

Abstract
In recent years, there has been substantial progress in our knowledge of the molecular pathways by which stem cell factor SALL4 regulates the embryonic stem cell (ESC) properties, developmental events, and human cancers. This review summarizes recent advances in the biology of SALL4 with a focus on its regulatory functions in normal and leukemic hematopoiesis. In the normal hematopoietic system, expression of SALL4 is mainly enriched in the bone marrow hematopoietic stem/progenitor cells (HSCs/HPCs), but is rapidly silenced following lineage differentiation. In hematopoietic malignancies, however, SALL4 expression is abnormally re-activated and linked with deteriorated disease status in patients. Further, SALL4 activation participates in the pathogenesis of tumor initiation and disease progression. Thus, a better understanding of SALL4’s biologic functions and mechanisms will facilitate development of advanced targeted anti-leukemia approaches in future.
Keywords: Pluripotency, Hematopoietic stem and progenitor cells, Apoptosis, Transgenic, Wnt/β-catenin signaling, Mixed-lineage leukemia, Chromatin modification
Background
SALL4 is one of four human homologues (SALL1, 2, 3, and 4) of the Drosophila region-specific homeotic gene spalt (sal) [1–4]. SALL4 encodes multiple Cys2His2 zinc finger (C2H2-ZF) domain-containing transcription factor that either activates or represses gene transcription depending on cell context. In mammals, expression of SALL4 has been primarily detected in ESCs and adult tissue “stem-like” cell populations, where it acts as a core controller regulating cell “stemness” in developmental events and also in tumor growth [5–9]. To date, aberrant SALL4 expression in humans has been observed in over 10 types of solid tumors and in several common types of leukemia (see review [8–11]), and SALL4 has been considered a biomarker for the diseases. In addition, studies suggest that SALL4 may be a useful therapeutic target in combating human leukemia in clinic [10, 11]. For these reasons, it will be important to understand, how SALL4 as a stem cell factor exerts its effects in different cell contexts, and how we can effectively translate our gained knowledge into treatment breakthroughs in future.
Sall4 stem cell gene
The stem cell factor SALL4 is required for ESC pluripotency and for early embryonic development. It is an essential component of the ‘stemness’ regulatory circuit involving OCT4, SOX2, NANOG and other factors in maintaining ESC self-renewal and pluripotency [12–17]. In ESCs, a well-controlled SALL4/OCT4 transcription regulatory loop balances proper expression dosage of SALL4 and OCT4; and like OCT4, reduction of SALL4 results in re-specification of ESCs to the trophoblast lineage [18–20]. SALL4 is also a critical regulator in reprogramming of somatic cells to pluripotency. Shu et al. recently reported that GATA family members GATA4 and GATA6 can substitute for OCT4 in mouse somatic reprogramming, and identified SALL4 as a major target gene of the GATA members, serving as a bridge linking the lineage-specifying GATA family to the pluripotency circuit [21]. In fact, ectopic expression of SALL4, NANOG, ESRRB, and LIN28 [22], or the combination of SALL4, SALL1, UTF1, NANOG and MYC [23] in embryonic fibroblasts reprogram them into induced pluripotent stem cells (iPSCs) in the absence of OCT4.
In murine development, significant zygotic SALL4 transcription occurs at as early as the 4-cell stage. At the blastocyst stage, SALL4 expression becomes enriched in the inner cell mass (ICM) and the trophectoderm [12, 24–26]. Reduction of SALL4 in oocytes and ESCs results in early embryo defects, and disruption of both Sall4 alleles causes embryonic lethality during peri-implantation [27–29]. SALL4 is also expressed in extraembryonic endoderm (XEN) cells, where it participates in cell fate decision by simultaneously activating key pluripotency maintaining factors and silencing endoderm lineage-associated factors such as GATA6, GATA4, and SOX17 [30, 31]. During subsequent stages, heterozygous disruption of Sall4 allele leads to multi-organ malformations including limb and heart defects, which model human disease [27, 29]. It has been reported that TBX5, a gene encoding a T-box transcription factor, regulates SALL4 expression in the developing forelimb and heart, and interacts with SALL4 to synergistically regulate downstream gene expression [28, 29, 32].
In both humans and mice, SALL4 proteins exist at least in three isoforms termed A, B and C, with A and B isoforms being the most studied [32–36]. In ESCs, the SALL4 proteins are sequestered into the nuclear foci and bind to heterochromatin, where they participate in chromatin structure remodeling during transcription repression [37, 38]. SALL4 has also been shown to interact with the histone H3 lysine 36 (H3K36me3)-specific methyltransferase Wolf-Hirschhorn syndrome candidate 1 (WHSC1), which affects histone modification and thus regulate the expression of their target genes [39–41].
Functions of Sall4 and its regulated networks in normal hematopoiesis
Given SALL4’s prominent roles in ESCs, development, and its specific expression patterns in the hematopoietic system, our research group previously investigated SALL4’s functions in the self-renewal of HSCs/HPCs. We demonstrated that the SALL4 isoforms are robust stimulators for human CD34+ HSCs/HPCs expansion [42, 43]. Of note, we reported that the SALL4-driven ex vivo expansion of HSCs/HPCs is dependent on excessive special cytokines, and does not affect mature colony formation in colony-forming unit (CFU) assays [42]. In another study, Tatetsu et al. report that ex vivo culture of mobilized peripheral blood CD34+ cells with histone deacetylase (HDAC) inhibitors leads to expansion of a CD34+ CD90+ population, and SALL4 is identified as a key transcription factor responsible for the process [44]. In mouse model studies, forced overexpression of SALL4 in Lineage− Sca-1+c-Kit+ (LSK) bone marrow (BM) cells likewise leads to sustained cell proliferation, as well as enhanced marrow-repopulating potential [45]. Transcripts assays showed that the increased HSC/HPC growth was associated with upregulation of multiple HSC regulatory genes including HOXB4, NOTCH1, BMI1, RUNX1, CMYC, MEIS1 and NF-YA [45]. Further, in a myeloid progenitor cell line (32D cells), overexpression of SALL4 blocked granulocyte-colony stimulating factor (G-CSF)-induced granulocytic differentiation, and permitted expansion of undifferentiated cells in the presence of defined cytokines [42, 45]. Thus, the SALL4 isoforms stimulate HSC/HPC proliferation by activating important self-renewal regulators and simultaneously inhibiting differentiation. Recently, Mossahebi-Mohammadi et al. reported that the CD133+ umbilical cord blood HSCs/HPCs are also efficiently expanded following SALL4 lentivirus transduction, and the SALL4-expanded CD133+ cells retain self-renewal and differentiation capacities with no chromosomal aberrations [46]. In another study, it is further reported that SALL4 in CD34+ cells was downregulated by microRNAs miR-15b and miR-219-5p, and inhibition of miR-15b, −which activates SALL4 expression, significantly increased the number of CD34+ HSCs/HPCs in culture [47].
In elucidating the SALL4 regulated networks, Gao et al. sorted human CD34+ BM cells and performed chromatin immunoprecipitation followed by microarray hybridization (ChIP-on-chip), together with gene expression assays [48]. These works identified that CD34, RUNX1, HOXA9, and PTEN are SALL4-directed target genes. In particular, HOXA9 is being characterized as a major SALL4 target in hematopoiesis. Moreover, down-regulation of SALL4 or HOXA9 expression in CD34+ cells results in similar effects, i.e., reduced in vitro myeloid colony formation and impaired in vivo engraftment [48]. In another study [49], the polycomb complex protein BMI-1 as a critical SALL4 downstream target has been documented. Chromatin immunoprecipitation coupled with quantitative PCR (ChIP-qPCR) in 32D cells reveals that SALL4 binds to a specific region of Bmi-1 gene promoter, and heterozygous disruption of Sall4 allele significantly reduced BMI-1 expression in BM cells. Further, in transgenic mice that constitutively overexpress human SALL4B, there is up-regulated expression of BMI-1, and the levels of BMI-1 in these mice increase as they progress from normal to preleukemic (myelodysplastic syndrome [MDS]) and leukemic (acute myeloid leukemia [AML]) stages.
