

Abstract
The Cdc48 ATPase and its cofactors Ufd1/Npl4 (UN) extract polyubiquitinated proteins from membranes or macromolecular complexes, but how they perform these functions is unclear. Cdc48 consists of an N-terminal domain that binds UN and two stacked hexameric ATPase rings (D1 and D2) surrounding a central pore. Here, we use purified components to elucidate how the Cdc48 complex processes substrates. After interaction of the polyubiquitin chain with UN, ATP hydrolysis by the D2 ring moves the polypeptide completely through the double ring, generating a pulling force on the substrate and causing its unfolding. ATP hydrolysis by the D1 ring is important for subsequent substrate release from the Cdc48 complex. This release requires cooperation of Cdc48 with a deubiquitinase, which trims polyubiquitin to an oligoubiquitin chain that is then also translocated through the pore. Together, these results lead to a new paradigm for the function of Cdc48 and its mammalian ortholog p97/VCP.
Graphical abstract
INTRODUCTION
The yeast Cdc48 ATPase and its metazoan ortholog p97 (or VCP) are critical components of many ubiquitin-dependent cellular pathways that require the segregation of individual proteins from binding partners or membranes (for review, see Buchberger, 2013; Meyer and Weihl, 2014; Xia et al., 2016). This ATPase is present in all eukaryotic cells and is essential for their viability. The function of Cdc48/p97 is best understood in endoplasmic reticulum (ER)-associated protein degradation (ERAD), in which it extracts polyubiquitinated, misfolded proteins from the ER and transfers them to the proteasome for degradation (Christianson and Ye, 2014). Cdc48/p97 also functions in mitochondrion-associated protein degradation (Taylor and Rutter, 2011), ribosomal quality control (Verma et al., 2013), and the extraction of chromatin-bound proteins (Franz et al., 2016; Ramadan et al., 2007). Consistent with this central role in protein quality control, mutations in human p97 cause several neurodegenerative proteopathies (Chapman et al., 2011; Kimonis et al., 2008). Despite its biological significance, the mechanism of Cdc48/p97 action is poorly understood.
Cdc48 belongs to the AAA+ family of ATPases (ATPases associated with various cellular activities), whose members use ATP hydrolysis to exert force on macromolecules (Erzberger and Berger, 2006; Sauer and Baker, 2011). Many of the AAA proteins form hexamers with either a single or double ring of ATPase domains (type I and II ATPases, respectively). Cdc48 is a type II ATPase consisting of an N-terminal (N) domain, two tandem AAA domains (D1 and D2) separated by a short linker, and a flexible C-terminal tail (DeLaBarre and Brunger, 2003). The AAA domains form two stacked rings surrounding a central pore. Both ATPase rings hydrolyze ATP (Chou et al., 2014; Ye et al., 2003), but their roles in substrate processing are unknown. ATP hydrolysis in the D1 ring seems to move the N domains from a so-called “up-conformation” in the ATP state to a position co-planar with the D1 ring in the ADP-bound state (“down-conformation”) (Banerjee et al., 2016). The function of this N domain movement is unclear.
Cdc48 cooperates with a large number of protein cofactors that provide pathway selectivity and fine-tune substrate processing. One of the most important cofactors is the Ufd1/Npl4 heterodimer (UN), an essential complex that participates in many Cdc48-dependent processes, including ERAD (Ye et al., 2004). UN binds to the N domain of Cdc48 and recruits polyubiquitinated substrates to the ATPase (Ye et al., 2003). In a reconstituted in vitro system of partial ERAD reactions, a polyubiquitinated, misfolded protein could be extracted from the membrane in a UN- and Cdc48- dependent manner (Stein et al., 2014). However, most experiments with the Cdc48/p97 complex have been performed with intact cells, the complexity of which makes it impossible to obtain mechanistic insight. Dissection of Cdc48 function has also been hampered by a lack of suitable in vitro substrates. The only reported experiments employed non-ubiquitinated proteins (Baek et al., 2011; DeLaBarre et al., 2006), which are not the principal substrates in vivo. Cdc48’s physiological substrates are generally polypeptides modified with K48-linked polyubiquitin chains, which also serve as a major targeting signal for the proteasome (Pickart, 2000). In vitro experiments with the proteasome have employed polypeptides decorated with K63-linked polyubiquitin chains (Nathan et al., 2013), but these substrates are not appropriate for the more specific Cdc48 ATPase complex (Ye et al., 2003).
One of the most important questions is how the Cdc48 ATPase can “pull” on a substrate, thereby releasing it from a protein complex or membrane. Some AAA ATPases, including the Clp ATPases in bacteria, the archaeal Cdc48 homologs, and the 19S proteasome, use a translocation mechanism (for review, see White and Lauring, 2007). In these cases, central loops with conserved aromatic residues are thought to contact a polypeptide chain and move in response to ATP hydrolysis, thereby dragging the substrate through the pore. Archaeal Cdc48 and some mammalian p97 mutants can also translocate polypeptides into associated 20S proteasomes under certain conditions (Barthelme and Sauer, 2012; 2013), implying movement through the central pore. However, archaeal Cdc48 undergoes conformational changes that have never been observed with eukaryotic homologs (Huang et al., 2016), and wild type yeast Cdc48 and mammalian p97 are thought not to use a translocation process (Banerjee et al., 2016; DeLaBarre and Brunger, 2005). A mechanism that does not involve translocation is also employed by Cdc48’s closest relative, the NEM-sensitive fusion protein (NSF) (Zhao et al., 2015). NSF is thought to bind the SNARE complex and the cofactor αSNAP with its N domains, and to use ATP hydrolysis to unwind the supercoiled SNARE proteins without polypeptide passage through the central pore. A major argument against a translocation mechanism for Cdc48/p97 is that its central pore is very narrow or occluded in crystal or cryo-EM structures. These observations have led to the proposal of several alternative mechanisms. In one model, a substrate transiently enters the D2 ring or moves through the D2 ring, exiting between the D1 and D2 rings (DeLaBarre and Brunger, 2005). Other models postulate that the polypeptide chain inserts only shallowly into the D1 ring (for review, see Xia et al., 2016). Finally, it has been suggested that the large, nucleotide-dependent conformational changes of the N domains are sufficient to move a polypeptide chain (Schuller et al., 2016). In each of these cases, it is unclear how a continuous pulling force could be generated, although such a force would likely be required to extract a subunit from a tight multimeric complex or a multi-spanning protein from the ER membrane. Clearly, experiments are needed to test whether a translocation mechanism or any of the alternative models can explain the function of Cdc48.
Another important question concerns the mechanism by which a polyubiquitinated substrate is released from the Cdc48 complex and passed on to downstream components, such as the proteasome. There are at least three proposed models. In the first, a ubiquitinated substrate is transferred by ubiquitin-binding shuttling factors to the proteasome (Richly et al., 2005). In the second, the substrate is completely deubiquitinated, processed by Cdc48, and then re-ubiquitinated to enable shuttling factor and proteasome binding (for review, see Liu and Ye, 2012). In the third, substrate is only partially deubiquitinated, leaving sufficient ubiquitin moieties for interaction with downstream components. A candidate for a deubiquitinase involved in Cdc48-dependent reactions is Otu1 (called YOD1 in mammals). Otu1 binds via its UBX-like domain to the N domain of Cdc48, an interaction that stimulates its deubiquitination activity (Ernst et al., 2009; Stein et al., 2014). Expression of an enzymatically inactive Otu1 mutant blocks Cdc48 function in vivo and leads to the accumulation of polyubiquitinated proteins on the Cdc48/p97 complex. However, the precise role of Otu1 has yet to be clarified. Specifically, it is unclear whether it deubiquitinates substrates before they are processed by Cdc48 and thus acts as a negative regulator (Rumpf and Jentsch, 2006), or synergistically cooperates with Cdc48.
Here, we have elucidated the mechanism of Cdc48 from S. cerevisiae with purified components that include Cdc48 itself, its UN cofactor, and model substrates bearing K48-linked polyubiquitin chains. We demonstrate that polyubiquitinated substrate first binds to UN. Following ATP binding to the D1 ring, Cdc48 unfolds the substrate by passing it through the central pore, starting from the D1 side and moving it all the way through the D2 ring. This translocation process relies on ATP hydrolysis by the D2 domain. We show that subsequent substrate release from Cdc48 requires hydrolysis of ATP in D1 and partial trimming of the ubiquitin chain by the deubiquitinase Otu1. The remaining ubiquitin molecules follow the substrate through the central pore and are released on the D2 side of the pore. Our results establish a model for protein extraction and segregation that is broadly applicable to the various Cdc48-dependent quality control systems.
RESULTS
Generation of a purified Cdc48 substrate
Cdc48’s substrates in ERAD and other pathways commonly carry K48-linked polyubiquitin chains. To obtain a well-defined substrate modified with a single ubiquitin chain of this linkage, we employed a system based on the N-end rule pathway. This pathway degrades proteins with certain N-terminal amino acids, such as arginines (Chau et al., 1989). The substrate used for our studies is produced as a fusion protein (Figure 1A), consisting of an N-terminal SUMO tag, a 43 amino acid degron sequence that has been shown to be polyubiquitinated at a lysine residue (Chau et al., 1989), and superfolder GFP (sfGFP). The fusion protein was expressed in E. coli and purified on the basis of an N-terminal His-tag. The SUMO tag was then cleaved off with the SUMO-specific protease Ulp1. The cleavage product contains an N-terminal arginine, which renders it a substrate for Ubr1, an E3 ubiquitin ligase of the N-end rule pathway (Bartel et al., 1990). To facilitate detection of the substrate, a fluorescent dye was attached to a cysteine close to the N-terminus. The labeled protein was then incubated with a mixture of purified ubiquitin-activating enzyme (Uba1), ubiquitin-conjugating enzyme (Ubc2), ubiquitin ligase (Ubr1; for purity of the proteins, see Figure S1A), ubiquitin, and ATP. The polyubiquitinated substrate was purified on streptavidin beads via a C-terminal streptavidin binding peptide (SBP) tag and eluted from the resin by cleavage with 3C protease.
Figure 1. Cdc48/UN complex interacts with a polyubiquitinated substrate.
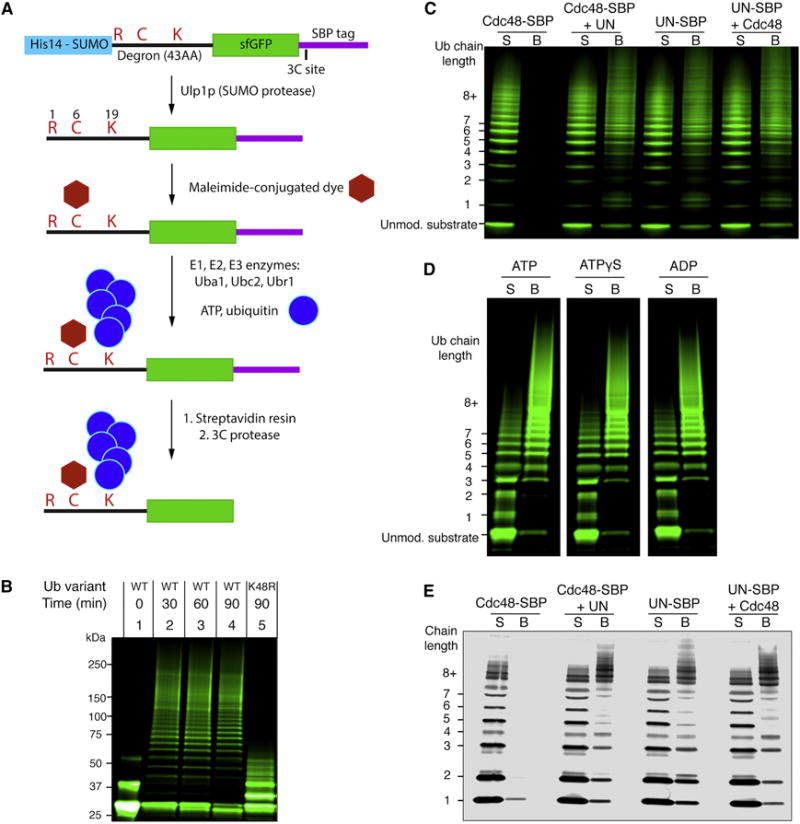
(A) Scheme for the synthesis and purification of polyubiquitinated superfolder GFP (sfGFP) a Cdc48 substrate containing an N-end degron; see text for details.
