

Abstract
We identify a previously unresolved, unrecognized, and highly stable conformation of the Protein Kinase A (PKA) regulatory subunit RIα. This conformation, which we refer to as the “Flipback” structure, bridges conflicting characteristics in the crystallographic structures and solution experiments of the PKA RIα heterotetramer. Our simulations reveal a hinge residue in the B/C helix that is conserved through all isoforms of RI. Brownian dynamics simulations suggest that the Flipback conformation plays a role in cAMP association to the A domain of R subunit.
Keywords: Molecular Dynamics, Brownian Dynamics, Protein Kinase A, PKA, cAMP
Graphical Abstract
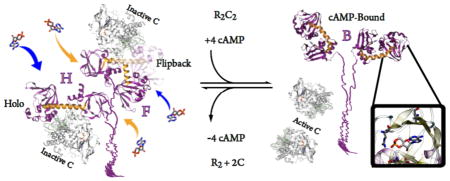
Protein Kinase A (PKA) is a ubiquitous eukaryotic kinase that modulates protein function through targeted phosphorylation. An ancient cellular second-messenger, cAMP, modulates the activity of PKA in a diverse set of biological processes; from synaptic plasticity1 to cardiac signaling2. Inactive PKA exists as a heterotetramer (R2C2), where two catalytic (C) subunits are maintained in the inactive state by binding to the regulatory (R) subunit homodimer.3 In response to extracellular signals like adrenaline4, adenylyl cyclase activity leads to an increase in cAMP. When four molecules of cAMP bind to the cyclic-nucleotide binding domains (CBD) of R dimer, a global conformational change in R occurs, releasing two active C subunits, free to phosphorylate protein targets. This activation cycle is the generalized mechanism for all non-redundant forms of R (RIα, RIβ, RIIα, RIIβ) and C (Cα, Cβ, Cγ). PKA subtypes are expressed in every cell and encoded by separate genes, differing in structure, activity, and cellular localization.5, 6 Thus, elucidating the structural organization of PKA complexes and their relationship to activation by cAMP is important for the development of novel therapeutics and the understanding of PKA’s fundamental biochemistry.
Structural biologists have elucidated PKA isoforms in various stages of the activation cycle. Although the heterotetrameric (R2C2) structure of the Type-IA Protein Kinase, RIα, was never fully resolved, it has been the subject of several studies and models. Su et al. crystallized RIα, in complex with two molecules of cAMP.7 This structure of the PKA R subunit at the end of the activation cycle, known to many as the “Bound” conformation or “B conformation” (RBound), reveals the amino acid residues important for coordinating cAMP. Each PKA R subunit has two cyclic-nucleotide binding domains (CBD-A and CBD-B) joined by a helical moiety known as the B/C helix. The CBD is a conserved sensor of cAMP and composed of non-contiguous alpha-helices and beta barrel subdomains. At the N-terminus, a 310helix-loop region (N3A motif8) is followed by a beta sandwich containing the cAMP binding site, and a terminal helical region (B and C-terminal helices). The cAMP-bound structure has served as an invaluable resource for understanding cAMP activation through molecular simulations9 and experiments.
The first structure of the RC “holoenzyme” heterodimer featured the C subunit in complex with RIα CBD-A (RAC).10 Point mutations made at key cAMP-interacting residue, Arg333R in CBD-B, led to the crystallization of the dual-domain R subunit with the C subunit11, RCHolo. The R subunit “Holoenzyme” conformation or “H conformation”, RHolo, is different from the cAMP-bound structure, “Bound” or “B conformation”, RBound (Figure 1). The Bound conformation is globular, with CBD contacts resulting from a bent B/C/C’ helix. In B conformation, the B/C helix breaks at L233 and Y244 (Figure 1A), bringing N3AB in contact with CBD-A. In RCHolo structure, the R subunit wraps around C subunit (Figure 1B), and the CBDs are separated by an extended B/C helix. In the absence of C subunit, The H form is stabilized by cAMP analogs.12 The cAMP bound, “B” conformation cannot physically accommodate C subunit because N3AB interacts with C in RCHolo while N3AB interacts with CBD-A in RBound(Figure 1).
FIGURE 1. Comparison of known RIα conformations.

