

Summary
How tumor-infiltrating T lymphocytes (TILs) adapt to the metabolic constrains within the tumor microenvironment (TME) and to what degree this affects their ability to combat tumor progression remain poorly understood. Using mouse melanoma models, we report that CD8+ TILs enhance PPAR-α signaling and catabolism of fatty acids (FAs) when simultaneously subjected to hypoglycemia and hypoxia. This metabolic switch partially preserves CD8+ TILs’ effector functions although co-inhibitor expression increases during tumor progression regardless of their antigen specificity. Further promoting FA catabolism improves the CD8+ TIL’s ability to slow tumor progression. PD-1 blockade delays tumor growth without changing TIL metabolism or functions. It synergizes with metabolic reprogramming of T cells to achieve superior antitumor efficacy and even complete cures.
Keywords: Melanoma, tumor microenvironment, CD8+ T cells, TILs, co-inhibitors, hypoxia, HIF-1α, hypoglycemia, fatty acid catabolism, fenofibrate
INTRODUCTION
Despite recent progress in cancer immunotherapy, cures remain rare. Transfer of ex vivo expanded TILs may affect tumor regression, but vaccines that induce such T cells have largely been ineffective (Dalgleish, 2011). This has been linked to exhaustion of tumor antigen (TA)-specific CD8+ TILs characterized by enhanced expression of co-inhibitors and loss of functions following chronic antigen stimulation (Mueller and Ahmed, 2009). Treatment with immune checkpoint inhibitors can improve TIL functions and is yielding promising results in patients (Zou et al., 2016).
Upon activation T cells enhance glycolysis to support energy and biomass production. Tumor cells also use glycolysis (Hamanaka and Chandel, 2012), which may lead to glucose (Glc) depletion within the TME. T cells with limited access to Glc switch to oxidative phosphorylation (OXPHOS). Although many nutrients fuel OXPHOS, it requires oxygen (O2), which due to insufficient blood supply can become limiting within tumors. TILs therefore face dual metabolic jeopardy, which may impair the efficacy of T cell-mediated cancer therapy.
How CD8+ TILs metabolically adapt to the nutrient and O2-limited TME and how these adjustments affect their tumor-killing functions are not well understood. In this study, we investigated the effects of metabolic challenges within the TME on CD8+ T cells and designed interventions that may allow for better preservation of CD8+ TIL functions.
RESULTS
CD8+ T cells become functionally impaired within the TME regardless of their antigen specificity
To test if the fate of CD8+ TILs depends on continued antigen stimulation, we used two vaccines in a transplantable mouse melanoma model. AdC68-gDMelapoly (Figure 1A) is an adenovirus (Ad)-based vaccine that elicits melanoma-associated antigen (MAA)-specific CD8+ T cell responses mainly to the Trp-1455-463 epitope (Zhang and Ertl, 2014). AdC68-gDE7 stimulates CD8+ T cells to human papilloma virus (HPV) E7 (Lasaro et al., 2008). We vaccinated mice bearing 3-day B16BrafV600E tumors and normal mice with AdC68-gDMelapoly mixed with AdC68-gDE7. Vaccination delays tumor progression (Figure 1B). Both MAA- and E7-specific CD8+ T cells accumulate within tumors, where they contract more rapidly than in the periphery (Figure 1C). Numbers of MAA-specific CD8+ TILs decline by >90% between day 10 and 30 after vaccination while E7-specific CD8+ TILs decline by ~80%. This prominent reduction in MAA-specific CD8+ TIL numbers occurs although they proliferate before entering tumors or within the TME (Figure 1D). Nevertheless, proliferation decreases over time despite continued presence of their antigens (not shown). MAA- and E7-specific CD8+ T cells from day 14 and, to a more pronounced extent, day 30 tumors increase expression of PD-1 and LAG-3 (Figure 1E). During tumor progression, frequencies of CD8+ T cells producing the effector molecules granzyme B (GrmB), perforin and interferon (IFN)-γ decline faster in tumors than in spleens and by day 30 after challenge CD8+ TILs lose polyfunctionality (Figure 1F). These data suggest that TILs regardless of their antigen specificity differentiate towards functional exhaustion during tumor progression, contesting the notion that sustained antigen stimulation is required to drive TIL exhaustion (Bucks et al., 2009). Ad vectors persist at low levels and thus maintain high frequencies of functional effector CD8+ T cells (Tatsis et al., 2007). Accordingly, most vaccine-induced MAA- (Figure 1G) and E7- (not shown) specific CD8+ T cells remain CD44hiCD62LloCD127loKLRG1hi during tumor progression. Levels of T-bet and Eomes decrease in specific T cells from spleens and tumors over time, presumably reflecting differentiation driven by reduced antigenic load in spleens and progression towards exhaustion in tumors (Zhu et al., 2010).
Fig 1. CD8+ TILs become functionally impaired.

(A) AdC68-gDMelapoly transgene. (B) B16BrafV600E tumor growth curves (Co [AdC68-gD], n=6; Vaccine, n=18). (C) Numbers of Trp-1 (MAA)- and E7-tetramer (tet)+CD8+ T cells/106 live mononuclear cells in spleens (Spl) and tumors of mice that had or had not been received tumor cells before vaccination (n=10/group). (D) % BrdU incorporation into antigen-specific CD8+ T cells from Spl and tumors. (E) Mean fluorescent intensity (MFI) and histograms for PD-1 and LAG-3 on CD8+ T cells from Spl (open histogram) and tumors (grey histogram) at 14 or 30 days after tumor challenge. (F) Left: Relative % antigen-specific CD8+ T cells from Spl and tumors producing GrmB, perforin or IFN-γ comparing day 30 to 14 samples. Right: % specific CD8+ T cells at day 30 from Spl or tumors producing 3 or 2 of the tested functions. The same functions are displayed throughout the manuscript unless indicated otherwise. * bottom to top: significant differences in 2 and 3 functions. Bottom: flow plot showing TIL functions: day 14 (grey) vs. 30 (black) overlays; control: Spl and TILs stimulated with irrelevant peptide. (G) Differentiation markers on MAA-specific T cells from Spl and tumors: as day 14 (black) vs. 30 (grey) overlays (left) or histograms (right). Gating control: Naive CD8+ T cells. (E–G) Numbers in plots: average % marker+ cells for the entire group; stars next to top number indicate significant differences between groups. This applies to all flow plots in the manuscript. (D–G) n=5/group. Data show mean with SEM. Data represent 2 (B, D, F, G) or 3 assays (C, E). For all figures, *p ≤0.05 – 0.01, ** p≤0.01–0.001, *** p ≤0.001–0.0001, **** p≤0.0001. See also Figure S1.
To determine whether exhaustion of TILs occurs independent of vaccination, we transfused mice bearing 5-day B16OVA tumors with OT-1 splenocytes, which carry a transgenic T cell receptor to SIINFEKL, the immunodominant epitope of ovalbumin (OVA). Few OT-1 T cells infiltrate day 14 tumors; more are detected in day 30 tumors. OT-1 TILs from day 30 tumors compared to those from spleens or day 14 tumors increase PD-1 and LAG-3 and decrease CD62L (Figure S1A) and IFN-γ production (Figure S1B), suggesting that in both models TILs become exhausted. This process may not be solely driven by chronic antigen stimulation (Bucks et al., 2009) as shown with vaccine-induced CD8+ TILs.
Metabolism dictates cellular fate and could affect CD8+ TILs independent of their antigen specificity. During tumor progression MAA- and E7-specific CD8+ TILs lose mitochondrial membrane potential (MMP) and increase levels of mitochondrial reactive oxygen species (MROS) (Figure 2A). MMPloMROShi MAA- and to a lesser extent E7-specific CD8+ TILs become prevalent in day 30-tumors (Figure 2B), while corresponding CD8+ T cells from spleens remain largely MMPhiMROSlo. We controlled the specificities of mitochondrial stains (Figure 2C) and confirmed that changes in MMP and MROS levels are not due to cell size differences (Figure 2D). Electron microscopy shows that mitochondria within CD44+CD8+ TILs from advanced tumors lose the typical rod-like structure as described (Siska et al., 2017). They display poorly defined membranes and cristae compared to those in splenic T cells (Figure 2E), confirming that CD8+ TILs experience mitochondrial stress within growing tumors.
Figure 2. CD8+ TILs increasingly experience metabolic stress.

(A) MFI of MMP and MROS in specific CD8+ T cells from Spl and tumors. (B) Left: MMP over MROS of CD8+ T cells from Spl and tumors. (−) not significant or (*) significant differences left to right are for populations from top to bottom in legend. Right: flow plots of specific T cells from day 30 tissues and naive T cells (Co). (C) Controls for DioC6 and Mitosox Red stains on CD8+ TILs. CD44+CD8+ TILs and E7-specific CD8+ TILs were treated with FCCP or oligomycin to collapse the MMP; with Mito-TEMPO to reduce MROS, or with oligomycin to induce MROS. (D) MAA- or E7-specific CD8+ T cell size comparison between those from Spl and tumors isolated from mice bearing day 14 or 30 tumors. Representative flow plots show cell subsets overlay upon gating on FSC over SSC. (E) Transmission electron micrographs of CD44+CD8+ T cells from day 30 Spl and tumors. Samples pooled from 15 mice. Data show mean - SEM; representative of 3 (A–C) or 2 assays (D).
Hypoxia through HIF-1α increases LAG-3 expression and impairs T cell functions
MAA- and E7-specific CD8+ TILs are subjected to hypoxia during tumor progression as shown by enhanced expressions of HIF-1α, the main transcriptional regulator in the cells’ response to hypoxia, and its downstream target Glut1, which facilitates Glc uptake (Figure 3A).
Figure 3. Effect of HIF-1α knockdown on TA-specific CD8+ TILs.

(A) MFI and representative histograms of HIF-1α and Glut1 in/on specific CD8+ T cells from day 30 Spl (open) and tumors (grey) (n=5 mice/group). (B) Experimental set-up: vaccine model. (C) MFI and flow plot of HIF-1α in MAA-specific CD8+ TILs transduced with scrambled (Co, open histogram) or HIF-1α shRNA lentivector (grey histogram). (D) MFI of PD-1 and LAG-3. (E) Left: % transduced MAA-specific CD8+ TILs producing individual functions. Right: Polyfunctionality. % transduced cells producing 3, 2 or 1 of the 3 tested functions. For all figures: * within (): significant difference in sum of responses. * without () left to right: significant difference in 3, 2, and 1 functions. (C–E) n=5–7 mice/group. (F) Experimental set up: OT-1 transfer model. (G) Tumor growth in mice that received transduced OT-1 T cells. (H) MFI of PD-1, LAG-3 and T-bet on/in donor transduced OT-1 CD8+ TILs. (I) Left: % transduced donor OT-1 CD8+ TILs producing individual factors. Right: flow plots show overlays of HIF-1α shRNA (blue) and Co (red) transduced factor-producing OT-1 CD8+ TILs. (J) Heatmap of transcript levels comparing HIF-1α shRNA over Co transduced OT-1 CD8+ TILs. (K) MFI of Glut1, PPAR-α and BODIPY C16 uptake. (*): p=0.05. (G–K) n=5–8 mice/group. Experiments were repeated twice. Data show mean - SEM. See also Figures S2, S3.
To determine how hypoxia affects CD8+ T cells, we stimulated them in vitro for 4 days under normoxia (21% O2) or for the last 16 hr under hypoxia (1% O2, Figure S2A). Hypoxia reduces blast formation (Figure S2B) and increases expression of HIF-1α and Glut1 (Figure S2C). The O2 consumption rate (OCR), a measure of OXPHOS, decreases under hypoxia while the extracellular acidification rate (ECAR), a measure of glycolysis, increases (Figure S2D). The T cells’ MMP decreases and MROS increases, leading to a rise in MMPloMROShi CD8+ T cells (Figure S2E) as in vaccine-induced TILs from late-stage tumors. Hypoxia reduces PD-1 but augments LAG-3 (Figure S2F), suggesting both are modulated upon HIF-1α activation. CD8+ T cells cultured under hypoxia decrease T-bet (Figure S2F), production of effector molecules and polyfunctionality (Figure S2G). A previously described protocol (Doedens et al., 2013), in which CD8+ T cells after initial activation are maintained at a more resting stage in IL-2 and exposed to hypoxia (Figure S2H), has no effect on blast formation (Figure S2I) or PD-1, although LAG-3 increases and T-bet decreases (Figure S2J). GrmB production increases while production of other effector molecules and polyfunctionality decline (Figure S2K). As vaccine-induced MAA-specific CD8+ T cells are unlikely to rest before infiltrating the TME’s hypoxic areas, we used the protocol of continuous CD8+ T cell activation for subsequent experiments.
