lower than ABCB1 and ABCG2 [14], which may be an indication for its specific role in blood–CSF barrier. A discussion of ABCC4 is outside of the scope of this review. In the following sections, we will highlight the expression, localization, and clinical relevance of these transporters at the BBB.
1.2.1. ABCB1 Expression at the BBB
ABCB1 was the first member of the ABC protein family to be identified at the BBB. ABCB1 was first implicated at the BBB by immunocytochemical and immunohistochemical studies of ABCB1 in human and rat brain tissues [15,16]. Notably antibodies against ABCB1 only detected it at the brain capillaries but not capillaries of non-CNS tissues. In mouse and rat, there are two ABCB1 isoforms, encoded by Abcb1a and Abcb1b genes with Abcb1a being the predominant gene expressed at the BBB. Mouse ABCB1A’s amino acid sequence and gene regulation parallels human ABCB1. In mouse, the Abcb1a transcript was one of the most enriched transcripts in brain ECs compared to pericytes, and its selective expression in brain ECs compared to liver and lung ECs makes it an excellent BBB marker. In situ hybridization revealed that Abcb1a expression is detected as early as E10.5 of mouse embryos in the neural tube [17]. In human brain microcapillaries, ABCB1 protein was detected by mid-gestation using immunohistochemistry. Adults might have greater ABCB1 amounts. For example, a quantitative Western blot analysis showed that ABCB1 protein amount in the BBB of adult rats was about five-fold higher than that of the 9- and 17-day-old rats [18,19]. Accordingly, mouse Abcb1a mRNA expression showed an increased expression in adult animals compared to post-natal pups [3]. Finally, ABCB1 function in adult mouse was greater than post-natal animals as determined by administration of an ABCB1 inhibitor, cyclosporine A coupled with the ABCB1 substrate, digoxin. Specifically, the brain to blood/plasma ratio, at 2 h post-administration, were 80–90% lower in the adult brain compared to the postnatal day 1 (P1) brain [20]. Abcb1a-deficient mice showed no difference, regardless of the age indicating that the increased transport activity, measured by digoxin, was entirely due to ABCB1 and not another transporter in adult mice.
1.2.2. Cellular Localization of ABCB1
Initially, ABCB1’s function at the BBB was only speculative because the immunohistochemical analysis had a limited ability to establish ABCB1 subcellular localization in the brain EC. This problem was resolved by immunoelectron microscopy with a monoclonal antibody against ABCB1 that localized it to the luminal membrane of human brain capillaries. Confocal immunofluorescence microscopy using a different monoclonal antibody also selectively stained ABCB1 at the luminal side of the brain ECs in rat, lending further support for its putative role in providing a transport barrier function at the BBB. Further, Beaulieu and colleagues fractionated brain capillaries in an effort to physically localize ABCB1. The luminal membrane from brain capillaries was enriched with ABCB1 [21]. To achieve enrichment, the endothelial luminal membrane was coated with a cationic colloidal silica, allowing selective isolation of the luminal membrane due to a difference in the density. A known capillary luminal membrane protein, glucose transporter 1 (GLUT1), was also enriched in the luminal membrane fraction, thus confirming the purity of the isolation. These studies provided strong evidence that ABCB1 is highly expressed at the BBB on the luminal side, suggesting it pumps toxic compounds back into the blood.
1.2.3. ABCB1 Function at the BBB and Clinical Importance
The Abcb1a-deficient mice provided definitive evidence that the function of ABCB1 at the BBB was to restrict drug penetration into the brain. Under normal housing conditions, the Abcb1a-deficient mice were phenotypically normal, in both appearance and behavior, until an inadvertent exposure to ivermectin, an agent that is routinely used to eradicate mites, proved lethal. With ABCB1, ivermectin is not a neurotoxin, but in its absence, ivermectin brain levels climb to nearly 100-fold higher in the Abcb1a-deficient mice compared to wild-type mice [22]. Subsequent study showed that ivermectin is transported by both mouse and human ABCB1. Since the serendipitous discovery of ABCB1 critical role in protecting the brain from ivermectin, many more drugs have been shown to be extruded from the brain by ABCB1. Incidentally, an inbred strain of CF1 (Carworth Farms) mice harbor an Abcb1a mutation that renders ABCB1 non-functional [23,24] and a Collie subpopulation has a non-functional ABCB1 due to a −4 bp deletion producing a frameshift and a premature stop codon [25]. These animals were also hypersensitive to ivermectin, indicating functional conservation of ABCB1. Ivermectin has been used to treat humans for a worm infection, referred to as African river blindness; however, it has not been determined if humans that are highly sensitive to this treatment have loss of ABCB1 function. The impact of loss of ABCB1 function in human BBB has not been extensively studied. The exome variant server (http://evs.gs.washington.edu/, accessed on 8 August 2017) shows over 70 single nucleotide polymorphisms (SNPs) in ABCB1 that result in missense mutations. It is conceivable that some of these SNPs result in non-functional ABCB1. Perhaps future studies will use this SNP data as a starting point to prospectively identify individuals with a defective ABCB1 that might be susceptible to drug-induced CNS toxicities.
1.2.4. ABCG2 Expression at the BBB
ABCG2, which was first named the breast cancer resistance protein (BCRP) for its high expression in an anthracycline resistant breast cancer cell line, is an ABC transporter expressed at the BBB. In human brain, ABCG2 transcript and protein was detected [26], and, importantly, immunohistochemistry showed it primarily localized to the luminal side of brain capillaries. This places ABCG2 in the same location as ABCB1 in the BBB, suggesting a similar role in exporting compounds toxic to the CNS.
ABCG2 expression in the developing brain EC of humans is detected early (mid-gestation-22 weeks), and in mid-gestation for mouse and rat (E12.5), but the amount does not appear to increase during development, unlike ABCB1. Although, Abcg2 transcripts increase in brain ECs from adult mice compared to post-natal pups the changes are non-significant [3]. Furthermore, functional assessment of ABCG2 revealed no difference in ABCG2-mediated prazocin transport between adult rats and P21 pups [27]. These results suggest, unlike ABCB1, that there is no developmental upregulation of ABCG2 and that it maintains peak function even early during development.
1.2.5. ABCG2 Function at the BBB; Redundancy with ABCB1?
ABCB1 and ABCG2 might have redundant roles at the BBB, as suggested by, brain capillaries from Abcb1a-deficient mice expressing ~3-fold higher Abcg2 transcripts compared to wild-type mouse brain capillaries [28]. We suggest that such compensation by ABCG2 provides protection of the fetal brain before ABCB1 expression reaches its peak in adulthood. Recently, positron emission tomography (PET) and radiotracers for tariquidar and elacridar revealed functional interplay between ABCB1 and ABCG2 at the human BBB [29]. The CNS distribution of these ABCB1 and ABCG2 inhibitors was assessed in healthy volunteers that differed in ABCG2 function (one group harbored an allele (c.421 C > A) that is functional impaired, the other group was ABCG2 wildtype). Administration of an ABCB1 inhibitor alone did not change the brain distribution of the radiotracers, whereas brain accumulation in the volunteers harboring the c.421 SNP was greater than the volunteers who are wild type for ABCG2. These results demonstrated that for some drugs ABCB1 and ABCG2 are functionally redundant in humans. Moreover, these results demonstrated that inhibition of both transporters is required to achieve effective CNS levels when drugs are substrates for both transporters. Furthermore, these results revealed the potential risk for neurotoxicity among individuals with a non-functional ABCG2 allele.
1.3. Regulation of ABC Transporters at the BBB
Ligand-activated receptors and inflammatory pathways are among those pathways that can elicit a rapid change in expression of drug-efflux transporters at the BBB. Additionally, studies have also suggested that gender affects expression and activity of both ABCB1 and ABCG2. Understanding how these transporters are regulated will provide insight into additional strategies capable of altering BBB integrity and improve the delivery of drugs to the brain.
1.3.1. Ligand-Activated Receptor Signaling
The nuclear receptors are a group of ligand-activated receptors that are activated by endogenous ligands (e.g., hormones and steroids) and xenobiotics [30]. In some cases, ligand binding to the receptor induces a conformational change that initiates receptor translocation to the nucleus where it forms a complex with various co-activators or co-repressors to either induce or repress transcription of target genes [30]. A subset of these nuclear receptors is expressed at the BBB and can regulate ABCB1 and ABCG2 levels.
PXR, arylhydrocarbon receptor (AhR), and constitutive androstene receptor (CAR) can affect both human and mouse ABCB1 expression at the BBB. The antibiotic rifampicin is a potent ligand for human PXR, which binds DNA elements referred to as DR4 (direct-repeat with a 4 base pair spacer). Abcb1 has three DR4 (AG(G/T)TCA) motifs approximately 8 kb upstream of its transcription start site [31]. In transgenic rodents expressing the human PXR, induction by specific PXR activators resulted in elevated Abcb1 mRNA and transport activity [32,33]. This could suggest activation of PXR might be an approach to increase ABCB1-mediated BBB function.
The nuclear receptor constitutive androstane receptor (CAR) is activated by phenobarbital (PB) through a novel kinase cascade, but can also be activated by ligands such as the mouse specific CAR ligand: 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP) or the human CAR ligand: 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxine (CITCO). CAR activation, by either of the aforementioned mechanisms increases Abcb1 and Abcg2 mRNA and transport activity in rat and mouse brain capillaries in vitro and ex vivo [34]. AhR also functions in rat BBB and is capable of increasing ABCB1 and ABCG2 expression [35]. Induction of AhR using 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), an organic pollutant, increased expression and transport activity of ABCB1 and ABCG2 in the brain capillary membrane and was subsequently found to reduce brain accumulation of verapamil, an ABCB1 substrate [35].
The estrogen receptor has been implicated in regulating Abcg2 transcription, specifically estrogen receptor β. Rat brain capillaries exposed to the estrogen receptor ligand, 17β-estradiol showed decreased ABCG2 levels and transport activity in an ex vivo rodent [36]. These results highlight the role of metabolites and xenobiotics in regulating drug-efflux transporters. A caveat is that most of these studies were conducted in rodent models and these pathways might not be conserved in humans. Incidentally, upregulation of ABCB1 and ABCG2 levels and function have been observed by some therapeutic agents (Table 1). For example, taxol has been shown to upregulate ABCB1 by PXR [37]. It remains to be determined how many of these agents (Table 1), use nuclear and/or hormone receptors, to upregulate ABCB1 and ABCG2.
Table 1.
List of chemotherapeutic reagents currently being used for treatment of gliomas and medulloblastoma.