Very recently, SALL4’s functions in normal hematopoiesis have been further explored using conditional gene targeting approaches [50]. Surprisingly, wild type Sall4f/f/CreERT2 mice that are treated with tamoxifen, or Vav-Cre-mediated (hematopoietic-specific) Sall4−/− mice are all healthy and display no significant hematopoietic defects, which is in contrast to previous human CD34+ cell studies. Reasons for this discrepancy have not been fully addressed. To be noted however, some genes may exert aberrant functions only when cells encounter transplantation or replicative stress (see review [51]), and some Vav-Cre knockout models may demonstrate hematopoietic defects at late stages [52]. Thus it may be necessary to perform serial transplantation and/or stress induction (such as 5-fluorouracil injury) assays with SALL4-deficient cells to fully clarify SALL4 effect and mechanisms in maintaining normal HSCs.
Functions of Sall4 and its regulated networks in leukemia
SALL4 as a diagnostic marker in human leukemia
Aberrant expression of SALL4 has been detected in MDS patients and its expression levels are correlated with disease progression [34, 53]. Also, SALL4 and BMI-1 share a similar expression pattern in the patients with both expressions increase in high-grade morphology and high International Prognostic Scoring System (IPSS) score cases [53]. In addition, higher SALL4 protein levels are associated with the complex karyotype (equal to or more than three aberrant karyotypes), and SALL4 appears to be involved in the DNA damage response in patients [54], as supported by their roles in the ESC system [38]. In AML cases, immunohistochemistry staining reveals that SALL4 proteins are present in nearly all the patient samples that are examined (n = 81, subtypes M1 to M5, the French-American-British [FAB] classification) [34], and SALL4 expression in patients with complex karyotype is significantly higher than that in MDS patients with normal karyotype [54]. Similarly, variable transcript levels of SALL4A and SALL4B expression are reported in pediatric AML patients [55]. In other leukemia cases, aberrant SALL4 expression has been reported in ALK positive anaplastic large cell lymphoma (ALK+ ALCL) [56], B cell acute lymphocytic leukemia (B-ALL), −most prominently in B-ALL patients with TEL-AML1 translocation, which is the most common genetic abnormality in pediatric B-ALL [57, 58]. SALL4 expression is also detected in patient samples from blastic stage of chronic myeloid leukemia (CML), as contrast to the chronic phase, and CML patients who have achieved complete remission or those who have tyrosine kinase inhibitor resistance [59–61]. Lastly, constitutive SALL4 expression has been documented in various human myeloid and lymphoid leukemia cell lines including KG1a (AML-M0), KASUMI-1 (AML-M2), HL-60 (AML- M2/M3), NB-4 (AML M3), THP1 (AML-M5), TEX (FUS/TLS-ERG oncogene immortalized AML), K562 (CML/AML M6), RPMI-8226 (myeloma), LAMA84 (CML-acute phase), BV173 (B-ALL), REH (ALL), NALM6 (ALL), 697 (pre-B ALL), BLIN-1 (pre-B ALL) and JURKAT (T cell leukemia) [34, 58, 61–65].
Interestingly, the SALL4 expression appears to be selectively enriched in leukemia side-population (SP) cells (defined by low Hoechst 33,342 blue/red fluorescence intensity), −a fraction specifically involved in cancer initiation and drug resistance [66], which supports the SALL4 expression features observed in above clinical cases, and suggests a role of SALL4 in maintaining the leukemia “initiating or stem” cell (LIC/LSC) populations.
Leukemogenesis induction by SALL4B in a transgenic model
In 2006, our research group reported the first SALL4-mediated leukemogenesis model [34]. We investigated transgenic mice that overexpress human SALL4A or SALL4B proteins, controlled by a universal CMV promoter. All the SALL4B mice from 6 founders developed MDS-like features at 2 months of age, and 9 of them (53%) progressed to AML. By contrast, none of the SALL4A mouse exhibits leukemia formation during the test period [34, 67]. These groups of studies suggest that SALL4B, but not SALL4A, is a novel oncogene in inducing leukemogenesis. As an adding note, however, while forced overexpression of either SALL4A or SALL4B isoform fails to transform primary BM cells, nor induces leukemia formation in transplanted mice [43, 67, 68], one may deduce that in the SALL4B transgenic mice, an abnormal “niche” resulting from aberrant SALL4B expression in various mouse tissues throughout the mouse development may play a role, and/or SALL4B may bear oncogenic potential during the dysregulated hematopoiesis. Further in-depth studies are needed to elucidate the associated mechanisms.
Role of SALL4 in mixed lineage leukemia (MLL)- rearranged (MLL-r) leukemogenesis
Considering the strong association of SALL4 expression with tumor initiation and leukemia survival, our group recently explored its effects in the pathogenesis of leukemia mediated by MLL-AF9, −one of the most common MLL-r oncoproteins found in leukemia patients [69–71]. MLL-r leukemias have been very challenging in therapy and associated with poor outcomes [70, 72, 73]. Further, a previous study has shown that the MLL wild type protein physically interacts with SALL4 in regulating HOXA9 expression [74]. We report that SALL4 expression is essentially required for MLL-AF9-mediated myeloid transformation in primary BM cells. Cre recombinase or shRNA mediated SALL4 knockdown in MLL-AF9-transformed cells induced apoptotic death and cell cycle arrest at G1. Consistently, disruption of both Sall4 alleles in transplanted mouse models completely prevented leukemia initiation and also attenuated pre-existing disease progression [50]. These studies support that the SALL4 pathway may not only be used as a useful biomarker or therapeutic target, proper inhibition of the SALL4 pathway may also effectively prevent disease initiation in patients at early stages.
SALL4 as a therapeutic target in human leukemic cells
SALL4 as a key regulator of survival has been widely documented in different subtypes of leukemias and in various human cancers [8, 10, 54, 75, 76]. In these malignancies, downregulation of SALL4 prominently causes increased apoptosis and cell cycle arrest [50, 62, 75–78]. In AMLs, aberrant SALL4 has also been shown to block all-trans retinoic acid (ATRA)-mediated myeloid differentiation in both ATRA-sensitive and ATRA-resistant AML subtypes [62]. Thus, these studies support that SALL4 maintains leukemic growth by protecting their proper proliferation and also inhibiting their differentiation. Not surprisingly, in recent years, SALL4-targeted anti-leukemia strategies have gained increasing interest and been elaborately explored (see review [11]). In a study by Gao et al., a SALL4- derived peptide blocking its protein-protein interaction with the nucleosome remodeling and histone deacetylation (NuRD) complex results in notable leukemic cell death, but causes no cytotoxic effects on normal CD34+ HSCs/HPCs [64]. Gao et al., also reported that SALL4 knockdown in combination with BCL-2 inhibitor treatment increased the apoptotic AML cells to 2 to 3 fold as compared to cells treated with each alone [77]. Our research group reported that while SALL4 and its interacting epigenetic factor LSD1 inhibited ATRA-induced granulocytic differentiation, co-inhibition of SALL4, LSD1, plus ATRA treatment severely disrupted ATRA-resistant AML cell growth, and blocked HL60 AML xenograft tumors by ~91%, while the treated mice exhibit no signs of illness [62, 79]. Considering that Sall4 depletion in mice minimally affects adult hematopoiesis [50], these separate works collectively suggest that the SALL4 regulation-targeted approaches, which induce leukemia apoptosis and differentiation but spare normal cells, likely represent clinically effective and novel anti-leukemia strategies.