(B) Dye-labeled sfGFP bearing the N-end degron was incubated with ubiquitination machinery, ATP, and the indicated ubiquitin variant. Samples were analyzed by SDS-PAGE and fluorescence scanning. The unmodified substrate appears as two bands due to incomplete denaturation in SDS.
(C) The indicated proteins were immobilized through a SBP tag on streptavidin beads and incubated with ubiquitinated sfGFP. Supernatants (S) and bound material (B) were analyzed by SDS-PAGE and fluorescence scanning.
(D) As in (C), with Cdc48 and SBP-tagged UN used in each condition. The indicated nucleotide was included in all steps following purification. Irrelevant lanes of the gel have been removed for display (white spaces).
(E) As in (C), but with free fluorescently labeled K48-linked polyubiquitin chains instead of polyubiquitinated sfGFP.
See also Figure S1.
The substrate contained ubiquitin chains of different lengths (Figure 1B; lanes 2–4). Essentially all chains are composed of K48 linkages, as most high-molecular weight chains failed to form when wild type ubiquitin was replaced with the ubiquitin mutant K48R (Figure 1B; lane 5). Mass spectrometry suggested that the remaining modification is monoubiquitination at several sites in the sfGFP moiety (Figure S1B). Analysis of tryptic peptides derived from the polyubiquitinated substrate also suggested that K63-linked chains occurred in only low abundance (three orders of magnitude lower than K48 linkages) and that polyubiquitin chains were attached almost exclusively at a single lysine in the degron segment preceding sfGFP (Figure S1B).
Substrate binding to the Cdc48 complex
To test substrate binding to the Cdc48 complex, polyubiquitinated sfGFP substrate (Ub(n)-GFP) was incubated with SBP-tagged S. cerevisiae Cdc48 ATPase, which was purified after expression in E. coli and immobilized on streptavidin beads. The incubation was performed in the presence or absence of the UN cofactor from S. cerevisiae, which was also expressed in E. coli and purified as a heterodimer (Figure S1A). Consistent with previous studies (Stein et al., 2014), substrate binding was only observed in the presence of the cofactor (Figure 1C). Substrate molecules with chains of five or more ubiquitin moieties bound more efficiently than those with shorter chains. The UN complex alone was capable of substrate binding, as shown by immobilizing the cofactor on streptavidin beads through an SBP tag on Ufd1 (Figure 1C). The extent of substrate binding was comparable in the presence of ATP, ATPγS, and ADP (Figure 1D). Similar results were obtained when the binding of free K48-linked ubiquitin chains was tested (Figure 1E). These chains were synthesized with purified Uba1, the ubiquitin-conjugating enzyme Ubc1 (Rodrigo-Brenni et al., 2010), and ubiquitin that was labeled with a fluorescent dye at a cysteine appended to the N-terminus. Again, the UN complex was responsible for all interaction with the Cdc48 complex (Figure 1E). Taken together, these results indicate that substrate is recruited to the Cdc48 complex through an interaction between the polyubiquitin chain and the UN cofactor.
The effect of substrate on Cdc48 ATPase activity
To evaluate the effect of substrate binding on Cdc48 activity, we measured the rate of steady state ATP hydrolysis. UN reduced the intrinsic ATPase rate of Cdc48 by a factor of about two (Figure 2A). When saturating Ub(n)-sfGFP was added to the Cdc48/UN complex, the ATP hydrolysis rate was stimulated approximately 5-fold. The substrate had no stimulatory effect in the absence of the UN complex (Figure 2A), and no stimulation was seen with non-ubiquitinated sfGFP (Figure 2B). Free K48-linked polyubiquitin chains stimulated ATP hydrolysis to the same extent as polyubiquitinated sfGFP (Figure 2B). Stimulation was dependent on the length of the ubiquitin chains, with mono- and di-ubiquitin having negligible effects, and penta-ubiquitin producing the highest rate of hydrolysis (Figure 2C). K63-linked penta-ubiquitin, by contrast, had no effect on ATP hydrolysis (Figure S2A).
Figure 2. Ubiquitin stimulates Cdc48 ATPase activity for substrate unfolding.
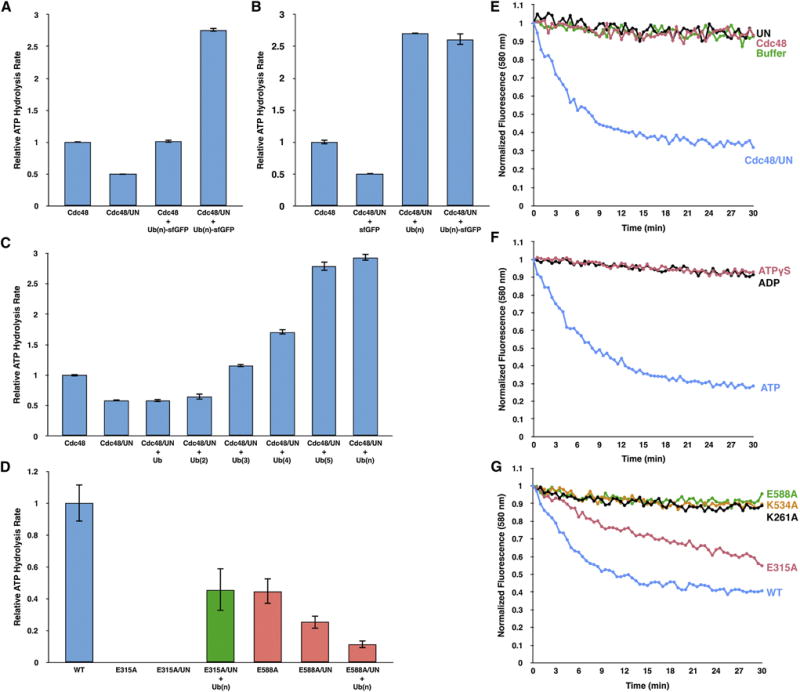
(A) ATP hydrolysis rates were determined with the indicated combinations of purified proteins and substrate (Cdc48; UN; Ub(n)-GFP). Substrate was included at 5-fold excess over the cofactor to maximize occupancy. The rates were normalized with respect to that of Cdc48 alone. Shown are the means and standard deviations of three experiments.
(B) As in (A), but also with free K48-linked polyubiquitin chains (Ub(n)), carrying up to ~15 ubiquitin moieties.
(C) ATPase stimulation with free K48-linked ubiquitin chains of increasing length.
(D) As in (B), but with Cdc48 Walker B mutations in D1 (E315A; green bars) or D2 (E588A; red bars). E315A and E588A can bind, but not hydrolyze, ATP in D1 and D2, respectively (Walker B mutations).
(E) Irradiated, fluorescent Eos, consisting of two polypeptide segments, was polyubiquitinated, as described for sfGFP (Figure 1A). This substrate (Ub(n)-Eos) was incubated with an ATP-regenerating system and excess of the indicated proteins. After addition of ATP, Eos fluorescence was followed over time.
(F) As in (E), but with Cdc48 and UN present in all conditions, and the nucleotide varied as indicated.
(G) As in (F), but with UN and an ATP regenerating system present in all conditions. Cdc48 or its point mutants were included as indicated. K261A and K534A are defective in ATP binding to the D1 and D2 ATPases, respectively (Walker A mutations), and E315A and E588A can bind, but not hydrolyze, ATP in D1 and D2, respectively (Walker B mutations).
See also Figure S2.
To test how substrate affects each of Cdc48’s two ATPase domains, we generated mutations designed to prevent ATP hydrolysis, but not ATP binding, in either the D1 (E315A) or D2 (E588A) domains (mutations in the Walker B motifs). A Cdc48 mutant in which D2 is the only active ATPase (E315A) eliminated ATP hydrolysis in the absence or presence of the UN complex (Figure 2D). However, the addition of both polyubiquitin and UN restored ATP hydrolysis to about half the level seen with wild type Cdc48 alone. Thus, substrate stimulates ATP hydrolysis in the D2 domain.
A mutant in which D1 is the only active ATPase (E588A) had reduced baseline activity, which was further suppressed by the addition of UN (Figure 2D). Interestingly, addition of polyubiquitin resulted in an even more pronounced decrease in ATP hydrolysis (Figure 2D). Thus, substrate inhibits the ATPase activity of the D1 domain.
Similar results were obtained with more commonly used Walker B mutations, in which E315 or E588 are replaced by Gln. The E315Q mutation in the D1 domain did not entirely abolish ATP hydrolysis, but stimulation by the simultaneous presence of UN and polyubiquitin was still observed (Figure S2B). Likewise, the E588Q mutation in the D2 domain resulted in a high baseline ATP hydrolysis rate, which was suppressed by the presence of cofactor and substrate. Thus, despite the fact that the conservative Gln mutations do not completely abolish ATPase activity, substrate still stimulates the D2 domain and slows the D1 domain. The effect on D2 is dominant, explaining why substrate stimulates the overall ATPase activity of the wild type protein.
Cdc48 unfolds substrate
The stimulatory effect of substrate on ATPase activity raised the possibility that Cdc48 actively unfolds the polypeptide chain. To test this possibility, we replaced the sfGFP moiety of the substrate (Figure 1A) with the monomeric fluorescent protein mEos3.2 (Zhang et al., 2012). This protein undergoes a peptide backbone cleavage when irradiated with near-UV light, producing a complex of two fragments. If Cdc48 unfolds this complex, the constituent polypeptides are separated and fluorescence is lost; the two chains cannot re-associate, rendering any unfolding irreversible (Glynn et al., 2012). In contrast, the sfGFP substrate used initially has the ability to refold, which counteracts unfolding by Cdc48 and therefore allows only a small decrease in fluorescence (Figure S2C).
The Eos substrate was polyubiquitinated and subjected to gel filtration to enrich for molecules carrying 5–10 ubiquitin moieties (Ub(n)-Eos). When incubated with Cdc48, UN, and an ATP regenerating system, Ub(n)-Eos was indeed unfolded, as evidenced by a decrease in fluorescence over time (Figure 2E). Unfolding did not occur when either Cdc48 or UN was omitted. The reaction was dependent on ATP hydrolysis, as no decrease in fluorescence was observed with ATPγS or ADP (Figure 2F).
Unfolding occurred with a Cdc48 molecule in which only the D2 ATPase is active (Walker B mutation E315A), although the rate of unfolding was slower than with wild type Cdc48 (Figure 2G). On the other hand, Cdc48 mutants in which only the D1 ATPase is active (E588A mutation) or which cannot bind ATP in either the D1 or D2 domain (Walker A mutations K261A or K534A) did not show any unfolding activity. The Walker B mutants E315Q and E588Q both exhibited residual unfoldase activity (Figure S2D), consistent with their higher ATPase rates. Taken together, these data indicate that Cdc48 uses the energy of ATP hydrolysis to unfold substrates. Unfolding requires the UN cofactor, as well as ATP binding, but not hydrolysis, in the D1 domain, and both ATP binding and hydrolysis in the D2 domain.