(A) RBound:cAMP2; The Bound or B conformation, RIα (purple ribbon) with two molecules of cAMP (licorice). (B) RHolo:C; The Holo or H conformation, RIα (purple ribbon) in complex with the C subunit (white ribbon). (C) RFlipback:C; The Flipback conformation or F conformation, an MD-derived metastable state, aligned with Cα subunit (white ribbon). B/C helices are shown in gold ribbon. In light purple, the N3A motifs of A and B (N3AA and N3AB) are shown. The C subunit is shown with ATP (licorice). Blue surface representation highlights the H/D exchanging regions of the C subunit measured by HDXMS.
Despite extensive efforts, the structure of full-length (RAB), wild-type Type-IA (RIα) PKA heterotetramer remains elusive. Structural models of the tetramer are available,13 but are not fully consistent with structure and dynamics of PKA in solution. Specifically, the heterodimeric mutant R333K crystal structure has a larger R/C interface than described by hydrogen/deuterium exchange mass spectrometry (HDXMS) (Figure 1B).14–16
Here, we use molecular simulation techniques to make sense of discordant experimental findings from X-ray crystallography, scattering and HDXMS experiments. We present a novel conformation of the regulatory subunit that resolves these disparities, the “Flipback” or “F conformation”. Finally, we use electrostatic descriptions of the biomolecules to understand cAMP association offering a role for Flipback in the regulation mechanism of PKA RIα.
To understand the flexibility of the apo WT R subunit, we performed all-atom molecular dynamics (MD) simulations starting from H conformation in the absence of C subunit and cAMP. We simulated, WT Holo R in five 200ns replicates using the AMBER17 force field in NTP ensemble at 310K. MD of WT-RHolo reveals a very flexible B/C helix as observed in other simulations18, 19. Mutagenesis of B/C helix residues recently showed pronounced effects on PKA activation.14
Our MD simulations reveal a unique, stable conformation of R subunit (Figure 1C, Figure S1), which we call the “Flipback” conformation or “F form”, RFlipback. Like RBound, RFlipback features inter-domain (CBD-A/B) interactions and a B/C helix break. However, RFlipback breaks in the opposite direction, with interactions between alternate CBDs and N3A motifs. RBound uses contacts between N3AB and CBD-A while RFlipback uses N3AA to interact with CBD-B (Figure 1B and 1C). In RFlipback, the B/C helix breaks at Gly235R early in the trajectory (~20ns), bringing the CBDs in contact for the rest of the simulation in a stable conformation (Figure S1). Mutations limiting the flexibility of the B/C helix (G235P) result in poor C subunit binding20 promoting activation. Residues 230–238 of B/C helix exhibit nearly equal Hydrogen-bond propensities in the Holoenzyme, cAMP-Bound, and cAMP-free forms,15 suggesting the B/C helix is equally flexible in all structures. Gly235 is conserved amongst all forms of RI.21 Thus, a helical break at this position may be important in the activation of other Type-I PKA R subunits.
When RFlipback is aligned with CBD-A of RCHolo, it is apparent that Flipback conformation can accommodate C subunit, unlike RBound. We created a PKA R2C2 model using F conformation (R2C2Flipback). As Flipback and Holo are the only known conformations that can accommodate C subunit, we were curious to understand how conformational changes in R affect cAMP association. The diffusion of cAMP, a polar molecule, is likely influenced by long-range electrostatic forces which are estimated computationally by Brownian Dynamics (BD) simulation methods. 22–24
Using the existing13 and newly constructed models of PKA heterotetramer and heterodimer (R2C2Holo, R2C2Flipback and RCHolo, RCFlipback) we examine the relative rate of cAMP encounter to CBD-A and CBD-B via BD simulations to determine association rates (kassociation). An “encounter complex” is formed when a specified distance between a set of atoms is reached. We chose three conserved residues to define encounter complexes in CBD-A/B: Val184/300, Glu200/324, and Arg209/333 (Figure S2, Table S1). Using BrownDye,25 we compare the effect of Holo and Flipback on cAMP association tetrameric and heterodimeric in conformations.