SALL4 regulated pathways in leukemia
ChIP-on-chip assays with a promyelocytic leukemic cell line, NB4 reveal that SALL4-binding genes are involved in more than 30 different signaling pathways [75]. Most prominent of these pathways are WNT/β-catenin, apoptosis, NOTCH signaling, the polycomb complex protein BMI-1, PTEN, and nuclear factor-kB. Further, SALL4 expression in these cells represses important apoptosis-inducing genes (CARD9, CARD11, CYCS, LTA, TNF, TP53, PTEN) but promotes apoptosis suppressor genes (BMI-1, BCL2, DAD1, TEGT, XIAP). In SALL4B transgenic mouse studies, the SALL4A and SALL4B isoforms have been found to bind β-catenin protein, and interaction of these factors synergistically activates the WNT/β-catenin pathway, −which plays critical roles in controlling LSC self-renewal [34, 80–82]. The interaction domain between SALL4 and β-catenin was not determined. However, it was deduced that the C-terminal portion of SALL4 may play a role, based on binding data between SALL1 and β-catenin [8, 83]. Intriguingly, in a recent study by Kode et al., transgenic activation of a mutated β-catenin allele in murine osteoblasts, −one of the key HSC/HPC niches in BM [84, 85], induced MDS and AML in mice at very early ages via dysregulated NOTCH ligand JAG1 [86–88]. Since SALL4B transgenic mice also develop MDS/AML, and SALL4B-overexpressed BM cells do not induce leukemia formation in transplanted mice, an interesting question will be in the SALLB mouse, if/how SALL4B potentially activates β-catenin signaling, which synergistically dysregulate the HSC/HPC osteoblastic niche and thus promote leukemogenesis. Further detailed transgenic studies are required to address this probability.
ChIP assays with sequencing (ChIP-Seq) assays with mouse MLL-AF9 transformed leukemic cells are also performed [50], which identified that SALL4 binds to 451 genes including MLL-AF9 targets (Meis1, Hoxa9), MLL-r leukemia-related genes (Cebpα, Id2, Elf1, Evl, Flt3, Nf1, Tal1, Tcf7l1, Nkx2–3), HOX factors (Hoxa-9, −10, −11, −13), Notch ligand Jag2, and Wnt/β-catenin regulator Wnt7b. Further, loss of SALL4 downregulated the expression levels of Bmi1, cMyb, Runx1, Meis1, and HOX factors. In the same MLL-AF9 transformed cells, mRNA microarrays assays following early Sall4 deletion identified upregulated genes including cell cycle inhibitors Cdkn1a (p21), Trp53inp1; HSC/HPC colony-forming repressor Slfn2; and hematopoietic differentiation markers Col5a1, Fyb, Irf8 and Pira6. In contrast, downregulated genes included multiple TGFβ signaling components Tgfβ2, Tgfβ3 and Tgfβr3; genes related to chemo-resistance or leukemia aggressiveness such as Thbs1, Tgm2, Ambp, and the AF9 regulator Sgk1, −which negatively regulates the DOT1A-AF9 complex-mediated transcriptional repression [50, 89]. Taken together, these studies suggest that SALL4 may regulate MLL-AF9 mediated leukemogenesis via multiple HSC/HPC- and LSC-related signaling pathways.
To date, upstream regulators of SALL4 expression in leukemia remain poorly understood, although OCT4, GATA4, GATA6, STAT3, TBX5, the WNT/β-catenin signaling factors, and micorRNAs such as miR-98, miR-33b and miR-294 are reported in other cell systems [8–10, 76, 90, 91]. Of note, DNA methylation modifications of the SALL4 promoter may play a role, −studies using B-ALL cell lines and patient samples detected hypomethylation of the SALL4 CpG islands spanning the exon1-intron1 region [57], which has also been observed in MDS and AML patients, and this hypomethylation correlates with high SALL4 expression [92, 93]. Some other studies have documented SALL4-repressing compounds, such as apigenin, matrine, indole and its derivative 2-(1-((2, 4-Aril)imino)-2,2,2-trifluoroethyl) phenyl-1H Indole-3- carbaldehyde (TFPHC) are documented [10, 94]. Such works suggest that the SALL4 regulatory pathway may be potentially disrupted by clinically applicable drugs, and more such related ongoing works are expected to promote translation of cutting-edge SALL4 knowledge into clinical practice in future.
Epigenetic mechanisms involved in Sall4’s regulatory functions in normal and leukemic hematopoiesis
Multiple chromatin modification regulators have been identified involving in SALL4’s regulatory functions. So far the reported SALL4-interacting epigenetic factors include: DNA methyltransferases DNMT-1, −3A, -3B, −3 L, methyl-CpG-binding domain 2 protein (MBD2) [95]; NuRD complex that contains histone deacetylases HDAC1/2 [64, 96]; H3K4 methyltransferase MLL1 [74, 97]; H3K79 methyltransferase DOT1L [50, 98]; H3K36 methyltransferase WHSC1 [39, 99], and lysine-specific histone demethylase LSD1/KDM1A [50, 62, 79]. All of these are critical regulators in normal blood development and are frequent targets for dysregulation in hematological malignancies [100–103]. Of note, the SALL4 proteins seem to interact with these epigenetic factors at different sites. For example, while the amino-terminal 174 amino acid sequence of SALL4 is critical for SALL4-DNMT1 or -HDAC interaction, it seems less relevant to SALL4-LSD1 interaction. This is important in designing protein-protein interaction based anti-SALL4 strategies. Noteworthily, clinical epigenetic remedies inhibiting such SALL4-interacting epigenetic factors have been shown effective in treating leukemia [104–108]. Indeed, in MLL-AF9-mediated mouse AML studies, inhibition of either SALL4, DNMT1, LSD1, or DOT1L completely blocked leukemia initiation and significantly delayed disease progression in vivo [109–111].
By dynamically recruiting each specific epigenetic factor, SALL4 expression can directly affect DNA and histone methylation/acetylation status at important genes that control hematopoietic differentiation, apoptosis, tumor induction or suppression. For example, in NB4 AML cells transduced with lentiviral SALL4, there was an overall increased percentage of DNA methylation (a range of 1.2 to 2-fold) at various CpG sites of tumor suppression gene PTEN promoter, which co-relates with a down-regulated gene transcription [95]. In mouse BM LSK cells, overexpression of the SALL4 isoforms also induces increased percentage of methylation (1.2 to 6- fold) at the CpG sites of early B-cell factor 1 (Ebf1) promoter, as well as the Sall4 gene promoter itself, which facilitates an undifferentiated cellular status [95]. Similarly, SALL4 overexpression or Cre-induced Sall4 gene deletion affected LSD1 binding and altered H3K4me2 levels at the promoter regions of tumor necrosis factor (Tnf) and differentiation-related genes Ebf1, Gata1 (up to ~300 and ~700 fold changes), which are associated with relevantly altered gene transcription levels [79]. Further, in 32D myeloid progenitor cells following lentiviral SALL4 transduction, the H3K4me3 and H3K79me2/3 levels at SALL4-occupied regions of Bmi1 promoter were increased [49]. Additionally, expression levels of SALL4 protein affect H3K4me3 and H3K79me3 amounts at promoter regions of Meis1 and multiple HOX family genes in normal or leukemic BM cells, as we and others reported [48, 50, 74]. Also, in the functional study by Gao et al., while SALL4 interacts with the HDAC complex, and silences PTEN promoter via reduced acetylation of histone H3 at its binding sites, the SALL4-derived peptide blocks this interaction and leads to reactivated PTEN expression, which induces leukemia cell death that can be rescued by a PTEN-specific inhibitor [64, 96].