| Drug | Type of Drug/Cancer | Transporter Implicated in Drug Resistance | Model System |
|---|---|---|---|
Temozolomide (TMZ) 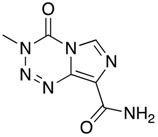
|
Alkylating agent GBM, MB [38] | ABCB1 | Mdr1a−/− mice accumulated higher TMZ in the brain compared to Mdr1a+/+ mice [39] (Mdrla = Abcb1a). |
| ABCB1 | GBM patients with C/C variant at amino acid 1236 of ABCB1 respond better to TMZ than patients with C/T and T/T variants, which results in higher ABCB1 expression [40]. | ||
| ABCG2 | GBM cell lines (U251, A271), cell lines derived from primary tumors (MZ-327, MZ-18), and cell lines from recurrent grade IV tumors (MZ-256, MZ-304) were treated with TMZ. Cell survival was measured by MTT and trypan blue assays. Cells treated with reversan, which inhibits both ABCB1 and ABCG2 showed increased sensitivity to TMZ [41]. | ||
| ABCG2 | GBM cells line (A172, U87, and U373) and neurospheres derived from primary GBM showed increased sensitivity to TMZ in the presence of melatonin. Melatonin treatment affects ABCG2 level via promoter methylation [42]. | ||
Procarbazine 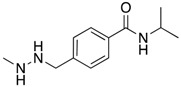
|
Alkylating agent GBM and MB | ABCB1 | Primary tumor GBM cells treated with nimodipine, which blocks ABCB1, showed increased sensitivity to procarbazine in an MTT assay [43]. |
Lomustin/CCNU 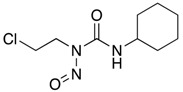
|
Alkylating agent GBM and MB | None reported in GBM/MB context | - |
Carmustin/BCNU 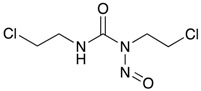
|
Alkylating agent GBM | ABCB1 | GBM cancer stem cell line (U87CS), established by growing U87 GBM cell line in neuronal stem cell condition, showed over 8-fold increase in ABCB1 expression and exhibited greater resistance to BCNU [44]. |
Cyloposphamide 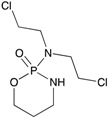
|
Alkylating agent MB | None reported in GBM/MB context | - |
Carboplatin 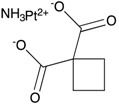
|
Platinum-based drugs. GBM, MB | ABCB1 | GBM cancer stem cell line (U87CS), established by growing U87 GBM cell line in neuronal stem cell condition, showed over 8-fold increase in ABCB1 expression and exhibited greater resistance tow carboplatin [44]. |
Cisplatin 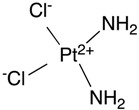
|
Platinum-based drugs. GBM, MB | None reported in GBM/MB context | - |
Etoposide 
|
Topoisomerase inhibitor | ABCB1 ABCG2 |
Patients treated with cyclosporine A, which can inhibit both ABCB1 and ABCG2, had increased systemic exposure to etoposide [45] GBM cancer stem cell line (U87CS), established by growing U87 GBM cell line in neuronal stem cell condition, showed over 8-fold increase in ABCB1 expression and exhibited greater resistance to etoposide [44]. |
| ABCB1 | ABCB1 expression is associated with high-risk MB in large patient cohorts. | ||
| ABCB1 | MB cell lines (DAOY, MED1, MED4, MED4R, MED5R, MED6) express high level of ABCB1. Treatment of these cells with etoposide in combination with ABCB1 inhibitors vardenafil or verapamil sensitizes cells to etoposide measured by clonogenic assay [38]. | ||
Topotecan 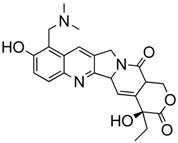
|
Topoisomerase inhibitor MB | ABCG2 | Inhibition of ABCG2 in Group 3 MB tumorspheres by FTC increased sensitivity of topotecan measured by cell titer glow assay and annexin V staining for apoptotic cells [46]; ABCG2 inhibition with Ko143 increased cytotoxicity of topotecan in Group 3 MB preclinical animal model [46]. |
Irinotecan 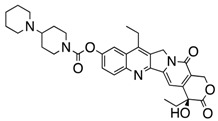
|
Topoisomerase inhibitor MB and GBM | ABCB1 | CPT-11/SN-38 brain accumulation was found to be higher in Mdr1a+/− and Mdr1−/− mice compared to Mdr1a+/+ mice [39]. |
| ABCB1 | Sensitivity towards irinotecan increased in U118, U87, and SK72 GBM cell lines when ABCB1 was inhibited with pitavastatin as measured by alamar blue assay [47]. ABCB1 mRNA level increased with irinotecan concentration [47]. | ||
Mitoxanthrone 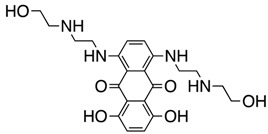
|
Topoisomerase inhibitor MB and GBM | ABCG2 | SF395 human GBM cell line treated with mitoxanthrone resulted in ABCG2 being duplicated in mitoxanthrone-resistant clone [48]. |
| ABCG2 | Group 3 tumorspheres treated with ABCG2 inhibitor, FTC, showed about 3-fold increase in sensitivity to mitoxanthrone [46]. | ||
Vinblastine 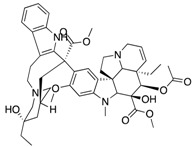
|
Vinca alkaloid/Anti-tubulin MB and GBM | ABCB1 | Mdr1a−/− mice showed 20-fold higher vinblastine level compared to Mdr1a+/+. The most striking difference in drug level was found in the brain [22]. |
Vincristine 
|
Vinca alkaloid/Anti-tubulin MB and GBM | ABCB1 | Rats injected with vincristine showed elevated level of ABCB1 in the brain. Transport activity, monitored by radioactive tracer, 99 mTc-sestamibi, was subsequently found to increase 4-fold 24 h post vincristine treatment [49]. |
Paclitaxel (Taxol) 
|
Anti-microtubule Glioma | ABCB1 | Oral administration of ABCB1 inhibitor, zosuquidar, in mice, increased penetration of paclitaxel in the brain [50]. |
| ABCB1 | Valspodar, an ABCB1 inhibitor, increased accumulation of paclitaxel in the brain of nude mice. Paclitaxel, in combination with valspodar, decreased tumor volume by 90% in nude mice bearing U118 MG glioblastoma [51]. | ||
Veliparib (ABT-888) 
|
PARP1/2 inhibitor GBM | ABCB1 ABCG2 |
Tumors from spontaneous high grade glioma model Pten; p16Ink4a/p19Arf; K-Rasv12 were isolated and implanted into nude mice which then received single dose of TMZ, ABT-888, or both with or without elacridar, a dual inhibitor of ABCB1 and ABCG2. Elacridar enhanced brain penetration of ABT-888. Co administration of elacridar enhances efficacy of TMZ and ABT-888 reduced glioblastoma tumor burden [52]. |
Lomeguatrib (O6Benzylguanine/O6BG) 
|
MGMT inhibitor GBM | ABCB1 | O6BG can compete with Rhodamine 123 and pheophorbide A, substrates of ABCB1 and ABCG2 respectively, in uptake assays, indicating O6BG is a substrate of ABCB1 and ABCG2. |
| ABCG2 | Human glioblastoma GBP61 cells treated with verapamil and Ko143, which inhibit both ABCB1 and ABCG2 increased toxicity of O6BG and TMZ treatment [53]. | ||
Dasatinib 
|
BCR-ABL/SRC kinase/Tyrosine kinase inhibitor (TKI) GBM | ABCB1 ABCG2 |
Abcb1a/b−/−, Abcg2−/−, Abcb1a/b−/−; Abcg2−/− mice received dasatinib by I.P and orally. Mice lacking both ABCB1 and ABCG2 showed higher accumulation in the brain than WT. Comparable level of dasatinib was observed in WT mice treated with ABCB1/ABCG2 dual inhibitor, elacridar and Abcb1a/b−/−; Abcg2−/− mice [54] |
| ABCB1 ABCG2 |
mPDGFβ-induced de novo model of murine GBM induced in WT and Abcb1a/b−/−; Abcg2−/− (KO) mice were given 15 mg/kg dasatinib by oral gavage. Dasatinib level was double in both brain and tumor of KO mice compared to WT. KO mice treated with dasatinib also survived longer than WT [55]; In glioma cell lines from humans and murine models, treatment with elacridar (dual ABCB1 and ABCG2) inhibitor sensitizes cells to dasatinib [55] | ||
Sunitinib 
|
VEGFR, Flk1, PDGFR-α/β inhibitor Tyrosine kinase inhibitor (TKI) GBM | ABCB1 ABCG2 |
MDCK-II cells overexpressing human ABCB1 and ABCG2 showed transport of sunitinib. Addition of elacridar inhibited sunitinib transport. Brain accumulation of sunitinib was found to be significantly high in Abcb1a/b−/−; Abcg2−/− mice compared to WT. Elacridar oral administration in WT cells yields similar level of brain sunitinib concentration to Abcb1a/b−/−; Abcg2−/− mice [56] |
| ABCB1 ABCG2 |
Sunitinib was administered to WT, Abcb1a/b−/−, Abcg2−/−, Abcb1a/b−/−; Abcg2−/− mice by IP infusion. Brain/plasma ratio was found highest in Abcb1a/b−/−; Abcg2−/− mice. Treatment with elacridar (dual ABCB1/ABCG2) inhibitor gave a brain/plasma ratio similar to Abcb1a/b−/−; Abcg2−/− mice [57]. Zosuquidar (ABCB1 specific inhibitor) only moderately increased sunitinib brain concentration while and Ko143 (ABCG2 inhibitor) had no effect [57]. | ||
Sorafenib 
|
Tyrosine kinase inhibitor (TKI) GBM | ABCB1 ABCG2 |
WT, Abcb1a/b−/−, Abcg2−/−, Abcb1a/b−/−; Abcg2−/− mice were given sorafenib. Sorafenib accumulation did not increase in the brain of ABCB1-lacking mice, but increased moderately in Abcg2−/−, and most significantly in Abcb1a/b−/−; Abcg2−/− mice. Elacridar (dual ABCB1 and ABCG2 inhibitor) increased brain exposure to sorafenib similar to double knockout mice [58]. |
ABC (ATP-Binding Cassette), GBM (Glioblastoma), MB (Medulloblastoma), MTT (3-(4,5-dimethylthiazol-2-yl)-2,-5-diphenyltetrazolium bromide), BCNU (Bis-chlorethyl-nitrosourea), CCNU (chloroethyl-cyloexyl-nitrosourea), CPT (camptothecin), SN-38 (7-ethyl-10-hydroxycamptothecin, active metabolite of irinotecan), FTC (Fumitremorgin C), ABT (Abbott, now Abbvie), WT (wildtype), mPDGFβ (mouse platelet-derived growth factor beta).
Regulation of ABC transporters at the BBB by developmental pathways also occurs. For example, Wnt signaling appears important as ABCB1 has 7 consensus TCF/LEF binding motifs (CCTTTGA/TA/T) within its promoter. Although speculative, there is concordance between activation of the Wnt pathway and the BBB expression of ABCB1, which suggests that ABCB1 developmentally increased in the BBB to protect nascent CNS development. The Wnt pathway also modulates ABCB1 in other non-BBB ECs [17]. In intestinal EC lines, TCF4, a transcription factor of Wnt signaling, binds the human ABCB1 gene promoter and activates transcription [59]. In the BBB, ABC transporter regulation by the Wnt pathway is conserved in brain ECs from both rats and humans (hCMEC/D3). Blocking the Wnt pathway “destruction complex” (composed of GSK3, Axin, PP2A, and CK1α) using various GSK3 inhibitors, activated the Wnt pathway, in both model systems producing upregulation of ABCB1 mRNA, protein, and a concomitant increase in transport activity [60]. Conversely, shutting off the canonical Wnt pathway with the Wnt inhibitor, Dkk-1 (Dickkopf-related protein 1) strongly reduced ABCB1 and ABCG2 expression. Subsequent studies, using chromatin immunoprecipitation (ChiP) demonstrated that β-catenin bound to the ABCB1 promoter upon activation of canonical Wnt signaling by WntA, correlated with increased ABCB1 mRNA in hCMEC/D3 cells [61]. Furthermore, a non-canonical Wnt signaling pathway mediated by the RhoA/RhoA kinase (RhoAK) might regulate ABC transporters at the BBB as constitutive activation of RhoAK influenced β-catenin mediated transcription of ABCB1 [61]. Together these results suggest Wnt signaling modulates ABCB1 expression and function through both canonical and non-canonical pathways. Modulation of the Wnt pathway might be an approach to alter the function of the BBB (discussed in Section 2.2.4).
Another pathway important for BBB maintenance (described above) is the SHH pathway. A direct role for SHH signaling regulating ABC transporters at the BBB has not been demonstrated. However, some model systems suggest SHH modulates ABC transporters. For instance, activation of the SHH pathway in squamous cell carcinoma, and esophageal and prostate carcinoma cell lines all increased the mRNA and activity of both ABCB1 and ABCG2, as measured by transport using their respective substrates: docetaxel and methotrexate [62]. Likewise, in diffuse large B-cell lymphoma, SHH modulated ABCG2 expression through its main transcriptional activator, GLI1 [63]. A GLI1 binding site is in the ABCG2 promoter and GLI1 knockdown resulted in a reduction in ABCG2 amount [64]. More definitive evidence for SHH signaling modulation of drug efflux transporters at the BBB awaits further study.
1.3.2. Inflammatory Signaling Pathways
In addition to ligand–receptor signaling described above, stress and inflammatory signaling pathways have been reported to modulate the drug-efflux transporters at the BBB. Stress induces secretion of inflammatory cytokines that result in a response mediated by neuroprotective proteins such as nuclear factor erythroid 2-related factor 2 (Nrf2), and nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB). However, the reported effects of stress and inflammatory cytokines on ABC transporters at the BBB are complicated, and depend on the model system and exposure time used.
ECs release inflammatory cytokines during cell injury. To mimic an insult with cytokines, rat brain ECs treated with the pro-inflammatory cytokines, tumor necrosis factor α (TNF-α) and interleukin-1β (IL-1β) exhibited a rapid decline in BBB integrity. This decline in BBB integrity was reversible, in contrast to interleukin-6, appears to permanently damage BBB integrity [65]. Subsequent study in an immortalized rat brain capillary EC line, GPNT, treated with TNF-α from 2 to 96 h showed induction of Abcb1 mRNA over time, but resulted in no induction of ABCB1 protein [66], however, ABCB1 function measured by vinblastine accumulation, gradually increased over the 96 h treatment interval. A caveat: cell lines might not recapitulate primary cells because in another study, using primary rat brain capillaries, showed that Abcb1 mRNA rapidly decreased after TNF-α exposure without altering ABCB1 protein level. This effect was reversible as the activity remained suppressed for 3 h post-treatment, but then gradually recovered [67]. The restoration of the protein during the recovery phase was attributed to activation of the NF-κB via the TNF-R1/NOS/PKC (Nitric oxide signaling/Protein kinase C) pathway.
1.3.3. Gender Bias and Age
Depending upon which transporter is studied, sexual dimorphism in transporter expression exists. However, in many cases, the mechanisms are unknown. Nonetheless, understanding how gender affects expression and function of these transporters may have important pharmacological and toxicological implications. For example, if one gender expresses lower transporter levels, possibly making this gender more prone to drug-induced side effects and toxicity, dosing adjustments may be necessary.
To our knowledge, gender variation in ABC transporters at the BBB has not been reported. However, in non-CNS tissues, gender variation is well known. For example, hepatic ABCB1 was found to be 2.4-fold lower in females compared to males [68]. Conversely, ABCB1 protein levels are higher in female rats compared to male [69]. In rat, kidney ABCB1 level is affected by the menstrual cycle where the highest ABCB1 protein level correlated with the maximum progesterone level. The changes in expression might be related to transcriptional activation or suppression by steroid hormone receptors. For example, testosterone administration reduced ABCB1 protein expression in the liver that resulted in a reduction in hepatic doxorubicin clearance [70]. For ABCG2, hepatic ABCG2 protein levels are higher in male mice compared to female [71]. Sex-dependent physiological states (e.g., pregnancy, lactation, or virginity mice) were not found to contribute to the sexual dimorphism in one study [71]. In rats, Abcg2 mRNA was higher in female brain and in male kidney.
Age also impacted BBB drug-efflux protein expression. Reduced expression and function of ABC transporters, at the BBB, has been a proposed factor in susceptibility to age-associated diseases, such as neurodegenerative diseases. Positron emission tomography (PET) was used to measure uptake of the ABCB1 substrate 11C verapamil in the brain of 35 healthy men and women. Verapamil accumulated to higher levels in the older male cohort (ages 55–70 years) compared to young (ages 20–30 years) and middle age (40–50 years) [72]. This suggested ABCB1 function at the BBB declined with age only in men. In contrast, there was no association between age and verapamil accumulation in women.
No age-related study on the BBB function of human ABCG2 has been reported to date. Nonetheless, studies on other animal models and tissues may still provide valuable insights. Monitoring hepatic Abcg2 mRNA level, in rats, from birth to Day 60, revealed Abcg2 mRNA peaked at seven days postnatal, followed by gradual decrease to Day 60 [73]. On Days 180 and 540, Abcg2 mRNA was further decreased by 4–5 fold relative to Day 60. Overall, females tended to express higher Abcg2 mRNA compared to males. This trend was not apparent at the protein level as males showed higher ABCG2 expression overall.
1.3.4. ABC Transporters and the Blood Brain Barrier in Cancer
The blood–brain barrier is altered in pathologies of the CNS including neurodegenerative diseases, epilepsy, brain cancers and metastatic brain tumors. Growing evidence indicates transporters at the BBB limit drug delivery to the brain, yet among treatments that target ABC transporters at the BBB, many of them have not been as successful as expected [74,75,76]. A possible explanation for this limited success is that drug resistant CNS tumors, expressing upregulated ABC transporters play a significant role in the diminished therapeutic response.
Another important factor to consider is that the function of the BBB might be modified by tumor cells in the micro-environment. The TJ proteins occludin-1 and 3 are downregulated at the BBB in metastatic colon cancer and glioblastoma cells, resulting in a leaky BBB [77]. One potential mechanism to account for leaky BBB: tumors cells modify the vasculature by secreting angiogenic factors, such as VEGF (vascular endothelial growth factor) to induce formation of new blood vessels, which may not share the non-leaky properties of BBB. Another impediment to therapy is that tumor cells can, by expressing export transporters, form a barrier, the blood–tumor barrier (BTB) [78,79,80]. Consistent with this, human brain tumors of various grades and metastatic brain tumors express ABCB1 to create a BTB [81,82].