HIF-1α positively correlates with expression of LAG-3 on TILs or CD8+ T cells subjected to hypoxia (Figure S3A). To determine whether HIF-1α directly promotes LAG-3 expression, we first knocked down (KD) HIF-1α using three lentivectors that express different HIF-1α-silencing short hairpin RNAs (shRNAs) or a scrambled shRNA. Each lentivector that reduces HIF-1α in activated CD8+ T cells also reduces Glut1 and LAG-3, increases GrmB, and decreases perforin production (Fig S3B–D), suggesting the results are not due to off-target effects. We used HIF-1α shRNA lentivector E1676 for in vitro studies and for the vaccine model. E1676 transduces ~ 34% of activated CD8+ T cells (Figure S3E) and achieves 20% and 60% reduction of HIF-1α transcripts under normoxia or hypoxia, respectively (Figure S3F). Under hypoxia HIF-1α KD decreases LAG-3 but not PD-1 on CD8+ T cells (Figure S3G), and improves production of GrmB and IFN-γ (Figure S3H).
To study how HIF-1α signaling affects CD8+ TILs, we activated CD8+ T cells in vitro and, after lentivector transduction, transferred them into tumor-bearing, AdC68-gDMelapoly-vaccinated mice (Figure 3B). We analyzed the transferred T cells ~3 weeks later from similar sized tumors (Figure S3I). HIF-1α KD, which remains stable after cell transfer (Figure 3C), reduces the MAA-specific CD8+ TILs’ expression of LAG-3 without affecting PD-1 (Figure 3D), and improves their effector functions including polyfunctionality (Figure 3E).
To further study the effect of HIF-1α on CD8+ TILs, we knocked down HIF-1α in activated OT-1 CD8+ T cells with a mixture of the three HIF-1α shRNA lentivectors. Approximately 75% of cells are transduced (Figure S3J). They show 50% reduction in HIF-1α transcripts (Figure S3K) and protein expression, combined with modest decreases in surface Glut1 (Figure S3L). HIF-1α KD reduces levels of transcripts encoding enzymes of glycolysis and with a delay increases transcripts for factors involved in the tricarboxylic acid (TCA) cycle and FA catabolism (Figure S3M).
Transduced Thy1.1+ OT-1 cells were transferred into mice bearing 5-day B16OVA tumors (Figure 3F). Tumor progression is delayed in mice transferred with HIF-1α shRNA-treated OT-1 CD8+ T cells (Figure 3G). TILs from day 20 tumors show comparable differentiation between the two groups (Figure S3N) and HIF-1α KD remains stable (Figure S3O). HIF-1α KD reduces LAG-3 without changing PD-1 on TILs. It increases T-bet (Figure 3H) and effector functions (Figure 3I). Levels of transcripts indicative of glycolysis decrease; those for the TCA cycle increase (Figure 3J). Accordingly, TILs with HIF-1α KD decrease Glut1 but enhance PPAR-α, a key transcription factor that promotes FA catabolism and uptake of FAs (Figure 3K). These data show that HIF-1α KD in TA-specific TILs reduces glycolysis and promotes OXPHOS. They further suggest that HIF-1α-driven increases in glycolysis might be counterproductive to T cell functions within an O2 and Glc -depleted TME.
Activated CD8+ T cells lacking both Glc and O2 enhance FA catabolism
Not only O2 but also Glc declines within the TME during tumor progression (Figure 4A). We studied the collective effects of hypoglycemia and hypoxia on CD8+ T cells by stimulating them in vitro in Glc-medium with the glycolysis inhibitor 2-deoxy-D-glucose (2-DG) or replacing Glc with galactose (Gal). Cells were cultured under normoxia or short-term hypoxia. Either condition decreases ECAR and augments the OCR/ECAR ratio (Figure S4A), suggesting increased energy production through OXPHOS. Cells cultured with 2-DG or Gal rather than Glc increase PD-1, indicating that OXPHOS is linked to high PD-1 expression on activated CD8+ T cells (Figure S4B). Compared to cells cultured with Glc, those with limited Glc decrease T-bet (Figure S4C) and lose functions and polyfunctions (Figure S4D). Polyfunctions of CD8+ T cells with limited Glc supply are better preserved if cells are also subjected to hypoxia, suggesting that cells may more readily cope with hypoglycemia with increased HIF-1α signaling.
Figure 4. Limited access to Glc and O2 forces CD8+ TILs to enhance FA catabolism.

(A–G) Vaccine model. (A) Glc concentrations in plasma, early and late stage B16BrafV600E tumors (n=3). (B) Relative transcript levels: Upper: CD8+ T cells in Gal- or Glc+2-DG- vs. Glc-medium under hypoxia. Middle: specific CD8+ TILs from day 30 vs. 14 tumors. Lower: specific CD8+ T harvested from day 90 vs. 14 Spl (n=3). Color code below 3rd and 4th row: comparisons between in vitro and in vivo samples. (C) Experimental set-up: 13C6-Glc in vivo tracing. (D) Intensity of glycolysis metabolites in CD44+CD8+ TILs. (E) Experimental set-up: 13C16-palmitate in vivo tracing. (F) Intensity of FA metabolites in CD44+CD8+ TILs. (G) Relative contribution of 13C6-Glc- and 13C16- palmitate-derived carbons to citrate and malate, calculated by dividing labeling carbon numbers from 13C16-palmitate by the numbers from 13C6-Glc. Data of CD44+CD8+ T cells from day 30 tumors are normalized to those from day 14 tumors (left) or day 30 Spl (middle); Spl data: day 30 normalized to 14 (right). (D, F, G) n=2–3, pooled from ~30 mice/sample. (H–I) OT-1 transfer model. (H) Normalized contribution of 13C16-palmitate to TCA cycle metabolites and L-palmitoylcarnitine in CD44+CD8+ TILs from day 30 vs. 14 tumors or day 30 Spl. (I) Intensities of FA metabolites. (H–I) n=2, pooled from ~20 mice/sample. (D, F, G–I) Data show as mean values. (J–L) Bar graphs: Relative intensity of free FA species in tumor interstitial fluid, dashed lines show ratio of 1. Pie charts: Abundance of different FA species. Total numbers: combined FA intensity. (J–K) day 30 over 14 B16BrafV600E tumors (J, n=3) or PDX melanomas (K, n = 5). (L) Human melanoma metastases vs. human sera (n = 4). (D, F, G–L) Representative of two assays. (A, J–L) mean - SEM. See also Figures S4, S5.
To study the metabolic pathways used by Glc- and O2-starved CD8+ T cells, we measured transcripts for factors that participate in nutrient consumption and energy production (Figure S4E, S4F, 4B). Upon short-term hypoxia CD8+ T cells stimulated under hypoglycemia rather than in Glc-medium decrease transcripts for factors of glycolysis, TCA cycle, ROS detoxification and electron transport chain (ETC). They show increased transcripts of PPAR-α and downstream molecules involved in FA uptake, triglyceride (TG) turnover, peroxisomal and mitochondrial FA catabolism. This pattern is mirrored by MAA- and E7-specific CD8+ TILs from day 30 vs. 14 tumors, indicating that metabolically stressed CD8+ TILs increasingly rely on FA catabolism. Changes in transcripts during tumor progression are not driven by TIL differentiation towards a more resting stage, as they are distinct from those in vaccine-induced splenic CD8+ T cells tested at 90 vs. 14 days after vaccination.
To directly measure effects of Glc and O2 deprivation on CD8+ T cell metabolism, we analyzed the intensity of metabolites by lipid chromatography-mass spectrometry (LC-MS). Metabolites of FA catabolism, i.e., acetylcarnitine, palmitoylcarnitine, and the ketone body 3-hydroxybutyrate increase in cells stimulated in Gal-medium and this is further enhanced under hypoxia (Figure S5A). 13C6-Glc/Gal and 13C16-palmitate isotope labeling in vitro show that CD8+ T cells activated with limited Glc and/or O2 compared to those activated in Glc-medium under normoxia have reduced carbohydrate-derived TCA cycle metabolites (Figure S5B), while more FA-derived carbons are incorporated into acetyl-CoA, TCA cycle metabolites and amino acids (Figure S5C), suggesting that FAs are increasingly used for energy and biomass production. This metabolic switch is supported by the cells’ higher FA uptake (Figure S5D) and enhanced oxidation of endogenous and exogenous FAs (Figure S5E).
We characterized the metabolism of CD8+ T cells in vivo by isotope labeling. Mice bearing 3-day tumors were vaccinated with AdC68-gDMelapoly and AdC68-gDE7. They were given 13C6-Glc or 13C16-palmitate 14 or 30 days later. The intensity of glycolysis intermediates declines (Figure 4C, 4D) while acylcarnitine species and ketone bodies increase in CD44+CD8+ T cells from day 30 compared to day 14 tumors (Figure 4E, 4F). The relative incorporation of 13C16-palmitate-dervied carbons into TCA cycle metabolites increases while that of carbons derived from 13C6-Glc declines comparing TILs from late to early stage tumors or T cells from tumors to spleens (Figure 4G), confirming that TILs progressively enhance FA catabolism. In splenic CD44+CD8+ T cells the relative contribution of FA-derived carbon to TCA cycle metabolites remains stable or decreases over time (Figure 4G).
In the OT-1 cell transfer model, activated CD8+ TILs from advanced tumors compared to those from day 14 tumors or day 30 spleens display markedly higher levels of 13C16-FA-derived carbon incorporation into TCA cycle metabolites and palmitoylcarnitine (Figure 4H). In addition, the intensities of palmitoylcarnitine and ketone bodies are significantly higher in OT-1 TILs from day 30 B16OVA tumors (Figure 4I), further validating that the metabolic switch towards enhanced FA catabolism during tumor progression is not limited to vaccine-induced TILs. We confirmed that sample processing does not affect the profile of isotopic labeling in T cells, as shown by the nearly identical 13C16-FA-derived carbon incorporation into TCA cycle metabolites from lymph node (LN) samples snap frozen on dry ice or processed into single cell suspensions and kept on ice for the time needed for sample processing (Figure S5F).
The metabolic switch of TILs is facilitated by enhanced uptake of FAs (Figure S5G) and increased expression of the FA β-oxidation (FAO) rate-limiting enzyme Cpt1a (Figure S5H) in vaccine-induced CD8+ TILs from late-stage tumors. This switch is not caused by their differentiation towards memory as evidenced by low levels of CD62L (Figure S5I). The enhanced FA catabolism in TILs is further supported by increasingly high abundance of free FAs within the interstitial fluid of B16BrafV600E tumors (Figure 4J). Increases of different FAs in the tumor interstitial fluid during tumor progression appears to be common as it is also observed in melanoma patient-derived xenografts (PDX) from NSG mice (Figure 4K) and in fresh human melanoma metastases that were tested against human sera (Figure 4L). We also compared different FA species within the interstitial fluid of PDX tumors to those within solid organs or serum and found that a number of FA species are markedly higher within the tumors (Figure S5J).