Evidence suggests that SALL4 may compensatively recruit different epigenetic regulators. For example, LSD1 has been shown to contribute to global DNA methylation via stabilizing DNMT1 [112], while in the absence of LSD1 proteins, SALL4 seems to compensatively recruit DNMT1 or DNMT3L to downstream genes (such as EBF1, TNF) and modulate their expression [79]. Also, co-inhibition of DNMTs and HDACs can synergistically block SALL4’s regulatory effects in cultured cells, which induces differentiation and cell growth arrest in human leukemia [113]. In addition, LSD1 has been shown to interact with the SALL4/CDX4 (a master regulator of the HOX genes) circuit and affect granulocytic/erythroid maturation [114, 115]. SALL4 also appears to confer cell resistance to DNA double-stranded break (DSB)-induced cytotoxicity by collaborating with HDAC-1 and -2 [38, 116].
It is important to note that SALL4’s regulation of specific epigenetic modification programs is strictly dependent on the cellular contexts. Analysis of the ChIP-on-chip data from NB4 AML cells and normal CD34+ HSCs/HPCs reveals distinct SALL4 binding patterns between these two cell types, which reflect a cell type-specific epigenetic signature and SALL4 function [48, 75]. This finding is supported by ChIP-Seq data analysis of ESCs and extra-embryonic endoderm cell studies [30]. In ESCs, the SALL4-bound genomic loci are largely enriched for activating marker H3K4me3, which indicates an association of SALL4 with non-repressed genes. In XEN cells, however, the SALL4-binding loci display significantly less H3K4me3 enrichment. Instead, over 60% of these regions are either accompanied with H3K27me3 or lacking both H3K4me3 and H3K27me3, −the “epi-markers” frequently associated with gene repression. In our MLL-AF9 leukemia model studies [50], SALL4 appears to recruit both DOT1L and LSD1 to specific downstream target genes and modulate their H3K79me2/3 and H3K4me3 levels, thereby maintaining proper gene expression and leukemic survival (see Fig. 1). As reported in previous studies, however, some non-MLL-r human AMLs may not rely on DOT1L-regulated H3K79 methylation, and DOT1L recruitment to MLL-AF9 is further associated with the level of leukemic transformation [117–120]. Thus, we may anticipate that in normal hematopoiesis and such different subtypes of human leukemias, SALL4 should differentially interact with such epigenetic factors during regulation of gene expression, thereby exerting a disease/subtype–dependent regulatory effect. In-depth functional and epigenetic studies are required to prove this assumption.
Fig. 1.
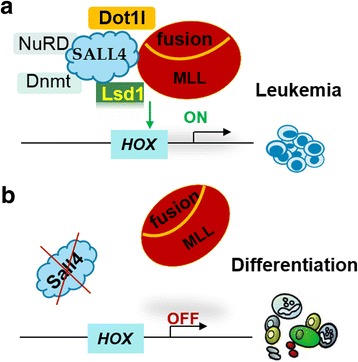
A tentative model for SALL4-regulated epigenetic mechanism in MLL-r leukemia. a Through interacting with MLL fusion oncoprotein, DOT1L, LSD1 and other epigenetic factors, SALL4 may act as a “guide” in modulating the expression levels of specific downstream gene targets. b SALL4-targetd strategies can disrupt MLL fusion oncoprotein function via dysregulated recruitment of essential epigenetic factors
Conclusions
This review summarized recent advances in the biology of the stem cell factor SALL4 with a focus on its regulatory functions in normal and leukemic hematopoiesis. In recent years, there have been gains in our understanding of SALL4-regulated molecular mechanisms and SALL4-targeted strategy in killing tumor cells. Understanding how SALL4 mechanisms maintain normal HSCs/HPCs vs. leukemic cells will facilitate development of newer, more efficient anti-leukemia strategies.
Acknowledgments
I would like to thank all my colleagues who have provided support in related research program. I would also like to thank Kimberly Macellaro, PhD, a member of the Baylor College of Medicine Michael E. DeBakey Department of Surgery Research Core Team, for her editorial assistance.
Funding
This project was supported by American Cancer Society Research Scholar Grant RSG-12-216-01-LIB (to J. Y.).
Availability of data and materials
All data generated in the study are included in the present article.
Author contributions
J.Y. wrote the paper.
Abbreviations
- ALK+ ALCL
ALK positive anaplastic large cell lymphoma
- AML
Acute myeloid leukemia
- ATRA
All-trans retinoic acid
- B-ALL
B cell acute 112 lymphocytic leukemia
- BM
Bone marrow
- C2H2-ZF
Cys2His2 zinc finger
- CFU
Colony-forming unit
- ChIP-on-chip
Chromatin immunoprecipitation followed by microarray hybridization
- ChIP-PCR
Chromatin immunoprecipitation coupled with quantitative PCR
- ChIP-Seq
ChIP assays with sequencing
- CML
Chronic myeloid leukemia
- Ebf1
Early 229 B-cell factor 1
- ESC
Embryonic stem cell
- FAB
The French-109 American-British classification
- G-CSF
Granulocyte-colony stimulating factor
- HDAC
Histone deacetylase
- HSPs/HPCs
Hematopoietic stem/progenitor cells
- ICM
Inner cell mass
- iPSCs
Induced pluripotent stem cells
- IPSS
International Prognostic Scoring System
- LIC/LSC
Leukemia “initiating or stem” cell
- LSK
Lineage- Sca-1+ c-kit+
- MBD2
Methyl-CpG-binding domain 2 protein
- MDS
Myelodysplastic syndrome
- MLL-r
Mixed lineage leukemia (MLL)-rearranged
- NuRD
Nucleosome remodeling and deacetylase
- SP
Side-population
- TFPHC
2-(1-((2, 4-Aril)imino)-205 2,2,2-trifluoroethyl) phenyl-1H Indole-3- carbaldehyde
- Tnf
Tumor necrosis factor
- WHSC1
Wolf-Hirschhorn syndrome candidate 1
- XEN
Extraembryonic endoderm
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The author declares that he has no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kohlhase J, Schuh R, Dowe G, Kuhnlein RP, Jackle H, Schroeder B, Schulz-Schaeffer W, Kretzschmar HA, Kohler A, Muller U. Isolation, characterization, and organ-specific expression of two novel human zinc finger genes related to the drosophila gene spalt. Genomics. 1996;38 [DOI] [PubMed]
- 2.Eildermann K, Aeckerle N, Debowski K, Godmann M, Christiansen H, Heistermann M, Schweyer S, Bergmann M, Kliesch S, Gromoll J, et al. Developmental expression of the pluripotency factor sal-like protein 4 in the monkey, human and mouse testis: restriction to premeiotic germ cells. Cells Tissues Organs. 2012;196(3):206–220. doi: 10.1159/000335031. [DOI] [PubMed] [Google Scholar]
- 3.Sweetman D, Munsterberg A. The vertebrate spalt genes in development and disease. Dev Biol. 2006;293(2):285–93. [DOI] [PubMed]
- 4.de Celis JF, Barrio R. Regulation and function of Spalt proteins during animal development. Int J Dev Biol. 2009;53(8–10):1385–1398. doi: 10.1387/ijdb.072408jd. [DOI] [PubMed] [Google Scholar]
- 5.Yang J, Liao W, Ma Y. Role of SALL4 in hematopoiesis. Curr Opin Hematol. 2012;19 [DOI] [PubMed]
- 6.Kohlhase J, Heinrich M, Schubert L, Liebers M, Kispert A, Laccone F, Turnpenny P, Winter RM, Reardon W. Okihiro syndrome is caused by SALL4 mutations. Hum Mol Genet. 2002;11(23):2979–2987. doi: 10.1093/hmg/11.23.2979. [DOI] [PubMed] [Google Scholar]
- 7.Kohlhase J: SALL4-Related Disorders. In: GeneReviews(R). edn. Edited by Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mefford HC, Stephens K, Amemiya A, Ledbetter N. Seattle (WA); 1993.
