

Abstract
Epidemiological studies indicate that light-moderate alcohol (ethanol) consumers tend to have reduced risks of cognitive impairment and progression to dementia during aging. Exploring possible mechanisms, we previously found that moderate ethanol preconditioning (MEP, 20-30 mM) of rat brain cultures for several days instigated neuroprotection against β-amyloid peptides. Our biochemical evidence implicated the NMDA receptor (NMDAR) as a potential neuroprotective “sensor”, specifically via synaptic NMDAR signaling. It remains unclear how ethanol modulates the receptor and its downstream targets to engender neuroprotection. Here we confirm with deconvolution microscopy that MEP of rat mixed cerebellar cultures robustly increases synaptic NMDAR localization. Phospho-activation of the non-receptor tyrosine kinases Src and Pyk2, known to be linked to synaptic NMDAR, is also demonstrated. Additionally, the preconditioning enhances levels of an antioxidant protein, peroxiredoxin 2 (Prx2), reported to be downstream of synaptic NMDAR signaling, and NMDAR antagonism with memantine (earlier found to abrogate MEP neuroprotection) blocks the Prx2 elevations. To further link Prx2 with antioxidant-based neuroprotection, we circumvented the ethanol preconditioning-NMDAR pathway by pharmacologically increasing Prx2 with the naturally-occurring cruciferous compound, 3H-1,2-dithiole-3-thione (D3T). Thus, D3T pretreatment elevated Prx2 expression to a similar extent as MEP, while concomitantly preventing β-amyloid neurotoxicity; D3T also protected the cultures from hydrogen peroxide toxicity. The findings support a mechanism that couples synaptic NMDAR signaling, Prx2 expression and augmented antioxidant defenses in ethanol preconditioning-induced neuroprotection. That this mechanism can be emulated by a cruciferous vegetable constituent suggests that such naturally-occurring “neutraceuticals” may be useful in therapy for oxidative stress-related dementias.
Keywords: alcohol, preconditioning, neuroprotection, NMDA receptor, peroxiredoxin 2, deconvolution microscopy
1. Introduction
Dementia is a leading cause of disability in aging populations and interventions are presently lacking. Non-heritable dementias including most of Alzheimer's disease (AD) have complex etiologies; however, certain underlying pathophysiological mechanisms appear to be common among them. These include free radical/oxidative stress-related neuronal damage (Uttara et al., 2009) and misregulation of NMDA type glutamate receptors (NMDARs) (Butterfield and Pocernich, 2003). The latter, being intimately involved with memory formation and retention (Nabavi et al., 2014), has obvious implications in dementia (Lin et al., 2014), but the former, although easily detected, has divergent or poorly understood manifestations. An approach to combating redox stress-related neuronal damage combinatorially with NMDAR modulation may prove useful in the treatment of oxidative stress-related dementias, as NMDAR blockers to date have shown limited clinical efficacy (Olivares et al., 2012).
A growing number of epidemiological studies indicate that low-to-moderate (social) alcohol consumption by older individuals is associated with a reduced risk of cognitive impairment and/or dementia (Neafsey and Collins, 2011). Unsurprisingly, the mechanisms at play affording such protection are debatable. Our laboratories have attempted to model aspects of ethanol neuroprotection in vitro (Belmadani et al., 2004; Mitchell et al., 2009; Sivaswamy et al., 2010). Consequently, when preconditioned for ∼6 days with experimentally moderate ethanol concentrations (20-30 mM), rat hippocampal-entorhinal cortical slice cultures and mixed cerebellar cultures are significantly neuroprotected from dementia-related proteins.
