

Abstract
Objective
To test the hypothesis that degeneration of the substantia nigra pars compacta (SNc) precedes that of the cholinergic basal forebrain (BF) in Parkinson disease (PD) using new multispectral structural magnetic resonance (MR) imaging tools to measure the volumes of the SNc and BF.
Design
Matched case-control study.
Setting
The Athinoula A. Martinos Imaging Center at the McGovern Institute for Brain Research, Massachusetts Institute of Technology (MIT), and the Massachusetts General Hospital/MIT Morris Udall Center of Excellence in Parkinson Disease Research.
Patients
Participants included 29 patients with PD (Hoehn and Yahr [H&Y] stages 1–3) and 27 matched healthy control subjects.
Main Outcome Measures
We acquired multiecho T1-weighted, multiecho proton density, T2-weighted, and T2-weighted fluid-attenuated inversion recovery (FLAIR) sequences from each participant. For the SNc, we created a weighted mean of the multiple echoes, yielding a single volume with a high ratio of contrast to noise. We visualized the BF using T2-weighted FLAIR images. For each participant, we manually labeled the 2 structures and calculated their volumes.
Results
Relative to the controls, 13 patients with H&Y stage 1 PD had significantly decreased SNc volumes. Sixteen patients with H&Y stage 2 or 3 PD showed little additional volume loss. In contrast, the BF volume loss occurred later in the disease, with a significant decrease apparent in patients having H&Y stage 2 or 3 PD compared with the controls and the patients having H&Y stage 1 PD. The latter group did not differ significantly from the controls.
Conclusion
Our results support the proposed neuropathological trajectory in PD and establish novel multispectral methods as MR imaging biomarkers for tracking the degeneration of the SNc and BF.
PARKINSON DISEASE (PD), A NEUrodegenerative disorder that causes motor, cognitive, and psychiatric symptoms, is typically characterized by a loss of nigrostriatal dopaminergic neurons in the substantia nigra pars compacta (SNc), accompanied by the aggregation of Lewy bodies throughout the brainstem.1,2 While denervation of dopaminergic nigrostriatal projections may explain the cardinal motor symptoms of PD, as evidenced by the dramatic motor improvement associated with dopamine therapy,3 abnormalities beyond the SNc4,5 may underlie the serious and potentially debilitating nonmotor features, including cognitive and memory impairments and progression to dementia.6–10 Notably, the cholinergic basal forebrain (BF) also degenerates in PD11,12 and could contribute to nonmotor deficits.
A prominent hypothesis regarding the neuropathological progression of PD posits that Lewy body deposition commences in the enteric and peripheral nervous systems, before appearing in the brainstem, and then progresses rostrally to the midbrain, forebrain, and neocortex.13 Motor symptom onset is believed to become clinically significant between Braak stages 3 and 4. Although this staging scheme is based on careful neuropathological examination of a large sample of postmortem specimens, definitive confirmation of the hypothesis is lacking, with some researchers questioning the general usefulness of the Braak classification scheme.14,15 More direct measures of the timing and progression of neuronal de- of generation in these areas would help to resolve this debate. In particular, according to this staging scheme, pathological changes in the SNc should precede the degeneration of the more rostral BF.13,16
Indirect support for this hypothesis comes from positron emission tomography studies17–19 that document altered cholinergic neurotransmission in patients with mild PD. Still, an in vivo comparison of the morphologic structure of the SNc and BF in the early stages of PD is lacking because reliable magnetic resonance (MR) imaging– based biomarkers have been unavailable for visualizing these structures.20 Although a few methods for imaging the SNc and BF have met with some success,21–24 morphologic structure was examined only in separate study groups using data collected at different times. The present study tested the hypothesis that the degeneration in the SNc precedes that in the BF. We used new multi-spectral structural MR imaging sequences (T2-weighted, T2-weighted fluid-attenuated inversion recovery [FLAIR], multiecho T1-weighted, and multiecho proton density) to visualize and measure disease-related changes in these structures in a single sample of patients with PD and control subjects. These images provide a valuable window on the subcortical structures that have been implicated in PD but are not readily visible on conventional MR imaging.
METHODS
PARTICIPANTS
We enrolled 29 patients (11 female; mean [SD] age, 65.3 [8.8] years) with idiopathic PD (Hoehn and Yahr [H&Y] stages 1–3) and 27 matched healthy control subjects (11 female; mean [SD] age, 63.7 [7.2] years). Their mean (SD) years of education were 17.1 (2.0) among the patients and 17.8 (2.4) among the controls. The mean (SD) Mini-Mental State Examinations cores were 28.0 (1.6) among the patients and 28.6 (1.7) among the controls.