- 8.Tatetsu H, Kong NR, Chong G, Amabile G, Tenen DG, Chai L. SALL4, the missing link between stem cells, development and cancer. Gene. 2016;584 [DOI] [PMC free article] [PubMed]
- 9.Xiong J. SALL4: engine of cell Stemness. Current gene therapy. 2014; [DOI] [PubMed]
- 10.Zhang X, Yuan X, Zhu W, Qian H, Xu W. SALL4: an emerging cancer biomarker and target. Cancer Lett. 2015;357(1):55–62. doi: 10.1016/j.canlet.2014.11.037. [DOI] [PubMed] [Google Scholar]
- 11.Wang F, Zhao W, Kong N, Cui W, Chai L. The next new target in leukemia: the embryonic stem cell gene SALL4. Mol Cell Oncol. 2014;1(4):e969169. doi: 10.4161/23723548.2014.969169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elling U, Klasen C, Eisenberger T, Anlag K, Treier M. Murine inner cell mass-derived lineages depend on Sall4 function. Proc Natl Acad Sci U S A. 2006;103(44):16319–16324. doi: 10.1073/pnas.0607884103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang J, Tam WL, Tong GQ, Wu Q, Chan HY, Soh BS, Lou Y, Yang J, Ma Y, Chai L, et al. Sall4 modulates embryonic stem cell pluripotency and early embryonic development by the transcriptional regulation of Pou5f1. Nat Cell Biol. 2006;8(10):1114–1123. doi: 10.1038/ncb1481. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Rao S, Chu J, Shen X, Levasseur DN, Theunissen TW, Orkin SH. A protein interaction network for pluripotency of embryonic stem cells. Nature. 2006;444(7117):364–368. doi: 10.1038/nature05284. [DOI] [PubMed] [Google Scholar]
- 15.Yang J, Chai L, Fowles TC, Alipio Z, Xu D, Fink LM, Ward DC, Ma Y. Genome-wide analysis reveals Sall4 to be a major regulator of pluripotency in murine-embryonic stem cells. Proc Natl Acad Sci U S A. 2008;105 [DOI] [PMC free article] [PubMed]
- 16.Wu Q, Chen X, Zhang J, Loh YH, Low TY, Zhang W, Sze SK, Lim B, Ng HH. Sall4 interacts with Nanog and co-occupies Nanog genomic sites in embryonic stem cells. J Biol Chem. 2006;281 [DOI] [PubMed]
- 17.Tan MH, KF A, Leong DE, Foygel K, Wong WH, Yao MW. An Oct4-Sall4-Nanog network controls developmental progression in the pre-implantation mouse embryo. Mol Syst Biol. 2013;9:632. doi: 10.1038/msb.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J, Tam WL, Tong GQ, Wu Q, Chan HY, Soh BS, Lou Y, Yang J, Ma Y, Chai L. Sall4 modulates embryonic stem cell pluripotency and early embryonic development by the transcriptional regulation of Pou5f1. Nat Cell Biol. 2006;8 [DOI] [PubMed]
- 19.Yang J, Gao C, Chai L, Ma Y. A novel SALL4/OCT4 transcriptional feedback network for pluripotency of embryonic stem cells. PLoS One. 2010;5(5):e10766. doi: 10.1371/journal.pone.0010766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nosi U, Lanner F, Huang T, Cox B. Overexpression of trophoblast stem cell-enriched MicroRNAs promotes trophoblast fate in embryonic stem cells. Cell Rep. 2017;19(6):1101–1109. doi: 10.1016/j.celrep.2017.04.040. [DOI] [PubMed] [Google Scholar]
- 21.Shu J, Zhang K, Zhang M, Yao A, Shao S, Du F, Yang C, Chen W, Wu C, Yang W, et al. GATA family members as inducers for cellular reprogramming to pluripotency. Cell Res. 2015;25(2):169–180. doi: 10.1038/cr.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buganim Y, Markoulaki S, van Wietmarschen N, Hoke H, Wu T, Ganz K, Akhtar-Zaidi B, He Y, Abraham BJ, Porubsky D, et al. The developmental potential of iPSCs is greatly influenced by reprogramming factor selection. Cell Stem Cell. 2014;15(3):295–309. doi: 10.1016/j.stem.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mansour AA, Gafni O, Weinberger L, Zviran A, Ayyash M, Rais Y, Krupalnik V, Zerbib M, Amann-Zalcenstein D, Maza I, et al. The H3K27 demethylase Utx regulates somatic and germ cell epigenetic reprogramming. Nature. 2012;488(7411):409–413. doi: 10.1038/nature11272. [DOI] [PubMed] [Google Scholar]
- 24.Xu K, Chen X, Yang H, Xu Y, He Y, Wang C, Huang H, Liu B, Liu W, Li J, et al. Maternal Sall4 is indispensable for epigenetic maturation of mouse oocytes. J Biol Chem. 2017;292(5):1798–1807. doi: 10.1074/jbc.M116.767061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakaki-Yumoto M, Kobayashi C, Sato A, Fujimura S, Matsumoto Y, Takasato M, Kodama T, Aburatani H, Asashima M, Yoshida N, et al. The murine homolog of SALL4, a causative gene in Okihiro syndrome, is essential for embryonic stem cell proliferation, and cooperates with Sall1 in anorectal, heart, brain and kidney development. Development. 2006;133(15):3005–3013. doi: 10.1242/dev.02457. [DOI] [PubMed] [Google Scholar]
- 26.Miller A, Gharbi S, Etienne-Dumeau C, Nishinakamura R, Hendrich B: Transcriptional control by Sall4 in blastocysts facilitates lineage commitment of inner cell mass cells. bioRxiv 2017.
- 27.Warren M, Wang W, Spiden S, Chen-Murchie D, Tannahill D, Steel KP, Bradley A. A Sall4 mutant mouse model useful for studying the role of Sall4 in early embryonic development and organogenesis. Genesis. 2007;45(1):51–58. doi: 10.1002/dvg.20264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harvey SA, Logan MP. sall4 acts downstream of tbx5 and is required for pectoral fin outgrowth. Development. 2006;133(6):1165–1173. doi: 10.1242/dev.02259. [DOI] [PubMed] [Google Scholar]
- 29.Koshiba-Takeuchi K, Takeuchi JK, Arruda EP, Kathiriya IS, Mo R, Hui CC, Srivastava D, Bruneau BG. Cooperative and antagonistic interactions between Sall4 and Tbx5 pattern the mouse limb and heart. Nat Genet. 2006;38(2):175–183. doi: 10.1038/ng1707. [DOI] [PubMed] [Google Scholar]
- 30.Lim CY, Tam WL, Zhang J, Ang HS, Jia H, Lipovich L, Ng HH, Wei CL, Sung WK, Robson P. Sall4 regulates distinct transcription circuitries in different blastocyst-derived stem cell lineages. Cell Stem Cell. 2008;3 [DOI] [PubMed]
- 31.Oikawa T, Kamiya A, Kakinuma S, Zeniya M, Nishinakamura R, Tajiri H, Nakauchi H. Sall4 regulates cell fate decision in fetal hepatic stem/progenitor cells. Gastroenterology. 2009;136(3):1000–1011. doi: 10.1053/j.gastro.2008.11.018. [DOI] [PubMed] [Google Scholar]
- 32.Bohm J, Heinritz W, Craig A, Vujic M, Ekman-Joelsson BM, Kohlhase J, Froster U. Functional analysis of the novel TBX5 c.1333delC mutation resulting in an extended TBX5 protein. BMC Med Genet. 2008;9:88. doi: 10.1186/1471-2350-9-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rao S, Zhen S, Roumiantsev S, McDonald LT, Yuan GC, Orkin SH. Differential roles of Sall4 isoforms in embryonic stem cell pluripotency. Mol Cell Biol. 2010;30(22):5364–5380. doi: 10.1128/MCB.00419-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma Y, Cui W, Yang J, Qu J, Di C, Amin HM, Lai R, Ritz J, Krause DS, Chai L. SALL4, a novel oncogene, is constitutively expressed in human acute myeloid leukemia (AML) and induces AML in transgenic mice. Blood. 2006;108 [DOI] [PMC free article] [PubMed]
- 35.Uez N, Lickert H, Kohlhase J, de Angelis MH, Kuhn R, Wurst W, Floss T. Sall4 isoforms act during proximal-distal and anterior-posterior axis formation in the mouse embryo. Genesis. 2008;46(9):463–477. doi: 10.1002/dvg.20421. [DOI] [PubMed] [Google Scholar]