Tumors of embryonal origin, which includes (non)-small cell lung cancer (NSCLC/SCLC), melanoma, lung, and breast cancers, exhibit a high frequency of brain metastases [83]. Some metastatic brain tumors arising from primary NSCLC/SCLC, breast, and melanoma cells exhibit low ABCB1 expression, which correlates with increased permeability and drug uptake. For instance, paclitaxel accumulated to a greater extent in the metastatic gliomas, areas with low ABCB1 levels, compared to primary glioma as measured by MRI (magnetic resonance imaging) [83]. In brain tumor arising from metastatic breast cancer models, uptake of ABCB1 substrates paclitaxel and doxorubicin was higher than normal brain. Unexpectedly, the cytotoxic effects of these drugs remained relatively low [84]. Studies in metastatic tumors suggest that ABC transporters presence at the BBB and/or BTB of the primary tumors remain a major contributor of drug resistance and that residual ABC transporters function at the BTB is sufficient to limit drugs’ cytotoxic effects.
In the following sections, we review the potential contributions of ABCB1 and ABCG2 in gliomas and medulloblastoma. We will attempt to highlight the disease etiology and progression with respect to ABC transporters levels and functions and whether they affect treatment outcomes.
2. ABC Transporters in Central Nervous System (CNS) Tumors
2.1. Gliomas
2.1.1. Prognosis, Treatment, Cells of Origin
The progenitor cells of glial tumors can be from either the brain or the spinal cord. These include astrocytes that support neuron function, oligodendrocytes that myelinate neurons, microglia that remove dead neurons and pathogens, and ependymal cells that secrete CSF. Astrocytes and microglia contribute to the maintenance and function of the BBB as described earlier. The BBB function is compromised in certain gliomas. Gliomas are histologically classified into the following subtypes: astrocytoma, oligodendrocytoma, oligodendroglioma, and glioblastoma [85]. Gliomas are also divided into four grades based on their cell of origin, each with a different prognosis. Grade I glioma is called pilocytic astrocytoma, typically seen in pediatric population and is relatively benign. Grade II glioma, also called low-grade glioma, includes astrocytoma, oligodendroglioma, and mixed oligoastrocytoma, typically occurring in young adults. Grade II glioma can evolve into a more aggressive tumor. Grade III glioma, called malignant glioma, includes anaplasticastrocytoma, anaplastic with recurrent properties and oligodendroglioma, and anaplastic mixed oligoastrocytoma. These tumors have invasive properties and are more aggressive than grade II tumors. Grade IV glioma, referred to as glioblastoma multiforme (GBM) is the most aggressive and common primary brain tumor in adults. Treatment regimens for gliomas start by the surgical resection of as much tumor possible, followed by radiation therapy to eradicate remaining tumor cells. In grade III glioma, chemotherapy may be given after radiation therapy, whereas in GBM, due to its aggressive nature, chemotherapy is given in combination with radiation therapy. Recently, the 2016 World Health Organization Classification of tumors of CNS further categorized gliomas into subgroups based on distinct molecular signatures (e.g., IDH (isocitrate dehydrogenase)) mutation status that underlie tumorigenesis [85].
2.1.2. Glioblastoma
Glioblastoma is the most lethal form of brain tumor in adults and about 50% of patients treated with the alkylating agent temozolomide (TMZ) are refractory to this treatment. In one of the largest phase II trials enrolling 162 anaplastic astrocytoma and glioblastoma patients, TMZ treatment resulted in a six-month overall survival rate of 75% with a median overall survival of 13.6 months, a marked improvement over the expected 6–9 months median survival. The overall response rate for this trial was 62% [86]. Furthermore, glioblastoma patients TMZ proved to be better than procarbazine monotherapy at first relapse [87]. In 2005, after a number of clinical trials found TMZ significantly increased survival, it was combined with radiotherapy to become the standard of care for glioblastoma treatment. Comparative analysis of large datasets, available from the Surveillance, Epidemiology, and End Results (SEER) Program, showed that TMZ addition to the radiation therapy regimen prolonged median survival times from 12.0 months to 14.2 months [88]. Prior to this modification, glioblastoma survival had not improved between 1980s and 2001. While TMZ has had a major impact on glioblastoma survival, improving elderly patient survival still remains a challenge. Their median survival is less than 15 months and many patients experience recurring tumors. In addition to O6−methylguanine methyltransferase (MGMT) status, treatment-refractory glioma stem cells (GSCs) are thought to be responsible for tumor recurrence. Compared to non-stem glioma cells, GSCs are more resistant to both chemotherapy and radiation. The resistance mechanisms include reduced cellular accumulation of chemotherapeutics, due to an increase in ABC efflux transporter expression, lack of or aberrant DNA repair mechanisms such as an increase in MGMT, which repairs TMZ-induced DNA damage, and upregulation of anti-apoptotic signals [89].
2.1.3. Glioblastoma and ABCB1, ABCG2
In certain glioblastomas, high expression of ABCB1 and ABCG2 has been reported to associate with poor prognosis. A likely explanation is that transporters that limit drug accumulation are upregulated in the tumor cells thereby limiting drug uptake. Conceivably, ABCG2 might also have a biological role in tumor progression by affecting tumor stem cells. It is worth noting that ABCG2, while mainly discussed here for its function at the BBB as a drug transporter, is highly expressed in many stem cell compartments including GSCs. GSCs are considered to be more resistant to TMZ treatment; therefore, GSCs that survived the treatment are likely the cause of the recurrence often seen in patients.
We took advantage of the publicly available databases to evaluate whether ABCB1 and ABCG2 genes were altered, by mutation or amplification, in CNS tumors, glioblastoma and medulloblastoma (the most common pediatric CNS tumor- will be described in details later). Interrogating the cBioportal (http://www.cbioportal.org/, accessed on 23 July 2017) and PeCan data portals (https://pecan.stjude.org/home, accessed on 23 July 2017), 10 mutations in ABCB1 and three in ABCG2 genes in were identified in glioblastoma alone (Figure 2 and Figure 3). Although the functional impact of these mutations has not been formally tested, it is worth noting that one mutation in ABCB1 produces a frameshift and an early stop codon, suggesting that this particular allele is an inactivating mutation. Interestingly, most of the mutations are clustered in the nucleotide-binding domains (NBDs) of both ABCG2 and ABCB1 (Figure 2 and Figure 3). ABC transporters use the binding and hydrolysis of ATP molecules at NBDs as the energy to induce both the conformational changes that facilitate substrate translocation and that re-set the transporters back to their original conformation. Mutations in the NBD can de-stabilize ABC transporters. Further studies are needed to determine the impact of these mutations on expression and transport function of these proteins. cBioportal also compiled data on the genomic amplification status of these genes. For ABCB1, as much as ~4% of the glioblastoma patients carried mutations and another 2.5% of the patients carried amplification of the gene (Figure 4A, TCGA). TCGA is the largest dataset available (500+ samples) so this might account for its power to discover genomic alterations compared to other studies. Unlike ABCB1, alterations in the ABCG2 gene rarely appear, with less than 1% of patients carrying either mutations or amplification.
Figure 2.

ABCB1 mutations found in gliomas and medulloblastoma patients. (Top) Patient mutations in various regions of ABCB1 protein. Mutations are color-coded based on cancer and mutation type. Black (Lower-grade glioma), green (glioblastoma), purple (medulloblastoma). Brown (missense mutation), red (frameshift/deletion). (Bottom) ABCB1 patient mutations mapped on mouse ABCB1 (PDB (Protein Data Bank): 49qh). Spheres highlight the α-carbon of the corresponding amino acid. Disordered regions containing mutation sites that were not visible in the structure are represented as dashed lines in red. TMD, transmembrane domain; NBD, nucleotide-binding domain.
Figure 3.

ABCG2 mutations found in gliomas and medulloblastoma patients. (Top) Patient mutations in various regions of ABCG2 protein. Mutations are color-coded based on cancer and mutation type. Black (Lower-grade glioma), green (glioblastoma), purple (medulloblastoma). Brown (missense mutation), red (frameshift/deletion). (Bottom) ABCG2 patient mutations mapped on human ABCG2 (PDB (protein data bank): 5nj3). Spheres highlight the α-carbon of the corresponding amino acid. Disordered regions containing mutation sites that were not visible in the structure are represented as dashed lines in red. G2 and G2’ refer to individual monomer polypeptides of dimeric ABCG2.
Figure 4.

BBB ABC transporters and gliomas. (A) Frequency of ABCB1; and (B) frequency of ABCG2 mutations and amplifications depicted glioma patient cohort data deposited in cBioportal. ABCB1 RNA expression data curated from the Oncomine database in normal tissue as described and different subtypes of gliomas plotted on Tukey plots from: (C) Shai [90]; (D) French [91]; and (E) Lee studies [92]. (F) DNA copy number levels of ABCB1 in brain and glioma samples in TCGA study. (G) Overall survival of glioma patients with ABCB1 DNA copy number higher (red) or lower (blue) than median is shown. (H) DNA copy number levels from Astrocytoma in Kotliarov study (Oncomine) for patients who were alive or dead at three-year follow up time point. (I) Overall survival of glioma patients with ABCG2 DNA copy number higher (red) or lower (blue) than median. p values were examined by 2-Way ANOVA for (C) and by two-tailed student t-test for (D–F,H), *, p < 0.05; **, p < 0.01; ****, p < 0.0001. Tukey plot whiskers (fences) extend to the most extreme data point that is no more than 1.5 times the interquartile range. The individually plotted values are outliers and the boxed limits cover the lower and upper quartiles with a line representing the median.
Neither ABCB1 nor ABCG2 expression was detected in the brain parenchyma under normal conditions, specifically astrocytes and glial cells, which are cells of origin for some gliomas. Interestingly, ABCB1 mRNA levels were higher in some gliomas than the control brain (Oncomine.org, accessed on 23 July 2017; Figure 4C–E) [90,91,92]. This increase in mRNA corresponded with a higher DNA copy number (TCGA dataset, Figure 4F). Of note, higher than median ABCB1 DNA copy number was also associated with significantly shorter survival in this dataset (Figure 4G). In another dataset from astrocytoma (the Kotliarov study [93]) patients who died by three-year follow-up had a significantly higher DNA copy number (Figure 4H). Although there was no difference in ABCG2 copy number in normal brain and glioma samples in the TCGA study, higher than median ABCG2 copy number was still significantly associated with poor overall survival (Figure 4I). It is important to note that mRNA levels of these genes were not significantly associated with overall survival status.
2.1.4. ABCB1 and ABCG2: Impact upon Temozolomide (TMZ) Therapy
The alkylating agent TMZ is the drug of choice in treating glioblastoma and the contribution of ABCB1 and ABCG2 to its efficacy has been widely studied. One study found increased TMZ accumulation in the tumors of mouse GBM model when the dual ABCB1/ABCG2 inhibitor, elacridar was used or in mice that were deficient for both genes, suggesting that TMZ is extruded from the brain by these transporters [52]. They also found that ABCG2 was likely to provide additional protection due to its high expression in tumor capillaries (blood–tumor barrier).
ABCG2 protein has been detected in both mouse models of glioblastoma and human glioblastoma samples by IHC (Immunohistochemistry) [52]. However, the intensity of immune-staining was heterogeneous, which might reflect the GSCs, noted for their high expression of ABCG2. Importantly, GSCs reportedly are refractory to TMZ treatment compared to non-GSC glioblastoma cells. Another study evaluated ABCG2 status in 33 human primary glial tumors from patients with refractory epilepsy [94]. Very robust ABCG2 expression was detected in anaplastic astrocytoma and glioblastoma patient samples by Western blot and modest level in grade II astrocytoma. Unexpectedly, IHC revealed only ABCG2 staining in the tumors microvessels, but not in the tumor cells. The authors proposed that the apparent increase in tumor ABCG2 expression was actually due to increased expression in the tumor vasculature, not the tumor. However, these studies overlooked the possibility that a rare GSC expressing ABCG2 can affect treatment outcome. In glioblastoma cell lines, TMZ treatment increases the side population (SP) [95], a phenotype characterized by high ABCG2 function and an enrichment of stem cells. Incidentally, TMZ has also been reported to increase ABCB1 expression and transcription via EGFR (Epidermal Growth Factor Receptor). These SP cells were more resistant to TMZ suggesting that ABCG2 might have a role in TMZ sensitivity. Expression of ABCG2 appears related to survival in glioblastoma patients. In a study of 50 human glioblastoma patients high ABCG2 levels were associated with much worse survival with an adjusted hazard ratio of 2.35 [96]. Furthermore, treating the patient-derived glioblastoma tumor spheres with an ABCG2 inhibitor reduced self-renewal of these cells suggesting that ABCG2 is important for maintaining their stemness [97]. Despite these findings, some studies showed that ABCG2 knockdown does not affect TMZ sensitivity in glioma cell lines. This discrepancy in the role of ABCG2 in affecting TMZ sensitivity might be due to inherent differences between in vivo and in vitro model systems.
In contrast to ABCG2, ABCB1 protein amounts in 60 CNS tumor samples, including several gliomas, revealed no significant change in expression compared to the normal brain parenchyma. However, some TMZ-resistant glioblastoma cells exhibit increased levels of ABCB1 and ABCG2. In these cells, the SHH pathway was upregulated as a result of PTCH1 repression by the microRNA, miR-9. Expression of miR-9, in U87 and T98G glioblastoma cell lines, was associated with increased amounts of ABCB1 and ABCG2 mRNA and protein [98]. This microRNA might mediate ABCB1 and ABCG2 upregulation. Strikingly, a link between ABCB1 and glioblastoma treatment efficacy came from an analysis of ABCB1 SNPs in TMZ-treated glioblastoma patients. In this cohort of 112 patients, three SNPs C1236T, G2677T, and C3435T were assessed for an association with the two-year overall survival [40]. Multivariate analysis revealed that the C1236 genotype was a predictive marker for survival. Patients with the 1236C/C genotype displayed a better two-year survival (37%), than patients with T/C and T/T, which had only 8% and 10% chance of survival, respectively (p = 0.02). The C1236T is reported to be an activating SNP, where ABCB1 transport function was increased for several chemotherapeutics.
2.1.5. Improving Glioblastoma Therapy by Altering Blood–Tumor Barrier (BTB) and BBB/ABC Transporter Function
As outlined above (and listed in Table 1), growing evidence highlights how glioblastoma treatment outcomes, in patients and model systems, are impacted by ABC transporters at the BBB. As such, inhibition of these transporters is an attractive strategy to improve therapeutic efficacy. Given the prominence and substrate overlap between the two major BBB ABC transporters discussed here, dual inhibition of ABCB1 and ABCG2 might be necessary to fully block drug exit from the CNS. For example, the ABCB1 specific inhibitor, zosuquidar (LY335979) enhanced sunitinib brain concentration in mice, but not to the same degree as the dual ABCB1/ABCG2 inhibitor, elacridar [57]. Elacridar was also effective in increasing brain exposure of dasatinib, a tyrosine kinase inhibitor, currently in clinical trials in glioblastoma cell lines and preclinical models [55]. In lower-grade glioma preclinical model, elacridar enhanced the brain penetration and efficacy of TMZ and PARP inhibitor ABT-888 (Abbott, now AbbVie) [52].