We tested whether CD8+ TILs isolated from melanoma patients display enhanced reliance on FA catabolism. Lymphocytes from blood of healthy donors or resected melanoma metastases were stained and gated onto naive, effector, effector memory (TEM) and central memory (TCM) CD8+ T cell subsets (Figure 5A). Compared to circulating CD8+ T cells, CD8+ TILs show increases in TEMs and decreases in naive and effector cells (Figure 5B). We analyzed each T cell subset for PD-1 expression and parameters indicative of FA catabolism. Antigen-experienced TILs show higher PD-1 levels than CD8+ T cells from blood (Figure 5C). TEM and TCM CD8+ TILs express higher levels of PPAR-α and Cpt1a compared to the corresponding peripheral blood mononuclear cell (PBMC) populations. Moreover, all TIL subsets show enhanced FA uptake. Collectively, our data suggest that a metabolic switch towards enhanced FA catabolism also occurs in TILs of melanoma patients.
Figure 5. Metabolism of T cells in human samples.

(A) Gating strategy for human CD8+ T cells subsets. Numbers show % of cells in each subset. (B) Pie charts show distribution of CD8+ T cell subsets within CD8+ T cells isolated from blood of healthy donors (n=14) or metastatic tumors of melanoma patients (n=9). p<0.0001 for differences in numbers of TEM cells normalized to 106 live CD8+ T cells. Numbers within pie charts show mean percentages. (C) Upper: % of cells positive for PD-1, Cpt1a, PPAR-α and FA uptake (BODIPY C16). Data show comparison for CD8+ T cell subsets isolated from blood vs. tumors. Lower: Representative histograms comparing markers of CD8+ TEM from blood vs. tumors. Data show mean - SEM.
Treatment with α-PD-1 slows tumor progression without changing CD8+ TIL metabolism or functions
In clinical trials, monoclonal (m) antibodies (Ab) to PD-1 can delay tumor progression (Hamid et al., 2013; Larkin et al., 2015). As in our model CD8+ TILs increase PD-1 over time, we tested if treatment with an anti-PD-1 mAb (α-PD-1) affects their metabolism or functions. Tumor-bearing mice were vaccinated and treated with α-PD-1 or an isotype control. α-PD-1 reduces PD-1 staining when the same antibody (29F.1A12) is used for treatment and detection but not upon staining with Ab RMP1-30, which is directed to a different PD-1 epitope (Figure 6A). α-PD-1 treatment enhances Phospho-Akt levels (Figure 6A), indicating it blocks the binding of PD-L1/PD-L2 to PD-1 (Patsoukis et al., 2013) without affecting the differentiation (Figure S6), metabolism (Figure 6B) or functions of TA-specific CD8+ TILs (Figure 6C). It delays tumor progression in vaccinated, unvaccinated or immune-deficient NSG mice (Figure 6D).
Figure 6. Metabolism and effector functions of CD8+ TILs are independent of PD-1.

(A) MFI of PD-1 tested with PD-1 Ab clone 29F.1A12 (same clone as α-PD-1 treatment Ab) or RMP1-30 and Phospho-Akt on/in specific CD8+ TILs from day 30 tumors (n=5–7). Iso: isotype control mAb. Lower: Flow plots for MAA-specific TILs. (B) Normalized 13C6-Glc or 13C16-palmitate contribution to TCA cycle metabolites and the intensity of ketone bodies in CD44+CD8+ T cells from day 30 tumors comparing α-PD-1- to Iso-treated mice. n=2 or 3, pooled from 25 mice/sample, shown as mean values. (C) % specific CD8+ TILs from day 30 tumors producing 3, 2 or 1 of the 3 tested functions (Iso: n=11; α-PD-1: n=15; data pooled from two assays). (D) B16BrafV600E tumor growth in mice that received Iso or α-PD-1 (unvaccinated: n=5; vaccinated: n=13; NSG: n=4). Arrows: red (vaccine); black (Ab treatment). (E) PD-1 and PD-L1 on B16BrafV600E tumor cells grown in vivo. Iso (grey), specific Ab (open). (F) PD-L1 KD in B16BrafV600E cells. (G) In vivo assay comparing survival of PD-L1hiCTVhi vs. PD-L1loCTVlo tumor cells in vaccinated or unvaccinated mice treated with Iso or α-PD-1 mAb. Bar graph: ratio of recovered live PD-L1hi over PD-L1lo B16BrafV600E cells (n=5). Right: histograms. (H) PD-1 and PD-L1 on B16OVA tumor cells grown in vivo. (I) PD-L1 KD in B16OVA cells. (J) Normalized survival of PD-L1hiCTVhi vs. PD-L1loCTVlo B16OVA cells in Iso or α-PD-1-treated mice that were transferred with activated OT-1 cells and representative histograms (n=5). (A, C, D, G, J) Data show mean with SEM (representative of 2 experiments). See also Figures S6.
B16BrafV600E tumor cells express low levels of PD-1 but are highly positive for PD-L1 (Figure 6E). To assess whether α-PD-1 treatment induces tumor cell death in a PD-L1-dependent manner (Azuma et al., 2008), we knocked-down PD-L1 in tumor cells (Figure 6F). PD-L1hi or PD-L1lo tumor cells were mixed in a 1:1 ratio and injected intraperitoneally (i.p.) into naive C57Bl/6 mice or mice that had been vaccinated 14 days earlier with Ad-gDMelapoly. Mice of each group were treated with α-PD-1 or isotype Ab prior to tumor cell challenge. Cells were isolated from the peritoneal cavity 1 day later. In isotype-treated unvaccinated or vaccinated mice, most of the recovered tumor cells are PD-L1hi (Figure 6G), suggesting that PD-L1 provides tumor cells with a survival advantage. Upon α-PD-1 treatment both PD-L1hi and PD-L1lo cells are recovered at equal levels, indicating the gain in survival of PD-L1hi cells requires back signaling through PD-1. B16OVA tumors are also PD-1lo but positive for PD-L1 (Figure 6H). After PD-L1 KD (Figure 6I), equal numbers of PD-L1hi and PD-L1lo B16OVA cells were transferred into α-PD-1- or isotype Ab-treated mice that also received in vitro activated OT-1 CD8+ T cells. The PD-L1hi tumor cells again show a survival advantage in the isotype Ab-treated group, which is abrogated in presence of α-PD-1 (Figure 6J). Therefore, in our models PD-1 blockade reduces tumor progression in a T cell-independent, PD-L1-dependent manner.
Enhanced reliance on FA catabolism is essential to maintain CD8+ TIL functions
To further assess the impact of FA catabolism on CD8+ T cell functions, we stimulated CD8+ T cell in presence of fenofibrate (FF), a PPAR-α agonist that increases FA catabolism, or etomoxir (ETO), an irreversible inhibitor of Cpt1 that decreases mitochondrial FAO (Figure S7A). Addition of the drugs does not affect CD8+ cells survival under the chosen experimental conditions (Figure S7B). In vitro FF-treated cells stimulated in Glc or Gal-medium compared to diluent-treated cells increase FA catabolism as shown by their transcriptional profile (Figure S7C) and increased FA uptake (Figure S7D). ETO decreases OCR of CD8+ T cells stimulated in either Glc- or Gal-medium (Figure S7E). OCR declines most in cells cultured with Gal under hypoxia, again confirming their increased reliance on FAO. Under hypoxia, PD-1 expression increases with FF but decreases with ETO (Figure S7F). FF increases while ETO decreases functions and polyfunctionality of CD8+ T cell cultured under hypoglycemia and hypoxia (Figure S7G).
To assess how increased FA catabolism affects CD8+ TILs, we vaccinated CD90.2+ mice congenic for CD45, and treated them with FF (CD45.1 mice) or diluent (CD45.2 mice). Splenocytes were mixed at a 1:1 ratio of MAA-specific CD8+ T cells from the 2 sets of donors and transferred into vaccinated, tumor-bearing CD90.1+ recipients (Figure 7A). Prior to transfer, lymphocytes from FF-treated mice show significantly increased 13C16-palmitate catabolism and higher ketone body intensity compared to those from controls (Figure S7H, S7I), validating the FF effect in vivo. FF-treated splenocytes show enhanced OCR, which is blocked by ETO, further confirming that FF enhances FAO (Figure S7J). Donor-derived MAA- and E7-specific CD8+ T cells from mice treated with FF or diluent show comparable expression of differentiation markers, indicating that FF does not overtly affect memory formation (Figure S7K). FF-treated cells concurrently increase PD-1 and T-bet (Figure S7L) and show trends towards higher functions (Figure S7M). Donor-derived specific CD8+ TILs were analyzed 3 weeks after transfer (Figure 7B). Compared to diluent-treated CD44+CD8+ TILs, those treated with FF show enhanced transcripts for factors involved in FA catabolism (Figure 7C). Both MAA- and E7-specific FF-treated CD8+ TILs show trends of increased PD-1 (Figure 7D), and their frequencies and functions are significantly higher compared to those of controls (Figure 7E). Upon transfer of splenocytes from FF- or diluent-treated mice into separate cohorts of tumor-bearing hosts, the former significantly delay tumor progression (Figure 7F), confirming that enhanced FA catabolism improves CD8+ TILs’ functions.
Figure 7. Promoting FA catabolism improves CD8+ TIL functions and works in synergy with PD-1 blockade to delay tumor growth.

(A–F) Vaccine model. (A) Experimental design. (B) % donor and host CD8+ T cells from tumors of recipients as bar graphs (left) or flow plot (right). (C) Transcript levels in donor-derived FF-vs. Dil-treated CD44+CD8+ TILs (n=3–4/group). (D) MFIs of PD-1 on specific donor CD8+ TILs. (E) Left: % specific donor CD8+ TILs producing 3, 2, or 1 of the 3 tested functions; * from bottom to top: differences in 3-1 factors; Right: Representative plots overlays of FF- (red) and Dil- (blue) treated MAA-specific donor TILs. (F) B16BrafV600E tumor weight at necropsy. (D–F, n=5). (G–I) PD-1 blockade combined with FF treatment. (G) Experimental design. (H) Left: tumor volume at day 30; Right: tumor growth curves. (I) Functions of donor CD8+ TILs. * bottom to top or right to left: differences cells producing 3, 2, or 1 of the 3 tested functions. Flow plots: factor-producing MAA-specific TILs from mice that received FF-treated cells, α-PD-1 (red) overlaid with Iso (blue)-treated cells. (H, I) Iso: n=6; α-PD-1: n=7. Studies were repeated twice. (J–L) OT-1 transfer model. (J) MFI of PD-1 on FF- or Dil-treated donor-derived activated OT-1 TILs. (K) Left: % donor-derived OT-1 TILs producing GrmB or IFN-γ. Middle: % cells producing 3, 2, or 1 of the 3 tested functions. Right: flow plots. (J, K) n=8 mice/group; data represent 2 experiments. (L) B16OVA tumor growth in mice received naive (n=24), Dil- or FF-treated activated OT-1 T cells (n=8/group). Data show mean - SEM. See also Figure S7.
To test if FF-induced increases in PD-1 affect CD8+ TIL functions, we fed vaccinated donor mice with FF or diluent. Upon transfer of splenocytes into separate groups of tumor-bearing mice, we treated the recipients with α-PD-1 or the isotype control (Figure 7G). Both FF treatment of donors and α-PD-1 treatment of recipients delay tumor progression; they act synergistically and together completely prevent tumor growth in 50% of vaccinated mice (Figure 7H). α-PD-1 reduces PD-1 staining with 29F.1A12 on donor cells and this is not affected by FF (Figure S7N). PD-1 blockade only has subtle effects on functions of MAA-specific CD8+ TILs derived from either set of donor mice (Figure 7I) but significantly increases frequencies of monofunctional E7-specific CD8+ TILs derived from FF treated donors. This may partially reflect the smaller tumor sizes in α-PD-1 treated mice, which might primarily benefit bystander TILs.
In the cell transfer model, treatment of OT-1 cells with FF or diluent during their in vitro stimulation confirms that the PPAR-α agonist promotes FA catabolism as evidenced by increases in transcripts of factors involved in this process (Figure S7O). FF-treated OT-1 T cells augment FA uptake and PPARα expression (Figure S7P). FF treatment does not change T cell differentiation but significantly enhances PD-1 (Figure S7Q). It increases GrmB but decreases IFN-γ production (Figure S7R). Within day 20 tumors, FF- or diluent-treated donor-derived OT-1 TILs show comparable PD-1 (Figure 7J). Frequencies of GrmB+ and/or IFN-γ+ donor CD8+ TILs increase upon FF pre-treatment (Figure 7K) and tumor progression is markedly delayed (Figure 7L).