NMDAR activation has been linked as a sensor in other neuroprotective preconditioning modalities such as with brief ischemia or NMDA itself (Severino et al., 2011). Neuroprotection from moderate ethanol preconditioning (MEP) requires NMDAR activity early (first 2-3 days of exposure) and correlates with increased NMDAR subunit expression and biochemical indicators of synaptically-localized NMDAR complexes (Mitchell et al., 2009). Additionally, certain kinases whose activities are known to modulate NMDAR surface expression, activity and synaptic localization are also enhanced by MEP, including protein kinase C (PKC) and focal adhesion kinase (FAK) (Sivaswamy et al., 2010). Moreover, recent studies have demonstrated that synaptic NMDAR activation is associated with neuroprotective or prosurvival mechanisms (Hardingham and Bading, 2003; Hardingham, 2006), in part by upregulating thiol-based neuronal antioxidant defenses (Papadia et al., 2008). One such enzyme is the neuronally-expressed peroxiredoxin (Prx) isoform Prx2, which is linked to neuroprotection from ischemia-induced oxidative stress (Gan et al., 2012).
Here we sought to determine whether synaptic NMDA receptor signaling was involved in acquisition of the neuroprotective phenotype by MEP, and if this mechanism resulted in augmented neuronal antioxidant defenses. Our results show that MEP upregulates activity of the non-receptor tyrosine kinases linked to synaptic NMDAR localization, Src and Pyk2. Furthermore, ethanol preconditioning results in robust increases in synaptically-localized NMDAR subunits and concomitant NMDAR-dependent elevations in the expression of Prx2. Importantly, we show that this pathway can be circumvented by a cruciferous compound, 3H-1,2-dithiole-3-thione (D3T), which mimics MEP upregulation of Prx2 and provides neuroprotection from both β-amyloid peptide and general oxidative stress (H2O2). It is possible that a better understanding this neuroprotective pathway might lead to novel therapeutics for redox-stress-related dementias.
2. Results
2.1. MEP increases phospho-activation of Src and Pyk2
Ethanol preconditioning results in the upregulation of NMDAR subunits, increased NR2BpY1472/NR2B (Mitchell et al., 2009), as well as activation of PKC isozymes and FAK (Sivaswamy et al., 2010). Synaptic localization of NMDARs has been shown to be dependent on the activity of the non-receptor tyrosine kinases Src (Yu et al., 1997) and Pyk2 (Huang et al., 2001). In order to determine if these two kinases were involved in the observed increases in NMDAR subunits early in the MEP protocol, we assayed their activation due to phosphorylation in mixed cerebellar cultures after 48h of ethanol preconditioning. As shown in Fig. 1A and quantified in 1B, ethanol pre-exposure results in strong Src-family and Pyk2 phospho-activation, consistent with reports of their actions in upregulation of synaptically localized NMDARs.
Figure 1. Phosphoactivation of Src-family and Pyk2 non-receptor tyrosine kinases by MEP.
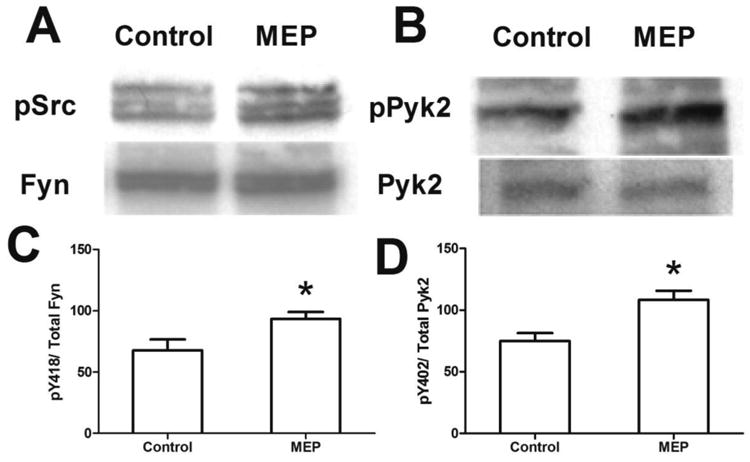
Cerebellar cultures (10DIV) were treated with 30mM ethanol (labeled MEP) or control media for 48 h. A) Protein was extracted and subjected to immunoblot analyses for anti-p-Src (Tyr416) and anti-p-Pyk2 (Tyr402). Blots were then stripped and probed with corresponding non-phosphorylated antibodies. B. Quantification of A. Data represents %phospho/total (mean ± sem); *p<0.05 vs. control (n= 4). NOTE: anti-Fyn was used as loading control for p-Src due to recognition of multiple p-Src family members.