In addition to utilizing dual ABCB1 and ABCG2 inhibitors, an assessment of how the BBB impacts a drug’s efficacy can guide the development of better therapeutics to improve clinical outcome. For example, Salphati and coworkers found that the PI3K (Phosphoinositide 3-kinase) inhibitor, GDC-0941, was effective in reducing the tumor volume in the U87 orthotopic glioblastoma mouse model, but not in the GS2 model [99]. The primary difference is that the former lacked an intact BBB, whereas in the latter the BBB was intact; and an important consideration was that GDC-0941 was a substrate of both ABCB1 and ABCG2 [99,100]. By improving the physicochemical properties of GDC-0941, such that it bypassed ABCB1 and ABCG2 efflux, GNE-317 (an improved version of GDC-0941) effectively decreased tumor growth in all glioblastoma models tested, regardless of the BBB integrity and function [99]. Most recently, the authors have demonstrated the effectiveness of GNE-317 for the treatment of brain metastasis arising from melanoma tumors [101]. Furthermore, tumors with an increased BTB permeability also responded well to these drugs, even if they were ABCB1 and ABCG2 substrates. Importantly, metastatic brain tumors with an intact BTB only responded to GNE-317 as it evades the BTB efflux transporters [101]. In cases where drug modification or ABC transporter inhibition is not feasible, alternative approaches might be used: either a drug delivery system such as nanoparticles or CNS-invading pathogens [102].
In total, these studies demonstrate that both BBB integrity and its resident transporters are the key obstacles that must be overcome to achieve efficacious treatment. Targeting the BBB integrity and improving the properties of inhibitors may aid in BBB penetration and improve clinical outcomes, not only for glioblastoma, but also metastatic brain tumors.
2.2. Medulloblastoma
2.2.1. Medulloblastoma Overview
Medulloblastoma (MB) is the most frequent malignant pediatric brain cancer, accounting for about 20% of all childhood brain tumors [103]. Historically, it was stratified based on histological features such as: desmoplastic/nodular, large-cell and anaplastic [104,105,106]. The advent of whole genome sequencing methods permitted the development of a molecular MB classification scheme, compromised of four distinct subgroups: Wingless (Wnt), sonic hedgehog (SHH), Group 3, and Group 4 [107,104]. Each subgroup exhibits signature molecular and transcriptomic features suggesting a distinct cell of origin for each subgroup [108]. These subgroups also have distinct histology, demographics, and clinical outcomes, the later due, in part, to different therapy.
MB therapy typically involves surgical resection, followed by cranio-spinal radiation and chemotherapy. MB can metastasize to the pial surface of the spinal cord and brain. MB subgroup does not typically change at recurrence. In two independent and distinct cohorts, the molecular signature, therefore, subgroup affiliations, of recurrent and metastatic MB tumors remained the same [109]. In contrast, recurring and metastatic gliboblastoma tumors frequently shifts to heterogeneous subclass of tumors [110,111]. MB patients with metastatic tumors are deemed high-risk and receive cranio-spinal radiation of the entire nervous system instead of just the primary tumor site. While frontline treatments of MB have largely been successful, with about 70% survival rate amongst average-risk patients, the long-term side effects include hearing loss, a result of both radiation treatment and platinum chemotherapy, and most severely, neurocognitive impairments. A study of 380, adult survivors of childhood MB and PNET (primitive neuroectodermal tumor) showed 37.4% incidence of hearing loss, and 72.3% of survivors experiencing problems of balance and coordination as well as tremors [112]. Lower educational attainment and social independence were also reported to be pronounced risks associated with current MB treatments [112]. Additional markers might also guide more effective treatment. For instance, β-catenin status is a marker for favorable of outcome in pediatric medulloblastoma [113,114]. For instance, pediatric cases with positive nuclear β-catenin staining exhibited significantly better survival than those with negative nuclear staining and positive cytoplasmic and membrane β-catenin staining. Such predictive markers could also guide treatment choice to reduce the adverse effects of conventional therapies [113]. While lowering the dosage can reduce the side effects of cranio-spinal radiation and chemotherapies on neurocognitive functions [115], the risk in a less intense therapy is a greater likelihood of an unfavorable outcome. Therefore, an improved understanding of the disease at the molecular level will help contribute to the refinement of the current therapies.
2.2.2. Etiology of Medulloblastoma
As embryonal tumors with primitive characteristics, MB have the ability to differentiate into multiple lineages [85]. About 30% of medulloblastoma tumors belong to the sonic hedgehog (SHH) subgroup. They arise as a result of over-proliferation of granule neuron progenitors (GNPs) [116,117]. The GNPs arise from a dorsal hindbrain structure called the rhombic lip [118]. They expand postnatally, receiving proliferative cues from the sonic hedgehog mitogen secreted by purkinje cells [119]. Expansion of GNPs is followed by their outward migration to the external germinal/granule layer (EGL) then inwardly along the radial fibers of Bergmann glial cells to form now differentiated neurons in the inner granule layer (IGL). The precise timing of GNPs expansion and migration ensure proper development of the cerebellum [120].
Aberrant SHH signaling was first linked to medulloblastoma as Gorlin’s syndrome patients, harboring a mutated PTCH1 gene, exhibited an increased incidence of medulloblastoma [121]. Heterozygous Ptch1 mice developed tumors on the surface of the cerebellum, which closely resemble human medulloblastoma [122]. GNP expansion occurs with precise spatiotemporal regulation. This cascade can be disrupted by PTCH1 inactivation by mutation or loss, SMO mutation, and GLI2 amplification, some of these are known drivers of SHH-MB [123]. Commitment to the GNP lineage is a crucial determinant of SHH-MB tumorigenesis as smoothened activation in astrocyte (Gfap+) or oligodendrocyte (Olig+) progenitors gave rise to MB with 100% penetrance and with similar histology and expression signatures as one derived from GNPs [124]. SHH-MB can also arise from GNPs derived from cochlear nuclei of the brain stem [125]. Complete loss of PTCH1 function is not immediately tumor-forming suggesting, temporal acquisition of genetic lesions contributes to the development of SHH-MB. Enforced expression of MYCN proto-oncogene (a known GLI1 target gene) in GNPs isolated from Trp53-null mice results in tumors that closely resemble Ptch1 sonic hedgehog tumors with increased penetrance [126,127].
In contrast, overexpression of c-MYC in GNPs (isolated from Trp53-null mice) enabled the development of the first engineered murine model that reproduced the human Group 3 medulloblastoma [127]. Enforced expression of a partially stabilized T58A MYC mutant with a dominant negative Trp53 in neural stem cells also faithfully recapitulated Group 3 medulloblastoma [128]. Interestingly, activation of GABAergic and photoreceptor pathways is a signature of Group 3 tumors [129,123]. MYC amplification occurs in about 10–20% of Group 3, the highest among the subgroups [104,105]. Interaction of MYC with a binding partner, MIZ1 (MYC-interacting zinc finger protein 1) is required for development of Group 3 tumors [130]. Expression of MYC V394D mutant, with attenuated binding to MIZ1, delayed disease onset and produced tumors with transcriptional profiles that are distinct from both Group 3 and SHH-MB.
The cells of origin for Group 4 are not known to date. Targeted expression of partially stabilized version of MYCN (T28A) in neural stem cells isolated from P0 cerebella resulted in tumors with a Group 4 signature [131,132]. At the transcriptional level, Group 4 tumors, are characterized by activation of neuronal and glutaminergic pathways [123,105]. Group 4 tumors show in 80% of the cases of aberration at i17q (isochromosome 17q). Mutations in chromatin modifiers are common among Group 3 and Group 4, suggesting a shared mechanism for transformation [129]. Efforts to better delineate Group 3 and Group 4 signatures include integration of genomic, transcriptomic and epigenomic analyses [133,134].
In contrast to Group 3 and 4 tumors, which arise from GNPs or neural stem cell populations, Wnt tumors arise from progenitors in the lower rhombic lip of the dorsal brainstem [108]. Similar to SHH tumors, Wnt MB result from constitutively active Wnt signaling. Activating mutations in the CTNNB1 gene have been mapped as a strong driver of Wnt-MB with a mutation rate of 85–95% [135]. Wnt tumors are very responsive therapeutically (see below).
2.2.3. Demographics, Treatments and Clinical Outcome
Medulloblastoma in general are more commonly found in males than females with an overall ratio of 1.5:1 [136,137]. Wnt and SHH tumors have balanced sex ratios whereas there Group 3 and 4 tumors has 2:1 male:female ratios [136,137]. The mechanisms underlying the slight gender bias and whether it contributes to clinical outcome remain unknown. Wnt tumors, which are rarely metastatic, account for about 10% of all MB tumors. Medulloblastoma affects infants, children, and adults. The overall 5 years survival rate for Wnt MB is 95% [113,117]. Although Wnt-MB have mutant TP53, it does not appear tocontribute to clinical outcome (overall 5 year survival rates of 90% and 97% for WT and mutated TP53 respectively) [138].
SHH tumors are more prevalent in infants and adults compared to children. The risk stratification of SHH tumors is influenced by their TP53 status with higher risk associated with p53 germline or somatic mutations. Multivariate analysis deemed TP53 mutation status as the most important risk factor in SHH-MB. Patients without TP53 mutations have 5-year overall survival rate of 81% compared to 41% for mutated TP53 high-risk patients [138]. PTCH1 and SUFU germline mutations are frequently found in infants. Surprisingly infants have the best prognosis compared to children and adults. However, TP53 mutation in children is frequently accompanied with GLI2 and MYCN amplification, conferring a poor clinical outcome. Adult SHH tumors carry the most mutational burden which includes PTCH, SMO, and TERT [139,133]. SHH tumors can metastasize, but recurrence is usually local [109]. FDA (Food and Drug Administration)-approved SMO inhibitors have been included in clinical trials for SHH-driven tumors and they show moderate efficacy, but often transient due to the rise of mutants resistant to SMO inhibition [140,141,142].
Group 3 tumors, which account for 25% of all MB tumors, are rarely found in adults. The overall 5 years survival rate of Group 3 patients is 50% [135]. The outcome depends on MYC status and whether metastases are present at the time of diagnosis with about 50% of cases at the time of diagnosis harboring metastases. Non-metastatic and absence of MYC amplification patients are considered standard risk with 75–90% survival while metastatic tumors, especially with MYC amplification considered very high risk (<50% survival) [143]. With faithful model systems available, promising therapeutics targets have been identified for this MB subgroup and are actively being pursued which includes bromodomain inhibitors, PI3K and histone deactylase (HDAC) inhibitors, pemetrexed, gemcitabine, and CDK (cyclin-dependent kinase) 4/6 inhibitors [144,145].
The remaining 35% of MB tumors belongs to Group 4, which affects all ages, but predominates in children age 3–16 with a slight gender bias towards males. About 35–40% of Group 4 metastatic cases are presented at diagnosis. Gain of chr17 or loss or chr11 is associated with a favorable outcome [117]. Non-metastatic Group 4 patients have a survival rate of 90% [146]. The lack of preclinical model for Group 4 tumors hinders the development of a targeted therapy.
Overall, the subclassification of medulloblastoma, using both clinical and molecular signatures, has proven beneficial in guiding therapy leading to improved prognosis. Nonetheless, a challenge remains for MB patients who do not respond to current therapies. This year, a number of groups proposed further refinements in the classification of medulloblastoma subtypes [134,147]. These in-depth stratifications can solve the heterogeneity within subgroups and is crucial in determining when de-escalation therapy might be beneficial; for example, in subtypes with an excellent prognosis such as Wnt and a subset of the SHH medulloblastoma. For MB patients, that are refractory to current therapies, an improved stratification might guide in the development and selection of therapies that target implicated disease driver pathways, an approach that should improve clinical outcomes.
2.2.4. ABC Transporters and Medulloblastoma
A major challenge to efficacious medulloblastoma therapy is identifying drugs that are not just effectious in cell line models, but also must also be capable of penetrating the blood–brain barrier (BBB). Furthermore, if a drug is able to cross the BBB, the next hurdle is to effectively penetrate the tumor (the blood–tumor barrier, BTB). Drug-efflux transporters, inherently expressed in tumors may restrict drug penetration or tumors acquire increased expression through therapy. Standard chemotherapeutics used in MB treatment include combinations of vincristine, carmustine, procarbazine, prednisone, cyclophosphamide, CCNU (chloroethyl-cyclohexyl-nitrosourea), cisplatin, and carboplatin, many of which are reported to be substrates of ABCB1 and/or ABCG2 in various cell lines and in vivo murine models [148,149] (Table 1). For example, Saridegib (IPI-926), an FDA-approved SMO inhibitor, showed survival advantage for an extremely aggressive SHH-MB mouse model (Ptc1-null GNPs) [150]. Nonetheless, a fraction of these tumors survived despite the initial response to saridegib. This impaired response was attributed to ABCB1, whose expression unexpectedly increased during the 6 weeks of saridegib treatment. Treatment of tumors with an ABCB1 inhibitor, verapamil, enhanced saridegib’s ability to reduce GLI1 level even after extended treatment periods, highlighting the role of ABCB1 in mediating tumor growth in this SHH-MB model. ABCB1 expression is also associated with high-risk MB. Cells lines established from primary and recurrent tumors display high ABCB1 expression and activity [38]. Inhibition of ABCB1 by verapamil and vardenafil sensitize to etoposide, implicating the role of ABCB1 in etoposide export in these medulloblastoma model systems [38].
In addition to high ABC transporter expression in tumors, gain of function mutations of these transporters might account for impaired responses to chemotherapy. To evaluate ABCB1 and ABCG2 for mutations in patients, we used the cBioportal platform with cohorts from four studies at the Broad, ICGC (international cancer genome consortium) , PCGP(pediatric cancer genome project), and Sickkids. Although some overlap occurs among the cohorts, somatic mutations in ABCG2 and ABCB1 are rare and only observed at a frequency of 1–2% (Figure 5). The somatic mutations in MB patients were mapped and the α-carbon of the mutant residue is highlighted in purple in the x-ray crystal and cryo-EM (electron-microscopy) structure of mouse ABCB1 and human ABCG2 respectively (Figure 2 and Figure 3). Intriguingly, R467W mutation in ABCB1 lies within the nucleotide-binding domain (NBD), which might alter the nucleotide-dependent activity of the transporter (Figure 2). In contrast, no mutation residing within the NBD was discovered in ABCG2. However, the S302Y mutation is present at the intervening region connecting the NBD and transmembrane domain (TMD) (Figure 3), whereas Q393K lies within the lipid bilayer interphase (Figure 3). Studies to determine whether these mutations affect ABCB1 and ABCG2 expression and function will provide valuable insight into the possible role of these ABC transporters in either the pathogenesis of MB or response to therapy.