Substrate passes through the central pore of Cdc48
One possible mechanism for the unfolding activity of Cdc48 is the translocation of the substrate through the central pore; the ubiquitin chain on the substrate would be bound to the N-domain associated UN cofactor, and the GFP moiety would be pulled through the D1 and D2 rings, from the “cis” to the “trans” side of the double ring. To test this idea, we used site-specific photo-crosslinking. The photo-reactive amino acid p-benzoylphenylalanine (Bpa) was introduced at several positions in the ATPase using amber codon suppression (Chin et al., 2003) (Figure 3A). Upon UV irradiation, Bpa forms covalent crosslinks to targets located within a distance of ~3Å from the selected positions (Dorman and Prestwich, 1994).
Figure 3. Substrate passes through the central pore of Cdc48.
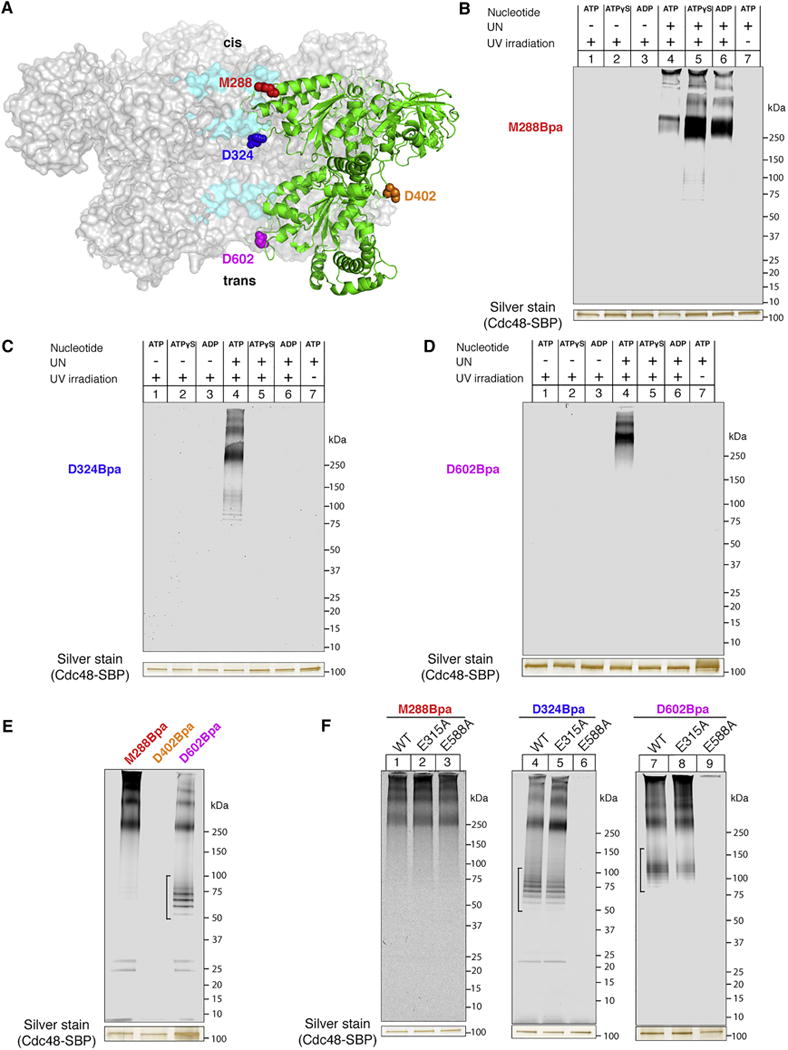
(A) Cdc48 positions used for introduction of the Bpa crosslinker. Sections of the central pore are highlighted in cyan, and a single Cdc48 monomer in the hexamer is shown in green. The Cdc48 model was generated based on PDB ID 3CF1.
(B) SBP-tagged Cdc48 (Cdc48-SBP) with Bpa at the M288 position (D1 pore loop) was incubated with dye-labeled, polyubiquitinated sfGFP and the indicated combinations of UN and nucleotide. After irradiation and pulldown with streptavidin beads, crosslinked species were detected by SDS-PAGE and fluorescence scanning. The silver-stained band of Cdc48-SBP serves as a loading control.
(C) As in (B), but with Bpa at the D324 position in the interior of the central pore.
(D) As in (B), but with Bpa at the D602 position in the D2 pore-2 loop.
(E) As in (B), but with Bpa at an external position (D402). For comparison, crosslinking was also performed with probes in the pore. UN and ATP were included in all conditions. It should be noted that some non-crosslinked substrate remained associated with Cdc48. We generally observed that in the presence of ATP, non-crosslinked material had a tendency to stick to Cdc48 (brackets; see also Figure 3F), likely because it was present in an unfolded conformation inside the pore.
(F) As in (B), but with Cdc48 mutants that contain the crosslinker and Walker mutations at the indicated positions. E315A, Walker B mutation in D1; E588A, Walker B mutation in D2. UN and ATP were included in all conditions.
See also Figure S3.
We first tested a position in the D1 pore loop, which is located at the cis side of the double ring, at the entrance of the central pore (M288; Figure 3B). A SBP-tagged version of Cdc48 that carries Bpa at this position was incubated with fluorescently labeled Ub(n)-sfGFP in the presence or absence of the UN complex and various nucleotides. After irradiation, Cdc48-SBP was recovered with streptavidin beads, which were then washed with high-salt buffer. The remaining bound material was subjected to SDS-PAGE and analyzed with a fluorescence scanner. Products of crosslinking between substrate and Cdc48 were only seen when the UN complex was present and the samples were irradiated (Figure 3B, lanes 4–6 versus lanes 1–3, 7). The major product corresponds to one Ub(n)-sfGFP molecule crosslinked to one Cdc48 molecule. Higher molecular weight bands likely contain additional crosslinked Cdc48 molecules. The crosslinks were most prominent in ATPγS and ADP (lanes 5, 6), but were also observed in the presence of ATP (lane 4). These results indicate that, in the presence of UN, substrate binds to the entrance of the D1 ring even without ATP hydrolysis. Crosslinking might be weaker in the presence of ATP because, under these conditions, the D1 subunits are not all in the same nucleotide state, which may result in only some pore loops being close to substrate, or because substrate can enter the pore and might move the pore loops into an unfavorable position for interaction.
Next, we incorporated Bpa at a position in the interior of the central pore (D324), immediately below the D1 constriction that has been proposed to block substrate access to the pore (Figures 3A, C). Crosslinks between substrate and Cdc48 were formed in the presence of ATP, but not ATPγS or ADP (Figure 3C, lane 4 versus lanes 5, 6). No crosslinks were observed in the absence of the UN complex or without irradiation (lanes 1–3, 7). These results show that Cdc48 uses the energy of ATP hydrolysis to move substrate molecules past the D1 opening of the pore.
Finally, we placed the photoreactive probe at the exit of the D2 ring (position D602), in the pore-2 loop located at the trans side of the double ring (Figures 3A, D). The pore-2 loop is positioned below the pore-1 loop, which contains the conserved aromatic residues responsible for substrate movement in other AAA proteins. Crosslinks were again formed in the presence of ATP, but not ATPγS or ADP, or when the UN complex or irradiation were omitted (Figure 3D). As expected, no crosslinks were observed when Bpa was placed on the exterior of the hexamer (position D402; Figures 3A, E). Crosslinking efficiency was higher for the M288 position (~20%) than for the D324 and D602 positions (~5%) (Figure S3), likely because only a fraction of the substrate bound to the cis side was translocated through the pore. Taken together, these data suggest that ATP hydrolysis allows the Cdc48 ATPase to pull the substrate all the way from the entrance of the D1 ring (cis side), through the central pore, to the trans side of the double-ring.
Consistent with its capacity to unfold Ub(n)-Eos, a Cdc48 mutant in which only the D2 ATPase is active (Walker B mutation E315A in the D1 ring) maintained its ability to form substrate crosslinks to the cis side (M288), interior (D324), and trans side (D602) of the pore (Figure 3F). In contrast, a mutant in which only the D1 ATPase is active (Walker B mutation E588A in the D2 ring) eliminated crosslinking to the interior and trans side of the pore, while maintaining crosslinking to the cis side (Figure 3F). Thus, ATP hydrolysis in D2 is required for pore entry and passage, but not for the initial interaction with the D1 pore entrance. These results confirm that the translocation and unfolding of substrates require only ATP binding, but not hydrolysis, in the D1 domain, whereas ATP hydrolysis in the D2 domain is obligatory for both activities.
Substrate moves entirely through the double-ring of Cdc48
To test whether a substrate molecule moves all the way through the double-ring of the Cdc48 ATPase, we attached a ring-shaped protease to the D2 ring; if a polypeptide chain emerges from the D2 ring, it should be degraded. We took advantage of the fact that the bacterial AAA protease FtsH contains tandem ATPase and protease domains, which form stacked hexameric rings (Ito and Akiyama, 2005). The FtsH protease domain from Aquifex aeolicus, which forms a hexamer on its own (Suno et al., 2006), was fused to the C-terminus of Cdc48, removing the last 10 residues of the flexible native C-terminal tail but otherwise leaving the ATPase unaltered (Figure 4A). As with FtsH, this hybrid construct (Cdc48-FtsH) is expected to degrade proteins that emerge from the trans side of the ATPase ring. For these experiments, we used polyubiquitinated mEos3.2 that had not been photo-converted, preserving an intact polypeptide. A single exposed cysteine near the C-terminus of Eos (C195 of the native sequence) was used to label the protein with a maleimide-conjugated dye. When this substrate was incubated with either UN or Cdc48-FtsH alone in the presence of ATP, no degradation was seen over a period of 20 min (Figure 4B). Addition of a large excess of the deubiquitinating enzyme Otu1 at the end of the incubation period generated unmodified substrate, with a minority of the substrate retaining 1–3 ubiquitin moieties. Importantly, when substrate was incubated with both UN and Cdc48-FtsH, it was degraded in a time-dependent manner. A stable proteolytic fragment of ~13 kDa appeared over time (Figure 4B, arrow head), which contains Cys238, to which the fluorescent dye is attached. The fragment likely corresponds to the C-terminus of the substrate, as shown by the identification of a peptide by mass spectrometry (Figure S4A), but the abundance of the fragment was too low to unambiguously define its boundaries. A comparison of the intensity of bands after Otu1 treatment indicated that this stable cleavage product accounts for approximately 60% of the total substrate signal, consistent with the efficiency of the unfolding reaction (Figure 2E). Mass spectrometry analysis of the full reaction mixture without further proteolytic processing identified peptides representative of the full substrate sequence (Figure S4A). Thus, the entire substrate polypeptide is moved through the D2 ring of Cdc48 into the FtsH ring. Consistent with this conclusion, no substrate degradation was observed when ATP was replaced with ATPγS or ADP (Figure 4C). Both substrate unfolding and positioning of the proteolytic sites at the D2 pore exit are required for this reaction, as no degradation was observed when wild type Cdc48 was incubated with the isolated FtsH protease domain (Figure S4B).
Figure 4. Substrate exits from the D2 side of the double ring ATPase.