2.2. Deconvolution microscopy shows increased synaptically-localized NMDAR subunits following MEP
Because the biochemical indications of synaptic NMDAR localization appear to be early in the MEP protocol, and because of the association of synaptic NMDAR activity with neuroprotective paradigms, we employed deconvolution microscopy to determine if MEP results in evident increases in synaptic NMDARs. Mixed cerebellar cultures stained with antibodies directed against the post-synaptic scaffolding protein PSD-95 and the NR2 subunit of the NMDAR (pan-NR2, GluN2A-C) were examined. As shown in Fig. 2A, MEP-treated cultures appeared to have elevated PSD-95 and NR2 fluorescence, consistent with the increased expression of these proteins at 2 and 6 days of MEP (Mitchell et al., 2009). To verify this observation, z-stack images of these cerebellar cultures were acquired and deconvolved (Freeman et al., 2013) in a blinded fashion to measure the intensity of PSD-95 and NR2 staining. An algorithm using Imaris image analysis software to identify putative synaptic sites based on PSD-95 immunoreactivity was then constructed (see Fig. 2B), and an arbitrary ratio of NR2 fluorescence to PSD-95 for each surface identified was derived. Based on this approach, we observed an increase in the NR2/PSD-95 intensities for MEP-treated cultures to 133±0.3% compared to control (Fig. 2C), revealing increased synaptic NMDAR content of ethanol-treated cultures. Further plotting the relative intensity of NR2 immunoreactivity for surfaces identified by PSD-95 for each putative synapse (Fig. 2D), the significant increase in the slope of MEP-treated neurons compared to control (MEP=1.37, red diamonds vs. control=0.85 green circles) was documented. Overall, our results confirm that MEP enhances synaptically-localized NMDAR complexes, consistent with their role in MEP's neuroprotective mechanism.
Figure 2. MEP augments synaptically-localized NMDARs.
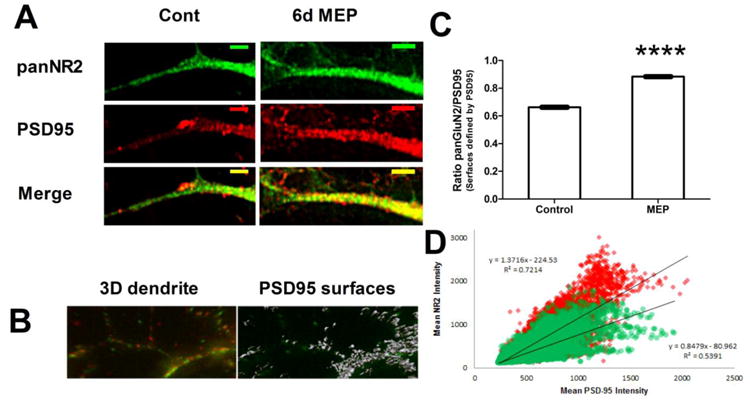
A) Representative dendrite of cerebellar cultures (10DIV) treated for 6d with media containing 30mM ethanol (MEP) or vehicle (control) as described. Cultures were fixed and stained with the indicated primary antibodies, and appropriate fluorophore-conjugated secondary antibodies. Images were collected on a Delta Vision deconvolution microscope and a representative z-plane is shown (scale bar = 5μm). B) Representative 3D image generated with Imaris 3.1 showing PSD-95 (red) and pan NR2 (green). Gray surfaces represent areas defined by PSD-95 staining and identified by Imaris algorithm for quantification. C) Arbitrary intensities were determined for each surface using Imaris, and the ratio of NR2/PSD-95 was calculated and plotted as mean ± sem. At least 20 neurons were imaged per group. ****p<0.0001 vs. control. D) Scatter plot of surfaces identified in C, with regression data showing increased slope of MEP-treated putative synapses. Red diamond=MEP, green circles = control.