- 36.Gassei K, Orwig KE. SALL4 expression in gonocytes and spermatogonial clones of postnatal mouse testes. PLoS One. 2013;8(1):e53976. doi: 10.1371/journal.pone.0053976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hobbs RM, Fagoonee S, Papa A, Webster K, Altruda F, Nishinakamura R, Chai L, Pandolfi PP. Functional antagonism between Sall4 and Plzf defines germline progenitors. Cell Stem Cell. 2012;10(3):284–298. doi: 10.1016/j.stem.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiong J, Todorova D, NY S, Kim J, Lee PJ, Shen Z, Briggs SP, Xu Y. Stemness factor Sall4 is required for DNA damage response in embryonic stem cells. J Cell Biol. 2015;208(5):513–520. doi: 10.1083/jcb.201408106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nimura K, Ura K, Shiratori H, Ikawa M, Okabe M, Schwartz RJ, Kaneda Y. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to wolf-Hirschhorn syndrome. Nature. 2009;460(7252):287–291. doi: 10.1038/nature08086. [DOI] [PubMed] [Google Scholar]
- 40.Kim JY, Kee HJ, Choe NW, Kim SM, Eom GH, Baek HJ, Kook H, Kook H, Seo SB. Multiple-myeloma-related WHSC1/MMSET isoform RE-IIBP is a histone methyltransferase with transcriptional repression activity. Mol Cell Biol. 2008;28(6):2023–2034. doi: 10.1128/MCB.02130-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu C, Yao X, Zhao L, Wang P, Zhang Q, Zhao C, Yao S, Wei Y. Wolf-Hirschhorn syndrome candidate 1 (whsc1) functions as a tumor suppressor by governing cell differentiation. Neoplasia. 2017;19(8):606–616. doi: 10.1016/j.neo.2017.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aguila JR, Liao W, Yang J, Avila C, Hagag N, Senzel L, Ma Y. SALL4 is a robust stimulator for the expansion of hematopoietic stem cells. Blood. 2011;118(3):576–585. doi: 10.1182/blood-2011-01-333641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liao W, Aguila JR, Yao Y, Yang J, Zieve G, Jiang Y, Avila C, Senzel L, Lai R, Xu D, et al. Enhancing bone marrow regeneration by SALL4 protein. J Hematol Oncol. 2013;6:84. doi: 10.1186/1756-8722-6-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tatetsu H, Wang F, Gao C, Ueno S, Tian X, Armant M, Federation A, Qi J, Bradner JE, Tenen DG, et al. SALL4 is a key factor in HDAC inhibitor mediated ex vivo expansion of human peripheral blood mobilized stem/progenitor CD34+CD90+ cells. Blood. 2014;124(21):1566–6.
- 45.Yang J, Aguila JR, Alipio Z, Lai R, Fink LM, Ma Y. Enhanced self-renewal of hematopoietic stem/progenitor cells mediated by the stem cell gene Sall4. J Hematol Oncol. 2011;4:38. doi: 10.1186/1756-8722-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mossahebi-Mohammadi M, Atashi A, Kaviani S, Soleimani M. Efficient expansion of SALL4-transduced umbilical cord blood derived CD133+hematopoietic stem cells. Acta Med Iran. 2017;55(5):290–296. [PubMed] [Google Scholar]
- 47.Akhavan Rahnama M, Movassaghpour AA, Soleimani M, Atashi A, Anbarlou A, Shams Asenjan K. MicroRNA-15b target Sall4 and diminish in vitro UCB-derived HSCs expansion. EXCLI J. 2015;14:601–610. doi: 10.17179/excli2014-687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao C, Kong NR, Li A, Tatetu H, Ueno S, Yang Y, He J, Yang J, Ma Y, Kao GS. SALL4 is a key transcription regulator in normal human hematopoiesis. Transfusion. 2013;53 [DOI] [PMC free article] [PubMed]
- 49.Yang J, Chai L, Liu F, Fink LM, Lin P, Silberstein LE, Amin HM, Ward DC, Ma Y. Bmi-1 is a target gene for SALL4 in hematopoietic and leukemic cells. Proc Natl Acad Sci U S A. 2007;104(25):10494–10499. doi: 10.1073/pnas.0704001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang L, Liu L, Gao H, Pinnamaneni JP, Sanagasetti D, Singh VP, Wang K, Mathison M, Zhang Q, Chen F, et al. The stem cell factor SALL4 is an essential transcriptional regulator in mixed lineage leukemia-rearranged leukemogenesis. J Hematol Oncol. 2017;10(1):159. doi: 10.1186/s13045-017-0531-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rossi L, Lin KK, Boles NC, Yang L, King KY, Jeong M, Mayle A, Goodell MA. Less is more: unveiling the functional core of hematopoietic stem cells through knockout mice. Cell Stem Cell. 2012;11(3):302–317. doi: 10.1016/j.stem.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Damnernsawad A, Kong G, Wen Z, Liu Y, Rajagopalan A, You X, Wang J, Zhou Y, Ranheim EA, Luo HR, et al. Kras is required for adult hematopoiesis. Stem Cells. 2016;34(7):1859–1871. doi: 10.1002/stem.2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang F, Guo Y, Chen Q, Yang Z, Ning N, Zhang Y, Xu Y, Xu X, Tong C, Chai L, et al. Stem cell factor SALL4, a potential prognostic marker for myelodysplastic syndromes. J Hematol Oncol. 2013;6(1):73. doi: 10.1186/1756-8722-6-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang F, Gao C, Lu J, Tatetsu H, Williams DA, Muller LU, Cui W, Chai L. Leukemic survival factor SALL4 contributes to defective DNA damage repair. Oncogene. 2016;35(47):6087–6095. doi: 10.1038/onc.2016.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Milanovich S, Peterson J, Allred J, Stelloh C, Rajasekaran K, Fisher J, Duncan SA, Malarkannan S, Rao S: Sall4 overexpression blocks murine hematopoiesis in a dose-dependent manner. Experimental hematology 2015, 43(1):53–64 e51–58. [DOI] [PMC free article] [PubMed]
- 56.Wang P, Zhang JD, Wu F, Ye X, Sharon D, Hitt M, McMullen TP, Hegazy SA, Gelebart P, Yang J, et al. The expression and oncogenic effects of the embryonic stem cell marker SALL4 in ALK-positive anaplastic large cell lymphoma. Cell Signal. 2012;24(10):1955–1963. doi: 10.1016/j.cellsig.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 57.Ueno S, Lu J, He J, Li A, Zhang X, Ritz J, Silberstein LE, Chai L. Aberrant expression of SALL4 in acute B cell lymphoblastic leukemia: mechanism, function, and implication for a potential novel therapeutic target. Exp Hematol. 2014;42(4):307–316. doi: 10.1016/j.exphem.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cui W, Kong NR, Ma Y, Amin HM, Lai R, Chai L. Differential expression of the novel oncogene, SALL4, in lymphoma, plasma cell myeloma, and acute lymphoblastic leukemia. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2006;19(12):1585–1592. doi: 10.1038/modpathol.3800694. [DOI] [PubMed] [Google Scholar]
- 59.Lu J, Ma Y, Kong N, Alipio Z, Gao C, Krause DS, Silberstein LE, Chai L. Dissecting the role of SALL4, a newly identified stem cell factor, in chronic myelogenous leukemia. Leukemia. 2011;25(7):1211–1213. doi: 10.1038/leu.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shen Q, Liu S, Hu J, Chen S, Yang L, Li B, Wu X, Ma Y, Yang J, Ma Y, et al. The differential expression pattern of the BMI-1, SALL4 and ABCA3 genes in myeloid leukemia. Cancer Cell Int. 2012;12(1):42. doi: 10.1186/1475-2867-12-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hupfeld T, Chapuy B, Schrader V, Beutler M, Veltkamp C, Koch R, Cameron S, Aung T, Haase D, Larosee P, et al. Tyrosinekinase inhibition facilitates cooperation of transcription factor SALL4 and ABC transporter A3 towards intrinsic CML cell drug resistance. Br J Haematol. 2013;161(2):204–213. doi: 10.1111/bjh.12246. [DOI] [PubMed] [Google Scholar]