To further study the effects of FA catabolism, we stimulated CD8+ T cells from PPAR-α knockout (KO) mice in vitro and compared them to those from wild type (WT) mice. Transcripts for most factors involved in the TCA cycle and lipid metabolism are higher in PPAR-α KO compared to WT CD8+ T cells when stimulated in Glc-medium under hypoxia (Figure 8A). This profile reverses in cells cultured in Gal-medium with low O2, suggesting that knocking out PPAR-α significantly decreases FA catabolism in Glc-deprived CD8+ T cells. PPAR-α KO CD8+ T cells cultured with limited Glc express less PD-1 than WT cells (Figure 8B) and exhibit lower functions and polyfunctions (Figure 8C). These data suggest that FA catabolism is required to maintain CD8+ T cell function when access to Glc is limited.
Figure 8. Inhibiting FA catabolism impairs CD8+ T functions while decreasing PD-1 expression.

(A) Transcript levels comparing CD8+ T cells from PPAR-α KO and WT mice stimulated in Glc- or Gal-medium under hypoxia (n=4). (B) Normalized MFIs for PD-1 on PPAR-α KO vs. WT activated CD8+ T cells cultured under different conditions (n=4–5). (C) Left: Normalized % PPAR-α KO vs. WT CD8+ T cells producing 3, 2, or 1 of the 3 tested functions (n=5). Right: Overlays of WT (blue) vs. PPAR-α KO (red) CD8+ T cells cultured in Gal-medium under normoxia. (D) Experimental design. (E) Comparison of transcript levels in CD44+CD8+ TILs from PPAR-α KO vs. WT donors (n=3 mice/group). (F) MFI of PD-1 on WT and PPAR-α KO donor CD8+ TILs with histograms. (G) Left: % donor-derived specific CD8+ TILs from the two groups producing 3, 2, or 1 of the 3 tested functions; Right: Overlays of factor-producing WT (blue) vs. PPAR-α (red) donor-derived TILs. (F, G) n=6/group. Data represent 2 assays, shown as mean - SEM. See also Figure S8.
We explored the effect of reduced FA catabolism on vaccine-induced TILs in an adoptive transfer system, in which splenocytes from vaccinated PPAR-α KO and WT mice were mixed at a 1:1 ratio of MAA-specific CD8+ T cells and co-transferred into tumor-bearing, vaccinated recipients (Figure 8D). Prior to transfer, expression of CD127 (Figure S8A) and other differentiation markers (not shown) are similar between the two T cell subsets. Similarly, functions and polyfunctionality of MAA-specific CD8+ T cells are comparable between the two groups, while E7-specfic CD8+ T cells are less abundant and polyfunctional in PPAR-α KO mice (Figure S8B). This difference may reflect that strength of T cell receptor signaling, which is lower for the E7 epitope, affects the T cells’ differentiation (Kaech and Cui, 2012) and thereby their metabolism (O’Sullivan et al., 2014). Within 3-week tumors, PPAR-α KO compared to WT donor-derived CD44+CD8+ TILs show a transcriptional profile indicative of reduced FA catabolism (Figure 8E). Vaccine-induced, donor-derived PPAR-α KO CD8+ TILs have less PD-1 (Figure 8F) and lower functions including polyfunctions compared to those of WT controls (Figure 8G). Collectively these data confirm that PPAR-α-promoted FA catabolism preserves effector functions of CD8+ TILs.
DISCUSSION
Within the TME CD8+ T cells experience hypoxia and must compete for nutrients especially Glc. Recent studies report that hypoglycemia within the TME impairs CD8+ T cells functions and reduces the efficacy of active immunotherapy (Chang et al., 2015; Ho et al., 2015). Our results show that metabolic stress within the TME impairs the performance of CD8+ TILs including bystander TILs, although TA-specific CD8+ TILs tend to be more affected, presumably for they continue to respond to stimulatory signals and may penetrate more deeply into tumors where nutrients and O2 are especially limiting.
Melanoma develops areas of hypoxia, which activates the HIF-1α pathway in cells of the TME. HIF-1α expression also rises upon T cell activation. In our study HIF-1α increases in both MAA-specific and bystander CD8+ TILs, pointing towards hypoxia as the underlying cause. The effect of hypoxia and HIF-1α on CD8+ T cells is controversial. Some studies show that O2 is required for T cell activation and effector functions, and HIF-1α functions as a negative regulator of T cell responses (Ohta et al., 2011; McNamee et al., 2013). Others using protocols in which CD8+ T cells were subjected to hypoxia during a resting period or testing cells lacking the von Hippel–Lindau tumor suppressor report that hypoxia or HIF-1α signaling increase functions (Doedens et al., 2013; Finlay et al., 2012). Our data agree with the former as they show reduced HIF-1α signaling improves the functions of activated CD8+ T cells experiencing hypoxia in vitro or within the TME, and increases the efficacy of TA-specific CD8+ T cells to delay tumor progression. These data imply that when Glc is limiting, promoting glycolysis and inhibiting OXPHOS by HIF-1α becomes detrimental to CD8+ TILs. LAG-3, which according to our data is up-regulated by HIF-1α, inhibits T cell expansion and effector functions (Grosso et al., 2007). The LAG-3 locus has several HIF-1α response elements ([A/G]CGTA), which may influence LAG-3 expression.
Hypoxia and hypoglycemia send opposing metabolic signals. The former promotes glycolysis while the latter forces cells to use OXPHOS, which can be fueled by various nutrients but requires O2. Cancer cells increase de novo lipogenesis (Menendez and Lupu, 2007) and recruit adipose progenitor cells. In our models including mouse and human melanomas, the abundance of free FA species increases during tumor progression, which could activate PPARα signaling in CD8+ TILs, facilitate their switch towards FA catabolism and preserve their effector functions. Energy production through FAO rather than glycolysis comes at a price; more O2 is needed to generate equivalent amounts of ATP and ROS production increases. Generating energy through FAO within a hypoxic TME may thus not be the only method by which CD8+ TILs maintain their functions. Ketone bodies are highly efficient fuels that require less O2 (Veech, 2004). They serve as a preferred energy source for cells of the nervous system subjected to hypoxia and hypoglycemia (Takahashi et al., 2014). Ketone bodies could be synthesized and secreted by other cells (Martinez-Outschoorn et al., 2012), or they could be produced by TILs directly as suggested by increased transcript levels of Bdh1, a key enzyme in ketone body metabolism, and enhanced intensities of ketone bodies in CD8+ TILs from late-stage tumors. We hypothesize that ketone bodies may serve as a nutrient source for CD8+ TILs, although this remains to be investigated further. We would also like to point out that levels of O2 differ within a tumor and as TILs can randomly migrate within the TME (Mrass et al., 2006) they may use FAO and ketone bodies alternatively depending on surrounding O2 levels.
In addition to contributing to energy production, FA catabolism could improve T cell functions through alternative pathways. When Glc is limited, FAs are used for amino acid synthesis thus promoting production of effector molecules. In addition, FAs are increasingly converted to acetyl-CoA, which can acetylate key enzymes in the TCA cycle and glycolysis pathways such as GAPDH. Acetylation increases GAPDH’s enzymatic activity and reduces its binding to the 3′UTR region of IFN-γ mRNA, thus enhancing IFN-γ production (Balmer et al., 2016) and T cell effector functions.
High expression of PD-1 is viewed to signal CD8+ T cell exhaustion and loss of effector functions. Our results suggest that high PD-1 expression is not inevitably linked to impaired T cell functions. Hypoxia-decreased PD-1 expression is associated with impaired functions, while FF-treated CD8+ T cells show a trend towards increased PD-1 expressions but their functions improve. PD-1 signaling inhibits TCR- and CD28-mediated activation of the PI3K/AKT/mTOR pathway, which in turn decreases glycolysis (Parry et al., 2005) and promotes lipolysis and FAO (Patsoukis et al., 2015). Enhanced PD-1 signaling in CD8+ TILs might thus facilitate the CD8+ TILs metabolic switch within a Glc-poor TME. In our model blockade of PD-1 after the initial phase of T cell activation affects neither metabolism nor effector functions of TILs. These results differ from those of a recent study in a mouse sarcoma model, which reports improved glycolysis and IFN-γ production by CD8+ TILs treated with α-PD-1 during their initial activation (Chang et al., 2015). We assume that these apparently opposing results reflect intrinsic differences in tumor models. Alternatively, differences in timing of treatment may affect the results. PD-1 blockade during the initial stages of T cell activation may allow them to better compete for Glc within a TME; once TILs have switched to FA catabolism they remain committed to this pathway regardless of PD-1 signaling.
Our data show that PD-1 blockade may promote MAA-specific CD8+ T cell infiltration into tumors (not shown). However, as α-PD-1 treatment also delays tumor progression in immune-deficient mice, we assume that it acts directly on tumors. The mouse melanoma cells in our models express low levels of PD-1 but are positive for PD-L1. In vivo PD-L1hi tumor cells have a survival advantage over PD-L1lo cells presumably due to back signaling upon binding to PD-1 expressed on T cells or innate immune cells. This ‘molecular shield’ is obstructed by the α-PD-1 mAb, which could thus enhance the tumor cells’ vulnerability to apoptosis or, in immunocompetent mice, indirectly improve TIL functions by increasing the tumor cells’ susceptibility to lytic enzymes (Azuma et al., 2008; Chen and Han, 2015).
Our results indicate that metabolic reprogramming of CD8+ T cells to increase energy production through FA catabolism prior to adoptive cell transfer might enhance the overall efficacy of cell therapy in patients with cancers characterized by low Glc content. We further show that this metabolic manipulations improves treatment outcome upon PD-1 blockade, which is in agreement with the synergistic effect of anti-PD-1 treatment with chemicals that activate mitochondrial functions (Chamoto et al., 2017). Similarly, other studies show that memory CD8+ T cells, which prefer FAO and OXPHOS for energy production, more efficiently slow tumor progression than effector cells (Crompton et al., 2015; Sukumar et al., 2013). In contrast, others report that increasing the TILs’ ability to use glycolysis improves their antitumor effect (Chang et al., 2015). Which metabolic manipulations are most suited to improve TIL-mediated tumor regression will likely depend on the nature of the tumor. Those with sufficient levels of Glc may benefit from CD8+ T cells with high glycolytic potential, while tumors with a hypoglycemic TME may best be combated by CD8+ T cells that favor FA catabolism.
Our in vivo studies focus on vaccine-induced CD8+ T cells or adoptively transferred TA-specific CD8+ T cells. The latter may serve as a model for in vitro expanded TILs or T cells with a chimeric antigen receptor, which upon adoptive transfer will encounter similar challenges within the TME and might thus benefit from metabolic reprogramming. Although most of our studies were conducted in mice, we confirm that T cells isolated from human melanoma metastases show evidence of enhanced FA catabolism, which could be fueled by the increased levels of FAs within tumor interstitial fluid. This indicates that our results are relevant for cancer patients. Our study invites clinical investigations into the use of metabolic manipulations to improve the outcome of cancer immunotherapy.