Figure 5.

BBB ABC transporters in medulloblastoma. (A) Frequency of ABCB1 and ABCG2 mutations in medulloblastoma patient cohorts data deposited in cBioportal (n = 37 for PCGP (pediatric cancer genome project, n = 46 for Sickkids, n = 92 for broad, n = 125 for ICGC (International Cancer Genome Consortium); Gene expression data from these cohorts were not available for analysis. (B) Expression data for mouse granule neuron progenitor and medulloblastoma tumors from publicly available dataset, GSE33199 [127] shown in a heatmap. (C) ABCB1 MYC binding site identified by ENCODE ChIP assays is highlighted in red circle (UCSC genome browser). MBL, medulloblastoma.
Transcriptional regulation of ABCB1 and ABCG2 is another avenue by which these transporters. Proto-oncogenes MYC and MYCN, both of which are commonly amplified in MB have displayed regulatory effects on ABCB1 and ABCG2 in various models. In CD34+ chronic myeloid leukemia (CML) hematopoietic progenitor cells, ABCG2 expression is positively regulated by MYC [151]. In neuroblastoma cohorts, ChIP analyses revealed that MYC and MYCN can bind at ABCG2 promoter, demonstrating a correlation between level of these transcription factors and ABCG2 mRNA [152]. In the context of medulloblastoma, work by Ingram and coworkers showed MYC and ABCG2 are highly expressed together at the mRNA and protein levels in 10 Gy radiation-resistant MB cells (DAOY and UW228 parental line) [153]. Whether MYC is the one responsible for increasing ABCG2 expression was not explored in this study. Nonetheless, qRT-PCR analysis of tumors resected from pediatric MB patients showed ABCG2 is commonly expressed in pediatric MB tumors and its expression is higher than normal cerebellum [153]. When we assessed publically available mouse medulloblastoma gene expression data [127], a similar trend revealed higher ABCG2 expression in the majority of tumors compared to the cell of origin for MB, the GNPs (Figure 5B). Specifically, high ABCG2 expression clustered with Group 3 tumors, which was engineered by enforced expression of MYC, likely hinting that MYC might be responsible for the high ABCG2 expression as has been previously reported in CML and neuroblastoma. Interestingly, ABCB1 expression was also high in Group 3 tumors (Figure 5B). MYC might also transcriptionally activate ABCB1 as a candidate MYC binding site is reported within the ABCB1 gene (Figure 5C). Overall, given that both of ABCB1 and ABCG2 are upregulated by MYC, MYCN, and Wnt signaling, it is conceivable that they could play a role in not just MB progression, but response to certain drug therapies and by altering their expression affect these regulators, to improve chemotherapeutic response.
2.2.5. Improving Medulloblastoma Therapy by Concurrently Altering the BTB and BBB by Targeting ABC Transporter Function
The low survival rate of Group 3 MB patients indicates an unmet need for an effective therapeutic intervention. Our group first reported that ABC transporter present in the tumor contributes to resistance to chemotherapy in medulloblastoma patients. Specifically, Abcg2 was found to be highly expressed in the murine model of Group 3, (developed enforced expression of MYC in GNPs isolated from Trp53−/−; Cdkn2c−/− mice) which has gene expression signatures that resemble the human counterpart [46]. RNA expression of ABCG2 is high in both human and murine Group 3 MB. These findings supported the idea that Group 3 MB has intrinsically high levels of ABCG2. In the Group 3 model, in vitro studies (with tumorspheres) suggested ABCG2 inhibition might improve the therapeutic response. ABCG2 functions at the plasma membrane where it exports topotecan, one of the drugs used for recurrent MB in Group 3 patients [154]. In vivo, we found that the combination of topotecan with the well-tolerated Ko143, a dual inhibitor of ABCB1 and ABCG2, reduced tumor growth by more than 240% compared to single agents treatment, translating to an increase in survival from 17 to 28 days [46].
A recent study showed altering BBB integrity could improve therapeutic endpoint [155]. Phoenix and coworkers observed significantly more frequent CNS hemorrhaging in Wnt subgroup tumors in both patients and a Wnt mouse model (Blbp-Cre+/−; Ctnnb1+/lox(ex3); Trp53+/flx mouse model) compared to other subgroups. Both human and mouse Wnt tumors had 3–4 times marker stainings that are indicative of aberrant vasculature and compromised BBB. Transcriptomic analysis of EC from Wnt tumors revealed downregulation of typical BBB endothelial cell markers such as CLDN5 (Claudin5) and SLC2A1 (Solute carrier family 2 member 1), which was confirmed by an accumulation of Tetramethylrhodamine-dextran(TMR-dextran) in Wnt tumors compared to SHH tumors, signifying that Wnt tumors have a leaky BBB. This BBB leakiness, specific to Wnt MB tumors, is a result of high levels of soluble Wnt pathway inhibitors, Dickkopf 1 (Dkk1) and Wnt inhibitor factor 1 (Wif1), both of which were absent in SHH and Group 3 tumors. The authors reasoned that the high level of Wnt inhibitor is a result of constitutive paracrine Wnt signaling induced by β-catenin mutation, secreting Wnt inhibitors as a negative feedback loop. Indeed, nuclear LEF1 was specifically absent in the endothelial cells of Wnt tumors, indicating Wnt signaling was not active within the tumor, but active everywhere else. To test the role of Wnt signaling in regulating BBB phenotype in medullobastoma, the authors enforced expression of Wnt inhibitors, Wif1 and Dkk1 in mouse SHH-MB model and enforced activation of Wnt signaling in Wnt MB tumor by expressing Wnt7a agonist. Restoration of endothelial Wnt signaling in Wnt-MB tumor restored BBB integrity. Inhibition of Wnt signaling in SHH tumor significantly increased BBB permeability. These manipulations translated into changes in drug penetrance. Tumor concentrations of vincristine were higher in Wnt tumors with the leaky BBB compared to the BBB from tumors engineered to express a Wnt7a agonist. Together, this study highlights the BBB as the major barrier for drug penetration into the brain parenchyma in medulloblastoma.
In the context of ABC transporters, endothelial cells isolated from Wnt-MB tumors exhibited significant reduction in Abcb1 mRNA [155]. As previously described, interplay exists between BBB, ABCG2 and Wnt signaling where β-catenin activation upregulates ABCG2 level. Since ABCB1 and ABCG2 are the major drug-efflux transporters at the BBB, that are highly expressed in Group 3 MB and MYCN-amplified SHH-MB (Figure 4), it would be beneficial to determine if these transporters are expressed at the tumor ECs of the various MB subgroups. Finally, given the availability of SHH and Group 3 preclinical models, pre-treatment or combination of ABCB1 and ABCG2 inhibitors with frontline therapies is worth investigating as it could increase penetration and cytotoxic effects of drugs and improve clinical outcomes (Figure 6).
Figure 6.

Schematic depicting the effect of ABCB1/ABCG2 inhibitors. (Left) Compromised BBB (tight junctions (TJ) impairment, etc.) in certain tumors, such as glioblastoma, increases drug penetration into brain parenchyma. However, ABC transporters (purple, ABCB1; and green, ABCG2) expressed in the tumor cells may extrude chemotherapeutic drugs, thus limiting the drugs effectiveness. (Right) Inhibition of ABC transporters with or without compromised BBB will not only increase drug accumulation in the brain parenchyma but also allow the drugs to accumulate in the tumor and tumor stem cells.
3. Perspective
Therapeutic targeting of CNS cancers remains a challenge. Both the BBB and the BTB have impeded success; our understanding of the latter is nascent and up until recently BTB has not been a point of focus because the BBB seemed to be such a prominent obstacle. Currently, we have only a rudimentary understanding of the BTB and which transporters contribute to therapeutic failure. This is a future avenue of exploration. An ideal current scenario, to advance therapy, might be combination therapy, using an inhibitor that blocked or suppressed the function of both ABCG2 and ABCB1 (depicted in Figure 6).
Acknowledgments
We thank Dan McNamara (SJCRH) for his expert assistance with PyMol to generate structural illustrations. We gratefully acknowledge the Schuetz lab members for their comments and suggestions. This work was supported by National Institutes of Health grants R01CA194057, R01CA194206, NCI P30 CA021745, CA21865, and ALSAC.
Author Contributions
Juwina Wijaya and Yu Fukuda contributed to data generation. John D. Schuetz, Juwina Wijaya and Yu Fukuda contributed to the writing of the manuscript. Juwina Wijaya and Yu Fukuda contributed equally to data generation and the writing of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Saunders N.R., Dreifuss J.-J., Dziegielewska K.M., Johansson P.A., Habgood M.D., Møllgård K., Bauer H.-C. The rights and wrongs of blood-brain barrier permeability studies: A walk through 100 years of history. Front. Neurosci. 2014;8:404. doi: 10.3389/fnins.2014.00404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Motoike T., Loughna S., Perens E., Roman B.L., Liao W., Chau T.C., Richardson C.D., Kawate T., Kuno J., Weinstein B.M., et al. Universal GFP reporter for the study of vascular development. Genesis. 2000;28:75–81. doi: 10.1002/1526-968X(200010)28:2<75::AID-GENE50>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 3.Daneman R., Zhou L., Agalliu D., Cahoy J.D., Kaushal A., Barres B.A. The Mouse Blood-Brain Barrier Transcriptome: A New Resource for Understanding the Development and Function of Brain Endothelial Cells. PLoS ONE. 2010;5:e13741. doi: 10.1371/journal.pone.0013741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Obermeier B., Daneman R., Ransohoff R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013;19:1584–1596. doi: 10.1038/nm.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daneman R., Agalliu D., Zhou L., Kuhnert F., Kuo C.J., Barres B.A. Wnt/β-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc. Natl. Acad. Sci. USA. 2009;106:641–646. doi: 10.1073/pnas.0805165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MacDonald B.T., Tamai K., He X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liebner S., Corada M., Bangsow T., Babbage J., Taddei A., Czupalla C.J., Reis M., Felici A., Wolburg H., Fruttiger M., et al. Wnt/β-catenin signaling controls development of the blood-brain barrier. J. Cell Biol. 2008;183:409–417. doi: 10.1083/jcb.200806024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alvarez J.I., Dodelet-Devillers A., Kebir H., Ifergan I., Fabre P.J., Terouz S., Sabbagh M., Wosik K., Bourbonnière L., Bernard M., et al. The Hedgehog Pathway Promotes Blood-Brain Barrier Integrity and CNS Immune Quiescence. Science. 2011;334:1727–1731. doi: 10.1126/science.1206936. [DOI] [PubMed] [Google Scholar]
- 9.Daneman R., Zhou L., Kebede A.A., Barres B.A. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donahue S., Pappas G.D. The fine structure of capillaries in the cerebral cortex of the rat at various stages of development. Am. J. Anat. 1961;108:331–347. doi: 10.1002/aja.1001080307. [DOI] [PubMed] [Google Scholar]
- 11.Van Horssen H.E., Witte J., Schreibelt E.M., de Vries G. Radical changes in multiple sclerosis pathogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 2011;1812:141–150. doi: 10.1016/j.bbadis.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 12.Olmez I., Ozyurt H. Reactive oxygen species and ischemic cerebrovascular disease. Neurochem. Int. 2012;60:208–212. doi: 10.1016/j.neuint.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 13.Pun P.B.L., Lu J., Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic. Res. 2009;43:348–364. doi: 10.1080/10715760902751902. [DOI] [PubMed] [Google Scholar]
- 14.Uchida Y., Ohtsuki S., Katsukura Y., Ikeda C., Suzuki T., Kamiie J., Terasaki T. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J. Neurochem. 2011;117:333–345. doi: 10.1111/j.1471-4159.2011.07208.x. [DOI] [PubMed] [Google Scholar]
- 15.Cordon-Cardo C., O’Brien J.P., Casals D., Rittman-Grauer L., Biedler J.L., Melamed M.R., Bertino J.R. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc. Natl. Acad. Sci. USA. 1989;86:695–698. doi: 10.1073/pnas.86.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thiebaut F., Tsuruo T., Hamada H., Gottesman M.M., Pastan I., Willingham M.C. Immunohistochemical localization in normal tissues of different epitopes in the multidrug transport protein P170: Evidence for localization in brain capillaries and crossreactivity of one antibody with a muscle protein. J. Histochem. Cytochem. 1989;37:159–164. doi: 10.1177/37.2.2463300. [DOI] [PubMed] [Google Scholar]
- 17.Qin Y., Sato T.N. Mouse multidrug resistance 1a/3 gene is the earliest known endothelial cell differentiation marker during blood-brain barrier development. Dev. Dyn. 1995;202:172–180. doi: 10.1002/aja.1002020209. [DOI] [PubMed] [Google Scholar]
- 18.Gazzin S., Strazielle N., Schmitt C., Fevre-Montange M., Ostrow J.D., Tiribelli C., Ghersi-Egea J.-F. Differential expression of the multidrug resistance-related proteins ABCb1 and ABCc1 between blood-brain interfaces. J. Comp. Neurol. 2008;510:497–507. doi: 10.1002/cne.21808. [DOI] [PubMed] [Google Scholar]