- 62.Liu L, Liu L, Leung E, Cooney AJ, Chen C, Rosengart TK, Ma Y, Yang J. Knockdown of SALL4 enhances all-trans retinoic acid-induced cellular differentiation in acute myeloid leukemia cells. J Biol Chem. 2015; [DOI] [PMC free article] [PubMed]
- 63.Yang F, Yao Y, Jiang Y, Lu L, Ma Y, Dai W. Sumoylation is important for stability, subcellular localization, and transcriptional activity of SALL4, an essential stem cell transcription factor. J Biol Chem. 2012;287(46):38600–38608. doi: 10.1074/jbc.M112.391441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gao C, Dimitrov T, Yong KJ, Tatetsu H, Jeong HW, Luo HR, Bradner JE, Tenen DG, Chai L. Targeting transcription factor SALL4 in acute myeloid leukemia by interrupting its interaction with an epigenetic complex. Blood. 2013;121 [DOI] [PMC free article] [PubMed]
- 65.Schenk T, Chen WC, Gollner S, Howell L, Jin L, Hebestreit K, Klein HU, Popescu AC, Burnett A, Mills K, et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med. 2012;18(4):605–611. doi: 10.1038/nm.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jeong HW, Cui W, Yang Y, Lu J, He J, Li A, Song D, Guo Y, Liu BH, Chai L. SALL4, a stem cell factor, affects the side population by regulation of the ATP-binding cassette drug transport genes. PLoS One. 2011;6 [DOI] [PMC free article] [PubMed]
- 67.Aguila JR, Liao W, Yang J, Avila C, Hagag N, Senzel L, Ma Y. SALL4 is a robust stimulator for the expansion of hematopoietic stem cells. Blood. 2011;118 [DOI] [PMC free article] [PubMed]
- 68.Yang J, Aguila JR, Alipio Z, Lai R, Fink LM, Ma Y. Enhanced self-renewal of hematopoietic stem/progenitor cells mediated by the stem cell gene Sall4. J Hematol Oncol. 2011;4 [DOI] [PMC free article] [PubMed]
- 69.Prange KHM, Mandoli A, Kuznetsova T, Wang SY, Sotoca AM, Marneth AE, van der Reijden BA, Stunnenberg HG, Martens JHA. MLL-AF9 and MLL-AF4 oncofusion proteins bind a distinct enhancer repertoire and target the RUNX1 program in 11q23 acute myeloid leukemia. Oncogene. 2017;36(23):3346–3356. doi: 10.1038/onc.2016.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Winters AC, Bernt KM. MLL-rearranged Leukemias-an update on science and clinical approaches. Front Pediatr. 2017;5:4. doi: 10.3389/fped.2017.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhu N, Chen M, Eng R, DeJong J, Sinha AU, Rahnamay NF, Koche R, Al-Shahrour F, Minehart JC, Chen CW, et al. MLL-AF9- and HOXA9-mediated acute myeloid leukemia stem cell self-renewal requires JMJD1C. J Clin Invest. 2016;126(3):997–1011. doi: 10.1172/JCI82978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marschalek R. MLL leukemia and future treatment strategies. Arch Pharm (Weinheim) 2015;348(4):221–228. doi: 10.1002/ardp.201400449. [DOI] [PubMed] [Google Scholar]
- 73.de Boer J, Walf-Vorderwulbecke V, Williams O. Focus: MLL-rearranged leukemia. Leukemia. 2013;27(6):1224–1228. doi: 10.1038/leu.2013.78. [DOI] [PubMed] [Google Scholar]
- 74.Li A, Yang Y, Gao C, Lu J, Jeong HW, Liu BH, Tang P, Yao X, Neuberg D, Huang G, et al. A SALL4/MLL/HOXA9 pathway in murine and human myeloid leukemogenesis. J Clin Invest. 2013;123(10):4195–4207. doi: 10.1172/JCI62891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang J, Chai L, Gao C, Fowles TC, Alipio Z, Dang H, Xu D, Fink LM, Ward DC, Ma Y. SALL4 is a key regulator of survival and apoptosis in human leukemic cells. Blood. 2008;112 [DOI] [PMC free article] [PubMed]
- 76.Zhou W, Zou B, Liu L, Cui K, Gao J, Yuan S, Cong N. MicroRNA-98 acts as a tumor suppressor in hepatocellular carcinoma via targeting SALL4. Oncotarget. 2016;7(45):74059–74073. doi: 10.18632/oncotarget.12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gao C, Kong NR, Chai L. The role of stem cell factor SALL4 in leukemogenesis. Crit Rev Oncog. 2011;16(1–2):117–127. doi: 10.1615/CritRevOncog.v16.i1-2.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li A, Jiao Y, Yong KJ, Wang F, Gao C, Yan B, Srivastava S, Lim GS, Tang P, Yang H, et al. SALL4 is a new target in endometrial cancer. Oncogene. 2015;34(1):63–72. doi: 10.1038/onc.2013.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu L, Souto J, Liao W, Jiang Y, Li Y, Nishinakamura R, Huang S, Rosengart T, Yang VW, Schuster M. Histone lysine-specific demethylase 1 (LSD1) protein is involved in Sal-like protein 4 (SALL4)-mediated transcriptional repression in hematopoietic stem cells. J Biol Chem. 2013;288 [DOI] [PMC free article] [PubMed]
- 80.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434(7035):843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 81.Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, Zon LI, Armstrong SA. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010;327(5973):1650–1653. doi: 10.1126/science.1186624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Heidel FH, Mar BG, Armstrong SA. Self-renewal related signaling in myeloid leukemia stem cells. Int J Hematol. 2011;94(2):109–117. doi: 10.1007/s12185-011-0901-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sato A, Kishida S, Tanaka T, Kikuchi A, Kodama T, Asashima M, Nishinakamura R. Sall1, a causative gene for Townes-brocks syndrome, enhances the canonical Wnt signaling by localizing to heterochromatin. Biochem Biophys Res Commun. 2004;319(1):103–113. doi: 10.1016/j.bbrc.2004.04.156. [DOI] [PubMed] [Google Scholar]
- 84.Yin T, Li L. The stem cell niches in bone. J Clin Invest. 2006;116(5):1195–1201. doi: 10.1172/JCI28568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Levesque JP, Helwani FM, Winkler IG. The endosteal 'osteoblastic' niche and its role in hematopoietic stem cell homing and mobilization. Leukemia. 2010;24(12):1979–1992. doi: 10.1038/leu.2010.214. [DOI] [PubMed] [Google Scholar]
- 86.Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, Khiabanian H, Lee A, Murty VV, Friedman R, et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature. 2014;506(7487):240–244. doi: 10.1038/nature12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weber JM, Calvi LM. Notch signaling and the bone marrow hematopoietic stem cell niche. Bone. 2010;46(2):281–285. doi: 10.1016/j.bone.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pajcini KV, Speck NA, Pear WS. Notch signaling in mammalian hematopoietic stem cells. Leukemia. 2011;25(10):1525–1532. doi: 10.1038/leu.2011.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang W, Xia X, Reisenauer MR, Rieg T, Lang F, Kuhl D, Vallon V, Kone BC. Aldosterone-induced Sgk1 relieves Dot1a-Af9-mediated transcriptional repression of epithelial Na+ channel alpha. J Clin Invest. 2007;117 [DOI] [PMC free article] [PubMed]