In addition, a complete set of multispectral data was collected from a “training sample” of healthy adults, including 5 younger participants (1 male and 4 female; mean [SD] age, 21.4 [3.8] years) and 6 older participants (3 male and 3 female; mean [SD] age, 64.3 [12.2] years). We used these training data to establish the MR imaging analysis protocols and anatomical definitions that we later applied to our study samples. All patients with PD were referred by neurologists in the Massachusetts General Hospital/MIT [Massachusetts Institute of Technology] Morris Udall Center of Excellence in Parkinson Disease Research. Neurologists established the diagnosis of idiopathic PD by clinical examination according to research diagnostic criteria.25 We obtained Unified Parkinson Disease Rating Scale (UPDRS) motor scores for 25 patients with PD; UPDRS scores were obtained when patients were optimally medicated (ie, in the on state); all but 2 patients with PD were being treated with anti-PD drugs at the time of the study. The patients and controls were between the ages of 50 and 85 years, had completed at least 12 years of schooling, and gave informed consent to participate in the study. Institutional review boards at the Massachusetts Institute of Technology and the Massachusetts General Hospital approved the project. Exclusion criteria were parkinsonian syndromes secondary to severe depression; reserpine or neuroleptic administration; progressive supranuclear palsy, multisystem atrophy, Lewy body disease, and the rigid form of Huntington disease; a Mini-Mental State Examination score below 26; a Beck Depression Inventory score above 18; history of a neurological disorder other than PD, cancer, or serious chronic underlying medical illness (such as serious cardiac disease); un treated hypertension; history of a psychiatric disorder; and any contraindication for MR imaging (claustrophobia, mechanical or electromagnetic implants, ferromagnetic or nonstatic metal implants, or tattoos with metal ink).
MR IMAGING DATA ACQUISITION
Magnetic resonance imaging data were acquired using a 3-T MR imaging system (Trio; Siemens) with a 12-channel matrix head coil. For each patient with PD and control subject, we collected a series of high-resolution (1-mm isotropic) multispectral data that included multiecho magnetization-prepared rapid gradient-echo with T1-weighting (eFigure 1A; http://www.jamaneuro.com), 3-dimensional (3D) T2-weighted turbo spin-echo (eFigure 1B), multiecho fast low-angle shot with proton density weighting (eFigure 1C), and 3D T2-weighted FLAIR turbo spin-echo (eFigure 1D) sequences. Each acquisition consisted of a 3D slab with 176 sagittal sections (1.0 mm thick). In-plane field of view was 256 mm sampled on a 256 × 256– pixel matrix, giving an in-plane resolution of 1.0 × 1.0 mm. For proton density weighting, the flip angle was 3°, and the repetition time (TR) was 20 milliseconds, during which 6 echoes were collected after a nonselective excitation. Echo times (TEs) were evenly spaced at 1.85, 3.85. 5.85, 7.85, 9.85, and 11.85 milliseconds. A proton density volume was generated from a weighted linear mean of acquisitions. For T1-weighting, we used a flip angle of 7°, TR of 2530 milliseconds, and inversion time of 1100 milliseconds, during which 4 echoes were obtained after nonselective excitation, with TEs of 1.61, 3.39, 5.17, and 6.95 milliseconds. A single T1-weighted volume was generated from a root mean square average of acquisitions. For T2-FLAIR weighting, we collected a 3D slab consisting of 176 sagittal sections (1.0 mm thick). The in-plane field of view was 256 mm sampled on a 256 × 256–pixel matrix, giving an in-plane resolution of 1.0 × 1.0 mm, with a TR of 3200 milliseconds and a TE of 444 milliseconds. For T2-FLAIR weighting, we obtained a 3D slab consisting of 176 sagittal sections (1.0 mm thick). The in-plane field of view was 256 mm sampled on a 256 × 256–pixel matrix, giving an in-plane resolution of 1.0 × 1.0 mm, with a TR of 6000 milliseconds, a TE of 494 milliseconds, and an inversion time of 2100 milliseconds. All sequences were matched at a bandwidth of 698 Hz per pixel. To minimize head motion artifact and fatigue, we used parallel acquisition (generalized autocalibrating partially parallel acquisitions) techniques and acquired each sequence twice for each participant during a single imaging session.
MULTISPECTRAL VISUALIZATION OF THE SNc AND BF
To differentiate the boundaries of the SNc from surrounding structures, we created a weighted mean of the 4 contrasts to obtain a single volume with an optimized ratio of contrast to noise for the SNc. Using the MR imaging data from our training sample, we obtained excellent contrast for the SNc, with coefficients for T1-weighting of 11, for T2-weighting of 25, for proton density weighting of 94, and for T2-FLAIR weighting of 19 (Figure 1A and B). To generate this multispectral weighted mean for each participant, we first performed motion correction and averaging of multiple acquisitions for each multispectral sequence. We then coregistered the 4 averaged, motion-corrected multispectral volumes for each participant using a linear rigid-body transformation with trilinear interpolation and then generated a weighted mean of the multi-spectral images using the optimal weighting coefficients. Morphometric details of the SNc were not detectable in our high-resolution T1-weighted images (eFigure 1A), but this structure was clearly apparent in the mean multispectral volume (Figure 1A and B, red arrows), emphasizing the contribution of the proton density weighting and T2-weighted data. The SNc was easily distinguished from the surrounding red nucleus, substantia nigra pars reticulata, and cerebral peduncles (Figure 1C).
Figure 1.