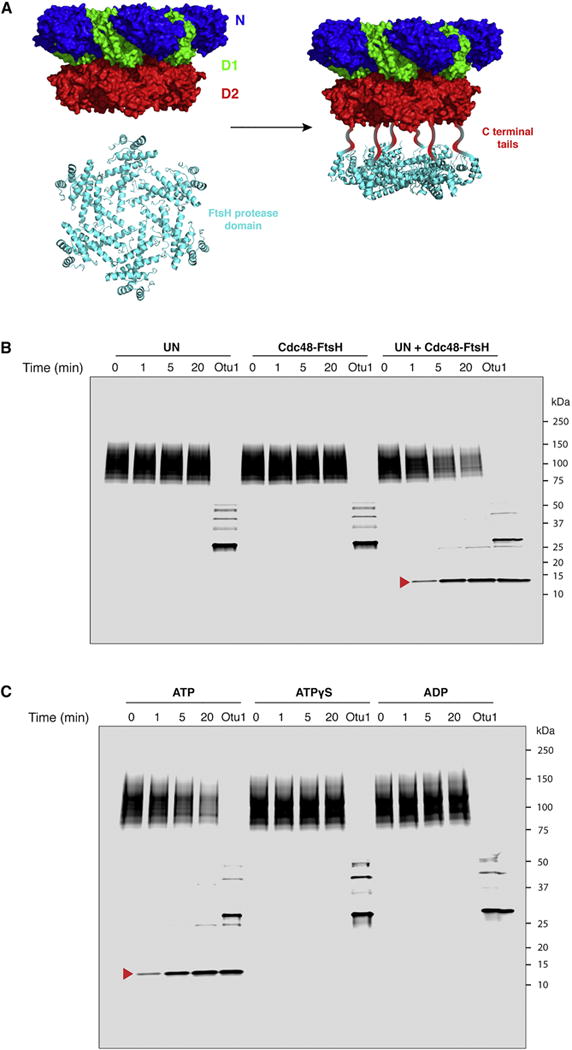
(A) The proteolytic domain of FtsH (PDB ID 2DI4) was fused to the C-terminal tail of the Cdc48 ATPase to produce a hybrid AAA protease (Cdc48-FtsH).
(B) Dye-labeled, polyubiquitinated Eos was incubated with an ATP regenerating system and the indicated proteins. Aliquots were removed at the indicated time points and analyzed by SDS-PAGE and fluorescence scanning. After the time course, a 50-fold excess of Otu1 was added to remove ubiquitin chains. The red arrow head indicates a stable fragment containing the fluorescent dye, which was generated when both Cdc48-FtsH and UN are present. The dye is attached to C238, near the C-terminus of the 275-residue substrate.
(C) As in (B), with Cdc48-FtsH and UN in all conditions, and the nucleotide varied as indicated.
See also Figure S4.
Substrate release requires ubiquitin chain trimming
If substrate is pulled through the central pore from the cis to the trans side, but the associated ubiquitin chain remains bound to the UN cofactor on the cis side, how is substrate released from the Cdc48 complex? Indeed, we found that substrate does not spontaneously dissociate, as labeled substrate remained bound over a 20-minute time course even in the presence of excess unlabeled substrate (Figure S5A, compare lanes 1–4 and 5–8). These experiments were performed in the presence of ATP, i.e. under conditions in which substrate translocation and unfolding occur. Thus, an additional mechanism is required for substrate release. Previous experiments have implicated the deubiquitinating (DUB) enzyme Otu1 in Cdc48 function (Stein et al., 2014); indeed, addition of Otu1 rapidly reversed the sfGFP fluorescence loss mediated by Cdc48 and UN, suggesting that deubiquitination liberates the substrate and allows it to refold (Figure S2C).
To further test the role of deubiquitination in substrate release, we immobilized preformed complexes of Ub(n)-sfGFP, Cdc48, and SBP-tagged UN on streptavidin beads. Addition of Otu1 resulted in the efficient release of substrate from the beads (Figure 5A, lanes 5–8 versus 1–4; Figure S5A, lanes 9–12 versus 1–4). Release was much slower with an Otu1 mutant that lacked the Cdc48-interacting UBX-like domain (Figure 5A, lanes 9–12), and no substrate dissociation was observed with a catalytically inactive Otu1 mutant (C120S) (Figure 5A, lanes 13–16).
Figure 5. Substrate release from the Cdc48 complex requires deubiquitination.
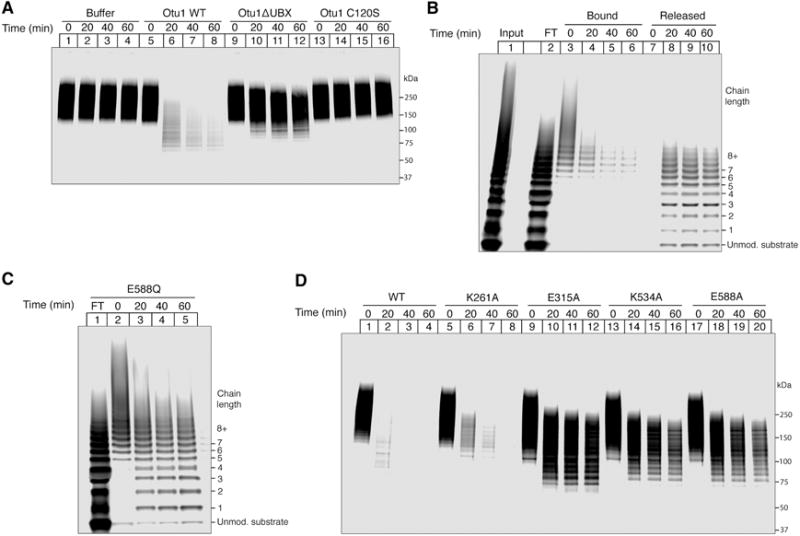
(A) Complexes of Cdc48, SBP-tagged UN, and dye-labeled, polyubiquitinated sfGFP were immobilized on streptavidin beads in the presence of ATP. After washing, the indicated Otu1 variants were added and samples of the bound material were analyzed at the indicated time points by SDS-PAGE and fluorescence scanning. WT, wild type; Otu1ΔUBX, Otu1 lacking the UBX domain; Otu1 C120S, catalytically inactive Otu1.
(B) As in (A), but with both bound and released material analyzed over time. Lane 1 shows the input material before incubation with streptavidin beads, and lane 2 the fraction that did not bind (flow-through; FT).
(C) As in (A), but with wild type Otu1 and the Walker B mutant E588Q in D2, which slowly hydrolyzes ATP.
(D) As in (C), but with other Walker mutations in D1 or D2. K261A, Walker A mutation in D1; E315A, Walker B mutation in D1; K534A, Walker A mutation in D2; E588A, Walker B mutation in D2.
See also Figure S5.
Analysis of the supernatants from the Otu1-mediated release reaction showed that only a small percentage of the dissociating substrate molecules were completely deubiquitinated; the majority contained short ubiquitin chains with up to about 10 ubiquitin moieties (Figure 5B, lanes 8–10). Some portion of these released species probably rebind to the Cdc48 complex, as they bear chains long enough to interact with UN (lanes 5 and 6). Interestingly, a similar pattern of oligoubiquitinated substrate molecules was retained by the Cdc48 complex when Cdc48 carried the Walker B mutation E588Q (Figure 5C), in contrast to the situation with wild type Cdc48, where Otu1 incubation resulted in essentially complete release of all substrate molecules (Figure 5B, lanes 3–6). Most of the ubiquitin chains retained by the mutant Cdc48 complex are too short to mediate the initial interaction with the complex (Figure 5C; compare lanes 1 and 3–5), suggesting that they represent pre-release intermediates. Thus, the slow ATPase rate in the D2 ring of the E588Q mutant must have delayed the release of Otu1-processed substrate molecules from the Cdc48 complex.
We next tested whether Otu1-mediated substrate release depends on the nucleotide bound to the D1 and D2 domains. A mutant in the Walker A motif of the D1 domain (K261A) allowed substrate release with similar kinetics as wild type Cdc48 (Figure 5D, lanes 5–8 versus 1–4). In contrast, a mutant in the Walker B motif of the D1 domain (E315A) had markedly slowed release kinetics (Figure 5D, lanes 9–12). This altered deubiquitination rate is not attributable to impaired assembly of the Otu1-Cdc48 complex, as Otu1 bound equivalently to wild-type Cdc48 and all Walker mutants (Figure S5B). Instead, given that the D1 domain is in the ATP bound state in the Walker B mutant and the N domains therefore adopt the “up-conformation”, Otu1 may not have full access to the ubiquitin chain. In the Walker A mutant, the N domain is likely in the “down- conformation”, allowing efficient deubiquitination by Otu1.
Both ATP binding and hydrolysis in the D2 domain are required for efficient substrate release by Otu1, as mutations in the Walker A (K524A) or B (E588A) motifs resulted in slow release kinetics (Figure 5D, lanes 13–16 and 17–20). These results suggest that a full cycle of ATP hydrolysis has to occur before D1 can hydrolyze ATP and return the N domains to the “down conformation”, in which the ubiquitin chains are accessible to Otu1.
Ubiquitin molecules can pass through the central pore
The retention of one or more ubiquitin moieties on released substrate molecules raised the possibility that not only an unmodified substrate segment, but also the oligoubiquitin chain, can be translocated through the central pore. We therefore tested the path of free fluorescent polyubiquitin chains through the Cdc48 ATPase with photo-crosslinking experiments.
As with Ub(n)-sfGFP, free polyubiquitin chains crosslinked to the cis side of the D1 ring (position M288) in all nucleotide states (Figure 6A). Again, the interaction was dependent on the presence of the UN complex. Crosslinking to the interior and trans positions (D324 and D602, respectively) was observed with ATP, but not with ATPγS or ADP (Figures 6B and 6C). These results are comparable to those obtained with Ub(n)-sfGFP (Figure 3) and indicate that Cdc48 can translocate at least one ubiquitin molecule, even in the absence of an associated substrate or flexible peptide region. Interestingly, penta-ubiquitin crosslinked only weakly to the position at the entrance of the D1 ring and not at all to positions inside the central pore (Figure 6D), although it can maximally stimulate ATPase activity and must therefore be able to bind to the Cdc48 complex (Figure 2C). Additional ubiquitin molecules in the chain seem to be required to allow efficient insertion of a polypeptide into the central pore of Cdc48.
Figure 6. Ubiquitin passes through the central pore of Cdc48.
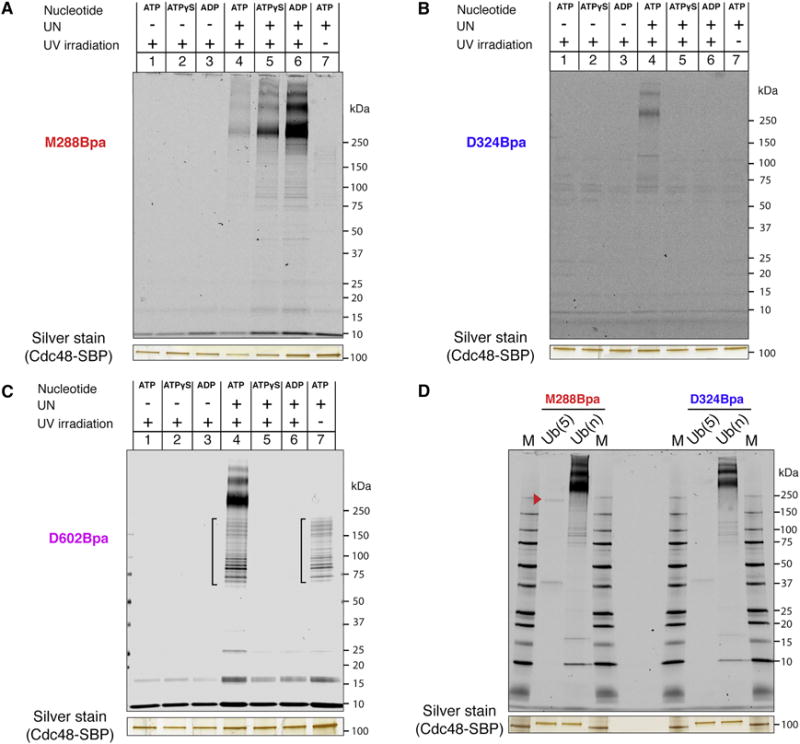
(A) SBP-tagged Cdc48 (Cdc48-SBP) with Bpa at the M288 position (D1 pore loop) was incubated with dye-labeled free, K48-linked polyubiquitin chains and the indicated combinations of UN and nucleotide. After irradiation and streptavidin pulldown, crosslinked species were detected by SDS-PAGE and fluorescence scanning. The silver-stained band of Cdc48-SBP serves as a loading control.