- 19.Gazzin S., Berengeno A.L., Strazielle N., Fazzari F., Raseni A., Ostrow J.D., Wennberg R., Ghersi-Egea J.-F., Tiribelli C. Modulation of Mrp1 (ABCc1) and Pgp (ABCb1) by Bilirubin at the Blood-CSF and Blood-Brain Barriers in the Gunn Rat. PLoS ONE. 2011;6:e16165. doi: 10.1371/journal.pone.0016165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goralski K.B., Acott P.D., Fraser A.D., Worth D., Sinal C.J. Brain Cyclosporin a Levels Are Determined by Ontogenic Regulation of Mdr1a Expression. Drug Metab. Dispos. 2005;34:288–295. doi: 10.1124/dmd.105.007427. [DOI] [PubMed] [Google Scholar]
- 21.Beaulieu É., Demeule M., Ghitescu L., Béliveau R. P-glycoprotein is strongly expressed in the luminal membranes of the endothelium of blood vessels in the brain. Biochem. J. 1997;326:539–544. doi: 10.1042/bj3260539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schinkel A.H., Smit J.J.M., van Tellingen O., Beijnen J.H., Wagenaar E., van Deemter L., Mol C.A.A.M., van der Valk M.A., Robanus-Maandag E.C., te Riele H.P., et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 1994;77:491–502. doi: 10.1016/0092-8674(94)90212-7. [DOI] [PubMed] [Google Scholar]
- 23.Lankas G.R., Cartwright M.E., Umbenhauer D. P-Glycoprotein Deficiency in a Subpopulation of CF-1 Mice Enhances Avermectin-Induced Neurotoxicity. Toxicol. Appl. Pharmacol. 1997;143:357–365. doi: 10.1006/taap.1996.8086. [DOI] [PubMed] [Google Scholar]
- 24.Umbenhauer D.R., Lankas G.R., Pippert T.R., Wise L.D., Cartwright M.E., Hall S.J., Beare C.M. Identification of a P-Glycoprotein-Deficient Subpopulation in the CF-1 Mouse Strain Using a Restriction Fragment Length Polymorphism. Toxicol. Appl. Pharmacol. 1997;146:88–94. doi: 10.1006/taap.1997.8225. [DOI] [PubMed] [Google Scholar]
- 25.Mealey K.L., Bentjen S.A., Gay J.M., Cantor G.H. Ivermectin sensitivity in collies is associated with a deletion mutation of the mdr1 gene. Pharmacogenetics. 2001;11:727–733. doi: 10.1097/00008571-200111000-00012. [DOI] [PubMed] [Google Scholar]
- 26.Cooray H.C., Blackmore C.G., Maskell L., Barrand M.A. Localisation of breast cancer resistance protein in microvessel endothelium of human brain. Neuroreport. 2002;13:2059–2063. doi: 10.1097/00001756-200211150-00014. [DOI] [PubMed] [Google Scholar]
- 27.Harati R., Benech H., Villégier A.S., Mabondzo A. P-Glycoprotein, Breast Cancer Resistance Protein, Organic Anion Transporter 3, and Transporting Peptide 1a4 during Blood–Brain Barrier Maturation: Involvement of Wnt/β-Catenin and Endothelin-1 Signaling. Mol. Pharm. 2013;10:1566–1580. doi: 10.1021/mp300334r. [DOI] [PubMed] [Google Scholar]
- 28.Cisternino S., Mercier C., Bourasset F., Roux F., Scherrmann J.-M. Expression, Up-Regulation, and Transport Activity of the Multidrug-Resistance Protein Abcg2 at the Mouse Blood-Brain Barrier. Cancer Res. 2004;64:3296–3301. doi: 10.1158/0008-5472.CAN-03-2033. [DOI] [PubMed] [Google Scholar]
- 29.Bauer M., Römermann K., Karch R., Wulkersdorfer B., Stanek J., Philippe C., Maier-Salamon A., Haslacher H., Jungbauer C., Wadsak W., et al. Pilot PET Study to Assess the Functional Interplay Between ABCB1 and ABCG2 at the Human Blood-Brain Barrier. Clin. Pharmacol. Ther. 2016;100:131–141. doi: 10.1002/cpt.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi Y. Orphan nuclear receptors in drug discovery. Drug Discov. Today. 2007;12:440–445. doi: 10.1016/j.drudis.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geick A., Eichelbaum M., Burk O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J. Biol. Chem. 2001;276:14581–14587. doi: 10.1074/jbc.M010173200. [DOI] [PubMed] [Google Scholar]
- 32.Bauer B.O., Hartz A.M.S., Fricker G., Miller D.S. Pregnane X Receptor Up-Regulation of P-Glycoprotein Expression and Transport Function at the Blood-Brain Barrier. [(accessed on 13 August 2017)]; doi: 10.1124/mol.66.3.. Available online: http://molpharm.aspetjournals.org. [DOI] [PubMed]
- 33.Bauer B., Yang X., Hartz A.M.S., Olson E.R., Zhao R., Kalvass J.C., Pollack G.M., Miller D.S. In vivo activation of human pregnane X receptor tightens the blood-brain barrier to methadone through P-glycoprotein up-regulation. Mol. Pharmacol. 2006;70:1212–1219. doi: 10.1124/mol.106.023796. [DOI] [PubMed] [Google Scholar]
- 34.Wang X., Sykes D.B., Miller D.S. Constitutive Androstane Receptor-Mediated Up-Regulation of ATP-Driven Xenobiotic Efflux Transporters at the Blood-Brain Barrier. Mol. Pharmacol. 2010;78:376–383. doi: 10.1124/mol.110.063685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang X., Hawkins B.T., Miller D.S. Aryl hydrocarbon receptor-mediated up-regulation of ATP-driven xenobiotic efflux transporters at the blood-brain barrier. FASEB J. 2011;25:644–652. doi: 10.1096/fj.10-169227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mahringer A., Fricker G. BCRP at the Blood–Brain Barrier: Genomic Regulation by 17β-Estradiol. Mol. Pharm. 2010;7:1835–1847. doi: 10.1021/mp1001729. [DOI] [PubMed] [Google Scholar]
- 37.Synold T.W., Dussault I., Forman B.M. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat. Med. 2001;7:584–590. doi: 10.1038/87912. [DOI] [PubMed] [Google Scholar]
- 38.Othman R.T., Kimishi I., Bradshaw T.D., Storer L.C., Korshunov A., Pfister S.M., Grundy R.G., Kerr I.D., Coyle B. Overcoming multiple drug resistance mechanisms in medulloblastoma. Acta Neuropathol. Commun. 2014;2:57. doi: 10.1186/2051-5960-2-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldwirt L., Beccaria K., Carpentier A., Farinotti R., Fernandez C. Irinotecan and temozolomide brain distribution: A focus on ABCB1. Cancer Chemother. Pharmacol. 2014;74:185–193. doi: 10.1007/s00280-014-2490-0. [DOI] [PubMed] [Google Scholar]
- 40.Schaich M., Kestel L., Pfirrmann M., Robel K., Illmer T., Kramer M., Dill C., Ehninger G., Schackert G., Krex D. A MDR1 (ABCB1) gene single nucleotide polymorphism predicts outcome of temozolomide treatment in glioblastoma patients. Ann. Oncol. 2009;20:175–181. doi: 10.1093/annonc/mdn548. [DOI] [PubMed] [Google Scholar]
- 41.Tivnan A., Zakaria Z., O’Leary C., Kogel D., Pokorny J.L., Sarkaria J.N., Prehn J.H. Inhibition of multidrug resistance protein 1 (MRP1) improves chemotherapy drug response in primary and recurrent glioblastoma multiforme. Front. Neurosci. 2015;9:218. doi: 10.3389/fnins.2015.00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martín V., Sanchez-Sanchez A.M., Herrera F., Gomez-Manzano C., Fueyo J., Alvarez-Vega M.A., Antolín I., Rodriguez C. Melatonin-induced methylation of the ABCG2/BCRP promoter as a novel mechanism to overcome multidrug resistance in brain tumour stem cells. Br. J. Cancer. 2013;108:2005–2012. doi: 10.1038/bjc.2013.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Durmaz R., Deliorman S., Uyar R., Işiksoy S., Erol K., Tel E. The effects of anticancer drugs in combination with nimodipine and verapamil on cultured cells of glioblastoma multiforme. Clin. Neurol. Neurosurg. 1999;101:238–244. doi: 10.1016/S0303-8467(99)00061-X. [DOI] [PubMed] [Google Scholar]
- 44.Nakai E., Park K., Yawata T., Chihara T., Kumazawa A., Nakabayashi H., Shimizu K. Enhanced MDR1 Expression and Chemoresistance of Cancer Stem Cells Derived from Glioblastoma. Cancer Investig. 2009;27:901–908. doi: 10.3109/07357900801946679. [DOI] [PubMed] [Google Scholar]
- 45.Lum B.L., Kaubisch S., Yahanda A.M., Adler K.M., Jew L., Ehsan M.N., Brophy N.A., Halsey J., Gosland M.P. Alteration of etoposide pharmacokinetics and pharmacodynamics by cyclosporine in a phase I trial to modulate modulate multidrug resistance. J. Clin. Oncol. 1992;10:1635–1642. doi: 10.1200/JCO.1992.10.10.1635. [DOI] [PubMed] [Google Scholar]
- 46.Morfouace M., Cheepala S., Jackson S., Fukuda Y., Patel Y.T., Fatima S., Kawauchi D., Shelat A.A., Stewart C.F., Sorrentino B.P., et al. ABCG2 Transporter Expression Impacts Group 3 Medulloblastoma Response to Chemotherapy. Cancer Res. 2015;75:3879–3889. doi: 10.1158/0008-5472.CAN-15-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang P.F., Mukthavavam R., Chao Y., Bharati S., Fogal V., Pastorino S., Cong X., Nomura N., Gallagher M., Vali S., et al. Novel anti-glioblastoma agents and therapeutic combinations identified from a collection of FDA approved drugs. J. Transl. Med. 2014;12 doi: 10.1016/j.canlet.2005.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rao V.K., Wangsa D., Robey R.W., Huff L., Honjo Y., Hung J., Knutsen T., Ried T., Bates S.E. Characterization of ABCG2 gene amplification manifesting as extrachromosomal DNA in mitoxantrone-selected SF295 human glioblastoma cells. Cancer Genet. Cytogenet. 2005;160:126–133. doi: 10.1016/j.cancergencyto.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 49.Balayssac D., Cayre A., Authier N., Bourdu S., Penault-Llorca F., Gillet J.P., Maublant J., Eschalier A., Coudore F. Patterns of P-glycoprotein activity in the nervous system during vincristine-induced neuropathy in rats. J. Peripher. Nerv. Syst. 2005;10:301–310. doi: 10.1111/j.1085-9489.2005.10308.x. [DOI] [PubMed] [Google Scholar]
- 50.Kemper E.M., van Zandbergen A.E., Cleypool C., Mos H.A., Boogerd W., Beijnen J.H., van Tellingen O. Increased Penetration of Paclitaxel into the Brain by Inhibition of P-Glycoprotein. Clin. Cancer Res. 2003;9:2847–2855. [PubMed] [Google Scholar]
- 51.Fellner S., Bauer B., Miller D.S., Schaffrik M., Fankhänel M., Spruss T., Bernhardt G., Graeff C., Färber L., Gschaidmeier H., et al. Transport of paclitaxel (Taxol) across the blood-brain barrier in vitro and in vivo. J. Clin. Investig. 2002;110:1309–1318. doi: 10.1172/JCI0215451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin F., de Gooijer M.C., Roig E.M., Buil L.C.M., Christner S.M., Beumer J.H., Eurdinger T.W., Beijnen J.H., van Tellingen O. ABCB1, ABCG2, and PTEN determine the response of glioblastoma to temozolomide and ABT-888 therapy. Clin. Cancer Res. 2014;20:2703–2713. doi: 10.1158/1078-0432.CCR-14-0084. [DOI] [PubMed] [Google Scholar]
- 53.Tomaszowski K.H., Schirrmacher R., Kaina B. Multidrug Efflux Pumps Attenuate the Effect of MGMT Inhibitors. Mol. Pharm. 2015;12:3924–3934. doi: 10.1021/acs.molpharmaceut.5b00341. [DOI] [PubMed] [Google Scholar]
- 54.Lagas J.S., van Waterschoot R.A.B., van Tilburg V.A.C.J., Hillebrand M.J., Lankheet N., Rosing H., Beijnen J.H., Schinkel A.H. Brain accumulation of dasatinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by elacridar treatment. Clin. Cancer Res. 2009;15:2344–2351. doi: 10.1158/1078-0432.CCR-08-2253. [DOI] [PubMed] [Google Scholar]
- 55.Agarwal S., Mittapalli R.K., Zellmer D.M., Gallardo J.L., Donelson R., Seiler C., Decker S.A., Santacruz K.S., Pokorny J.L., Sarkaria J.N., et al. Active efflux of Dasatinib from the brain limits efficacy against murine glioblastoma: Broad implications for the clinical use of molecularly targeted agents. Mol. Cancer Ther. 2012;11:2183–2192. doi: 10.1158/1535-7163.MCT-12-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tang S.C., Lagas J.S., Lankheet N.A.G., Poller B., Hillebrand M.J., Rosing H., Beijnen J.H., Schinkel A.H. Brain accumulation of sunitinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by oral elacridar and sunitinib coadministration. Int. J. Cancer. 2012;130:223–233. doi: 10.1002/ijc.26000. [DOI] [PubMed] [Google Scholar]
- 57.Oberoi R.K., Mittapalli R.K., Elmquist W.F. Pharmacokinetic assessment of efflux transport in sunitinib distribution to the brain. J. Pharmacol. Exp. Ther. 2013;347:755–764. doi: 10.1124/jpet.113.208959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lagas J.S., van Waterschoot R.A.B., Sparidans R.W., Wagenaar E., Beijnen J.H., Schinkel A.H. Breast cancer resistance protein and P-glycoprotein limit sorafenib brain accumulation. Mol. Cancer Ther. 2010;9:319–326. doi: 10.1158/1535-7163.MCT-09-0663. [DOI] [PubMed] [Google Scholar]
- 59.Yamada T., Takaoka A.S., Naishiro Y., Hayashi R., Maruyama K., Maesawa C., Ochiai A., Hirohashi S. Transactivation of the Multidrug Resistance 1 Gene by T-Cell Factor 4/β-Catenin Complex in Early Colorectal Carcinogenesis. Cancer Res. 2000;60:4761–4766. [PubMed] [Google Scholar]
- 60.Lim J.C., Kania K.D., Wijesuriya H., Chawla S., Sethi J.K., Pulaski L., Romero I.A., Couraud P.O., Weksler B.B., Hladky S.B., et al. Activation of β-catenin signalling by GSK-3 inhibition increases P-glycoprotein expression in brain endothelial cells. J. Neurochem. 2008;106:1855–1865. doi: 10.1111/j.1471-4159.2008.05537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pinzón-Daza M.L., Salaroglio I.C., Kopecka J., Garzòn R., Couraud P.-O., Ghigo D., Riganti C. The Cross-Talk between Canonical and Non-Canonical Wnt-Dependent Pathways Regulates P-Glycoprotein Expression in Human Blood–Brain Barrier Cells. J. Cereb. Blood Flow Metab. 2014;34:1258–1269. doi: 10.1038/jcbfm.2014.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sims-Mourtada J., Izzo J.G., Ajani J., Chao K.S.C. Sonic Hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene. 2007;26:5674–5679. doi: 10.1038/sj.onc.1210356. [DOI] [PubMed] [Google Scholar]