2.3. Enhanced Prx2 expression occurs in response to MEP
Synaptic NMDAR activity has been causally linked to neuroprotection from oxidative stress via upregulation of neuronal thiol-based redox systems. In order to determine if MEP mediates neuroprotection from pro-oxidant β-amyloid by a similar mechanism, we analyzed tissue extracts from control, MEP and MEP+memantine (which inhibits MEP neuroprotection at high concentrations of 50μM) -treated cultures by western blot for expression of peroxiredoxin 2 (Prx2). As depicted in Fig. 3A and quantified in 3B, MEP resulted in an increase in Prx2 expression to 178±42% control, which was completely blocked by memantine co-application. The result provides support for the contention that MEP treatment signals through synaptic NMDARs to increase neuronal antioxidant defenses.
Figure 3. MEP neuroprotection correlates with enhanced Prx2 expression that is NMDAR-dependent.
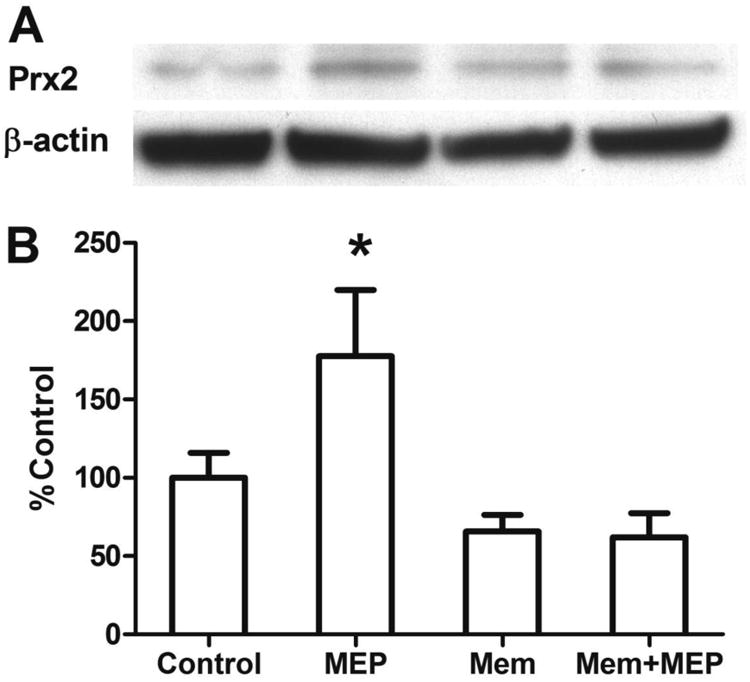
Cerebellar cultures (10DIV) were treated with 30mM ethanol (MEP) or control media for 6 days with and without memantine (MEM). Protein extracts were resolved by SDS-PAGE and immunoblots were probed for Prx2 and β-actin. A) Representative immunoblots; B) immunoblot quantification (mean ± sem); * p<0.05 vs. control, n=4-6.