(B) As in (A), but with Bpa at the D324 position in the interior of the central pore.
(C) As in (A), but with Bpa at the D602 position in the D2 pore-2 loop. Brackets, non-crosslinked material.
(D) As in (A), but with Bpa at positions M288 or D324 and either penta-ubiquitin ((Ub(5)) or polyubiquitin (Ub(n)). The arrowhead indicates a weak crosslink of Ub(5) to position 288. M, molecular weight markers.
DISCUSSION
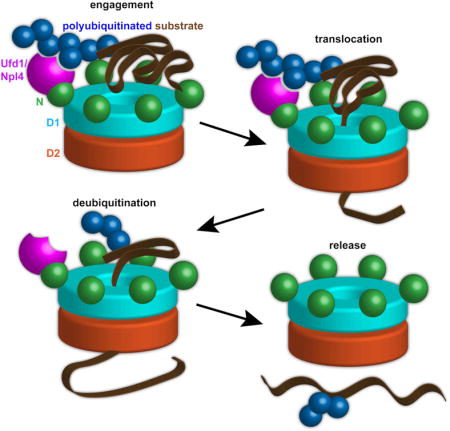
Our results clarify major aspects of the molecular mechanism of the Cdc48 ATPase. They lead to a model that can explain how Cdc48 and its mammalian ortholog p97/VCP function together with the conserved UN cofactor to disassemble protein complexes and extract proteins from membranes.
In the model, Cdc48 starts out with the D1 ATPases in the ADP-bound state (Figure 7, stage 1). The N domains are in the “down-conformation”, co-planar with the D1 ring. When the D1 domain binds ATP, the N domains move upwards (stage 2). The UN complex can bind to either conformation, inhibiting the overall ATPase activity in the absence of substrate. Substrate is initially bound to the Cdc48 complex exclusively through an interaction of the attached K48-linked polyubiquitin chain with the UN cofactor (stage 3). Most of the UN cofactor in the cell is probably bound to Cdc48, which is present at high concentrations, so free UN would not compete for substrate. Substrate binding to the Cdc48 complex reduces ATPase activity in the D1 domain, biasing it toward its ATP-bound state with the N domain in its “up-conformation”. The UN cofactor and D1 ring ATPases might form a composite binding surface that can locally denature substrate without energy input, generating an unfolded polypeptide loop that can reach into the central pore. Substrate binding also stimulates ATP hydrolysis in the D2 domain. This activity allows pore loops in the D2 ring to move and drag the substrate polypeptide through the central pore (stage 4). The pulling force exerted by the D2 ATPases results in the unfolding of the substrate (stage 5). During translocation, most of the polyubiquitin moiety remains on the cis side, bound to the UN cofactor. However, a portion of the ubiquitin chain can enter the central pore along with the substrate (stage 5). The final step is substrate release. Once D1 has hydrolyzed ATP, the N domains convert back to the “down-conformation”, allowing access of the polyubiquitin chain to the deubiquitinating enzyme Otu1 (stage 6). When the ubiquitin chain has been shortened sufficiently, its affinity for the UN complex is reduced or lost, and the remaining ubiquitin moieties are unfolded and pulled through the central pore (stage 7). The ubiquitin probably refolds rapidly after translocation (Sivaraman et al., 2001), although this needs to be tested by future experimentation.
Figure 7. Stages of substrate processing by the Cdc48 complex.
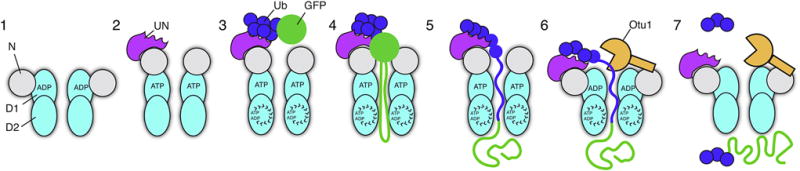
See text for details.
Our results argue against several alternative models proposed for Cdc48 function. We show that Cdc48 is an unfoldase that pulls the polypeptide substrate through the central pore, indicating that models of substrate segregation without unfolding are incorrect. Likewise, models in which the substrate inserts shallowly into only the D1 or D2 side of the pore are inconsistent with the observed interactions along the full length of the pore. The crosslinking data also invalidate models in which relative motions of the N domains displace substrates or substrates interact with the exterior of the ATPase.
Our data indicate that the D1 domain only needs to hydrolyze ATP a few times, or perhaps even just once, during the processing of a given substrate molecule, whereas the D2 domain hydrolyzes ATP many times. Consistent with this model, only D2 contains the canonical aromatic pore loop residues implicated in substrate binding and translocation. However, the two ATPase rings likely communicate with each other. ATP hydrolysis in D1 not only controls the position of the N domains, but also affects D2 activity, as substrate binding at the cis side stimulates the ATPase rate of D2. Inter-ring communication may also occur in the reverse direction, as our substrate release data indicate that the nucleotide state of D2 influences the activity of Otu1, which is bound to the N domain. It is also likely that the ATPase subunits influence one another within each ring when the subunits are in different nucleotide states. Because all members of the ring are forced to be in the same state in the presence of ADP or ATPγS, or when Walker motifs are mutated, our model is likely a simplification; in the presence of ATP, only some of the six subunits of the D1 or D2 rings may undergo the described nucleotide binding and hydrolysis events in a synchronous manner.
Several findings support the proposition that not only substrate but also some ubiquitin moieties are translocated by Cdc48 and released from the D2 side of the central pore. First, ubiquitin itself can serve as a translocation substrate, forming crosslinks to positions along the full length of the pore. Thus, at least one ubiquitin molecule completely traverses the double-ring ATPase. Second, a slowly hydrolyzing Cdc48 mutant retained oligoubiquitin chains, indicating that these are slowly translocated through the ATPase and released with delay. Third, fully released substrates are primarily oligoubiquitinated, rather than fully deubiquitinated. The last two points suggest that not only a single ubiquitin molecule, but also a short ubiquitin chain can be translocated through the central pore. We attempted to confirm ubiquitin translocation by incubating Ubn-Eos with Cdc48-FtsH but were unable to reproducibly detect ATP-dependent peptides by mass spectrometry, perhaps because FtsH does not efficiently cleave ubiquitin. It should be noted that the proteasome can also translocate ubiquitin under a variety of circumstances (Shabek and Ciechanover, 2010).
A translocation mechanism for Cdc48/p97 had previously been dismissed because the ATPase has a narrow central pore relative to other double-ring AAA ATPases of known structure, and its D1 pore is even occluded by a Zn2+ ion in a crystal structure (DeLaBarre and Brunger, 2005). However, our data now suggest that at least two polypeptide strands can be accommodated, as the ubiquitin attachment site on the substrate is likely moved into the pore. Our results thus imply that the pore diameter widens during translocation, although this has yet to be confirmed experimentally. A hairpin structure inside the central pore has been demonstrated for other AAA ATPases, and there is even evidence that three strands can be present (Burton et al., 2001). This mechanism differs from that of NSF, where the SNARE substrate is not translocated through the central pore, although the D1 ring has canonical pore residues(Zhao et al., 2015), and a single ATPase cycle disassembles the SNARE complex (Ryu et al., 2015).
In our model, Cdc48 activity is coupled with substrate deubiquitination. Indeed, free Otu1 has much lower deubiquitination activity than Otu1 bound to the Cdc48 complex (Stein et al., 2014). This stimulation is in part due to the UN cofactor, which may present bound K48-linked ubiquitin molecules to Otu1 in an appropriate conformation. In addition, our data suggest that deubiquitination is delayed until the substrate is at least partially translocated. The slow deubiquitination and substrate release exhibited by the D1 Walker B mutant indicate that Otu1 may not have full access to the UN-associated ubiquitin chain while the D1 domains are in the ATP-bound state and the N domains in the “up-conformation”. Movement of the N domain to the “down-conformation” by ATP hydrolysis in D1 would relieve this inhibition and permit full deubiquitination. Efficient deubiquitination also requires ATP binding and hydrolysis in D2, ensuring that substrate translocation commences before Otu1 can act. Furthermore, the repression of D1 ATP hydrolysis by substrate gives the D2 ring a chance to translocate a substantial portion of the polypeptide before deubiquitination occurs. Taken together, these mechanisms allow substrate binding, translocation, and release to occur in a defined order. Our results show that Otu1 cooperates with Cdc48 and argue against an earlier model in which Otu1 antagonizes Cdc48 function by preventing substrate recognition (Rumpf and Jentsch, 2006).
The overall mechanism of the Cdc48 complex resembles that of the 19S regulatory subunit of the proteasome, which also uses receptor proteins to bind polyubiquitin chains attached to a substrate and employs a translocation mechanism (for review, see Kish-Trier and Hill, 2013). As with Cdc48, full substrate movement through the central pore requires deubiquitination, a reaction performed by Rpn11 of the 19S subunit. In contrast to Otu1, which cleaves between ubiquitin moieties and does not fully remove a substrate-associated ubiquitin chain, Rpn11 cleaves off the entire chain. However, in both cases, deubiquitination cooperates with, rather than antagonizes, substrate processing by the ATPase complex. This is distinct from the function of other DUBs, such as the Ubp6 component of the 19S subunit, which serve as negative regulators.
In many cellular functions, such as ERAD, Cdc48 acts upstream of the proteasome. Cdc48 may be required in cases where a substrate does not have a flexible segment (Beskow et al., 2009), which is needed to initiate translocation into the 19S proteasomal subunit (Prakash et al., 2004). Indeed, in our experiments, the Cdc48 complex can translocate a polyubiquitin chain, which does not expose extended flexible segments. The exact reason why Cdc48, but not the proteasome, can deal with folded substrates remains to be clarified. The mechanism of substrate transfer from Cdc48 to the proteasome also needs further investigation. Most of the ubiquitin chains released by Cdc48 would be long enough to bind directly, or through the shuttling factors Rad23 or Dsk2, to the proteasome (Kim et al., 2004)(Medicherla et al., 2004), and chains that are too short could first be extended by the Cdc48-associated “E4 ligase” Ufd2 (Richly et al., 2005). Alternatively, some substrates may be transferred from Cdc48 into the 20S proteasome without the involvement of the 19S subunit (Barthelme and Sauer, 2013; Barthelme and Sauer, 2012). Regardless of the downstream events, our results show that Cdc48 pulls on substrate polypeptides and unfolds them, explaining how the ATPase complex disassembles protein complexes and extracts proteins from membranes.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tom Rapoport (tom_rapoport@hms.harvard.edu).
EXPERIMENTAL MODELS
Recombinant proteins, with the exception of Uba1 and Ubr1, were expressed in Escherichia coli BL21 (DE3) or BL21 (DE3) RIPL cells grown in Terrific Broth. Uba1 was expressed in Saccharomyces cerevisiae INVSc1, and Ubr1 was expressed in Saccharomyces cerevisiae BY4741; both strains were grown in YPD medium.
METHOD DETAILS
Purification of proteins
Uba1 was purified as described (Stein et al., 2014). Ubr1 was expressed in S. cerevisiae from the plasmid pFlagUBR1SBX, a gift from Alexander Varshavsky. The protein was purified by FLAG affinity chromatography from the yeast strain BY4741 according to a previously established protocol (Du et al., 2002).
Cdc48, Otu1, and Ufd1/Npl4 were purified as described (Stein et al., 2014). Point mutants, truncated constructs, SBP-tagged variants, and the Cdc48-FtsH chimera were purified by the procedures for their parent constructs, with the exception of constructs incorporating Bpa (see below).