Predicted BD rates are remarkably consistent with apparent kon from experiments:26 4.52 x 106 and 1.00 x 105 (CBD-B and CBD-A, respectively). The fastest rate of cAMP encounter to PKA tetramers is to R2C2Holo CBD-B (~107 M−1s−1) while CBD-A association is slowest (~105 M−1s−1, see Table 1). Domain-B preference in Holo validates the standing “gate-keeper” theory of PKA RIα, which holds that cAMP binds to CBD-B first.27 Domain B preference is neutralized in R2C2Flipback, where both CBD-A and CBD-B bind on the order of ~106 M−1s−1 (Table 1 and Scheme 1).
Table 1.
Comparison of cAMP association rates to cyclic nucleotide binding domains of PKA complexes
| cAMP binding Domain (CBD) | PKA Conformation | kassociation (M−1, s−1) |
|---|---|---|
| A | R2C2Holo | 3.07 x 105 |
| B | R2C2Holo | 2.57 x 107 |
| A | R2C2Flipback | 4.29 x 106 |
| B | R2C2Flipback | 4.16 x 106 |
| A | RCHolo | 2.50 x 104 |
| B | RCHolo | 2.72 x 104 |
| A | RCFlipback | 1.38 x 108 |
| B | RCFlipback | 1.98 x 106 |
SCHEME 1.

Activation mechanism of PKA RIα with different R conformations and relative association rates
Electrostatically, the systems differ in the distribution of charge on the surfaces of PKA complexes. Flipback has a more electropositive CBD-A than Holo (Figure S4), yielding a higher association rate by cAMP. The phenomenological preference for CBD-A of Flipback is most pronounced in heterodimers, with association rates two to four orders of magnitude higher in F. This difference in association rates is due to the very different electrostatic potential surfaces of the heterodimer vs. heterotetramer forms in H and F conformations (see Figure S4). From these results, we hypothesize that the Flipback conformation is important for cAMP association to CBD-A. It is not known if RC heterodimers are important players in the activation mechanism of PKA; but if they are, we predict that the Flipback conformation plays a role in CBD-A association.
The only resolved conformation of full-length R in complex with C necessitated the R333K mutation to stabilize CBD-B.11 However, it has been shown that R333K mutant (RCHolo) has a different solution structure than WT. Small-angle X-ray scattering (SAXS) of full-length WT heterodimer and heterotetramer28 exhibit a shouldering region; one not observed in symmetric SAXS p(r) distributions of mutant RC (R333K) and RAC, a heterodimer lacking the B domain.29
The Flipback heterotetramer structure corroborates observations from multiple solution experiments. First, the C subunit interface of the WT full-length holoenzyme measured by HDXMS is consistent with R/C interface of the R2C2Flipback structure, where the amides of residues 212–221C and 278–289C were reported to be unprotected.14 Second, structural models from scattering experiments suggest that conformational changes in R cause the release of one set of R/C contacts30, consistent with the Flipback conformation (Scheme 1). It has been shown that cAMP binding to CBD-B leads to increase H/D exchange at the B/C helix.31 It is likely that once binding to CBD-B, Flipback conformation is formed, leading to CBD-A association. Attempts to elucidate the heterotetrameric structure of PKA RIα with SAXS have proposed a Flipback-like conformation.28 Finally, the majority of R/C on RIα contacts result from interactions in CBD-A, consistent with Flipback. 10, 14–16, 21, 29, 32, 33
Flipback dynamics are consistent with other molecular simulations and models.27, 34 Guo et al. recently observed a flexible B/C helix when simulating apo-B form,9 opposite to the starting point of our H form simulations. A backward-bending B/C helix is observed in these simulations, in conformation resembling Flipback. This suggests that F is a state accessible from both B and H forms.
An interesting role for Flipback emerges when we consider the termination phase of PKA regulation. Phosphodiesterase enzyme (PDE) hydrolyzes cAMP to 5′-cAMP, regulating concentration of the second messenger. Computational docking and HDXMS determined that for PDE to bind RIα subunit, the B/C helix requires a complete reorganization.35 It is possible that Flipback is a binding partner of PDE, though further exanimation of this hypothesis is necessary.