STAR METHODS
KEY RESOURCES TABLE
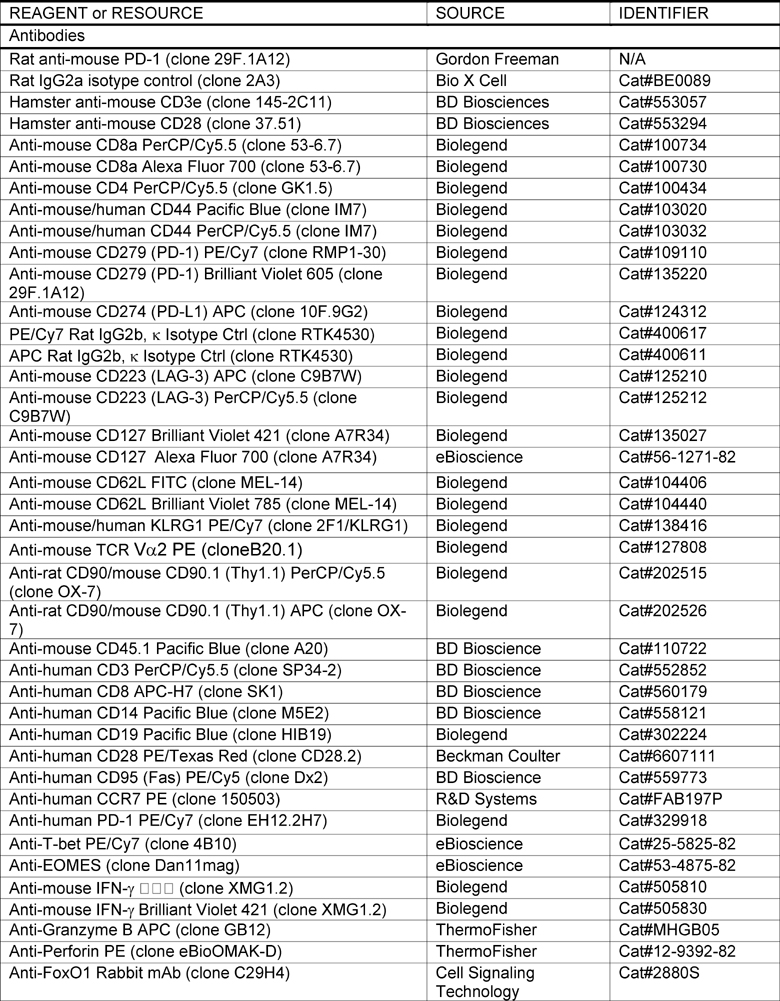
|
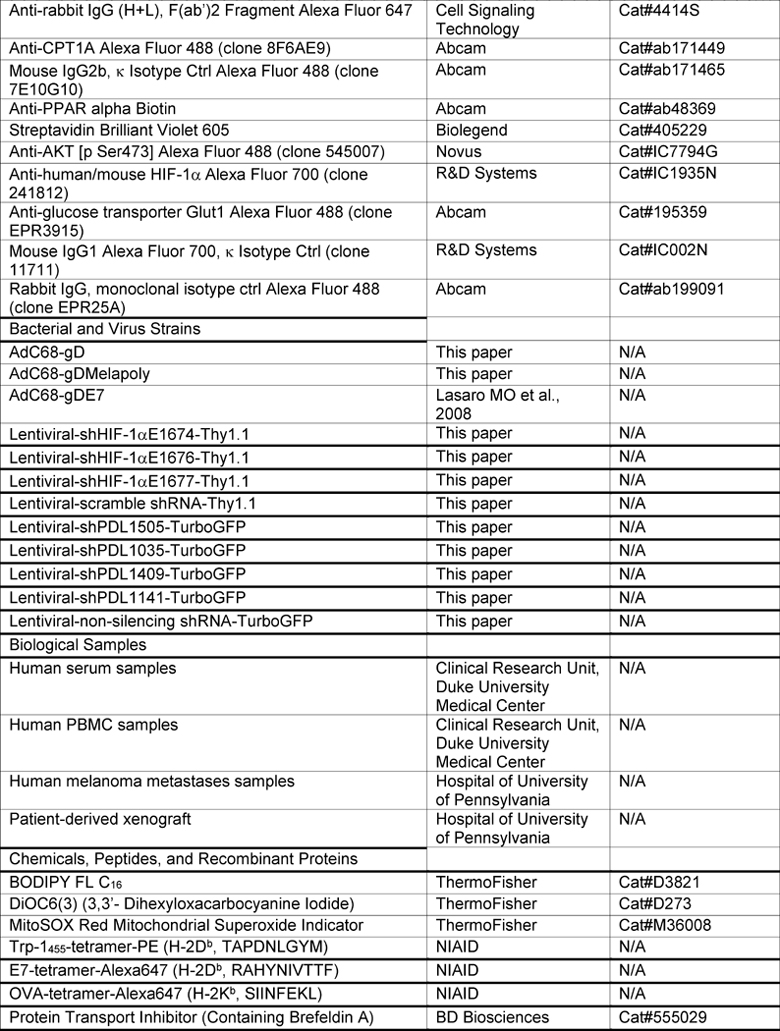
|
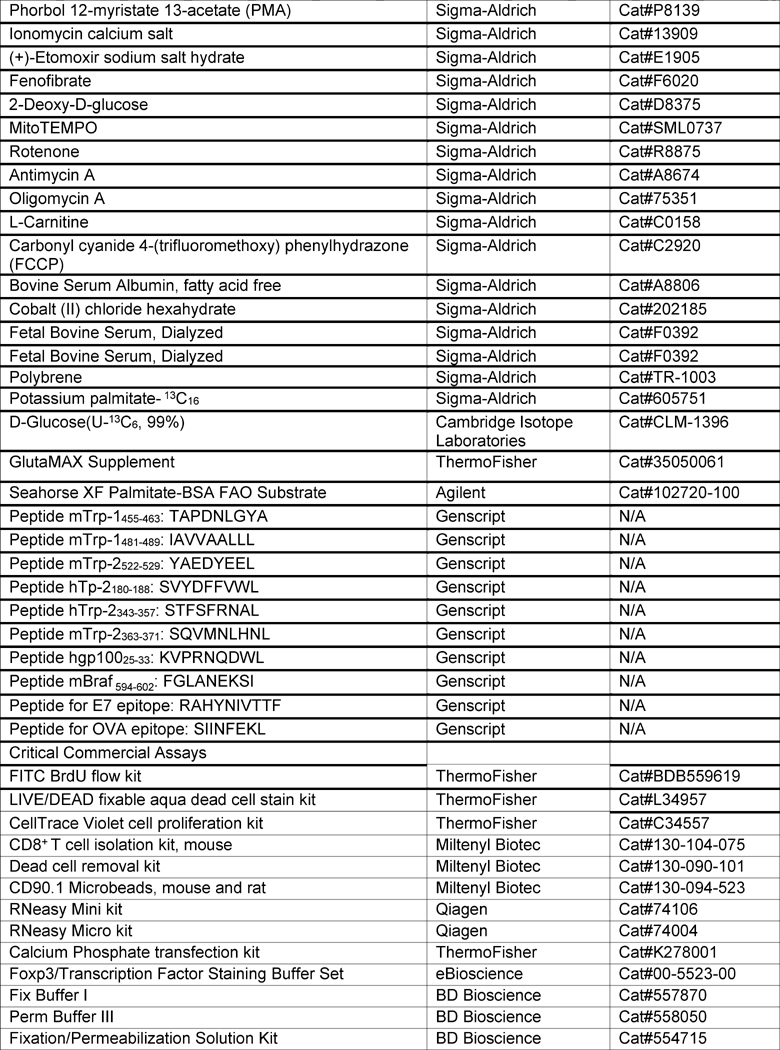
|
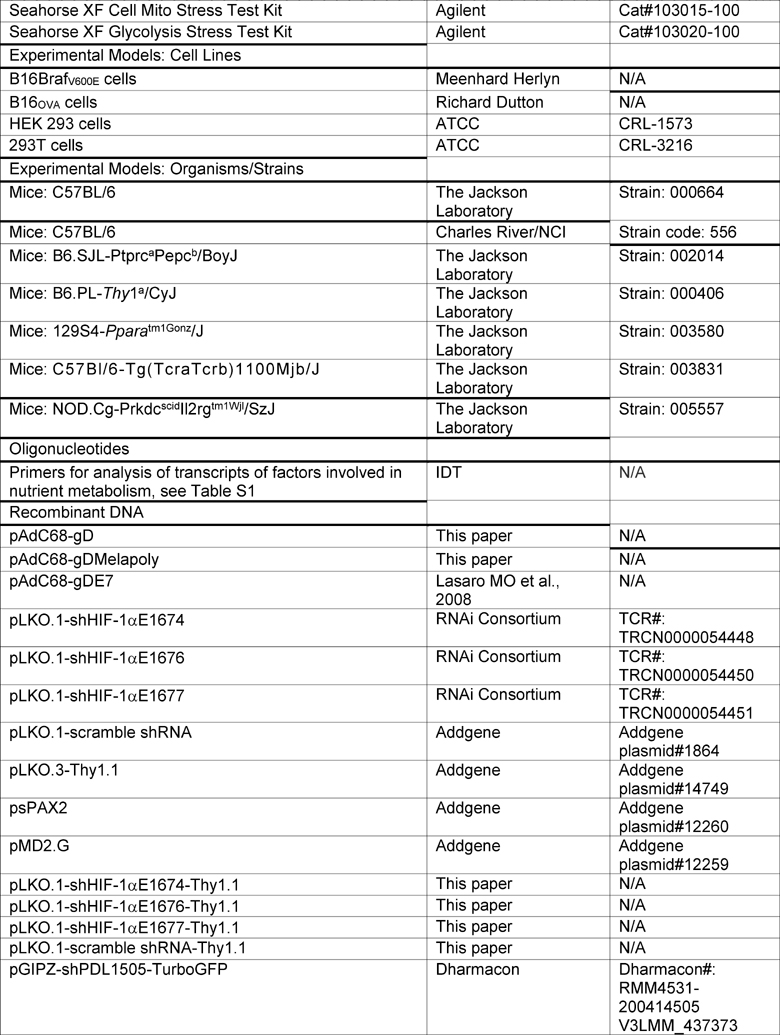
|
|
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled according to institutional rules by the Lead Contact, Hildegund Ertl (ertl@wistar.org).
EXPERIMENTAL MODEL AND SUGJECT DETAILS
Mice
All procedures were conducted following protocols approved by the Wistar IACUC. Female C57Bl/6, B6.SJL-PtprcaPepcb/BoyJ (B6 CD45.1+), B6.PL-Thy1a/CyJ (B6 CD90.1+), B6; 129S4-Pparatm1Gonz/J (B6 PPAR-α KO), C57Bl/6-Tg(TcraTcrb)1100Mjb/J (OT-1) and NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice (6–8 weeks) were purchased from Charles River, National Cancer Institute (NCI) or Jackson Laboratories and housed at the Wistar Institute Animal Facility. Groups of 5–80 C57BL/6 mice were vaccinated intramuscularly (i.m.) with 1010 virus particles [vp] of AdC68-gDMelapoly, 1011vp of AdC68-gDE7 or 1011vp AdC68-gD. For tumor challenge experiments B16BrafV600E cells (5×104 cells/mouse) or B16OVA cells (105 cells/mouse) were resuspended in PBS and injected subcutaneously (s.c.) into the right flank. For PDX model, NSG mice were injected s.c. with WM4231-2 cells (105 cells/mouse). The WM4231-2 cells were established from a surgically removed metastatic lesion from a patient with treatment-naive melanoma. Tumor growth was monitored by measuring the perpendicular diameters of tumors every other day. Depending on size early stage tumors were harvested 10–14 days after challenge (referred to as day 14) while late stage tumors were harvested 4–5 weeks after challenge (referred to as day 30). For adoptive transfer in the B16BrafV600E tumor model, 1×107 in vitro activated CD8+ T cells transduced with lentivectors were injected intravenously (i.v.) into recipient mice bearing 5 day-old tumors. For the OT-1 T cell transfer model, 2×106 OT-1 splenocytes or 106 OT-1 CD8+ T cells stimulated for 4–5 days in vitro were transferred i.v. into mice bearing 5 day-old B16OVA tumors.