2.4. D3T increases Prx2 and affords protection from oxidative insult
The cruciferous neutraceutical D3T has been shown to induce phase II enzyme expression by a mechanism that requires activity of the transcription factor Nrf2 (Munday and Munday, 2004). Therefore we hypothesized that if D3T could recapitulate the MEP-induced increase in Prx2 expression, it may circumvent ethanol exposure by acting downstream of NMDAR activation. As shown in Fig. 4A and quantified in 4B, 48h of D3T treatment enhanced the expression of Prx2 to a similar extent as MEP (141±9.0% of control), but was without effect on redox partners thioredoxin (125±11% of control), thioredoxin reductase (110±11% of control) and sulfiredoxin (103±8% of control). Moreover, as shown in Fig. 4C, similar to MEP neuroprotection, 48h D3T treatment completely ameliorated neurotoxicity induced by 24h exposure to β-amyloid. In order to determine the extent to which D3T affords neuroprotection from oxidative stress, control and D3T-treated cultures for 24h were challenged with increasing concentrations of H2O2. Fig. 4D shows that D3T treatment profoundly reduced H2O2-induced neurotoxicity in cerebellar cultures to such an extent that only the highest peroxide concentration (1mM) produced significant yet partial toxicity.
Figure 4. D3T increases Prx2 expression and affords protection against Aβ or oxidative stress (H2O2).
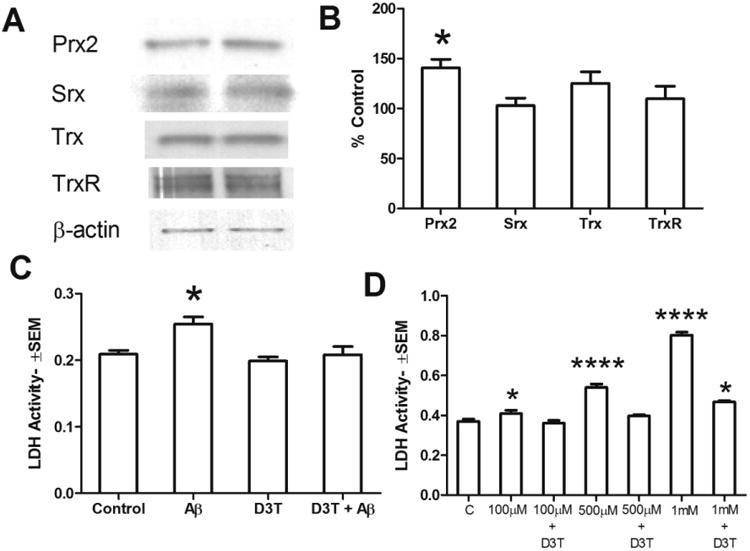
Cerebellar cultures (DIV10) were treated for 48h with 25μM D3T. A) Culture lysates were separated by SDS-PAGE as described and blotted for the indicated proteins; control (left) and D3T (right). B) Densitrometric quantification of A, represented as % control (mean ± sem), shows a significant elevation in Prx2 due to D3T; *p<0.05 vs. control, n=3. C) Following 48h in D3T-containing or control media, cultures were exposed to 25μM β-amyloid [Aβ 25-35]. D3T treatment inhibits neurotoxicity as assessed using media LDH activity (mean ± sem); *p<0.01 vs. control, n=6. D) Following 48h in D3T-containing or control media, cultures were treated for 24h with the indicated concentrations of H2O2 and culture media assayed for toxicity by LDH activity (mean ± sem). Strong protection by D3T from pro-oxidant H2O2 was evident even at the highest H2O2concentration (1mM); *p<0.01 vs. control, ****p<0.0001 vs. control, n=4-6.
3. Discussion
3.1
Previous research from our laboratories has shown that preconditioning brain cultures with moderate ethanol concentrations affords significant neuroprotection from neurotoxic proteins, including AD-related β-amyloid peptides (Belmadani et al., 2004; Collins et al., 2010; Mitchell et al., 2009). Neuroprotection requires the activity of the NMDAR and correlates with increased activation of NMDAR-associated enzymes, PKC and FAK (Sivaswamy et al., 2010). Our current results provide several insights to understanding ethanol's ability to engender a neuroprotective phenotype. Also, we provide a possibility concerning potential neuroprotection that circumvents the requirement of ethanol, with its well-known propensity for increased abuse and associated health problems (although moderate drinkers have additional cardiovascular protection). Specifically, 1) ethanol preconditioning of brain cultures is capable of increasing neuronal activity of non-RTKs associated with synaptic NMDAR localization; 2) this leads to enhanced synaptic NMDAR expression in vitro; 3) blockade of NMDAR activity inhibits this pathway; 4) Prx2 increases correlate with the protective phenotype; and 5) the requirement for ethanol can be circumvented by the cruciferous alkaloid D3T, resulting in sustained protection from β-amyloid as well as from severe oxidative insult with H2O2.