Our simulations, coupled with experimental data make a case for a viable, and stable Flipback conformation of PKA RIα that may play important roles in the cAMP regulatory mechanism. Our work reveals a new structure of the WT PKA R subunit, which supports observations from ensemble-averaged solution structures and experiments. BD suggests a role for RFlipback conformation in mechanism of PKA activation. We hope our findings will lead to a re-examination PKA; especially on differences between the conformations of WT and R333K mutants, and the role of structural ensembles in ligand binding and ultimately, signal transduction.
Supplementary Material
Acknowledgments
Funding Sources
This research was funded by a Fellowship from the Interfaces Graduate Training Program (NBIB T32 EB009380), and by the National Biomedical Computational Resource (NIH P41-GM103426) and NIH DP2 OD007237.
We especially acknowledge Dr. Alexandr Kornev for frequent and helpful discussions We acknowledge Drs. Jamie M. Schiffer, Susan S. Taylor, and Elizabeth Komives for their insights. Finally, we acknowledge Dr. Gary Huber and Dr. Lane W. Votapka for help and advice with BD simulations.
ABBREVIATIONS
- PKA
Protein Kinase A
- cAMP
cyclic adenosine monophosphate
- CBD
cyclic-nucleotide binding domain
- B
cAMP-Bound
- H
Holo
- F
Flipback
Footnotes
ASSOCIATED CONTENT
Detailed experimental methods; Figure S1, MD of the Flipback structure. Figures S2, S3 and Table S2 Encounter complex; Figure S4, Electrostatic profiles of Holo and Flipback. This material is available free of charge via the Internet at http://pubs.acs.org.”
Author Contributions
S.P.H. wrote the manuscript, analyzed MD, and performed and designed BD experiments. R.D.M. performed and designed MD experiments. R.E.A. directed the research. This manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
REA is a co-founder of, has equity interest in, and serves on the Scientific Advisory Board of Actavalon, Inc.
References
- 1.Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- 2.Kamp TJ, Hell JW. Circ Res. 2000;87:1095–1102. doi: 10.1161/01.res.87.12.1095. [DOI] [PubMed] [Google Scholar]
- 3.Johnson LN, Noble MEM, Owen DJ. Cell. 1996;85:149–158. doi: 10.1016/s0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- 4.Chasiotis D. Acta Physiol Scand. 1985;125:537–540. doi: 10.1111/j.1748-1716.1985.tb07752.x. [DOI] [PubMed] [Google Scholar]
- 5.Taylor SS, Zhang P, Steichen JM, Keshwani MM, Kornev AP. Bba-Proteins Proteom. 2013;1834:1271–1278. doi: 10.1016/j.bbapap.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taylor SS, Ilouz R, Zhang P, Kornev AP. Nat Rev Mol Cell Bio. 2012;13:646–658. doi: 10.1038/nrm3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Su Y, Dostmann WRG, Herberg FW, Durick K, Xuong NH, Teneyck L, Taylor SS, Varughese KI. Science. 1995;269:807–813. doi: 10.1126/science.7638597. [DOI] [PubMed] [Google Scholar]
- 8.Kornev AP, Taylor SS, Ten Eyck LF. P Natl Acad Sci USA. 2008;105:14377–14382. doi: 10.1073/pnas.0807988105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo C, Zhou HX. P Natl Acad Sci USA. 2016;113:E6776–E6785. doi: 10.1073/pnas.1610142113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim C, Xuong NH, Taylor SS. Science. 2005;307:690–696. doi: 10.1126/science.1104607. [DOI] [PubMed] [Google Scholar]