For in vivo treatment FF (100 mg/kg/day) in DMSO/PBS was given by oral gavage daily for 3 weeks. Control mice received diluent. For co-transfer experiments, splenocytes containing 105 MAA-specific CD8+ T cells from the experimental and control groups were mixed and transferred i.v. into CD90.1+ recipient mice. For FF-treated OT-1 CD8+ T cell transfer, FF was added for 48 hr at 25 μM on day 4 of OT-1 T cell activation. For PD-1 blockade experiments in NSG or unvaccinated C57BL/6 mice, α-PD-1 Ab (clone 29F.1A12) or the isotype control Ab (Iso, Clone: 2A3, Bio X Cell) were given as of day 3 after tumor cell challenge. In vaccinated C57BL/6 mice, α-PD-1 or Iso treatment was started 10 days after vaccination. The Abs were given i.p. every 3rd day at a dose of 200 μg/mouse.
Patient samples
Human studies were approved by the Institutional Review Board (IRB) of the Wistar Institute (protocol 21111257, protocol 2802240-4b). Informed consent was obtained from each patient before blood or tumor tissue collection. The human blood and serum samples were obtained from clinical research unit, Duke University (Durham, NC, USA) in accordance with the local IRB. Samples were from male and female Caucasians between the ages of 32–39. Lymphocytes from periphery blood were isolated by Ficoll-Paque PLUS (GE Healthcare Biosciences) gradient using standard procedures. The PDX sample and the fresh metastatic tumors from melanoma patients were obtained from melanoma patients at the Hospital of the University of Pennsylvania upon their informed consent under protocol 2802240-4b. To ensure patient confidentiality no information on age, gender or ethnicity were provided to the investigators of this study. Metastases were cut into small pieces, digested with Collagenase/DNAse I and filtered through 70 μm cell strainers to produce single cell suspension. TILs were purified by Percoll-gradient.
Cell lines
The B16BrafV600E cell line was derived from B16.F10 cells by transduction with the lentivector pLU-EF1a-mCherry expressing mouse BrafV600E. The B16OVA cell line was a gift from Dr. R. Dutton (Wadsworth Center, Albany, NY). HEK 293 cells were used to propagate vaccine vectors. 293T cells were used to produce lentivectors. Cells were grown in Dulbecco’s Modified Eagles medium (DMEM) supplemented with 10% fetal bovine serum (FBS).
METHOD DETAILS
Construction of recombinant adenoviral vectors and lentivectors
Molecular construction, rescue, purification and titration of the adenoviral vectors have been described (Zhang and Ertl, 2014). The Melapoly sequence is composed of eight CD8+ T cell epitopes from human (h) or mouse (m)Trp-2, mTrp-1, hgp100 and mBrafV600E fused into herpes simplex virus (HSV) glycoprotein (g)D. We design the spacers between each epitopes using PAPROCI, Netchop 3.1 and IEBD Analysis Resource. Briefly, gDMelapoly or gDE7 sequences were inserted into the E1-deleted AdC68 viral molecular clone using I-CeuI and PI-SceI restriction enzyme sites. The constructed plasmids were used to transfect HEK 293 cells by calcium phosphate (Invitrogen). Cells infected with Ad vectors were harvest 7–10 days later upon plague formation. Virus was expanded on HEK 293 cells by serial infections and harvested by three cycles of freeze thawing. Virus was purified from cell-free supernatant after the third cycle of thawing by cesium chloride density ultracentrifugation.
For production of lentivectors for HIF-1α KD, three pLKO.1 lentivectors containing shRNAs targeting different regions of HIF-1α or a pLKO.1 lentivector containing scrambled shRNA were obtained from the RNAi Consortium or Addgene. The selection marker Thy1.1 was cloned from the pLKO.3-Thy1.1 lentivector (Addgene) into each of the shRNA lentivectors. Lentivectors were generated using the 2nd generation lentivector package system (Addgene) by transfecting 293T cells with the packaging plasmid PsPAX2, the envelope plasmid PMD2.G and each of the shRNA-Thy1.1-expressing insert plasmids at a ratio of 3:1:1. Supernatants were collected 48 and 72 hr after transfection, spun at 3000rpm for 10 min and filtered through 0.45 μm filters. Lentivectors were concentrated by ultracentrifugation at 25,000 rpm, 4 °C for 2 hr. Vector pellets were incubated with PBS at 4 °C overnight before resuspension. Each lentivector, E1674, E1676 and E1677, was used to transduce activated CD8+ T cells independently for in vitro experiments, to control for potential off-target effect. E1676 was used in the vaccine model. A mixture of three lentivectors was used to transduce activated OT-1 cells in the OT-1 cell transfer model. For PD-L1 KD, four GIPZ lentiviral mouse CD274 shRNA constructs with TurboGFP and puromycin selection markers (Dharmacon) were used to produce lentivectors. A lentivector with scrambled non-silencing shRNA was used as control. The efficiency of PD-L1 KD was compared between different vectors. Two lentivectors shPDL1505-TurboGFP and shPDL1035-TurboGFP were independently used for downstream studies with B16BrafV600E cells; one was selected for studies with B16OVA cells. Transduction with PD-L1 shRNA vectors or the control vector did not affect the growth of B16OVA tumor cells in vitro.
In vitro stimulation of CD8+ T cells and drug treatments
Enriched CD8+ T cells from naive C57BL/6 mice were activated for 4 days in 6-well plates pre-coated with Ab to CD3 (5 μg/ml) and CD28 (1 μg/mL) (BD Bioscience). For some samples, cells were transferred for the last 16 hr to a hypoxia chamber. OT-1 splenocytes were stimulated with SIINFEKL peptide (2 μg/mL) and IL-2 (100 U/ml, Roche) for 48 hr. They were then maintained in medium containing IL-2 and split every other day. To study the impact of hypoxia on relatively resting CD8+ T cells, enriched CD8+ T cells were stimulated for 48 hr under normoxia. Cells were then removed from the plates, washed and replated in fresh medium with IL-2 (100 U/ml) for 96 hr, followed by culture in normoxia or hypoxia with IL-2 for another 36 hr before analysis. Cells were cultured in Roswell Park Memorial Institute (RPMI) medium without Glc (Life Technologies) supplemented with Glc (10 mM) or Gal (10 mM), 10% dialyzed FBS (Life Technologies), 20 mM HEPES, 2 mM Glutamax, 1 mM sodium pyruvate, 0.05 mM 2-mercaptoethanol and 1% penicillin-streptomycin. Hypoxia experiments were performed in a Thermo Napco series 8000WJ CO2 incubator equipped with nitrogen tank for O2 replacement. O2 level was kept at 1% during hypoxia experiments for the indicated time periods. In all assays cell viability was assessed before staining. Drugs and corresponding vehicle controls (all from Sigma) were added as follows: 2-deoxy-D-glucose (2-DG, 2 mM) or Fenofibrate (FF, 50 μM) for the entire culture period; Etomoxir (ETO, 200 μM) for the last 48 hr. FCCP (500 μM), Mito-TEMPO (500 μM) and oligomycin (1 mM) were used as positive and negative controls for mitochondrial stains. They were incubated with cells at 37 °C for 20 min before samples were stained with DiOC6 and MitoSOX Red. DMSO concentrations were kept below 0.2% for all culture conditions.
Lentivector transduction of CD8+ T cells
For in vitro experiments, 4×106 enriched CD8+ T cells were stimulated as described above for 24–28 hr. Freshly concentrated lentivectors were spun-inoculated into activated CD8+ T cells supplemented with polybrene (6 μg/ml, Santa Cruz) at 2000 rpm, 32°C for 2 hr. For tumor cell transduction, lentivectors were incubated with tumor cells in 1ml serum-free, antibiotic-free medium and spun at 2500 rpm, 30 °C for 30 min. One ml of complete medium was added 6 hr later. Cells were washed 20 hr after transduction and stimulated for another 40 hr under normoxia or part-time hypoxia. For in vivo adoptive transfer experiment in the vaccine model, recipient mice had been challenged with tumor cells 5 days earlier and had been vaccinated with AdC68-gDMelapoly 2 days earlier. CD8+ T cells from spleens of naive C57Bl/6 mice were purified by negative selection using magnetic beads (MACS, STEMCELL Technologies). Enriched CD8+ T cells were stimulated with anti-CD3/CD28 Ab for 24 hr prior to lentivector transduction. Cells were washed 20 hr after transduction and cultured for an additional 48 hr in medium with IL-2 (100 U/ml) before transfer. For in vivo assays in the adoptive cell transfer model, OT-1 splenocytes were stimulated with peptide and IL-2 for 24 hr prior to spin inoculation with lentivectors. 72 hr later, dead cells were removed and transduced OT-1 CD8+ T cells were further purified by positive selection using CD90.1 microbeads (Miltenyi Biotec) before cell transfer.
Isolation of lymphocytes from mice
PBMCs and lymphocytes from spleens and lymph nodes were harvested as described (Zhang and Ertl, 2014). Briefly, blood samples were collected by submandibular puncture and PBMCs were isolated by Histopaque (Sigma) gradient centrifugation. Single cell suspension was generated by mincing spleens and lymph nodes with mesh screens in Leibovitz’s L15 medium followed by passing cells through a 70 μm filter (Fisher Scientific). Red blood cells were lysed by 1x RBC lysis buffer (eBioscience). To obtain tumor-infiltrating lymphocytes, tumors were harvested, cut into small fragments and treated with 2 mg/ml Collagenase P, 1 mg/ml DNase I (all from Roche) and 2% FBS (Tissue Culture Biologicals) in Hank’s balanced salt solution (HBSS,1X, Thermo Fisher Scientific) under agitation for 1 hour. Tumor fragments were homogenized, filtrated through 70 μm strainers and lymphocytes were purified by Percoll-gradient centrifugation and washed with DMEM supplemented with 10% FBS. Pre-experiments were conducted to ensure that this treatment did not affect expression levels of any of the tested markers.
Ab, staining and flow cytometry
Cells were stained with a PE-labeled Trp-1-specific MHC class I (H-2Db) tetramer carrying the TAPDNLGYM peptide, an Alexa647-labeled HPV-16 E7-specific MHC class I (H-2Db) tetramer carrying the RAHYNIVTTF peptide or an Alexa647-labeled OVA-specific MHC class I (H-2Kb) tetramer carrying the SIINFEKL peptide (NIAID Tetramer Facility). Lymphocytes were stained with anti-CD8-PerCPCy5.5 or -Alexa700, CD4-PerCPCy5.5, CD44-PacBlue or PerCPCy5.5, LAG-3-APC or -PerCPCy5.5, PD-1-PE-Cy7 (clone RMP1-30) or -Brilliant violet (BV) 605 (clone 29F.1A12), CD127-BV421 or -Alexa700, CD62L-FTIC or BV785, KLRG1-PE-Cy7, CD45.1-Pacific Blue, Vα2-PE and Thy1.1-PerCPCy5.5 or -APC (all from Biolegend or eBioscience) and Amcyan fluorescent reactive dye (ThermoFisher). For human PBMC or TILs analysis, cells were stained with fluorochrome-labeled Ab to CD14/CD19 (dump gate), CD3, CD8, CD95, CD28 and CCR7 to identify subsets and with Ab to PD-1, PPAR-α, Cpt-1a and BODYPY FL C6. Levels of PD-1 and PD-L1 expression on tumor cells were determined with anti-PD-1-PE-Cy7 or anti-PD-L1-APC Ab used in comparison to isotype control Ab (Biolegend). For analysis of mitochondrial markers, cells were stained with MitoSOX Red (5μM, MROS) and DiOC6 (40nM, MMP) (ThermoFisher) at 37 °C for 30 min under normoxia or hypoxia. For fatty acid uptake experiments, cells stimulated under different conditions in vitro or isolated from spleen and tumors of mice bearing day 14 or day 30 tumors were immediately incubated with 1 μM BODIPY FL C16 (ThermoFisher) for 30 min at 37 °C. Cells were washed twice with cold PBS before surface staining. For staining of T-bet, Eomes, total FoxO1, Cpt1a or PPAR-α cells were first stained for surface markers, then fixed and permeabilized with Foxp3/Transcription factor staining buffer and stained with T-bet-PE-Cy7, Eomes-Alexa488 (all from eBioscience), primary Ab against FoxO1 (C29H4, Cell Signaling Technology), anti-Cpt1a-Alexa 488 Ab or mouse IgG2b Isotype Ab; or anti-PPAR-α Ab (Abcam). Total FoxO1 was further determined by anti-rabbit secondary Ab staining (Cell Signaling Technology). PPAR-α was further detected with Streptavidin-BV605 (Biolegend). Phorphorylated (p)Akt was detected by staining cells with BD Phosflow buffers (Fix Buffer I and Perm Buffer III) and Phospho-Akt (Novus, clone 545007) Ab to Akt phosphorylated at serine 473.