- 90.Lin Y, Liu AY, Fan C, Zheng H, Li Y, Zhang C, Wu S, Yu D, Huang Z, Liu F, et al. MicroRNA-33b inhibits breast cancer metastasis by targeting HMGA2, SALL4 and Twist1. Sci Rep. 2015;5:9995. doi: 10.1038/srep09995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Melton C, Judson RL, Blelloch R. Opposing microRNA families regulate self-renewal in mouse embryonic stem cells. Nature. 2010;463(7281):621–626. doi: 10.1038/nature08725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ma JC, Qian J, Lin J, Qian W, Yang J, Wang CZ, Chai HY, Li Y, Chen Q, Qian Z. Aberrant hypomethylation of SALL4 gene is associated with intermediate and poor karyotypes in acute myeloid leukemia. Clin Biochem. 2013;46(4–5):304–307. doi: 10.1016/j.clinbiochem.2012.11.018. [DOI] [PubMed] [Google Scholar]
- 93.Lin J, Qian J, Yao DM, Qian W, Yang J, Wang CZ, Chai HY, Ma JC, Deng ZQ, Li Y, et al. Aberrant hypomethylation of SALL4 gene in patients with myelodysplastic syndrome. Leuk Res. 2013;37(1):71–75. doi: 10.1016/j.leukres.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 94.Sheikhrezaei Z, Heydari P, Farsinezhad A, Fatemi A, Khanamani Falahati-pour S, Darakhshan S, Noroozi Karimabad M, Darekordi A, Khorramdelazad H, Hassanshahi G. A new indole derivative decreased SALL4 gene expression in acute Promyelocytic leukemia cell line (NB4). Iran Biomed J. 2017; [DOI] [PMC free article] [PubMed]
- 95.Yang J, Corsello TR, Ma Y. Stem cell gene SALL4 suppresses transcription through recruitment of DNA methyltransferases. J Biol Chem. 2012;287(3):1996–2005. doi: 10.1074/jbc.M111.308734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lu J, Jeong HW, Kong N, Yang Y, Carroll J, Luo HR, Silberstein LE, Yupoma, Chai L. Stem cell factor SALL4 represses the transcriptions of PTEN and SALL1 through an epigenetic repressor complex. PLoS One. 2009;4(5):e5577. doi: 10.1371/journal.pone.0005577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, Roeder RG. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13(8):713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- 98.Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011;25(13):1345–1358. doi: 10.1101/gad.2057811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Campos-Sanchez E, Deleyto-Seldas N, Dominguez V, Carrillo-de-Santa-Pau E, Ura K, Rocha PP, Kim J, Aljoufi A, Esteve-Codina A, Dabad M, et al. Wolf-Hirschhorn syndrome candidate 1 is necessary for correct hematopoietic and B cell development. Cell Rep. 2017;19(8):1586–1601. doi: 10.1016/j.celrep.2017.04.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rice KL, Hormaeche I, Licht JD. Epigenetic regulation of normal and malignant hematopoiesis. Oncogene. 2007;26(47):6697–6714. doi: 10.1038/sj.onc.1210755. [DOI] [PubMed] [Google Scholar]
- 101.Goyama S, Kitamura T. Epigenetics in normal and malignant hematopoiesis: an overview and update 2017. Cancer Sci. 2017;108(4):553–562. doi: 10.1111/cas.13168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ding LW, Sun QY, Tan KT, Chien W, Mayakonda A, Yeoh AEJ, Kawamata N, Nagata Y, Xiao JF, Loh XY, et al. Mutational landscape of pediatric acute lymphoblastic leukemia. Cancer Res. 2017;77(2):390–400. doi: 10.1158/0008-5472.CAN-16-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Feng Z, Yao Y, Zhou C, Chen F, Wu F, Wei L, Liu W, Dong S, Redell M, Mo Q, et al. Pharmacological inhibition of LSD1 for the treatment of MLL-rearranged leukemia. J Hematol Oncol. 2016;9:24. doi: 10.1186/s13045-016-0252-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wouters BJ, Delwel R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood. 2016;127(1):42–52. doi: 10.1182/blood-2015-07-604512. [DOI] [PubMed] [Google Scholar]
- 105.Gallipoli P, Giotopoulos G, Huntly BJ. Epigenetic regulators as promising therapeutic targets in acute myeloid leukemia. Ther Adv Hematol. 2015;6(3):103–119. doi: 10.1177/2040620715577614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bernt KM, Armstrong SA. Targeting epigenetic programs in MLL-rearranged leukemias. Hematology / the Education Program of the American Society of Hematology American Society of Hematology Education Program. 2011;2011:354–360. doi: 10.1182/asheducation-2011.1.354. [DOI] [PubMed] [Google Scholar]
- 107.Saygin C, Carraway HE. Emerging therapies for acute myeloid leukemia. J Hematol Oncol. 2017;10(1):93. doi: 10.1186/s13045-017-0463-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Song Y, Wu F, Wu J. Targeting histone methylation for cancer therapy: enzymes, inhibitors, biological activity and perspectives. J Hematol Oncol. 2016;9(1):49. doi: 10.1186/s13045-016-0279-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Trowbridge JJ, Sinha AU, Zhu N, Li M, Armstrong SA, Orkin SH. Haploinsufficiency of Dnmt1 impairs leukemia stem cell function through derepression of bivalent chromatin domains. Genes Dev. 2012;26(4):344–349. doi: 10.1101/gad.184341.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kuntimaddi A, Achille NJ, Thorpe J, Lokken AA, Singh R, Hemenway CS, Adli M, Zeleznik-Le NJ, Bushweller JH. Degree of recruitment of DOT1L to MLL-AF9 defines level of H3K79 di- and tri-methylation on target genes and transformation potential. Cell Rep. 2015;11(5):808–820. doi: 10.1016/j.celrep.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y, Ciceri F, Blaser JG, Greystoke BF, Jordan AM, et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 2012;21(4):473–487. doi: 10.1016/j.ccr.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 112.Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, Su H, Sun W, Chang H, Xu G, et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet. 2009;41(1):125–129. doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- 113.Thurn KT, Thomas S, Moore A, Munster PN. Rational therapeutic combinations with histone deacetylase inhibitors for the treatment of cancer. Future Oncol. 2011;7(2):263–283. doi: 10.2217/fon.11.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kerenyi MA, Shao Z, Hsu YJ, Guo G, Luc S, O'Brien K, Fujiwara Y, Peng C, Nguyen M, Orkin SH. Histone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturation. elife. 2013;2:e00633. doi: 10.7554/eLife.00633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Paik EJ, Mahony S, White RM, Price EN, Dibiase A, Dorjsuren B, Mosimann C, Davidson AJ, Gifford D, Zon LI. A Cdx4-Sall4 regulatory module controls the transition from mesoderm formation to embryonic hematopoiesis. Stem Cell Reports. 2013;1(5):425–436. doi: 10.1016/j.stemcr.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S, Jackson SP. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010;17(9):1144–1151. doi: 10.1038/nsmb.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20(1):66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bernt KM, Armstrong SA. A role for DOT1L in MLL-rearranged leukemias. Epigenomics. 2011;3(6):667–670. doi: 10.2217/epi.11.98. [DOI] [PubMed] [Google Scholar]
- 119.Chen CW, Armstrong SA. Targeting DOT1L and HOX gene expression in MLL-rearranged leukemia and beyond. Exp Hematol. 2015;43(8):673–684. doi: 10.1016/j.exphem.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated in the study are included in the present article.