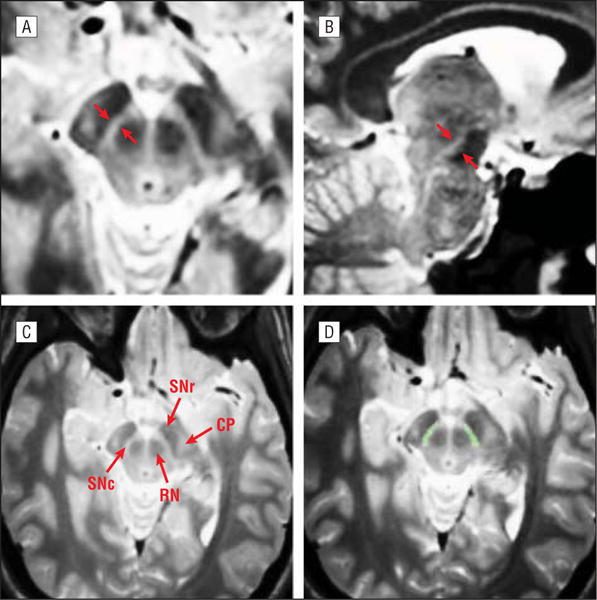
Control subject. A and B, Representative axial (A) and sagittal (B) views of the midbrain of a control subject’s multispectral weighted mean. The red arrows in A and B indicate the substantia nigra. C, Shown are the substantia nigra pars compacta (SNc), substantia nigra pars reticulata (SNr), red nucleus (RN), and cerebral peduncles (CP). D, Example of the same control subject’s manually delimited label of the SNc (green).
Blinded to diagnostic group, we next manually labeled the SNc in all PD and control brains (Figure 1D). The anatomical boundaries of the SNc were defined in consultation with 2 expert neuroanatomists (Thomas Kemper, MD, and John Hedreen, MD) using data from our training sample. The SNc was initially identified in axial sections through the midbrain at a plane where the red nucleus and cerebral peduncle were clearly apparent. The most superficial extent of the red nucleus was then identified, and this section was used as the starting point for each label. We labeled the SNc in axial sections, moving to the most inferior extent of the red nucleus. Labels were checked for accuracy in the sagittal and coronal planes.
Although previous investigations typically used T2-weighted images to visualize the BF,26 we found that the contrast obtained with our T2-FLAIR weighted images (Figure 2B) far exceeded that obtained with the T2-weighted images (Figure 2A). In particular, the inferior boundaries of the BF were more clearly defined in the T2-FLAIR weighted images because this contrast negated the hyperintensity typically associated with the cerebrospinal fluid. Furthermore, no additional boost in contrast was afforded by averaging the T2-FLAIR weighted images with any other multispectral data. Therefore, we manually labeled the BF on motion-corrected, averaged, and intensity-normalized T2-FLAIR weighted images. We used a labeling convention similar to that described by Muth et al,26 whereby the center of the label was identified as the coronal section in which the anterior commissure was most prominent. The BF was apparent as a narrow band just inferior to the globus pallidus and superior to the cerebrospinal fluid (Figure 2C). We traced the entire band in this section and in 2 others, one anterior to the initial section and one posterior to it.
Figure 2.
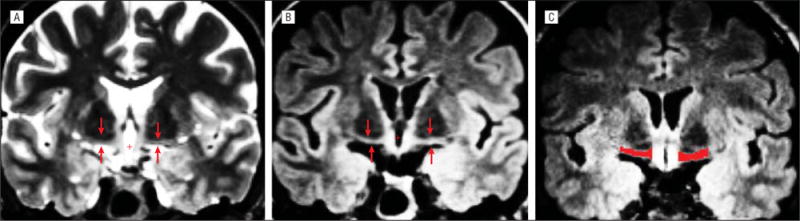
Coronal images from a control subject showing the basal forebrain at the level of the anterior commissure. A, T2-weighted image. B, T2–fluid-attenuated inversion recovery (FLAIR) weighted image clearly demonstrates the improved contrast for both the superior and inferior boundaries in T2-FLAIR weighted images. The red arrows in A and B indicate the basal forebrain. C, T2-FLAIR weighted image with a manually delimited label of the basal forebrain (red).
STATISTICAL ANALYSIS
To ensure that the patient and control groups were well matched, we calculated independent t tests to detect any group differences in age, years of education, and Mini-Mental State Examination scores. A χ2 test for independence tested for differences in the sex distribution. A multivariate general linear model tested for volumetric differences between the patients with PD and the controls. The dependent variables were the left and right hemisphere volumes of the SNc and BF. To uncover any dependence of the effects on disease stage, we divided participants into the following 3 groups: controls, patients with H&Y stage 1 PD, and patients with H&Y stages 2 and 3 PD (we combined stages 2 and 3 because of the few patients with stage 3 PD). All analyses included age, sex, years of education, and intracranial volume as covariates to preclude any confounding effects of minor differences in these measures. The estimate of intracranial volume was derived using a standard process implemented in the morphological processing stream (FreeSurfer; http://surfer.nmr.mgh.harvard.edu).27 To test for covariance in the degeneration of the SNc and BF, we calculated partial correlations, controlling for intracranial volume, between the left SNc and left BF and between the right SNc and right BF.
RESULTS
The patients with PD and the controls did not differ significantly in sex distribution (P=.83), age (P=.48), years of education (P=.26), or Mini-Mental State Examination scores (P=.23). Among the 13 patients with H&Y stage 1 PD, the mean (SD) UPDRS score was 9.2 (5.4), and the mean (SD) disease duration was 3.1 (1.4) years. Among the 16 patients with H&Y stage 2 or 3 PD, the mean (SD) UPDRS score was 19.7 (8.3), and the mean (SD) disease duration was 4.8 (2.9) years. The UPDRS motor scores for patients with H&Y stage 2 or 3 PD (mean [SD], 19.7 [8.3]) were significantly higher than those for patients with H&Y stage 1 PD (mean [SD], 9.2 [5.4]), confirming the expected increase in motor symptom severity (P=.002). The disease duration was longer in patients with H&Y stage 2 or 3 PD than in patients with H&Y stage 1 PD, although the difference did not reach statistical significance (P=.06).