For HIF-1α and Glut staining, in ex vivo assays mice were perfused immediately after euthanasia with Hank’s balanced salt solution (HBSS) and heparin (10 units/ml) and then with 1 mM Cobalt (II) chloride (COCl2, Sigma-Aldrich) diluted in PBS. For both ex vivo and in vitro experiments, lymphocyte isolation and staining before fixation were performed in medium containing 200 μM COCl2. Cells were stained with Abs to cell surface markers for 30 min. Cells were fixed and permeabilized using the FoxP3 buffer kit, and stained for HIF-1α with anti-HIF-1α-Alexa700 Ab (R&D), or anti-Glut1-Alexa488 (Abcam) and the corresponding isotype control antibodies.
For intracellular cytokine staining (ICS) of ex vivo stimulated lymphocytes, ~106 cells per samples were cultured in DMEM containing 2% FBS and Golgiplug (Fisher Scientific, 1.5 μl/ml) for 6 hr with either a peptide pool (5 ug/ml for each peptide) including all CD8+ T cell epitopes expressed by gDMelapoly (mTrp-1455-463: TAPDNLGYA, mTrp-1481-489: IAVVAALLL, mTrp-2522-529: YAEDYEEL, hTp-2180-188: SVYDFFVWL, hTrp-2343-357: STFSFRNAL, mTrp-2363-371: SQVMNLHNL, hgp10025-33: KVPRNQDWL, mBraf 594-602: FGLANEKSI); the E7 peptide: RAHYNIVTTF or SIINFEKL peptide (all from Genscript) or a rabies virus glycoprotein control peptide. For ICS performed with CD8+ T cells activated in vitro, ~106 cells were transferred to 96 well plates in the original medium and stimulated with PMA (50 ng/ml), ionomycin (2 μg/ml) and Golgiplug for 4 hr under normoxia or hypoxia. Cells were stained with Ab to IFN-γ (APC or BV421), GrmB (APC, Life Technologies) and perforin (PE, eBioscience) after fixation/permeabilization (BD Pharmingen). Cells were analyzed by an LSRII (BD Biosciences). Data were analyzed with FlowJo (TreeStar).
T cell assays
CD8+ T cells were analyzed by tetramer staining to assess phenotypes or by intracellular cytokine staining (ICS) following a 5-hour period of peptide stimulation in presence of GolgiPlug (BD Biosciences). The dominant CD8+ T response elicited by the AdC68-gDMelapoly vector is directed against the Trp-1455 epitope (~90% of MAA-specific CD8+ T cell response), which was assessed by tetramer staining. All peptides carrying the tumor antigen-specific epitopes of the vaccine inserts were used for ICS. For easier reference in the text we refer to both types of AdC68-gDMelapoly-induced CD8+ T cells as MAA-specific CD8+ T cells.
In vivo tumor cell survival assay
Mice were treated with isotype control- or α-PD-1 Ab (200 μg/mouse) 4 and 1 days before tumor cell challenge. Some of the mice had either been vaccinated with AdC68-gDMelapoly 14 days earlier, or been injected i.p. with in vitro activated OT-1 CD8+ T cells (105 cells/mouse) 4–6 hr before tumor cell challenge. PD-L1hi (scrambled shRNA lentivector transduced) or PD-L1lo (PD-L1 shRNA lentivectors transduced) B16BrafV600E tumor cells or B16OVA cells were sorted based on lentivector GFP expression and passaged for several rounds in presence of puromycin to create transduced stable cell lines. Transduced tumor cells were labeled with high (2 μM) or low (0.2 μM) levels of CellTrace Violet (CTV), respectively, mixed at 1:1 ratio and injected i.p. into recipient mice at a dose of 106 cells/tumor cell subset. 1-day later mice were euthanized. Cells were collected by vigorously rinsing the peritoneal cavity with a trypsin solution. Cells were washed and stained with Ab to CD3, CD14 and CD19 as dump gates and with mAb to PD-L1. The relative recovery of CTVhiPD-L1hi vs. CTVloPD-L1lo cell subsets in each treatment groups was determined by flow cytometry.
BrdU proliferation assay
Mice were injected i.p. with of BrdU (1.5–2 mg/mouse) and fed water-containing BrdU at a concentration of 0.8 mg/ml on days 9, 19 or 29 after vaccination; they were euthanized the next day and lymphocyte samples were analyzed for BrdU incorporation. Cells were first stained for surface markers and then for intracellular BrdU (1:50 dilution) according to the manufacture’s instruction (ThermoFisher).
Extracellular Flux Analysis and FAO assay
OCR and ECAR for CD8+ T cells stimulated under different conditions were measured with XF24 and XF96 Extracellular Flux Analyzers (Seahorse Bioscience). Hypoxia samples were prepared in a hypoxia chamber under 1% O2. Dead cells were removed by a dead cell removal kit (Miltenyi Biotec) and live cells were pre-incubated with 100 μM COCl2 before being removed from the hypoxia chamber and entered into the Seahorse analyzer. To determine the contribution of FAO to OCR, 200 μM ETO was added 15 min before the Seahorse analysis. Briefly after repeated measures of basal respiration and lactate production, 1 μM OM was added to measure ATP leakage by OCR and glycolytic capacity by ECAR. 1.5 μM FCCP was then added to measure maximal respiration by OCR followed by addition of 100 nM Rotenone and 1 μM Antimycin A to determine spare respiratory capacity by OCR and then 100 mM 2-DG to determine glycolytic reserve by ECAR. For measuring oxidation of exogenous and endogenous FAs, cells activated in either Glc- or Gal-medium for 3 days were washed and transferred to substrate-limited Glc- or Gal-medium for overnight stimulation. Substrate limited media contained 0.5 mM Glc or Gal, 1 mM GlutaMAX, 0.5 mM carnitine (all form Sigma) and 1% dialyzed FBS. Samples were treated with either ETO or vehicle control 15 min before the assay. Palmitate: BSA or BSA was added just before the assay. The contributions of FAO to OCR was calculated as follows: Basal respiration due to exogenous FA oxidation= Basal Palm:BSA-ETO OCR rate – basal BSA-ETO OCR rate - OCR due to uncoupling by FFA; uncoupling by FFA= after OM injection, Palm:BSA-ETO OCR rate - BSA-ETO rate. Basal OCR due to endogenous FAs consumption = basal BSA-ETO OCR rate - basal BSA+ETO OCR rate.
Lipid and Glc concentration measurement in tumor or tissue interstitial fluid
Interstitial fluid from kidney, heart and mouse or human melanoma samples were collected by centrifugation as described (Wiig et al., 2003) and snap frozen on dry ice. Mice were perfused with HBSS with 10 U/ml heparin before tissue collection. Concentrations of free FA species were determined by LC-MS. Absolute concentrations of Glc were measured by LC-MS upon adding 13C6-Glc as the internal standard.
Isotopic labeling in vitro
For 13C6- Glc/Gal tracing in vitro, cells were cultured in Glc-free RPMI medium with 10 mM 13C6-Glc/Gal (Sigma or Cambridge) for 4 days. For 13C16-palmitate tracing in vitro, cells were stimulated for 3 days in Glc- or Gal-medium. On the night of day 3, some samples were transferred to 1% O2 for overnight culture. 13C16-palmitate (Sigma) was first dissolved in 100% ethanol at 200 mM and conjugate to fatty acid-free BSA at a 5:1 molar ratio to a final concentration of 8 mM-13C16-palmitate-BSA by vortexing at 37 °C for 3–4 hr with sonication. On day 4, samples were pelleted and replated in fresh medium with 10% delipidated FBS (Cocalico Biologicals) and 400 μM 13C16-palmitate-BSA. Hypoxia samples were returned to 1% O2. All samples were cultured for another 4 hr. Dead cells were removed. Samples were pelleted at 4000 rpm for 5 min. All collection procedures were conducted at 4 °C. Cell numbers in each sample were determined. Metabolism was quenched and metabolites were extracted by adding 1 ml −80 °C 80:20 methanol: water per million cells. After 20 min of incubation on dry ice, samples were centrifuged at 10000 g for 5 min. Insoluble pellets were re-extracted with 0.5 ml −80 °C 80: 20 methanol: water on dry ice. The supernatants from two rounds of extraction were combined, dried under N2, resuspended in 100 μl water per million cells. Metabolites were normalized to cell numbers.
Isotope labeling in vivo
Tumor-bearing mice were fasted for 16 hr. 13C6-Glc (Cambridge Isotope laboratories) diluted in PBS was given i.p. to mice at 2 g/kg. Spleens and tumors were collected 30 min later. 13C16-potassium palmitate (Sigma-Aldrich) was conjugated to FA-free BSA (6:1 molar ratio) and given to mice at ~0.35 g/kg by oral gavage. 1 hour later 13C16-palmitate-BSA was given i.v. at 125 mg/kg. Spleens and tumors were collected 30 min later and cells were isolated on ice. To assess the potential effects of sample processing on lymphocyte metabolism, lymph nodes (LN) were snap frozen on dry ice immediately upon isolation. In the same experiments, single cell suspensions were prepared from other LNs of the same mice and incubated at 4 °C for 2–2.5 hr prior to the metabolic analyses. CD44+CD8+ T cells from pooled spleen or tumor samples were stained and sorted at 4 °C. Metabolites were extracted with −80 °C 80: 20 methanol: water, dried under N2 and resuspended in water at 100 mg tissue/ml or 106 cells/100 μl.
LC-MS Instrumentation and method development
Glycolytic and TCA metabolites were analyzed by reversed-phase ion-pairing chromatography coupled with negative-mode electrospray-ionization high-resolution MS on a stand-alone orbitrap (Thermo)(Lu et al., 2010). Carnitine species were analyzed by reversed-phase ion pairing chromatography coupled with positive-mode electrospray-ionization on a Q Exactive hybrid quadrupole-orbitrap mass spectrometer (Thermo); Liquid chromatography separation was achieved on a Poroshell 120 Bonus-RP column (2.1 mm ×150 mm, 2.7 μm particle size, Agilent). The total run time is 25 min, with a flow rate of 50 μl/min from 0 min to 12 min and 200 μl/min from 12 min to 25 min. Solvent A is 98: 2 water: acetonitrile with 10 mM amino acetate and 0.1% acetic acid; solvent B is acetonitrile. The gradient is 0–70% B in 12 min. All isotope-labeling patterns were corrected for natural 13C-abundance.
Electron Microscopy
CD44+CD8+ T cells were sorted from spleens and TILs of mice bearing day 30 tumors. They were fixed with 2.5% glutaraldehyde, 2.0% paraformaldehyde in 0.1 M sodium cacodylate buffer, pH 7.4, overnight at 4 °C. After washes in buffer, the samples were post-fixed in 2.0% osmium tetroxide for 1 hour at room temperature (RT), and then washed again in buffer followed by distilled H2O. After dehydration through a graded ethanol series, the samples were infiltrated and embedded in EMbed-812 (Electron Microscopy Sciences). Thin sections were stained with lead citrate and examined with a JEOL 1010 electron microscope fitted with a Hamamatsu digital camera and AMT Advantage image capture software.