- 63.Singh R.R., Kunkalla K., Qu C., Schlette E., Neelapu S.S., Samaniego F., Vega F. ABCG2 is a direct transcriptional target of hedgehog signaling and involved in stroma-induced drug tolerance in diffuse large B-cell lymphoma. Oncogene. 2011;30:4874–4886. doi: 10.1038/onc.2011.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gaillard P.J., van der Sandt I.C.J., Voorwinden L.H., Vu D., Nielsen J.L., de Boer A.G., Breimer D.D. Astrocytes Increase the Functional Expression of P-Glycoprotein in an In Vitro Model of The Blood-Brain Barrier. Pharm. Res. 2000;17:1198–1205. doi: 10.1023/A:1026406528530. [DOI] [PubMed] [Google Scholar]
- 65.De Vries H.E., Blom-Roosemalen M.C.M., van Oosten M., de Boer A.G., van Berkel T.J.C., Breimer D.D., Kuiper J. The influence of cytokines on the integrity of the blood-brain barrier in vitro. J. Neuroimmunol. 1996;64:37–43. doi: 10.1016/0165-5728(95)00148-4. [DOI] [PubMed] [Google Scholar]
- 66.Théron D., de Lagerie S.B., Tardivel S., Pélerin H., Demeuse P., Mercier C., Mabondzo A., Farinotti R., Lacour B., Roux F., et al. Influence of tumor necrosis factor-α on the expression and function of P-glycoprotein in an immortalised rat brain capillary endothelial cell line, GPNT. Biochem. Pharmacol. 2003;66:579–587. doi: 10.1016/S0006-2952(03)00340-X. [DOI] [PubMed] [Google Scholar]
- 67.Hartz A.M.S., Miller D.S., Carolina N. Tumor Necrosis Factor α and Endothelin-1 Increase P-Glycoprotein Expression and Transport Activity at the Blood-brain Barrier. Mol. Pharmacol. 2007;71:667–675. doi: 10.1124/mol.106.029512. [DOI] [PubMed] [Google Scholar]
- 68.Schuetz E.G., Furuya K.N., Schuetz J.D. Interindividual variation in expression of P-glycoprotein in normal human liver and secondary hepatic neoplasms. J. Pharmacol. Exp. Ther. 1995;275:1011–1018. [PubMed] [Google Scholar]
- 69.Bebawy M., Chetty M. Gender Differences in P-Glycoprotein Expression and Function: Effects on Drug Disposition and Outcome. Curr. Drug Metab. 2009;10:322–328. doi: 10.2174/138920009788498996. [DOI] [PubMed] [Google Scholar]
- 70.Suzuki T., Zhao Y.L., Nadai M., Naruhashi K., Shimizu A., Takagi K., Takagi K., Hasegawa T. Gender-related differences in expression and function of hepatic P-glycoprotein and multidrug resistance-associated protein (Mrp2) in rats. Life Sci. 2006;79:455–461. doi: 10.1016/j.lfs.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 71.Merino G., van Herwaarden A.E., Wagenaar E., Jonker J.W., Schinkel A.H. Sex-Dependent Expression and Activity of the ATP-Binding Cassette Transporter Breast Cancer Resistance Protein (BCRP/ABCG2) in Liver. Mol. Pharmacol. 2005;67:1765–1771. doi: 10.1124/mol.105.011080. [DOI] [PubMed] [Google Scholar]
- 72.Van Assema D.M.E., Lubberink M., Boellaard R., Schuit R.C., Windhorst A.D., Scheltens P., Lammertsma A.A., van Berckel B.N.M. P-Glycoprotein Function at the Blood–Brain Barrier: Effects of Age and Gender. Mol. Imaging Biol. 2012;14:771–776. doi: 10.1007/s11307-012-0556-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhu Q.-N., Hou W.-Y., Xu S.-F., Lu Y.-F., Liu J. Ontogeny, aging, and gender-related changes in hepatic multidrug resistant protein genes in rats. Life Sci. 2017;170:108–114. doi: 10.1016/j.lfs.2016.11.022. [DOI] [PubMed] [Google Scholar]
- 74.Leonard G.D., Fojo T., Bates S.E. The role of ABC transporters in clinical practice. Oncologist. 2003;8:411–424. doi: 10.1634/theoncologist.8-5-411. [DOI] [PubMed] [Google Scholar]
- 75.Tamaki A., Ierano C., Szakacs G., Robey R.W., Bates S.E. The controversial role of ABC transporters in clinical oncology. Essays Biochem. 2011;50:209–232. doi: 10.1042/bse0500209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Falasca M., Linton K.J. Investigational ABC transporter inhibitors. Expert Opin. Investig. Drugs. 2012;21:657–666. doi: 10.1517/13543784.2012.679339. [DOI] [PubMed] [Google Scholar]
- 77.Wolburg H., Wolburg-Buchholz K., Kraus J., Rascher-Eggstein G., Liebner S., Hamm S., Duffner F., Grote E.-H., Risau W., Engelhardt B. Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol. 2003;105:586–592. doi: 10.1007/s00401-003-0688-z. [DOI] [PubMed] [Google Scholar]
- 78.Bhowmik A., Khan R., Ghosh M.K. Blood brain barrier: A challenge for effectual therapy of brain tumors. Biomed Res. Int. 2015;2015 doi: 10.1155/2015/320941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Van Tellingen O., Yetkin-Arik B., de Gooijer M.C., Wesseling P., Wurdinger T., de Vries H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updates. 2015;19:1–12. doi: 10.1016/j.drup.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 80.Tóth K., Vaughan M.M., Peress N.S., Slocum H.K., Rustum Y.M. MDR1 P-glycoprotein is expressed by endothelial cells of newly formed capillaries in human gliomas but is not expressed in the neovasculature of other primary tumors. Am. J. Pathol. 1996;149:853–858. [PMC free article] [PubMed] [Google Scholar]
- 81.Fattori S., Becherini F., Cianfriglia M., Parenti G., Romanini A., Castagna M. Human brain tumors: Multidrug-Resistance P-glycoprotein expression in tumor cells and intratumoral capillary endothelial cells. Virchows Arch. 2007;451:81–87. doi: 10.1007/s00428-007-0401-z. [DOI] [PubMed] [Google Scholar]
- 82.Demeule M., Shedid D., Beaulieu E., del Maestro R.F., Moghrabi A., Ghosn P.B., Moumdjian R., Berthelet F., Béliveau R. Expression of multidrug-resistance P-glycoprotein (MDR1) in human brain tumors. Int. J. Cancer. 2001;93:62–66. doi: 10.1002/ijc.1306. [DOI] [PubMed] [Google Scholar]
- 83.Gerstner E.R., Fine R.L. Increased permeability of the blood-brain barrier to chemotherapy in metastatic brain tumors: Establishing a treatment paradigm. J. Clin. Oncol. 2007;25:2306–3212. doi: 10.1200/JCO.2006.10.0677. [DOI] [PubMed] [Google Scholar]
- 84.Lockman P.R., Mittapalli R.K., Taskar K.S., Rudraraju V., Gril B., Bohn K.A., Adkins C.E., Roberts A., Thorsheim H.R., Gaasch J.A., et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin. Cancer Res. 2010;16:5664–5678. doi: 10.1158/1078-0432.CCR-10-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Louis D.N., Perry A., Reifenberger G., von Deimling A., Figarella-Branger D., Cavenee W.K., Ohgaki H., Wiestler O.D., Kleihues P., Ellison D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016;131:803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 86.Agarwala S.S., Kirkwood J.M. Temozolomide, a Novel Alkylating Agent with Activity in the Central Nervous System, May Improve the Treatment of Advanced Metastatic Melanoma. Oncologist. 2000;5:144–151. doi: 10.1634/theoncologist.5-2-144. [DOI] [PubMed] [Google Scholar]
- 87.Yung W.K.A., Albright R.E., Olson J., Fredericks R., Fink K., Prados M.D., Brada M., Spence A., Hohl R.J., Shapiro W., et al. A phase II study of temozolomide vs. procarbazine in patients with glioblastoma multiforme at first relapse. Br. J. Cancer. 2000;83:588–593. doi: 10.1054/bjoc.2000.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Johnson D.R., O’Neill B.P. Glioblastoma survival in the United States before and during the temozolomide era. J. Neurooncol. 2012;107:359–364. doi: 10.1007/s11060-011-0749-4. [DOI] [PubMed] [Google Scholar]
- 89.Bleau A.M., Huse J.T., Holland E.C. The ABCG2 resistance network of glioblastoma. Cell Cycle. 2009;8:2936–2944. doi: 10.4161/cc.8.18.9504. [DOI] [PubMed] [Google Scholar]
- 90.Shai R., Shi T., Kremen T.J., Horvath S., Liau L.M., Cloughesy T.F., Mischel P.S., Nelson S.F. Gene expression profiling identifies molecular subtypes of gliomas. Oncogene. 2003;22:4918–4923. doi: 10.1038/sj.onc.1206753. [DOI] [PubMed] [Google Scholar]
- 91.French P.J., Swagemakers S.M.A., Nagel J.H.A., Kouwenhoven M.C.M., Brouwer E., van der Spek P., Luider T.M., Kros J.M., van den Bent M.J., Sillevis Smitt P.A. Gene Expression Profiles Associated with Treatment Response in Oligodendrogliomas. Cancer Res. 2005;65:11335–11344. doi: 10.1158/0008-5472.CAN-05-1886. [DOI] [PubMed] [Google Scholar]
- 92.Lee J., Kotliarova S., Kotliarov Y., Li A., Su Q., Donin N.M., Pastorino S., Purow B.W., Christopher N., Zhang W., et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 93.Kotliarov Y., Steed M.E., Christopher N., Walling J., Su Q., Center A., Heiss J., Rosenblum M., Mikkelsen T., Zenklusen J.C., et al. High-resolution global genomic survey of 178 gliomas reveals novel regions of copy number alteration and allelic imbalances. Cancer Res. 2006;66:9428–9436. doi: 10.1158/0008-5472.CAN-06-1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Aronica E., Gorter J.A., Redeker S., van Vliet E.A., Ramkema M., Scheffer G.L., Scheper R.J., van der Valk P., Leenstra S., Baayen J.C., et al. Localization of Breast Cancer Resistance Protein (BCRP) in Microvessel Endothelium of Human Control and Epileptic Brain. Epilepsia. 2005;46:849–857. doi: 10.1111/j.1528-1167.2005.66604.x. [DOI] [PubMed] [Google Scholar]
- 95.Chua C., Zaiden N., Chong K.-H., See S.-J., Wong M.-C., Ang B.-T., Tang C. Characterization of a side population of astrocytoma cells in response to temozolomide. J. Neurosurg. 2008;109:856–866. doi: 10.3171/JNS/2008/109/11/0856. [DOI] [PubMed] [Google Scholar]
- 96.Emery I.F., Gopalan A., Wood S., Chow K., Battelli C., George J., Blaszyk H., Florman J., Yun K. Expression and function of ABCG2 and XIAP in glioblastomas. J. Neurooncol. 2017;133:47–57. doi: 10.1007/s11060-017-2422-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bleau A.M., Hambardzumyan D., Ozawa T., Fomchenko E.I., Huse J.T., Brennan C.W., Holland E.C. PTEN/PI3K/Akt Pathway Regulates the Side Population Phenotype and ABCG2 Activity in Glioma Tumor Stem-like Cells. Cell Stem Cell. 2009;4:226–235. doi: 10.1016/j.stem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Munoz J.L., Rodriguez-Cruz V., Ramkissoon S.H., Ligon K.L., Greco S.J., Rameshwar P., Munoz J.L., Rodriguez-Cruz V., Ramkissoon S.H., Ligon K.L., et al. Temozolomide resistance in glioblastoma occurs by miRNA-9-targeted PTCH1, independent of sonic hedgehog level. Oncotarget. 2015;6:1190–1201. doi: 10.18632/oncotarget.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Salphati L., Heffron T.P., Alicke B., Nishimura M., Barck K., Carano R.A., Cheong J., Edgar K.A., Greve J., Kharbanda S., et al. Targeting the PI3K pathway in the brain—Efficacy of a PI3K inhibitor optimized to cross the blood-brain barrier. Clin. Cancer Res. 2012;18:6239–6248. doi: 10.1158/1078-0432.CCR-12-0720. [DOI] [PubMed] [Google Scholar]
- 100.Salphati L., Lee L.B., Pang J., Plise E.G., Zhang X. Role of P-glycoprotein and breast cancer resistance protein-1 in the brain penetration and brain pharmacodynamic activity of the novel phosphatidylinositol 3-kinase inhibitor GDC-0941. Drug Metab. Dispos. 2010;38:1422–1426. doi: 10.1124/dmd.110.034256. [DOI] [PubMed] [Google Scholar]
- 101.Osswald M., Blaes J., Liao Y., Solecki G., Gömmel M., Berghoff A.S., Salphati L., Wallin J.J., Phillips H.S., Wick W., et al. Impact of Blood-Brain Barrier Integrity on Tumor Growth and Therapy Response in Brain Metastases. Clin. Cancer Res. 2016;22:6078–6087. doi: 10.1158/1078-0432.CCR-16-1327. [DOI] [PubMed] [Google Scholar]
- 102.Banks W.A. From blood–brain barrier to blood–brain interface: New opportunities for CNS drug delivery. Nat. Rev. Drug Discov. 2016;15:275–292. doi: 10.1038/nrd.2015.21. [DOI] [PubMed] [Google Scholar]
- 103.Gajjar A.J., Robinson G.W. Medulloblastoma-translating discoveries from the bench to the bedside. Nat. Rev. Clin. Oncol. 2014;11:714–722. doi: 10.1038/nrclinonc.2014.181. [DOI] [PubMed] [Google Scholar]
- 104.Northcott P.A., Shih D.J.H., Peacock J., Garzia L., Morrissy A.S., Zichner T., Stütz A.M., Korshunov A., Reimand J., Schumacher S.E., et al. Subgroup-specific structural variation across 1000 medulloblastoma genomes. Nature. 2012;488:49–56. doi: 10.1038/nature11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jones D.T.W., Jäger N., Kool M., Zichner T., Hutter B., Sultan M., Cho Y.-J., Pugh T.J., Hovestadt V., Stütz A.M.T., et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100–105. doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Taylor M.D., Northcott P.A., Korshunov A., Remke M., Cho Y.-J., Clifford S.C., Eberhart C.G., Parsons D.W., Rutkowski S., Gajjar A., et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012;123:465–472. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Remke M., Hielscher T., Northcott P.A., Witt H., Ryzhova M., Wittmann A., Benner A., von Deimling A., Scheurlen W., Perry A., et al. Adult medulloblastoma comprises three major molecular variants. J. Clin. Oncol. 2011;29:2717–2723. doi: 10.1200/JCO.2011.34.9373. [DOI] [PubMed] [Google Scholar]