A multivariate general linear model revealed a significant main effect of group for the volumes of the left and right SNc and BF (F=3.5, P=.001) (Figure 3). Post hoc tests confirmed that volumes of the SNc were smaller on the left in the patients with H&Y stage 1 PD compared with the controls (P=.001); this difference did not reach statistical significance on the right (P=.08). The patients with H&Y stage 2 or 3 PD also showed significantly smaller volumes of the SNc compared with the controls on the left (P .001) and on the right (P=.02). The SNc volume did not differ significantly between the patients with H&Y stage 1 PD and the patients with H&Y stage 2 or 3 PD (P=.92 for the left and P=.75 for the right). A comparison of multispectral means from a 69-year-old woman with H&Y stage 2PDandamatchedcontrolsubjectdemonstratedtheclearly visible signal loss in the SNc of the patient (Figure 4). An additional analysis of the SNc volume loss as a function of the side of motor symptom onset is available in the eAppendix and eFigure 2.
Figure 3.
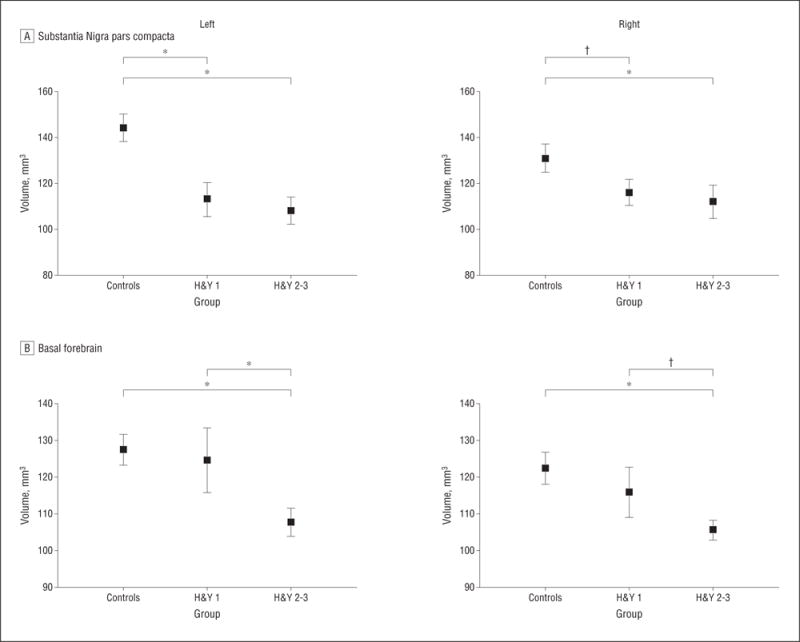
The mean volumes of the substantia nigra pars compacta (A) and basal forebrain (B) for the left and right hemispheres. Shown are values for control subjects, patients with Hoehn and Yahr stage 1 Parkinson disease (H&Y 1), and patients with Hoehn and Yahr stages 2 and 3 Parkinson disease (H&Y 2–3). Bars are means (SEs). *Denotes P ≤ .05; † denotes P ≤ .10.
Figure 4.
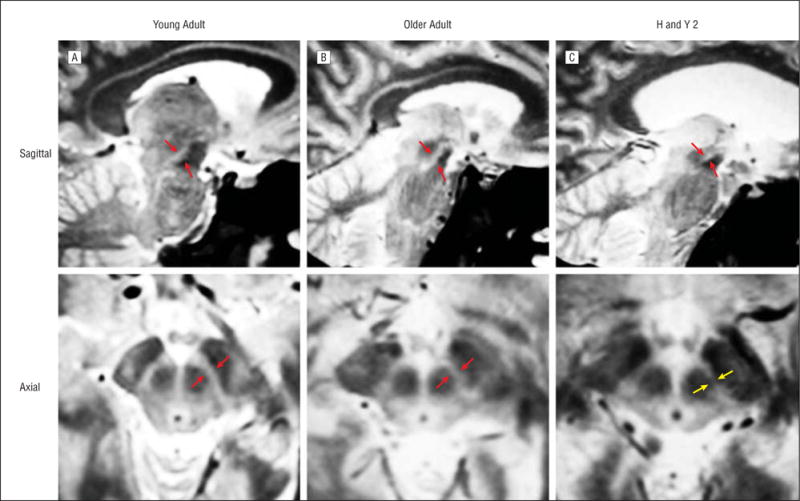
Multispectral weighted means showing the substantia nigra pars compacta (red arrows) in sagittal and axial views. A, Healthy young adult (21-year-old female). B, Healthy older control subject (69-year-old female). C, Age-matched and sex-matched patient with Hoehn and Yahr stage 2 Parkinson disease (H&Y 2) (69-year-old female). Signal loss is readily apparent in the Parkinson disease brain (yellow arrows).
In contrast to the results for the SNc, the BF volumes for the patients with H&Y stage 1 PD did not differ significantly from the controls on the left (P=.84) or on the right (P=.67). Consistent with the SNc results, the patients with H&Y stage 2 or 3 PD showed significantly reduced volumes bilaterally compared with the controls (P=.008 for the left and P=.01 for the right). In addition, relative to the patients with H&Y stage 1 PD, the patients with H&Y stage 2 or 3 PD had significantly reduced volumes of the BF on the left (P=.04), but the difference did not reach statistical significance on the right (P=.08).