Ubc1, Ubc2, sfGFP, mEos3.2, the FtsH proteolytic domain (residues 406–634 of the Aquifex aeolicus sequence) and Cys-ubiquitin (S. cerevisiae ubiquitin with a cysteine introduced at position 1) were expressed in E. coli BL21 DE3 RIPL cells with N-terminal His14-SUMO fusion tags (Frey and Gorlich, 2014). Cells were grown in Terrific Broth to an OD600 of 1. Protein expression was induced by the addition of 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 16 hrs at 18°C. Cells were harvested by centrifugation at 4,000 × g for 10 min and resuspended in lysis buffer (50 mM Tris pH 8, 200 mM NaCl, 30 mM imidazole). Phenylmethylsulfonyl fluoride (PMSF; 1 mM), a protease inhibitor cocktail, and DNase I (5 μg/mL) were added, and cells were lysed by sonication. Lysates were cleared by ultracentrifugation in a Ti45 rotor (Beckman) at 40,000 rpm for 30 min at 4°C. Supernatants were incubated with Ni-NTA resin for 90 min at 4°C. The resin was washed three times with 20 column volumes of buffer (50 mM Tris pH 8, 200 mM NaCl, 30 mM imidazole). Proteins were eluted twice with 10 mL elution buffer (50 mM Tris pH 8, 100 mM NaCl, 400 mM imidazole), and the eluates were combined and supplemented with 1 mM tris(2-carboxyethyl)phosphine (TCEP). His14-SUMO tags were removed by incubation with 2μM Ulp1 (SUMO protease) for 2 hrs at 4°C. Ubc1, Ubc2, sfGFP, and the FtsH protease were dialyzed against 50 mM Tris pH 8, 100 mM NaCl, and mEos3.2 was dialyzed against 50 mM HEPES pH 7, 50 mM NaCl. Proteins were loaded onto ion exchange columns (MonoQ 10/100 GL for Ubc1, Ubc2, sfGFP, and the FtsH protease; MonoS 10/100 GL for mEos3.2) equilibrated in the respective dialysis buffers, and eluted by linear gradients to 500 mM NaCl over 10 column volumes. Peak fractions were pooled, concentrated to 2 mg/mL or greater, snap frozen in liquid nitrogen, and stored at −80°C.
Cdc48 constructs for crosslinking experiments were expressed in E. coli BL21 DE3 cells harboring the plasmid pEVOL-pBpF, a gift from Peter Schultz (Addgene plasmid #31190) (Chin et al., 2002). Cells were grown in terrific broth without glycerol to an OD of 0.5. For induction, arabinose (0.02%), IPTG (0.1 mM), and p-benzoylphenylalanine (Bpa; 1 mM) were added and expression was carried out for 16 hrs at 18°C. Cell harvesting, lysis, and Ni-NTA purification was performed as described above, with 5 mM MgCl2 included in buffers throughout. After Ulp1 cleavage, proteins were diluted to an imidazole concentration of < 200 mM and applied to Ultra HBC streptavidin agarose resin (Gold Bio) for 2 hrs at 4°C. The resin was washed 3 times with 20 column volumes of buffer S (50 mM HEPES pH 7.5, 200 mM NaCl, 5 mM MgCl2). Proteins were eluted with 5 column volumes of buffer S containing 5 mM biotin, then dialyzed overnight (to remove biotin) against buffer S containing 0.5 mM TCEP. The protein solutions were supplemented with 10% glycerol, concentrated to 2 mg/mL or greater, snap frozen in liquid nitrogen, and stored at −80°C.
S. cerevisiae ubiquitin and H. sapiens ubiquitin chains of defined lengths were purchased from Boston Biochem.
The sequence inserted at the N terminus of sfGFP or mEos3.2 to facilitate ubiquitination by the N-end rule enzymes is as follows (note that the N-terminal arginine is exposed after SUMO cleavage):
RHGSG(C/S)GAWLLPVSLVKRKTTLAPNTQTASPPSYRALADSLMQ. For labeling of Eos, which already contains an exposed cysteine, Cys6 was changed to Ser to avoid dual labeling.
Labeling with fluorescent dyes
Prior to labeling, sfGFP, Eos, Cys-ubiquitin, and penta-ubiquitin chains were exchanged into buffer free of reducing agents (50 mM HEPES pH 7.5, 150 mM NaCl) by dialysis or PD-10 desalting columns. Proteins were incubated with a 5:1 molar excess of DyLight 800 maleimide, DyLight 680 maleimide, or Dylight 680 NHS ester for 1 hr at room temperature. Reactions were quenched by addition of 1 mM DTT (for maleimides) or 100 mM Tris (for NHS esters). Samples were then dialyzed or treated with dye-removal columns (Thermo Scientific).
Photoconversion of mEos3.2
The Eos protein (2 mg/mL) was placed in a 1.5 mL Eppendorf tube in an ice bath. A long-wave UV lamp (Blak-Ray) was positioned 5 cm from the tube, and the sample was irradiated for 2 hrs, with occasional mixing. Any precipitated protein was removed by filtration.
Ubiquitination of substrates
Ubiquitination of sfGFP or mEos3.2 was carried out as follows. Substrates (500 nM) were incubated with S. cerevisiae ubiquitin (100 μM), Uba1 (100 nM), Ubc2 (12 μM), Ubr1 (300 nM), and ATP (5 mM) for 60 to 90 min at 30°C in ubiquitination buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 10 mM MgCl2). Reaction mixtures were applied to Ultra HBC streptavidin agarose beads for 30 min at room temperature. The resin was washed 3 times with 20 column volumes of ubiquitination buffer, then resuspended in 5 column volumes of ubiquitination buffer containing 1 mM TCEP. Substrates were cleaved from the resin by incubation with 2 μM 3C protease for 2 hrs at room temperature. Substrates used for binding and release experiments, ATPase stimulation assays, and crosslinking were collected and snap frozen at this point.
Substrates used for unfolding and degradation experiments were concentrated after 3C protease cleavage to < 500 μL and applied to a gel filtration column (S200 10/300 GL) in ubiquitination buffer. Fractions were collected, analyzed by SDS-PAGE, supplemented with 50% glycerol, and stored at −20°C.
Synthesis of free polyubiquitin chains
Yeast ubiquitin (200 μM) was incubated with Uba1 (100 nM), Ubc1 (5 μM), and ATP (5 mM) for 2 hrs at 30°C in ubiquitination buffer. To generate fluorescently labeled chains, DyLight 680-conjugated Cys-ubiquitin was included at 5 μM. ATP was removed by dialysis into nucleotide-free ubiquitination buffer, and chains were snap frozen and stored at −80°C.
Binding and release assays
SBP-tagged Cdc48 or UN (200 nM) was applied to 10 μL magnetic streptavidin beads (Pierce) in a total of 250 μL buffer B (50 mM HEPES pH 7.5, 150 mM NaCl, 5 mM MgCl2, 0.5 mM TCEP, 0.5 mg/mL protease-free BSA, 2 mM nucleotide [ATP, ATPγS, or ADP]). When applicable, untagged Cdc48, UN, or Otu1 was included at 500 nM. Binding was carried out for 30 min at room temperature, followed by three washes and resuspension in 250 μL buffer B. Polyubiquitinated substrate or polyubiquitin chains (200 nM) were added and the binding reaction continued for another 30 min. Supernatants were recovered, and beads were again washed 3 times and eluted with BSA-free buffer B containing 5 mM biotin.
For Otu1 release assays, after the second set of washes, the beads with bound Cdc48, UN, and substrate were resuspended in 250 μL buffer B with ATP. Otu1 or its variants were added at 400 nM at time 0, and 50 μL of the reaction mix, including beads, was removed for each time point. Supernatants were recovered from the 50 μL samples, and beads were washed 3 times and eluted with buffer B containing 5 mM biotin. Supernatants and eluates were subjected to SDS-PAGE and scanned on an Odyssey imager (LI-COR).
The competition experiment with unlabeled substrate was performed as described for the Otu1 release assays, except that instead of Otu1, unlabeled substrate amounting to a 5-fold excess (1 μM) over the original fluorescently labeled input was added.
ATPase assays
ATPase rates were measured using an absorbance-based phosphate release assay (EnzChek, Thermo Fisher). Assays were performed at 30°C in ATPase buffer (50 mM HEPES pH 7.5, 50 mM NaCl, 10 mM MgCl2, 0.5 mM TCEP, 0.1 mg/mL protease-free BSA). As appropriate, reaction components included Cdc48 or its mutants (150 nM), Ufd/Npl4 (500 nM), and substrates, ubiquitin, or polyubiquitin (2.5 μM; the concentration of mixed polyubiquitin chains was calculated on the basis of an average chain length of 12). Proteins were pre-incubated for 10 min prior to the addition of 2 mM ATP. Absorbance (360 nm) was measured at 20 sec intervals in an M5 plate reader (Spectramax). The slope of the initial, linear portion of the curve was used to calculate the rate, and rates were normalized to that of wild type Cdc48. ATPase rates are reported as the average of three replicates, and error bars show one standard deviation.
Unfolding assays
Unfolding experiments were performed at 30°C in reaction buffer (50 mM HEPES pH 7.5, 130 mM KCl, 10 mM MgCl2, 0.5 mM TCEP, 0.5 mg/mL protease-free BSA). As appropriate, reaction components included Cdc48 or its mutants (200 nM), Ufd/Npl4 (500 nM), and polyubiquitinated mEos3.2 or sfGFP (50 nM). Proteins were pre-incubated for 10 min prior to the addition of an ATP regeneration system (2 mM ATP, 20 mM phosphocreatine, 100 μg/mL creatine kinase), ATPγS (2 mM), or ADP (2 mM). Fluorescence (excitation: 485 nm for GFP, 540 nm for Eos; emission: 516 nm for GFP, 580 nm for Eos) was measured at 30 sec intervals in an M5 plate reader (Spectramax) for 30 min. Fluorescence values were corrected by subtracting the measured fluorescence of polyubiquitinated mEos3.2 or sfGFP denatured in 6M guanidine-HCl.
Homology modeling
A structural model of yeast Cdc48 was calculated based on the p97 crystal structure (PDB: 3CF1) using the program SWISS-MODEL (Biasini et al., 2014). The structure was displayed using PyMol (Schrödinger, LLC).
Crosslinking assays
Crosslinking was performed in reaction buffer (50 mM HEPES pH 7.5, 130 mM KCl, 10 mM MgCl2, 0.5 mM TCEP, 0.5 mg/mL protease-free BSA). As appropriate, reaction components included Cdc48 or its mutants (200 nM), Ufd1/Npl4 (500 nM), and dye-labeled polyubiquitinated sfGFP or free polyubiquitin (1 μM). An ATP regeneration system (2 mM ATP, 20 mM phosphocreatine, 100 μg/mL creatine kinase), ATPγS (2 mM), or ADP (2 mM) were added. Reactions were assembled on ice, incubated at 30°C for 10 min, and transferred to individual wells of a black polystyrene plate. A long-wave UV lamp (Blak-Ray) was positioned 5 cm from the plate, and the samples were irradiated for 30 min. To prevent overheating, an ice-cold metal block was placed in contact with the bottom of the plate.
After irradiation, samples were diluted 10-fold in dissociation buffer (50 mM Tris pH 8, 800 mM KCl, 1% (v/v) Triton X-100, 1 mM EDTA, 0.5 mM TCEP) and incubated at room temperature for 5 min. Samples were then applied to 5 μL magnetic streptavidin beads (Pierce) equilibrated in dissociation buffer for 30 min at room temperature. Beads were washed three times, then eluted in 50 mM HEPES pH 7.5, 100 mM NaCl, 5 mM biotin. The eluted material was subjected to SDS-PAGE and the gel scanned on an Odyssey imager (LI-COR). For the experiment in Figure S3, the dissociation and pulldown steps were omitted, and only 200 nM substrate was used.