- 108.Gibson P., Tong Y., Robinson G., Thompson M.C., Currle D.S., Eden C., Kranenburg T.A., Hogg T., Poppleton H., Martin J., et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468:1095–1099. doi: 10.1038/nature09587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ramaswamy V., Remke M., Bouffet E., Faria C.C., Perreault S., Cho Y.J., Shih D.J., Luu B., Dubuc A.M., Northcott P.A., et al. Recurrence patterns across medulloblastoma subgroups: An integrated clinical and molecular analysis. Lancet Oncol. 2013;14:1200–1207. doi: 10.1016/S1470-2045(13)70449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Phillips H.S., Kharbanda S., Chen R., Forrest W.F., Soriano R.H., Wu T.D., Misra A., Nigro J.M., Colman H., Soroceanu L., et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 111.Sottoriva A., Spiteri I., Piccirillo S.G.M., Touloumis A., Collins V.P., Marioni J.C., Curtis C., Watts C., Tavaré S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA. 2013;110:4009–4014. doi: 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.King A.A., Seidel K., Di C., Leisenring W.M., Perkins S.M., Krull K.R., Sklar C.A., Green D.M., Armstrong G.T., Zeltzer L.K., et al. Long-term neurologic health and psychosocial function of adult survivors of childhood medulloblastoma/PNET: A report from the Childhood Cancer Survivor Study. Neuro Oncol. 2016;39:242. doi: 10.1093/neuonc/now242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ellison D.W., Onilude O.E., Lindsey J.C., Lusher M.E., Weston C.L., Taylor R.E., Pearson A.D., Clifford S.C. United Kingdom Children’s Cancer Study Group Brain Tumour Committee, β-Catenin status predicts a favorable outcome in childhood medulloblastoma: The United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J. Clin. Oncol. 2005;23:7951–7957. doi: 10.1200/JCO.2005.01.5479. [DOI] [PubMed] [Google Scholar]
- 114.Clifford S.C., Lusher M.E., Lindsey J.C., Langdon J.A., Gilbertson R.J., Straughton D., Ellison D.W. Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell Cycle. 2006;5:2666–2670. doi: 10.4161/cc.5.22.3446. [DOI] [PubMed] [Google Scholar]
- 115.Moxon-Emre I., Bouffet E., Taylor M.D., Laperriere N., Scantlebury N., Law N., Spiegler B.J., Malkin D., Janzen L., Mabbott D. Impact of craniospinal dose, boost volume, and neurologic complications on intellectual outcome in patients with medulloblastoma. J. Clin. Oncol. 2014;32:1760–1768. doi: 10.1200/JCO.2013.52.3290. [DOI] [PubMed] [Google Scholar]
- 116.Lee Y., Miller H.L., Jensen P., Hernan R., Connelly M., Wetmore C., Zindy F., Roussel M.F., Curran T., Gilbertson R.J., et al. A molecular fingerprint for medulloblastoma. Cancer Res. 2003;63:5428–5437. [PubMed] [Google Scholar]
- 117.Ellison D.W., Dalton J., Kocak M., Nicholson S.L., Fraga C., Neale G., Kenney A.M., Brat D.J., Perry A., Yong W.H., et al. Medulloblastoma: Clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011;121:381–396. doi: 10.1007/s00401-011-0800-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Álvarez-Buylla A., Ihrie R.A. Sonic hedgehog signaling in the postnatal brain. Semin. Cell Dev. Biol. 2014;33:105–111. doi: 10.1016/j.semcdb.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wechsler-Reya R.J., Scott M.P. Control of neuronal precursor proliferation in the cerebellum by sonic hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/S0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- 120.Hatten M.E., Alder J., Zimmerman K., Heintz N. Genes involved in cerebellar cell specification and differentiation. Curr. Opin. Neurobiol. 1997;7:40–47. doi: 10.1016/S0959-4388(97)80118-3. [DOI] [PubMed] [Google Scholar]
- 121.Muzio L.L. Nevoid basal cell carcinoma syndrome (Gorlin syndrome) Orphanet J. Rare Dis. 2008;3:32. doi: 10.1186/1750-1172-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Goodrich L.V., Milenković L., Higgins K.M., Scott M.P. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 123.Kool M., Koster J., Bunt J., Hasselt N.E., Lakeman A., van Sluis P., Troost D., Meeteren N.S., Caron H.N., Cloos J., et al. Integrated Genomics Identifies Five Medulloblastoma Subtypes with Distinct Genetic Profiles, Pathway Signatures and Clinicopathological Features. PLoS ONE. 2008;3:e3088. doi: 10.1371/journal.pone.0003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Schüller U., Heine V.M., Mao J., Kho A.T., Dillon A.K., Han Y.-G., Huillard E., Sun T., Ligon A.H., Qian Y., et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008;14:123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Grammel D., Warmuth-Metz M., von Bueren A.O., Kool M., Pietsch T., Kretzschmar H.A., Rowitch D.H., Rutkowski S., Pfister S.M., Schüller U. Sonic hedgehog-associated medulloblastoma arising from the cochlear nuclei of the brainstem. Acta Neuropathol. 2012;123:601–614. doi: 10.1007/s00401-012-0961-0. [DOI] [PubMed] [Google Scholar]
- 126.Zindy F., Uziel T., Ayrault O., Calabrese C., Valentine M., Rehg J.E., Gilbertson R.J., Sherr C.J., Roussel M.F. Genetic alterations in mouse medulloblastomas and generation of tumors de novo from primary cerebellar granule neuron precursors. Cancer Res. 2007;67:2676–2684. doi: 10.1158/0008-5472.CAN-06-3418. [DOI] [PubMed] [Google Scholar]
- 127.Kawauchi D., Robinson G., Uziel T., Gibson P., Rehg J., Gao C., Finkelstein D., Qu C., Pounds S., Ellison D.W., et al. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell. 2012;21:168–180. doi: 10.1016/j.ccr.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pei Y., Moore C.E., Wang J., Tewari A.K., Eroshkin A., Cho Y.J., Witt H., Korshunov A., Read T.A., Sun J.L., et al. An Animal Model of MYC-Driven Medulloblastoma. Cancer Cell. 2012;21:155–167. doi: 10.1016/j.ccr.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Robinson G., Parker M., Kranenburg T.A., Lu C., Chen X., Ding L., Phoenix T.N., Hedlund E., Wei L., Zhu X., et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43–48. doi: 10.1038/nature11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Vo B.T., Wolf E., Kawauchi D., Gebhardt A., Rehg J.E., Finkelstein D., Walz S., Murphy B.L., Youn Y.H., Han Y.-G., et al. The Interaction of Myc with Miz1 Defines Medulloblastoma Subgroup Identity. Cancer Cell. 2016;29:5–16. doi: 10.1016/j.ccell.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Swartling F.J., Grimmer M.R., Hackett C.S., Northcott P.A., Fan Q.-W., Goldenberg D.D., Lau J., Masic S., Nguyen K., Yakovenko S., et al. Pleiotropic role for MYCN in medulloblastoma. Genes Dev. 2010;24:1059–1072. doi: 10.1101/gad.1907510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Swartling F.J., Savov V., Persson A.I., Chen J., Hackett C.S., Northcott P.A., Grimmer M.R., Lau J., Chesler L., Perry A., et al. Distinct Neural Stem Cell Populations Give Rise to Disparate Brain Tumors in Response to N-MYC. Cancer Cell. 2012;21:601–613. doi: 10.1016/j.ccr.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Northcott P.A., Buchhalter I., Morrissy A.S., Hovestadt V., Weischenfeldt J., Ehrenberger T., Gröbner S., Segura-Wang M., Zichner T., Rudneva V.A., et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547:311–317. doi: 10.1038/nature22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cavalli F.M.G., Remke M., Rampasek L., Peacock J., Shih D.J.H., Luu B., Garzia L., Torchia J., Nor C., Morrissy A.S., et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell. 2017;31:737–754. doi: 10.1016/j.ccell.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Northcott P.A., Jones D.T.W., Kool M., Robinson G.W., Gilbertson R.J., Cho Y.-J., Pomeroy S.L., Korshunov A., Lichter P., Taylor M.D., et al. Medulloblastomics: The end of the beginning. Nat. Rev. Cancer. 2012;12:818–834. doi: 10.1038/nrc3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kool M., Korshunov A., Remke M., Jones D.T.W., Schlanstein M., Northcott P.A., Cho Y.-J., Koster J., Meeteren A.S., van Vuurden D., et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012;123:473–484. doi: 10.1007/s00401-012-0958-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Northcott P.A., Dubuc A.M., Pfister S., Taylor M.D. Molecular subgroups of medulloblastoma. Expert Rev. Neurother. 2012;12:871–884. doi: 10.1586/ern.12.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhukova N., Ramaswamy V., Remke M., Pfaff E., Shih D.J.H., Martin D.C., Castelo-Branco P., Baskin B., Ray P.N., Bouffet E., et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J. Clin. Oncol. 2013;31:2927–2935. doi: 10.1200/JCO.2012.48.5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Northcott P.A., Hielscher T., Dubuc A., Mack S., Shih D., Remke M., Al-Halabi H., Albrecht S., Jabado N., Eberhart C.G., et al. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol. 2011;122:231–240. doi: 10.1007/s00401-011-0846-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Robinson G.W., Orr B.A., Wu G., Gururangan S., Lin T., Qaddoumi I., Packer R.J., Goldman S., Prados M.D., Desjardins A., et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. 2015;33:2646–2654. doi: 10.1200/JCO.2014.60.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhai Y., Pierre D., Si R., Deng W., Ferrin P., Nilekar A.U., Peng G., Herron J.A., Bell D.C., Saltsburg H. Smoothened Mutation Confers Resistance to a Hedgehog Pathway Inhibitor in Medulloblastoma. Science. 2010;337:1633–1637. doi: 10.1126/science.1192449. [DOI] [PubMed] [Google Scholar]
- 142.Taipale J., Chen J.K., Cooper M.K., Wang B., Mann R.K., Milenkovic L., Scott M.P., Beachy P.A. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005–1009. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- 143.Ramaswamy V., Remke M., Bouffet E., Bailey S., Clifford S.C., Doz F., Kool M., Dufour C., Vassal G., Milde T., et al. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. 2016;131:821–831. doi: 10.1007/s00401-016-1569-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pei Y., Liu K.-W., Wang J., Garancher A., Tao R., Esparza L.A., Maier D.L., Udaka Y.T., Murad N., Morrissy S., et al. HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC-Driven Medulloblastoma. Cancer Cell. 2016;29:311–323. doi: 10.1016/j.ccell.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Morfouace M., Shelat A., Jacus M., Freeman B.B., Turner D., Robinson S., Zindy F., Wang Y.-D., Finkelstein D., Ayrault O., et al. Pemetrexed and Gemcitabine as Combination Therapy for the Treatment of Group3 Medulloblastoma. Cancer Cell. 2014;25:516–529. doi: 10.1016/j.ccr.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ramaswamy V., Remke M., Adamski J., Bartels U., Tabori U., Wang X., Huang A., Hawkins C., Mabbott D., Laperriere N., et al. Medulloblastoma subgroup-specific outcomes in irradiated children: Who are the true high-risk patients? Neuro Oncol. 2016;18:291–297. doi: 10.1093/neuonc/nou357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Schwalbe E.C., Lindsey J.C., Nakjang S., Crosier S., Smith A.J., Hicks D., Rafiee G., Hill R.M., Iliasova A., Stone T., et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: A cohort study. Lancet Oncol. 2017;18:958–971. doi: 10.1016/S1470-2045(17)30243-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rood B.R., MacDonald T.J., Packer R.J. Current treatment of medulloblastoma: Recent advances and future challenges. Semin. Oncol. 2004;31:666–675. doi: 10.1053/j.seminoncol.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 149.De Braganca K.C., Packer R.J. Treatment options for medulloblastoma and CNS primitive neuroectodermal tumor (PNET) Curr. Treat. Options Neurol. 2013;15:593–606. doi: 10.1007/s11940-013-0255-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lee M.J., Hatton B.A., Villavicencio E.H., Khanna P.C., Friedman S.D., Ditzler S., Pullar B., Robison K., White K.F., Tunkey C., et al. Hedgehog pathway inhibitor saridegib (IPI-926) increases lifespan in a mouse medulloblastoma model. Proc. Natl. Acad. Sci. USA. 2012;109:7859–7864. doi: 10.1073/pnas.1114718109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Porro A., Iraci N., Soverini S., Diolaiti D., Gherardi S., Terragna C., Durante S., Valli E., Kalebic T., Bernardoni R., et al. c-MYC Oncoprotein Dictates Transcriptional Profiles of ATP-Binding Cassette Transporter Genes in Chronic Myelogenous Leukemia CD34+ Hematopoietic Progenitor Cells. Mol. Cancer Res. 2011;9:1054–1066. doi: 10.1158/1541-7786.MCR-10-0510. [DOI] [PubMed] [Google Scholar]
- 152.Porro A., Haber M., Diolaiti D., Iraci N., Henderson M., Gherardi S., Valli E., Munoz M.A., Xue C., Flemming C., et al. Direct and coordinate regulation of ATP-binding cassette transporter genes by Myc factors generates specific transcription signatures that significantly affect the chemoresistance phenotype of cancer cells. J. Biol. Chem. 2010;285:19532–19543. doi: 10.1074/jbc.M109.078584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ingram W.J., Crowther L.M., Little E.B., Freeman R., Harliwong I., Veleva D., Hassall T.E., Remke M., Taylor M.D., Hallahan A.R., et al. ABC transporter activity linked to radiation resistance and molecular subtype in pediatric medulloblastoma. Exp. Hematol. Oncol. 2013;2:497–516. doi: 10.1186/2162-3619-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Stewart C.F., Iacono L.C., Chintagumpala M., Kellie S.J., Ashley D., Zamboni W.C., Kirstein M.N., Fouladi M., Seele L.G., Wallace D., et al. Results of a Phase II Upfront Window of Pharmacokinetically Guided Topotecan in High-Risk Medulloblastoma and Supratentorial Primitive Neuroectodermal Tumor. J. Clin. Oncol. 2004;22:3357–3365. doi: 10.1200/JCO.2004.10.103. [DOI] [PubMed] [Google Scholar]
- 155.Phoenix T.N., Patmore D.M., Boop S., Boulos N., Jacus M.O., Patel Y.T., Roussel M.F., Finkelstein D., Goumnerova L., Perreault S., et al. Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer Cell. 2016;29:508–522. doi: 10.1016/j.ccell.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]