COMMENT
The present study used new multispectral structural MR imaging tools to uncover a volumetric reduction of the SNc in the earliest stages of PD, followed by decreased BF volumes in later stages. By showing that the volume loss in the SNc precedes the degeneration in the BF, our results provide in vivo evidence in support of the staging scheme by Braak et al,13 adding credence to their proposed rostral-to-caudal progression of pathological change. These results underscore the findings of previous neuropathological studies in idiopathic PD showing the loss of cholinergic neurons in the BF12 and the degeneration of the SNc.1 The present article complements postmortem studies by establishing the temporal progression of the degeneration of these structures in living patients. Furthermore, the finding of decreased SNc volumes in the patients with H&Y stage 1 PD, with little additional volume loss in H&Y stages 2 and 3 PD, supports the view that cell loss in this area appears early in the disease course, with substantial SNc cell loss likely occurring before symptom onset.28 In contrast, the BF volume loss occurred later in the disease, with a significant decrease in volume in the patients with H&Y stage 2 or 3 PD relative to the controlsandthepatientswithH&Ystage1PD,whodidnot differ significantly from the controls.
DISEASE-RELATED CHANGES IN THE SNc
The present finding of significantly reduced SNc volumes in patients with H&Y stage 1 PD is consistent with the known neuropathological trajectory. Postmortem studies1,2,29 have established that PD is associated with a significant loss of dopaminergic neurons in the SNc, and dopamine therapy remains the gold standard treatment for motor symptoms.3 Furthermore, neuropathological studies2,30 indicate that a large percentage of dopaminergic neurons in the SNc is lost before symptom onset or formal diagnosis.
Because the borders of the SNc are almost impossible to visualize on conventional T1-weighted MR imaging, many previous researchers have attempted to use T2-weighted or proton density–weighted MR imaging to document nigral degeneration in PD. While some studies31–33 reported signal loss or reduced size of the SNc in the patients with PD compared with the controls, other studies34,35 failed to find disease-related changes. Newer methods, such as the use of MR sequences sensitive to neuromelanin36 and segmented inversion-recovery ratio imaging,37 have achieved greater success in documenting disease-related changes in the SNc. However, to our knowledge, the present study represents the first report of the SNc volume loss in patients with H&Y stage 1 PD.
BF DEGENERATION IN PD
Our T2-weighted FLAIR images provided superior contrast for the BF compared with that achieved in previous MR imaging investigations, enabling us to examine the morphologic structure in the same set of patients with PD and controls. Previous evidence of cholinergic degeneration in PD came directly from pathological studies12,38–40 and indirectly from positron emission tomography measurement of cholinergic markers.17,19,41,42
Magnetic resonance imaging studies of the BF have generally relied on T2-weighted images. One study23 reported a small, but significant, decrease in the thickness of the BF, measured in a single coronal section from a T2-weighted image, in cognitively intact patients with PD, as well as a more substantial reduction in patients with dementia. Another study43 showed decreased BF thickness in patients having dementia with Lewy bodies, but this study did not include a nondemented PD group. The volume of the BF has also been measured using T1-weighted images at high field strength,44 revealing a significant decrease in the volume of the BF between nondemented patients with PD and controls, with further volume loss in patients with dementia.45 The present finding of reduced BF forebrain volumes in patients with H&Y stage 2 or 3 PD is in accord with these previous investigations. To our knowledge, no other studies have examined differences between H&Y stage 1 PD and H&Y stages 2 and 3 PD, but the finding of more extreme cell and volume loss in patients with dementia is consistent with the trajectory of the volume loss reported herein.
COMPARISON OF THE SNc AND BF CHANGES IN PD
As the first direct in vivo comparison to date of the volumes of the SNc and BF in the same sample of patients with PD, our findings revealed that the volume loss in the SNc precedes the degeneration of the more rostral BF. These results are consistent with a proposed neuropathological trajectory whereby PD-related pathologic change begins in deep brain structures and progresses rostrally.13 Although some researchers have called into question the usefulness of the Braak neuropathological staging scheme,14,15 a longitudinal study46 of pathological progression revealed 3 distinct subgroups of patients, with the largest subgroup exhibiting pathological changes consistent with the Braak model. Differences in the patterns of neurodegeneration may represent subgroups of patients having PD with fundamentally distinct disease processes and trajectories of disease progression, and an emerging view is that heterogeneity may be the rule in PD. Notably, in our study, the volumes of the SNc and BF did not correlate significantly with each other. A larger sample with a broader distribution of disease stages, including patients with dementia, could reveal linked degeneration in these structures. Alternatively, our sample was possibly heterogeneous with respect to disease subgroups; therefore, a larger sample might reveal a subset of patients in whom the degeneration of these structures is more tightly linked.
In summary, we introduced powerful multispectral MR imaging tools to examine the temporal progression of the degeneration in the SNc and BF, as well as in other forebrain and neocortical structures. Although some aspects of the Braak neuropathological staging scheme remain a topic of debate and continued research, results from this study provide in vivo support for the Braak proposal of Lewy body pathological spread rostrally, affecting the SNc before the BF. A critical outstanding question is whether subgroups of patients exist who do not show this temporal progression of pathological change. If so, what are the frequencies and clinical ramifications? The development of new tools to measure subcortical brain structures in vivo such as those described herein will be essential to document expected anatomical changes across multiple structures. The future identification and characterization of PD subgroups will provide physicians and researchers with more focused therapeutic targets.