Degradation assays
Experiments were performed in reaction buffer plus zinc (50 mM HEPES pH 7.5, 130 mM KCl, 10 mM MgCl2, 0.5 mM TCEP, 0.5 mg/mL protease-free BSA, 200 nM ZnCl2). As appropriate, reaction components included the Cdc48-FtsH chimera (200 nM) or a mixture of wild-type Cdc48 and the free FtsH protease domain (200 nM each), Ufd1/Npl4 (500 nM), and polyubiquitinated, dye-labeled Eos (50 nM) that had not been photoconverted. An ATP regeneration system (2 mM ATP, 20 mM phosphocreatine, 100 μg/mL creatine kinase), ATPγS (2 mM), or ADP (2 mM) were added. Reactions were assembled on ice, incubated for 10 min, then transferred to 30°C. Aliquots were removed at appropriate time points, followed by addition of Otu1 (2.5 μM) and further incubation for 20 min, after which the final aliquot was removed. Samples were subjected to SDS-PAGE and scanned on an Odyssey imager (LI-COR). For mass spectrometry analysis, the deubiquitination step was omitted. Instead, reactions were quenched after 20 min by the addition of 50 mM EDTA and submitted for peptide identification without further processing.
QUANTIFICATION AND STATISTICAL ANALYSIS
Bar graphs shown in Figure 2 and Figure S2 display mean ± standard deviation of three experiments.
Supplementary Material
Figure S1. Purity of proteins used in this study, Related to Figure 1
(A) Purified proteins used in this study were subjected to SDS-PAGE and staining with Coomassie blue. Black stars indicate contaminating proteins. Degron-sfGFP and degron-Eos are substrates before being subjected to fluorescent labeling and ubiquitination. Cdc48-FtsH was run on a different gel; an irrelevant lane was removed from the gel on the left (white space). M, molecular weight markers.
(B) Tryptic peptides of the polyubiquitinated sfGFP substrate were analyzed by mass spectrometry, with a search for ubiquitin conjugation sites. Intensities of the peptides corresponding to ubiquitin chain linkage and ubiquitin conjugation sites are shown. Note that the predominant linkage is K48 and the predominant substrate ubiquitination site is K19. The # sign indicates the position of the isopeptide bond.
Figure S2. ATPase activity and substrate unfolding, Related to Figure 2
(A) ATP hydrolysis rates were determined with the indicated combinations of purified proteins and substrate. K48 and K63 indicate penta-ubiquitin with different linkages. The rates were normalized with respect to that of Cdc48 alone. Shown are the means and standard deviations of three experiments.
(B) As in (A), but with either wild type (WT) Cdc48 or Walker B glutamine mutants (Walker B mutation in D1 (E315Q; green bars) or D2 (E588Q; red bars)).
(C) Polyubiquitinated sfGFP (Ub(n)-sfGFP), generated as in Figure 1A, was incubated with an ATP regenerating system and excess of either Cdc48 alone or Cdc48 plus UN. After addition of ATP, GFP fluorescence was followed over time. One sample (yellow curve) received an equimolar amount of Otu1 at the indicated time point (arrow).
(D) Substrate unfolding assays with Ub(n)-Eos and Walker B glutamine mutants. Assays were performed as in Figure 2F.
Figure S3. Efficiency of substrate crosslinking, Related to Figure 3
SBP-tagged Cdc48 (Cdc48-SBP) with Bpa at the indicated positions was incubated with dye-labeled, polyubiquitinated sfGFP (Ub(n)-sfGFP), UN, and an ATP regenerating system. Where indicated, reactions were irradiated. The samples were then subjected to SDS-PAGE and fluorescence scanning. The silver-stained band of Cdc48-SBP serves as a loading control. The brackets indicate the positions of crosslinked products. Crosslinking efficiencies were estimated by comparing the fluorescence intensities of non-crosslinked and crosslinked species (~20% for M288, and ~5% for D324 and D602).
Figure S4. Substrate cleavage by the Cdc48-FtsH chimera, Related to Figure 4
(A) Dye-labeled, polyubiquitinated Eos (Ub(n)-Eos) was incubated with UN, the fusion of Cdc48 and FtsH (Cdc48-FtsH), and ATP, ATPγS, or ADP. One sample was directly analyzed by mass spectrometry. Sequences detected only in ATP are shown in red (15 unique peptides). Two unique peptides (shown in blue) were detected in all nucleotide states and correspond to part of the degron preceding Eos (underlined in black), which is likely flexible. Another sample was subjected to SDS-PAGE and the stable proteolytic fragment of ~13 kDa was trypsinized and analyzed by mass spectrometry. The identified peptide is underlined in green.
(B) Dye-labeled Ub(n)-Eos was incubated with an ATP regenerating system, UN, and either Cdc48-FtsH or a mixture of wild type Cdc48 and the isolated FtsH domain. Aliquots were removed at the indicated time points and analyzed by SDS-PAGE and fluorescence scanning. The arrow head indicates a stable fragment containing the fluorescent dye, which was generated with the Cdc48-FtsH fusion.
Figure S5. Otu1 binding to the Cdc48 complex, Related to Figure 5
(A) Complexes of Cdc48, SBP-tagged UN, and dye-labeled, polyubiquitinated sfGFP were immobilized on streptavidin beads in the presence of ATP. After washing, the indicated proteins were added and samples of the bound material were analyzed at the indicated time points by SDS-PAGE and fluorescence scanning. Where indicated, unlabeled, polyubiquitinated sfGFP was added in 5-fold excess over the original fluorescently labeled input.
(B) SBP-tagged UN and the indicated Cdc48 mutants were immobilized on streptavidin beads, and wild-type Otu1 was added. After washing, the bound material was recovered and subjected to SDS-PAGE and silver staining.
HIGHLIGHTS.
-
-
Cdc48 unfolds ubiquitinated substrates by translocation through its central pore
-
-
The two ATPase rings have distinct roles in translocation and substrate release
-
-
Substrate release requires cooperation between Cdc48 and a deubiquitinase
-
-
The remaining oligoubiquitin chain is translocated through the central pore
One sentence.
Polyubiquinated, misfolded substrates are extracted from membranes and macromolecular complexes by Cdc48 and its cofactors via a pulling force that causes substrate unfolding and are then released by the trimming of polyubiquitin chains to oligoubiquitin.
Acknowledgments
We thank Alex Stein for providing reagents, the Institute for Chemistry and Chemical Biology Longwood for use of a plate reader, and Dan Finley, Alex Stein, and Ryan Baldridge for critical reading of the manuscript. This work is supported by the NIH/NIGMS Award R01GM052586 and by award #T32GM007753 from the NIGMS. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. T.A.R. is a Howard Hughes Medical Institute Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
N.O.B. designed and performed the experiments. N.O.B. and T.A.R. interpreted the data and wrote the manuscript.
References
- Baek GH, Kim I, Rao H. The Cdc48 ATPase modulates the interaction between two proteolytic factors Ufd2 and Rad23. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:13558–13563. doi: 10.1073/pnas.1104051108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge RD, Rapoport TA. Autoubiquitination of the Hrd1 Ligase Triggers Protein Retrotranslocation in ERAD. Cell. 2016;166:394–407. doi: 10.1016/j.cell.2016.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Bartesaghi A, Merk A, Rao P, Bulfer SL, Yan Y, Green N, Mroczkowski B, Neitz RJ, Wipf P, et al. 2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science. 2016;351:871–875. doi: 10.1126/science.aad7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel B, Wünning I, Varshavsky A. The recognition component of the N-end rule pathway. The EMBO Journal. 1990;9:3179–3189. doi: 10.1002/j.1460-2075.1990.tb07516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthelme D, Sauer RT. Identification of the Cdc48•20S proteasome as an ancient AAA+ proteolytic machine. Science. 2012;337:843–846. doi: 10.1126/science.1224352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthelme D, Sauer RT. Bipartite determinants mediate an evolutionarily conserved interaction between Cdc48 and the 20S peptidase. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:3327–3332. doi: 10.1073/pnas.1300408110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beskow A, Grimberg KB, Bott LC, Salomons FA, Dantuma NP, Young P. A conserved unfoldase activity for the p97 AAA-ATPase in proteasomal degradation. Journal of Molecular Biology. 2009;394:732–746. doi: 10.1016/j.jmb.2009.09.050. [DOI] [PubMed] [Google Scholar]
- Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Research. 2014;42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchberger A. Roles of Cdc48 in regulated protein degradation in yeast. Subcell. Biochem. 2013;66:195–222. doi: 10.1007/978-94-007-5940-4_8. [DOI] [PubMed] [Google Scholar]
- Burton RE, Siddiqui SM, Kim YI, Baker TA, Sauer RT. Effects of protein stability and structure on substrate processing by the ClpXP unfolding and degradation machine. The EMBO Journal. 2001;20:3092–3100. doi: 10.1093/emboj/20.12.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman E, Fry AN, Kang M. The complexities of p97 function in health and disease. Mol Biosyst. 2011;7:700–710. doi: 10.1039/c0mb00176g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, Varshavsky A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–1583. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- Chin JW, Cropp TA, Anderson JC, Mukherji M, Zhang Z, Schultz PG. An expanded eukaryotic genetic code. Science. 2003;301:964–967. doi: 10.1126/science.1084772. [DOI] [PubMed] [Google Scholar]
- Chin JW, Martin AB, King DS, Wang L, Schultz PG. Addition of a photocrosslinking amino acid to the genetic code of Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:11020–11024. doi: 10.1073/pnas.172226299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TF, Bulfer SL, Weihl CC, Li K, Lis LG, Walters MA, Schoenen FJ, Lin HJ, Deshaies RJ, Arkin MR. Specific inhibition of p97/VCP ATPase and kinetic analysis demonstrate interaction between D1 and D2 ATPase domains. Journal of Molecular Biology. 2014;426:2886–2899. doi: 10.1016/j.jmb.2014.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JC, Ye Y. Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nature Structural & Molecular Biology. 2014;21:325–335. doi: 10.1038/nsmb.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLaBarre B, Christianson JC, Kopito RR, Brünger AT. Central pore residues mediate the p97/VCP activity required for ERAD. Molecular Cell. 2006;22:451–462. doi: 10.1016/j.molcel.2006.03.036. [DOI] [PubMed] [Google Scholar]
- DeLaBarre B, Brunger AT. Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nature Structural Biology. 2003;10:856–863. doi: 10.1038/nsb972. [DOI] [PubMed] [Google Scholar]
- DeLaBarre B, Brunger AT. Nucleotide dependent motion and mechanism of action of p97/VCP. Journal of Molecular Biology. 2005;347:437–452. doi: 10.1016/j.jmb.2005.01.060. [DOI] [PubMed] [Google Scholar]
- Dorman G, Prestwich GD. Benzophenone photophores in biochemistry. Biochemistry. 1994;33:5661–5673. doi: 10.1021/bi00185a001. [DOI] [PubMed] [Google Scholar]
- Du F, Navarro-Garcia F, Xia Z, Tasaki T, Varshavsky A. Pairs of dipeptides synergistically activate the binding of substrate by ubiquitin ligase through dissociation of its autoinhibitory domain. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:14110–14115. doi: 10.1073/pnas.172527399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst R, Mueller B, Ploegh HL, Schlieker C. The otubain YOD1 is a deubiquitinating enzyme that associates with p97 to facilitate protein dislocation from the ER. Molecular Cell. 2009;36:28–38. doi: 10.1016/j.molcel.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erzberger JP, Berger JM. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annual Review of Biophysics and Biomolecular Structure. 2006;35:93–114. doi: 10.1146/annurev.biophys.35.040405.101933. [DOI] [PubMed] [Google Scholar]