- 11.Kim C, Cheng CY, Saldanha SA, Taylor SS. Cell. 2007;130:1032–1043. doi: 10.1016/j.cell.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 12.Badireddy S, Gao YF, Ritchie M, Akamine P, Wu J, Kim CW, Taylor SS, Lin QS, Swaminathan K, Anand GS. Mol Cell Proteomics. 2011:10. doi: 10.1074/mcp.M110.004390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boettcher AJ, Wu JA, Kim C, Yang J, Bruystens J, Cheung N, Pennypacker JK, Blumenthal DA, Kornev AP, Taylor SS. Structure. 2011;19:265–276. doi: 10.1016/j.str.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anand GS, Hotchko M, Brown SH, Ten Eyck LF, Komives EA, Taylor SS. Journal of molecular biology. 2007;374:487–499. doi: 10.1016/j.jmb.2007.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anand GS, Hughes CA, Jones JM, Taylor SS, Komives EA. Journal of molecular biology. 2002;323:377–386. doi: 10.1016/s0022-2836(02)00919-1. [DOI] [PubMed] [Google Scholar]
- 16.Anand GS, Law D, Mandell JG, Snead AN, Tsigelny I, Taylor SS, Ten Eyck LF, Komives EA. P Natl Acad Sci USA. 2003;100:13264–13269. doi: 10.1073/pnas.2232255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salomon-Ferrer R, Gotz AW, Poole D, Le Grand S, Walker RC. J Chem Theory Comput. 2013;9:3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- 18.Pecora de Barros E, Malmstrom RD, Nourbakhsh K, Del Rio JC, Kornev AP, Taylor SS, Amaro RE. Biochemistry-Us. 2017;56:1536–1545. doi: 10.1021/acs.biochem.6b01152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malmstrom RD, Kornev AP, Taylor SS, Amaro RE. Nat Commun. 2015;6:7588. doi: 10.1038/ncomms8588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng CY. Chemistry. UC San Diego: 2009. Dissecting the allosteric regulation of PKA-I alpha activation. [Google Scholar]
- 21.Sjoberg TJ, Kornev AP, Taylor SS. Protein Sci. 2010;19:1213–1221. doi: 10.1002/pro.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Northrup SH, Allison SA, Mccammon JA. J Chem Phys. 1984;80:1517–1526. [Google Scholar]
- 23.Ermak DL, Mccammon JA. J Chem Phys. 1978;69:1352–1360. [Google Scholar]
- 24.Votapka LW, Amaro RE. PLoS computational biology. 2015:11. doi: 10.1371/journal.pcbi.1004381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huber GA, McCammon JA. Comput Phys Commun. 2010;181:1896–1905. doi: 10.1016/j.cpc.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herberg FW, Taylor SS, Dostmann WRG. Biochemistry-Us. 1996;35:2934–2942. doi: 10.1021/bi951647c. [DOI] [PubMed] [Google Scholar]
- 27.Boras BW, Kornev A, Taylor SS, McCulloch AD. The Journal of biological chemistry. 2014;289:30040–30051. doi: 10.1074/jbc.M114.568907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vigil D, Blumenthal DK, Heller WT, Brown S, Canaves JM, Taylor SS, Trewhella J. Journal of molecular biology. 2004;337:1183–1194. doi: 10.1016/j.jmb.2004.02.028. [DOI] [PubMed] [Google Scholar]
- 29.Cheng CY, Yang J, Taylor SS, Blumenthal DK. Journal of Biological Chemistry. 2009;284:35916–35925. doi: 10.1074/jbc.M109.059493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heller WT, Vigil D, Brown S, Blumenthal DK, Taylor SS, Trewhella J. Journal of Biological Chemistry. 2004;279:19084–19090. doi: 10.1074/jbc.M313405200. [DOI] [PubMed] [Google Scholar]
- 31.Moorthy BS, Badireddy S, Anand GS. Int J Mass Spectrom. 2011;302:157–166. [Google Scholar]
- 32.Hamuro Y, Anand GS, Kim JS, Juliano C, Stranz DD, Taylor SS, Woods VL. Journal of molecular biology. 2004;340:1185–1196. doi: 10.1016/j.jmb.2004.05.042. [DOI] [PubMed] [Google Scholar]
- 33.Huang LJ, Taylor SS. The Journal of biological chemistry. 1998;273:26739–26746. doi: 10.1074/jbc.273.41.26739. [DOI] [PubMed] [Google Scholar]
- 34.Boras BW, Hirakis SP, Votapka LW, Malmstrom RD, Amaro RE, McCulloch AD. Front Physiol. 2015;6:250. doi: 10.3389/fphys.2015.00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krishnamurthy S, Moorthy BS, Xiang LX, Shan LX, Bharatham K, Tulsian NK, Mihalek I, Anand GS. Biophys J. 2014;107:1426–1440. doi: 10.1016/j.bpj.2014.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.