Supplementary Material
Acknowledgments
Funding/Support: This research was supported by grants AG021525, DA022759-03, GM0007484, K23-AG22509, T32 GM007484, T32 MH082718, and T90 DA022759 from the National Institutes of Health and by the Health Sciences and Technology Martinos Catalyst Fund. The Athinoula A. Martinos Imaging Center at Massachusetts Institute of Technology’s McGovern Institute for Brain Research is supported in part by a generous gift from Pat and Lore McGovern.
Footnotes
Author Contributions: Study concept and design: Ziegler, Wonderlick, and Growdon. Acquisition of data: Ziegler, Wonderlick, Ashourian, Hansen, Young, Murphy, and Koppuzha. Analysis and interpretation of data: Ziegler, Wonderlick, Ashourian, Hansen, Young, Murphy, Koppuzha, and Corkin. Drafting of the manuscript: Ziegler, Wonderlick, and Corkin. Critical revision of the manuscript for important intellectual content: Ziegler, Wonderlick, and Corkin. Statistical analysis: Ziegler. Obtained funding: Corkin. Administrative, technical, and material support: Ashourian, Young, Koppuzha, and Growdon.
Conflict of Interest Disclosures: None reported.
Online-Only Material: The eFigures and eAppendix are available at http://www.jamaneuro.com.
Additional Contributions: Thomas Kemper, MD, and John Hedreen, MD, assisted in delineating anatomical boundaries, and Bradford C. Dickerson, MD, provided magnetic resonance imaging sequences.
Contributor Information
David A. Ziegler, Department of Brain and Cognitive Sciences, Massachusetts Institute of Technology, Cambridge.
Julien S. Wonderlick, Department of Brain and Cognitive Sciences, Massachusetts Institute of Technology, Cambridge.
Paymon Ashourian, Department of Brain and Cognitive Sciences, Massachusetts Institute of Technology, Cambridge.
Leslie A. Hansen, Department of Brain and Cognitive Sciences, Massachusetts Institute of Technology, Cambridge.
Jeremy C. Young, Department of Brain and Cognitive Sciences, Massachusetts Institute of Technology, Cambridge.
Alex J. Murphy, Department of Brain and Cognitive Sciences, Massachusetts Institute of Technology, Cambridge.
Cecily K. Koppuzha, Department of Brain and Cognitive Sciences, Massachusetts Institute of Technology, Cambridge.
John H. Growdon, Department of Neurology and Morris Udall Center of Excellence in Parkinson Disease Research, Massachusetts General Hospital, Boston.
Suzanne Corkin, Department of Brain and Cognitive Sciences, Massachusetts Institute of Technology, Cambridge.
References
- 1.Jellinger K. The pathology of Parkinson’s disease: recent advances. In: Galvez-Jimenez N, editor. Scientific Basis for the Treatment of Parkinson’s Disease. 2nd. New York, NY: Taylor & Francis; 2005. pp. 53–86. [Google Scholar]
- 2.Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991;114(pt 5):2283–2301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- 3.Poewe W. Treatments for Parkinson disease—past achievements and current clinical needs. Neurology. 2009;72(7 suppl):S65–S73. doi: 10.1212/WNL.0b013e31819908ce. [DOI] [PubMed] [Google Scholar]
- 4.Calabresi P, Picconi B, Parnetti L, Di Filippo M. A convergent model for cognitive dysfunctions in Parkinson’s disease: the critical dopamine-acetylcholine synaptic balance. Lancet Neurol. 2006;5(11):974–983. doi: 10.1016/S1474-4422(06)70600-7. [DOI] [PubMed] [Google Scholar]
- 5.Pillon B, Dubois B, Cusimano G, Bonnet AM, Lhermitte F, Agid Y. Does cognitive impairment in Parkinson’s disease result from non-dopaminergic lesions? J Neurol Neurosurg Psychiatry. 1989;52(2):201–206. doi: 10.1136/jnnp.52.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kensinger EA, Shearer DK, Locascio JJ, Growdon JH, Corkin S. Working memory in mild Alzheimer’s disease and early Parkinson’s disease. Neuropsychology. 2003;17(2):230–239. doi: 10.1037/0894-4105.17.2.230. [DOI] [PubMed] [Google Scholar]
- 7.Locascio JJ, Corkin S, Growdon JH. Relation between clinical characteristics of Parkinson’s disease and cognitive decline. J Clin Exp Neuropsychol. 2003;25(1):94–109. doi: 10.1076/jcen.25.1.94.13624. [DOI] [PubMed] [Google Scholar]
- 8.Sagar HJ, Sullivan EV, Gabrieli JD, Corkin S, Growdon JH. Temporal ordering and short-term memory deficits in Parkinson’s disease. Brain. 1988;111(pt 3):525–539. doi: 10.1093/brain/111.3.525. [DOI] [PubMed] [Google Scholar]
- 9.Brooks DJ, Pavese N. Imaging non-motor aspects of Parkinson’s disease. Prog Brain Res. 2010;184:205–218. doi: 10.1016/S0079-6123(10)84011-7. [DOI] [PubMed] [Google Scholar]