- Franz A, Ackermann L, Hoppe T. Ring of Change: CDC48/p97 Drives Protein Dynamics at Chromatin. Front Genet. 2016;7:73. doi: 10.3389/fgene.2016.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey S, Gorlich D. A new set of highly efficient, tag-cleaving proteases for purifying recombinant proteins. Journal of Chromatography A. 2014;1337:95–105. doi: 10.1016/j.chroma.2014.02.029. [DOI] [PubMed] [Google Scholar]
- Glynn SE, Nager AR, Baker TA, Sauer RT. Dynamic and static components power unfolding in topologically closed rings of a AAA+ proteolytic machine. Nature Structural & Molecular Biology. 2012;19:616–622. doi: 10.1038/nsmb.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R, Ripstein ZA, Augustyniak R, Lazniewski M, Ginalski K, Kay LE, Rubinstein JL. Unfolding the mechanism of the AAA+ unfoldase VAT by a combined cryo-EM, solution NMR study. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:E4190–E4199. doi: 10.1073/pnas.1603980113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Akiyama Y. Cellular functions, mechanism of action, and regulation of FtsH protease. Annu Rev Microbiol. 2005;59:211–231. doi: 10.1146/annurev.micro.59.030804.121316. [DOI] [PubMed] [Google Scholar]
- Kim I, Mi K, Rao H. Multiple interactions of rad23 suggest a mechanism for ubiquitylated substrate delivery important in proteolysis. Molecular Biology of the Cell. 2004;15:3357–3365. doi: 10.1091/mbc.E03-11-0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimonis VE, Fulchiero E, Vesa J, Watts G. VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: review of a unique disorder. Biochimica Et Biophysica Acta. 2008;1782:744–748. doi: 10.1016/j.bbadis.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Kish-Trier E, Hill CP. Structural biology of the proteasome. Annual Review of Biophysics. 2013;42:29–49. doi: 10.1146/annurev-biophys-083012-130417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress W, Mutschler H, Weber-Ban E. Both ATPase domains of ClpA are critical for processing of stable protein structures. The Journal of Biological Chemistry. 2009;284:31441–31452. doi: 10.1074/jbc.M109.022319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Huang C, Zhao G, Lennarz WJ. Interprotomer motion-transmission mechanism for the hexameric AAA ATPase p97. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:3737–3741. doi: 10.1073/pnas.1200255109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Ye Y. Roles of p97-associated deubiquitinases in protein quality control at the endoplasmic reticulum. Current Protein & Peptide Science. 2012;13:436–446. doi: 10.2174/138920312802430608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medicherla B, Kostova Z, Schaefer A, Wolf DH. A genomic screen identifies Dsk2p and Rad23p as essential components of ER-associated degradation. EMBO Reports. 2004;5:692–697. doi: 10.1038/sj.embor.7400164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H, Weihl CC. The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. Journal of Cell Science. 2014;127:3877–3883. doi: 10.1242/jcs.093831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan JA, Kim HT, Ting L, Gygi SP, Goldberg AL. Why do cellular proteins linked to K63-polyubiquitin chains not associate with proteasomes? The EMBO Journal. 2013;32:552–565. doi: 10.1038/emboj.2012.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM. Ubiquitin in chains. Trends in Biochemical Sciences. 2000;25:544–548. doi: 10.1016/s0968-0004(00)01681-9. [DOI] [PubMed] [Google Scholar]
- Prakash S, Tian L, Ratliff KS, Lehotzky RE, Matouschek A. An unstructured initiation site is required for efficient proteasome-mediated degradation. Nature Structural & Molecular Biology. 2004;11:830–837. doi: 10.1038/nsmb814. [DOI] [PubMed] [Google Scholar]
- Ramadan K, Bruderer R, Spiga FM, Popp O, Baur T, Gotta M, Meyer HH. Cdc48/p97 promotes reformation of the nucleus by extracting the kinase Aurora B from chromatin. Nature. 2007;450:1258–1262. doi: 10.1038/nature06388. [DOI] [PubMed] [Google Scholar]
- Richly H, Rape M, Braun S, Rumpf S, Hoege C, Jentsch S. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell. 2005;120:73–84. doi: 10.1016/j.cell.2004.11.013. [DOI] [PubMed] [Google Scholar]
- Rodrigo-Brenni MC, Foster SA, Morgan DO. Catalysis of lysine 48-specific ubiquitin chain assembly by residues in E2 and ubiquitin. Molecular Cell. 2010;39:548–559. doi: 10.1016/j.molcel.2010.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpf S, Jentsch S. Functional division of substrate processing cofactors of the ubiquitin-selective Cdc48 chaperone. Molecular Cell. 2006;21:261–269. doi: 10.1016/j.molcel.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Ryu JK, Min D, Rah SH, Kim SJ, Park Y, Kim H, Hyeon C, Kim HM, Jahn R, Yoon TY. Spring-loaded unraveling of a single SNARE complex by NSF in one round of ATP turnover. Science. 2015;347:1485–1489. doi: 10.1126/science.aaa5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer RT, Baker TA. AAA+ proteases: ATP-fueled machines of protein destruction. Annual Review of Biochemistry. 2011;80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- Schuller JM, Beck F, Lössl P, Heck AJR, Förster F. Nucleotide-dependent conformational changes of the AAA+ ATPase p97 revisited. FEBS Letters. 2016;590:595–604. doi: 10.1002/1873-3468.12091. [DOI] [PubMed] [Google Scholar]
- Shabek N, Ciechanover A. Degradation of ubiquitin: the fate of the cellular reaper. Cell Cycle. 2010;9:523–530. doi: 10.4161/cc.9.3.11152. [DOI] [PubMed] [Google Scholar]
- Sivaraman T, Arrington CB, Robertson AD. Kinetics of unfolding and folding from amide hydrogen exchange in native ubiquitin. Nature Structural Biology. 2001;8:331–333. doi: 10.1038/86208. [DOI] [PubMed] [Google Scholar]
- Stein A, Ruggiano A, Carvalho P, Rapoport TA. Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell. 2014;158:1375–1388. doi: 10.1016/j.cell.2014.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suno R, Niwa H, Tsuchiya D, Zhang X, Yoshida M, Morikawa K. Structure of the whole cytosolic region of ATP-dependent protease FtsH. Molecular Cell. 2006;22:575–585. doi: 10.1016/j.molcel.2006.04.020. [DOI] [PubMed] [Google Scholar]
- Taylor EB, Rutter J. Mitochondrial quality control by the ubiquitin-proteasome system. Biochem Soc Trans. 2011;39:1509–1513. doi: 10.1042/BST0391509. [DOI] [PubMed] [Google Scholar]
- Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. The EMBO Journal. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Oania RS, Kolawa NJ, Deshaies RJ. Cdc48/p97 promotes degradation of aberrant nascent polypeptides bound to the ribosome. eLife. 2013;2:e00308. doi: 10.7554/eLife.00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White SR, Lauring B. AAA+ ATPases: achieving diversity of function with conserved machinery. Traffic. 2007;8:1657–1667. doi: 10.1111/j.1600-0854.2007.00642.x. [DOI] [PubMed] [Google Scholar]
- Xia D, Tang WK, Ye Y. Structure and function of the AAA+ ATPase p97/Cdc48p. Gene. 2016;583:64–77. doi: 10.1016/j.gene.2016.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA. Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. The Journal of Cell Biology. 2003;162:71–84. doi: 10.1083/jcb.200302169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- Zhang M, Chang H, Zhang Y, Yu J, Wu L, Ji W, Chen J, Liu B, Lu J, Liu Y, et al. Rational design of true monomeric and bright photoactivatable fluorescent proteins. Nat Methods. 2012;9:727–729. doi: 10.1038/nmeth.2021. [DOI] [PubMed] [Google Scholar]
- Zhao M, Wu S, Zhou Q, Vivona S, Cipriano DJ, Cheng Y, Brunger AT. Mechanistic insights into the recycling machine of the SNARE complex. Nature. 2015;518:61–67. doi: 10.1038/nature14148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Purity of proteins used in this study, Related to Figure 1
(A) Purified proteins used in this study were subjected to SDS-PAGE and staining with Coomassie blue. Black stars indicate contaminating proteins. Degron-sfGFP and degron-Eos are substrates before being subjected to fluorescent labeling and ubiquitination. Cdc48-FtsH was run on a different gel; an irrelevant lane was removed from the gel on the left (white space). M, molecular weight markers.
(B) Tryptic peptides of the polyubiquitinated sfGFP substrate were analyzed by mass spectrometry, with a search for ubiquitin conjugation sites. Intensities of the peptides corresponding to ubiquitin chain linkage and ubiquitin conjugation sites are shown. Note that the predominant linkage is K48 and the predominant substrate ubiquitination site is K19. The # sign indicates the position of the isopeptide bond.
Figure S2. ATPase activity and substrate unfolding, Related to Figure 2
(A) ATP hydrolysis rates were determined with the indicated combinations of purified proteins and substrate. K48 and K63 indicate penta-ubiquitin with different linkages. The rates were normalized with respect to that of Cdc48 alone. Shown are the means and standard deviations of three experiments.
(B) As in (A), but with either wild type (WT) Cdc48 or Walker B glutamine mutants (Walker B mutation in D1 (E315Q; green bars) or D2 (E588Q; red bars)).
(C) Polyubiquitinated sfGFP (Ub(n)-sfGFP), generated as in Figure 1A, was incubated with an ATP regenerating system and excess of either Cdc48 alone or Cdc48 plus UN. After addition of ATP, GFP fluorescence was followed over time. One sample (yellow curve) received an equimolar amount of Otu1 at the indicated time point (arrow).
(D) Substrate unfolding assays with Ub(n)-Eos and Walker B glutamine mutants. Assays were performed as in Figure 2F.
Figure S3. Efficiency of substrate crosslinking, Related to Figure 3
SBP-tagged Cdc48 (Cdc48-SBP) with Bpa at the indicated positions was incubated with dye-labeled, polyubiquitinated sfGFP (Ub(n)-sfGFP), UN, and an ATP regenerating system. Where indicated, reactions were irradiated. The samples were then subjected to SDS-PAGE and fluorescence scanning. The silver-stained band of Cdc48-SBP serves as a loading control. The brackets indicate the positions of crosslinked products. Crosslinking efficiencies were estimated by comparing the fluorescence intensities of non-crosslinked and crosslinked species (~20% for M288, and ~5% for D324 and D602).
Figure S4. Substrate cleavage by the Cdc48-FtsH chimera, Related to Figure 4
(A) Dye-labeled, polyubiquitinated Eos (Ub(n)-Eos) was incubated with UN, the fusion of Cdc48 and FtsH (Cdc48-FtsH), and ATP, ATPγS, or ADP. One sample was directly analyzed by mass spectrometry. Sequences detected only in ATP are shown in red (15 unique peptides). Two unique peptides (shown in blue) were detected in all nucleotide states and correspond to part of the degron preceding Eos (underlined in black), which is likely flexible. Another sample was subjected to SDS-PAGE and the stable proteolytic fragment of ~13 kDa was trypsinized and analyzed by mass spectrometry. The identified peptide is underlined in green.
(B) Dye-labeled Ub(n)-Eos was incubated with an ATP regenerating system, UN, and either Cdc48-FtsH or a mixture of wild type Cdc48 and the isolated FtsH domain. Aliquots were removed at the indicated time points and analyzed by SDS-PAGE and fluorescence scanning. The arrow head indicates a stable fragment containing the fluorescent dye, which was generated with the Cdc48-FtsH fusion.
Figure S5. Otu1 binding to the Cdc48 complex, Related to Figure 5
(A) Complexes of Cdc48, SBP-tagged UN, and dye-labeled, polyubiquitinated sfGFP were immobilized on streptavidin beads in the presence of ATP. After washing, the indicated proteins were added and samples of the bound material were analyzed at the indicated time points by SDS-PAGE and fluorescence scanning. Where indicated, unlabeled, polyubiquitinated sfGFP was added in 5-fold excess over the original fluorescently labeled input.
(B) SBP-tagged UN and the indicated Cdc48 mutants were immobilized on streptavidin beads, and wild-type Otu1 was added. After washing, the bound material was recovered and subjected to SDS-PAGE and silver staining.