Gene expression analysis
Lymphocytes were isolated from spleens and tumors of mice at different time points and stained with dyes and Ab to live cells, CD8+, CD44+ and the Trp-1 and E7 tetramers. For co-adoptive transfer experiments, CD44+ CD8+ T donor cells of different origins were recovered from spleens and tumors of recipient mice upon Ab staining and sorting. Cells were sorted (Mono Astrios, Beckman Coulter) on ice for MAA- or E7-tet+CD44+CD8+ T cells into RLT lysis buffer (QIAGEN). For in vitro culture samples ~ 106 cells/sample were processed on ice to remove dead cells. For lentivector-mediated gene knockdown assays, transduced cells were further purified based on the selection marker using microbeads or cell sorting. RNA was isolated from purified cells using RNeasy Mini kits (Qiagen) and RNA concentrations were determined using Nanodrop (Thermo Scientific). cDNAs were obtained by reverse transcription using the high capacity cDNA reverse transcription kit (Life Technologies). Relative qRT-PCR analyses were performed using 7500 Fast Real-Time PCR system (Life Technologies). β-2 microglobulin or GAPDH were used as internal controls. Vector NTI was used for primers design (Table S1). Differences in transcript expression levels are visualized in heatmaps. Values were log transformed to show ratios of differences. Color scale was set as −2 (lower expression, deep blue) to 2 (higher expression, deep red).
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were conducted using GraphPad Prism 6 (GraphPad). Differences between 2 populations were calculated by Student’s t-test. Multiple comparisons between two groups were performed by multiple t-test with type I error correction. Differences among multiple populations were calculated by one- or two-way ANOVA. Differences in survival were calculated by Log-rank Mantel-Cox test. Differences between tumor growth curves were determined by repeated measures two-way ANOVA. Type I errors were corrected by Holm-Šídák method. Significance was set at p values of or below 0.05. For all figures, *p ≤0.05 – 0.01, ** p≤0.01–0.001, *** p ≤0.001–0.0001, **** p≤0.0001. Unless noted in the figure legend, all data are shown as mean +/− SEM. The n numbers for each experiment, as well as the numbers of experiments that has been repeated have been noted in the figure legends.
Supplementary Material
Significance.
Metabolic fitness is critical for cell functions. Dysregulated metabolism contributes to the exhaustion of TILs within the TME. If and how TILs adjust their metabolism to maintain functional remains to be explored. We show that hypoxia and hypoglycemia, two major metabolic challenges within the TME, impair CD8+ TILs through distinct pathways. Using two mouse melanoma models and human melanoma samples, our study shows that CD8+ TILs experiencing double metabolic jeopardy enhance PPAR-α signaling and fatty acid (FA) catabolism to preserve energy production and effector functions. Promoting FA catabolism with fenofibrate markedly improves their capacity to delay tumor growth. It synergizes with PD-1 blockade to efficiently boost the efficacy of melanoma immunotherapy.
Acknowledgments
We thank the NIH Tetramer Core Facility for tetramers; J. Faust, Y. Li, Drs. Z.Q. Xiang, A. Kossenkov, Y. Nefedova (Wistar Institute), R. Meade, X. Zuo and Dr. K. Foskett (U of PA) for help with reagents and equipment; Dr. Z. Schug for discussions and Drs. P. Matzinger and Y. Paterson for reviewing the manuscript. Funding: Wistar Vaccine Center Funding; NCI grants CA114046, CA174523, P50CA101942; DoD PRCRP CA1150619 and Dr. Miriam and Sheldon G. Adelson Medical Research Foundation.
Footnotes
AUTHOR CONTRIBUTIONS
YZ - design, experiments, data analyses, writing; RK - experiments; LL - metabolomics studies; XYZ- cloning design; AH, FF- assisted YZ; WG-D - cell culture; XWX, GK, LS, WX and RA - human samples collection; GF - α-PD-1 Ab; JR - metabolomics studies; GZ, MX, NS, CK, MH - PDX model; HE - design, data analyses, writing.
References
- Azuma T, Yao S, Zhu G, Flies AS, Flies SJ, Chen L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood. 2008;111:3635–3643. doi: 10.1182/blood-2007-11-123141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer ML, Ma EH, Bantug GR, Grählert J, Pfister S, Glatter T, Jauch A, Dimeloe S, Slack E, Dehio P, Krzyzaniak MA, King CG, Burgener AV, Fischer M, Develioglu L, Belle R, Recher M, Bonilla WV, Macpherson AJ, Hapfelmeier S, Jones RG, Hess C. Memory CD8+ T Cells Require Increased Concentrations of Acetate Induced by Stress for Optimal Function. Immunity. 2016;44:1312–1324. doi: 10.1016/j.immuni.2016.03.016. [DOI] [PubMed] [Google Scholar]
- Bucks CM, Norton JA, Boesteanu AC, Mueller YM, Katsikis PD. Chronic antigen stimulation alone is sufficient to drive CD8+ T cell exhaustion. J Immunol. 2009;182:6697–6708. doi: 10.4049/jimmunol.0800997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamoto K, Chowdhury PS, Kumar A, Sonomura K, Matsuda F, Fagarasan S, Honjo T. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proceedings of the National Academy of Sciences. 2017;114:E761–E770. doi: 10.1073/pnas.1620433114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJW, Tonc E, Schreiber RD, Pearce EJ, Pearce EL. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell. 2015:1–14. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015;125:3384–3391. doi: 10.1172/JCI80011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, Eil RL, Tran E, Hanada KI, Yu Z, Palmer DC, Kerkar SP, Michalek RD, Upham T, Leonardi A, Acquavella N, Wang E, Marincola FM, Gattinoni L, Muranski P, Sundrud MS, Klebanoff CA, Rosenberg SA, Fearon DT, Restifo NP. Akt Inhibition Enhances Expansion of Potent Tumor-Specific Lymphocytes with Memory Cell Characteristics. Cancer Res. 2015;75:296–305. doi: 10.1158/0008-5472.CAN-14-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgleish AG. Therapeutic cancer vaccines: Why so few randomised phase III studies reflect the initial optimism of phase II studies. Vaccine. 2011;29:8501–8505. doi: 10.1016/j.vaccine.2011.09.012. [DOI] [PubMed] [Google Scholar]
- Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, Johnson RS, Goldrath AW. Hypoxia-inducible factors enhance the effector responses of CD8+ T cells to persistent antigen. Nat Immunol. 2013;14:1173–1182. doi: 10.1038/ni.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, Panteleyev AA, Okkenhaug K, Cantrell DA. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Cell Biol. 2012;199:i8–i8. doi: 10.1083/JCB1996OIA8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosso JF, Kelleher CC, Harris TJ, Maris CH, Hipkiss EL, De Marzo A, Anders R, Netto G, Getnet D, Bruno TC, Goldberg MV, Pardoll DM, Drake CG. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J Clin Invest. 2007;117:3383–3392. doi: 10.1172/JCI31184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka RB, Chandel NS. Targeting glucose metabolism for cancer therapy. Journal of Experimental Medicine. 2012;209:211–215. doi: 10.1084/jem.20120162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, Zarour H, Joshua AM, Gergich K, Elassaiss-Schaap J, Algazi A, Mateus C, Boasberg P, Tumeh PC, Chmielowski B, Ebbinghaus SW, Li XN, Kang SP, Ribas A. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC, Kleinstein SH, Abel ED, Insogna KL, Feske S, Locasale JW, Bosenberg MW, Rathmell JC, Kaech SM. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell. 2015;162:1217–1228. doi: 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nature Reviews Immunology. 2012;12:749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, Carlino MS, Haanen JB, Maio M, Marquez-Rodas I, McArthur GA, Ascierto PA, Long GV, Callahan MK, Postow MA, Grossmann K, Sznol M, Dréno B, Bastholt L, Yang A, Rollin LM, Horak C, Hodi FS, Wolchok JD. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasaro MO, Tatsis N, Hensley SE, Whitbeck JC, Lin SW, Rux JJ, Wherry EJ, Cohen GH, Eisenberg RJ, Ertl HC. Targeting of antigen to the herpesvirus entry mediator augments primary adaptive immune responses. Nat Med. 2008;14:205–212. doi: 10.1038/nm1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Clasquin MF, Melamud E, Amador-Noguez D, Caudy AA, Rabinowitz JD. Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Anal Chem. 2010;82:3212–3221. doi: 10.1021/ac902837x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Sotgia F, Lisanti MP. Ketone body utilization drives tumor growth and metastasis. Cell Cycle. 2012;11:3964–3971. doi: 10.4161/cc.22137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamee EN, Korns Johnson D, Homann D, Clambey ET. Hypoxia and hypoxia-inducible factors as regulators of T cell development, differentiation, and function. Immunol Res. 2013;55:58–70. doi: 10.1007/s12026-012-8349-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- Mrass P, Takano H, Ng LG, Daxini S, Lasaro MO, Iparraguirre A, Cavanagh LL, von Andrian UH, Ertl HCJ, Haydon PG, Weninger W. Random migration precedes stable target cell interactions of tumor-infiltrating T cells. Journal of Experimental Medicine. 2006;203:2749–2761. doi: 10.1084/jem.20060710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SN, Ahmed R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc Natl Acad Sci USA. 2009;106:8623–8628. doi: 10.1073/pnas.0809818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan D, van der Windt GJW, Huang SCC, Curtis JD, Chang CH, Buck MD, Qiu J, Smith AM, Lam WY, DiPlato LM, Hsu FF, Birnbaum MJ, Pearce EJ, Pearce EL. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity. 2014;41:75–88. doi: 10.1016/j.immuni.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta A, Diwanji R, Kini R, Subramanian M, Ohta A, Sitkovsky M. In vivo T cell activation in lymphoid tissues is inhibited in the oxygen-poor microenvironment. Front Immunol. 2011;2:27. doi: 10.3389/fimmu.2011.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, Riley JL. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol Cell Biol. 2005;25:9543–9553. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P, Li L, Boussiotis VA. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nature Communications. 2015;6:6692. doi: 10.1038/ncomms7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsoukis N, Li L, Sari D, Petkova V, Boussiotis VA. PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol Cell Biol. 2013;33:3091–3098. doi: 10.1128/MCB.00319-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siska PJ, Beckermann KE, Mason FM, Andrejeva G, Greenplate AR, Sendor AB, Chiang YCJ, Corona AL, Gemta LF, Vincent BG, Wang RC, Kim B, Hong J, Chen CL, Bullock TN, Irish JM, Rathmell WK, Rathmell JC. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight. 2017;2:1–13. doi: 10.1172/jci.insight.93411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, Roychoudhuri R, Palmer DC, Muranski P, Karoly ED, Mohney RP, Klebanoff CA, Lal A, Finkel T, Restifo NP, Gattinoni L. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. 2013;123:4479–4488. doi: 10.1172/JCI69589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Iizumi T, Mashima K, Abe T, Suzuki N. Roles and regulation of ketogenesis in cultured astroglia and neurons under hypoxia and hypoglycemia. ASN Neuro. 2014;6:1759091414550997. doi: 10.1177/1759091414550997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsis N, Fitzgerald JC, Reyes-Sandoval A, Harris-McCoy KC, Hensley SE, Zhou D, Lin SW, Bian A, Xiang ZQ, Iparraguirre A, Lopez-Camacho C, Wherry EJ, Ertl HCJ. Adenoviral vectors persist in vivo and maintain activated CD8+ T cells: implications for their use as vaccines. Blood. 2007;110:1916–1923. doi: 10.1182/blood-2007-02-062117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veech RL. The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent Fatty Acids. 2004;70:309–319. doi: 10.1016/j.plefa.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Wiig H, Aukland K, Tenstad O. Isolation of interstitial fluid from rat mammary tumors by a centrifugation method. Am J Physiol Heart Circ Physiol. 2003;284:H416–24. doi: 10.1152/ajpheart.00327.2002. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ertl HCJ. The effect of adjuvanting cancer vaccines with herpes simplex virus glycoprotein D on melanoma-driven CD8+ T cell exhaustion. J Immunol. 2014;193:1836–1846. doi: 10.4049/jimmunol.1302029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Ju S, Chen E, Dai S, Li C, Morel P, Liu L, Zhang X, Lu B. T-bet and eomesodermin are required for T cell-mediated antitumor immune responses. J Immunol. 2010;185:3174–3183. doi: 10.4049/jimmunol.1000749. [DOI] [PubMed] [Google Scholar]
- Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4–328rv4. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.