- 10.Kehagia AA, Barker RA, Robbins TW. Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson’s disease. Lancet Neurol. 2010;9(12):1200–1213. doi: 10.1016/S1474-4422(10)70212-X. [DOI] [PubMed] [Google Scholar]
- 11.Javoy-Agid F, Taquet H, Ploska A, Cherif-Zahar C, Ruberg M, Agid Y. Distribution of catecholamines in the ventral mesencephalon of human brain, with special reference to Parkinson’s disease. J Neurochem. 1981;36(6):2101–2105. doi: 10.1111/j.1471-4159.1981.tb10843.x. [DOI] [PubMed] [Google Scholar]
- 12.Rogers JD, Brogan D, Mirra SS. The nucleus basalis of Meynert in neurological disease: a quantitative morphological study. Ann Neurol. 1985;17(2):163–170. doi: 10.1002/ana.410170210. [DOI] [PubMed] [Google Scholar]
- 13.Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 14.Halliday G, McCann H, Shepherd C. Evaluation of the Braak hypothesis: how far can it explain the pathogenesis of Parkinson’s disease? Expert Rev Neurother. 2012;12(6):673–686. doi: 10.1586/ern.12.47. [DOI] [PubMed] [Google Scholar]
- 15.Jellinger KA. Critical evaluation of the Braak staging scheme for Parkinson’s disease. Ann Neurol. 2010;67(4):550. doi: 10.1002/ana.21638. [DOI] [PubMed] [Google Scholar]
- 16.Shulman JM, De Jager PL, Feany MB. Parkinson’s disease: genetics and pathogenesis. Annu Rev Pathol. 2011;6:193–222. doi: 10.1146/annurev-pathol-011110-130242. [DOI] [PubMed] [Google Scholar]
- 17.Shimada H, Hirano S, Shinotoh H, et al. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology. 2009;73(4):273–278. doi: 10.1212/WNL.0b013e3181ab2b58. [DOI] [PubMed] [Google Scholar]
- 18.Shinotoh H, Hirano S. Emerging in vivo evidence of subcortical cholinergic dysfunction in Parkinsonian syndromes. Neurology. 2010;74(18):1406–1407. doi: 10.1212/WNL.0b013e3181dfc94e. [DOI] [PubMed] [Google Scholar]
- 19.Gilman S, Koeppe RA, Nan B, et al. Cerebral cortical and subcortical cholinergic deficits in parkinsonian syndromes. Neurology. 2010;74(18):1416–1423. doi: 10.1212/WNL.0b013e3181dc1a55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marek K, Jennings D. Can we image premotor Parkinson disease? Neurology. 2009;72(7 suppl):S21–S26. doi: 10.1212/WNL.0b013e318198df97. [DOI] [PubMed] [Google Scholar]
- 21.Hutchinson M, Raff U. Structural changes of the substantia nigra in Parkinson’s disease as revealed by MR imaging. AJNR Am J Neuroradiol. 2000;21(4):697–701. [PMC free article] [PubMed] [Google Scholar]
- 22.Oikawa H, Sasaki M, Tamakawa Y, Ehara S, Tohyama K. The substantia nigra in Parkinson disease: proton density–weighted spin-echo and fast short inversion time inversion-recovery MR findings. AJNR Am J Neuroradiol. 2002;23(10):1747–1756. [PMC free article] [PubMed] [Google Scholar]
- 23.Oikawa H, Sasaki M, Ehara S, Abe T. Substantia innominata: MR findings in Parkinson’s disease. Neuroradiology. 2004;46(10):817–821. doi: 10.1007/s00234-004-1257-4. [DOI] [PubMed] [Google Scholar]
- 24.Moon WJ, Kim HJ, Roh HG, Han SH. Atrophy measurement of the anterior commissure and substantia innominata with 3T high-resolution MR imaging: does the measurement differ for patients with frontotemporal lobar degeneration and Alzheimer disease and for healthy subjects? AJNR Am J Neuroradiol. 2008;29(7):1308–1313. doi: 10.3174/ajnr.A1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain. 2002;125(pt 4):861–870. doi: 10.1093/brain/awf080. [DOI] [PubMed] [Google Scholar]
- 26.Muth K, Schönmeyer R, Matura S, Haenschel C, Schröder J, Pantel J. Mild cognitive impairment in the elderly is associated with volume loss of the cholinergic basal forebrain region. Biol Psychiatry. 2010;67(6):588–591. doi: 10.1016/j.biopsych.2009.02.026. [DOI] [PubMed] [Google Scholar]
- 27.Buckner RL, Head D, Parker J, et al. A unified approach for morphometric and functional data analysis in young, old, and demented adults using automated atlas-based head size normalization: reliability and validation against manual measurement of total intracranial volume. Neuroimage. 2004;23(2):724–738. doi: 10.1016/j.neuroimage.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 28.Lees AJ. The Parkinson chimera. Neurology. 2009;72(7 suppl):S2–S11. doi: 10.1212/WNL.0b013e318198daec. [DOI] [PubMed] [Google Scholar]
- 29.Ma SY, Röytta¨ M, Rinne JO, Collan Y, Rinne UK. Correlation between neuromorphometry in the substantia nigra and clinical features in Parkinson’s disease using disector counts. J Neurol Sci. 1997;151(1):83–87. doi: 10.1016/s0022-510x(97)00100-7. [DOI] [PubMed] [Google Scholar]