3.2
Peroxiredoxins (Prx) are a ubiquitous family of antioxidant proteins that reduce and detoxify H2O2, peroxynitrite, and organic hydroperoxides. They are divided into three subclasses: typical 2-Cys (2-Cys refers to the catalytic site containing 2 cysteine residues, Prx1-4), atypical 2-Cys (Prx5), and 1-Cys (Prx6). Since first being characterized (Chae et al., 1994), this thiol-specific antioxidant family has rapidly gained prominence for its role in cytoprotection and disease. Prx isoforms have been shown to have aberrant expression in certain diseases such as AD, Parkinson's disease, Down's syndrome (Krapfenbauer et al., 2003), Creutzfeldt-Jacob disease (Krapfenbauer et al., 2002), and ALS (Kato et al., 2005). Furthermore, Prx2 is upregulated in AD brain and in two transgenic mouse models of AD, and transfection with Prx2 protects cultured cortical neurons from toxic β-amyloid (Yao et al., 2007). Over-oxidation of Prx2 in AD brain also has been reported (Cumming et al., 2007). Interestingly, Prx2 has been shown to mediate, at least in part, the neuroprotective effects of pituitary adenylate cyclase activating polypeptide in cultured cerebellar granule neurons (Botia et al., 2008). These authors further report that Prx2 expression is induced by both cyclic AMP and PKC stimulators, and also show the upregulation of Prx2 by ethanol after treatment of rat pups in vivo or cerebellar cultures in vitro (although the in vitro concentration used, 325 mM, was far above circulating levels in their in vivo study or, for that matter, in chronic alcoholism). The authors further speculate on the therapeutic potential of Prx2 for the treatment of some neurodegenerative diseases.
3.3
Extrasynaptic NMDAR activity has been linked to neurodegenerative processes involved in amyloidogenesis (Amadoro et al., 2006; Bordji et al., 2010). Although the clinically available NMDAR antagonist memantine is well tolerated, its therapeutic usefulness remains modest and it is only approved for moderate-to severe dementia. Its mechanism of action is that of an open channel blocker mainly at extrasynaptic NMDARs with high voltage dependence and rapid off rates (at clinically relevant concentrations of ∼1μM (Kornhuber and Quack, 1995)). Additionally, memantine has high affinity for α-7 nicotinic acetylcholine receptors (Aracava et al., 2005) that would seem disadvantageous for dementia patients, as cholinesterase inhibitors and cholinergic agonists have a degree of clinical efficacy for AD (Sugimoto, 2008). Moreover, extrasynaptic NMDARs may have physiological activity that is not necessarily degenerative (Papouin and Oliet, 2014). Therefore, as a sole approach to treating redox stress-related dementias, targeting extrasynaptic NMDARs will likely continue to show inadequate efficacy. However, circumventing synaptic NMDAR activity with chemical compounds such as D3T whilst engaging its downstream antioxidant defenses may prove useful for combating age-related dementias. Moreover, a combination of “new generation” extrasynaptic NMDAR allosteric inhibitors (Lipton, 2005), in addition to such agents may prove most effective (Rosini et al., 2008).