- 30.Greffard S, Verny M, Bonnet AM, et al. Motor score of the Unified Parkinson Disease Rating Scale as a good predictor of Lewy body–associated neuronal loss in the substantia nigra. Arch Neurol. 2006;63(4):584–588. doi: 10.1001/archneur.63.4.584. [DOI] [PubMed] [Google Scholar]
- 31.Duguid JR, De La Paz R, DeGroot J. Magnetic resonance imaging of the midbrain in Parkinson’s disease. Ann Neurol. 1986;20(6):744–747. doi: 10.1002/ana.410200618. [DOI] [PubMed] [Google Scholar]
- 32.Braffman BH, Grossman RI, Goldberg HI, et al. MR imaging of Parkinson disease with spin-echo and gradient-echo sequences. AJR Am J Roentgenol. 1989;152(1):159–165. doi: 10.2214/ajr.152.1.159. [DOI] [PubMed] [Google Scholar]
- 33.Antonini A, Leenders KL, Meier D, Oertel WH, Boesiger P, Anliker M. T2 relaxation time in patients with Parkinson’s disease. Neurology. 1993;43(4):697–700. doi: 10.1212/wnl.43.4.697. [DOI] [PubMed] [Google Scholar]
- 34.Adachi M, Hosoya T, Haku T, Yamaguchi K, Kawanami T. Evaluation of the substantia nigra in patients with Parkinsonian syndrome accomplished using multishot diffusion-weighted MR imaging. AJNR Am J Neuroradiol. 1999;20(8):1500–1506. [PMC free article] [PubMed] [Google Scholar]
- 35.Stern MB, Braffman BH, Skolnick BE, Hurtig HI, Grossman RI. Magnetic resonance imaging in Parkinson’s disease and parkinsonian syndromes. Neurology. 1989;39(11):1524–1526. doi: 10.1212/wnl.39.11.1524. [DOI] [PubMed] [Google Scholar]
- 36.Sasaki M, Shibata E, Tohyama K, et al. Neuromelanin magnetic resonance imaging of locus ceruleus and substantia nigra in Parkinson’s disease. Neuroreport. 2006;17(11):1215–1218. doi: 10.1097/01.wnr.0000227984.84927.a7. [DOI] [PubMed] [Google Scholar]
- 37.Minati L, Grisoli M, Carella F, De Simone T, Bruzzone MG, Savoiardo M. Imaging degeneration of the substantia nigra in Parkinson disease with inversion-recovery MR imaging. AJNR Am J Neuroradiol. 2007;28(2):309–313. [PMC free article] [PubMed] [Google Scholar]
- 38.Whitehouse PJ, Hedreen JC, White CL, III, Price DL. Basal forebrain neurons in the dementia of Parkinson disease. Ann Neurol. 1983;13(3):243–248. doi: 10.1002/ana.410130304. [DOI] [PubMed] [Google Scholar]
- 39.Arendt T, Bigl V, Arendt A, Tennstedt A. Loss of neurons in the nucleus basalis of Meynert in Alzheimer’s disease, paralysis agitans and Korsakoff’s disease. Acta Neuropathol. 1983;61(2):101–108. doi: 10.1007/BF00697388. [DOI] [PubMed] [Google Scholar]
- 40.Nakano I, Hirano A. Parkinson’s disease: neuron loss in the nucleus basalis without concomitant Alzheimer’s disease. Ann Neurol. 1984;15(5):415–418. doi: 10.1002/ana.410150503. [DOI] [PubMed] [Google Scholar]
- 41.Ruberg M, Rieger F, Villageois A, Bonnet AM, Agid Y. Acetylcholinesterase and butyrylcholinesterase in frontal cortex and cerebrospinal fluid of demented and non-demented patients with Parkinson’s disease. Brain Res. 1986;362(1):83–91. doi: 10.1016/0006-8993(86)91401-0. [DOI] [PubMed] [Google Scholar]
- 42.Mattila PM, Röyttä M, Lönnberg P, Marjamäki P, Helenius H, Rinne JO. Choline acetytransferase activity and striatal dopamine receptors in Parkinson’s disease in relation to cognitive impairment. Acta Neuropathol. 2001;102(2):160–166. doi: 10.1007/s004010100372. [DOI] [PubMed] [Google Scholar]
- 43.Hanyu H, Shimizu S, Tanaka Y, Hirao K, Iwamoto T, Abe K. MR features of the substantia innominata and therapeutic implications in dementias. Neurobiol Aging. 2007;28(4):548–554. doi: 10.1016/j.neurobiolaging.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 44.George S, Mufson EJ, Leurgans S, Shah RC, Ferrari C, deToledo-Morrell L. MRI-based volumetric measurement of the substantia innominata in amnestic MCI and mild AD. Neurobiol Aging. 2011;32(10):1756–1764. doi: 10.1016/j.neurobiolaging.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choi SH, Jung TM, Lee JE, Lee SK, Sohn YH, Lee PH. Volumetric analysis of the substantia innominata in patients with Parkinson’s disease according to cognitive status. Neurobiol Aging. 2012;33(7):1265–1272. doi: 10.1016/j.neurobiolaging.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 46.Halliday G, Hely M, Reid W, Morris J. The progression of pathology in longitudinally followed patients with Parkinson’s disease. Acta Neuropathol. 2008;115(4):409–415. doi: 10.1007/s00401-008-0344-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.