3.4
Figure 5 diagrams our findings of a neuroprotective mechanism with MEP for ∼2 days and ultimately 6 days. As depicted, results indicate that the ethanol preconditioning modulates tyrosine kinase signaling and enhances synaptic NMDAR localization, resulting in downstream elevation of thiol-based antioxidant Prx2 and subsequent neuroprotection from AD-related β-amyloid peptides. Moreover, although not represented in Figure 5, cruciferous D3T is capable of recapitulating MEP's effects by enhancing Prx2 levels. To an extent, this provides evidence that chemical compounds which circumvent NMDAR activation and enhance neuronal antioxidant capacity/Prx2 expression may prove useful in combating neurodegenerative diseases having an underlying redox stress component.
Figure 5. Summary diagram of MEP-mediated enhancement of synaptic NMDAR localization and upregulation of Prx2.
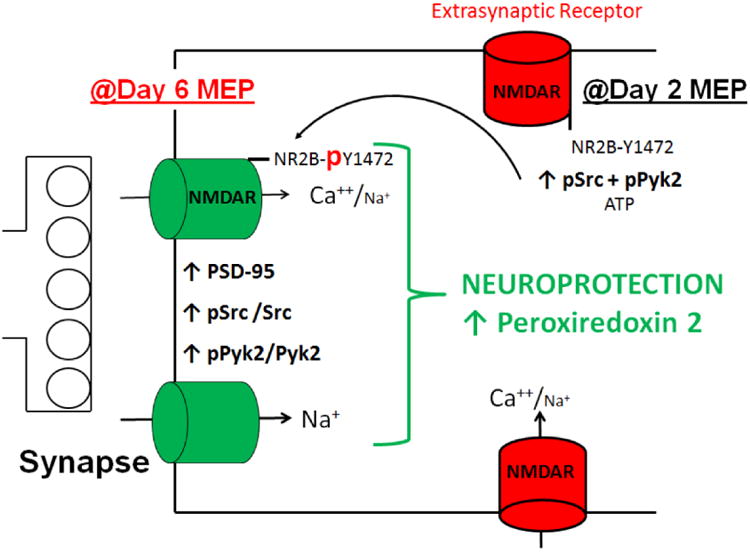
MEP increases phospho-activation of Src and Pyk2 by day 2 of treatment. These kinases are known to mediate synaptic localization of NMDARs by phosphorylation of their C-terminal regulatory domains, which occludes internalization by clathrin/AP2-mediated endocytosis. Increased expression of NMDAR subunits in combination with PSD-95 (Mitchell et al., 2009) results in enhanced synaptically-localized receptor complex. Synaptic NMDAR activity increases neuronal antioxidant defenses in the form of upregulating Prx2 and resultant neuroprotection. This pathway can be circumvented by D3T, which lends support to the speculation that naturally occurring phytochemicals in combination with extrasynaptic NMDAR blockers may act synergistically to combat redox stress-related neurodegenerative processes.
4. Experimental procedures
4.1. Chemicals and antibodies
Hoechst 33342, memantine, deoxyribonuclease I (DNAse), Tween 20, Triton-X 100, protease and phosphatase inhibitor cocktails, NAD-ADH reagent multiple test reagent (N 7160), penicillin/streptomycin, and D3T were from Sigma Co. (St. Louis MO). Neurobasal-A media, B27, trypsin and fetal bovine serum (FBS) were from Gibco (Carlsbad, CA). American Peptide (Sunnyvale, CA) provided β-amyloid [25-35]. Polyacrylamide gels were obtained from GenScript (Piscataway, NJ). Western blot buffers and enhanced chemiluminescence detection (ECL) solutions were from Pierce Chemical Co. (Rockford, IL). The antibodies obtained from Upstate Co. (Temecula, CA) were anti-NR1-CT (1:1000) and anti-NR2B (1:1000); from Chemicon International (Temecula, CA), PSD-95 (1:1000), NR2B-pY1472 (1:1000), neuronal specific nuclear antigen (NeuN, 1:1000) and glial acidic fibrillary protein (GFAP, 1:1000); and from Cell Signaling (Danvers, MA), actin (1:1000) and horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (1:1000-1:20,000).
4.2. Cerebellar mixed primary cultures
Modifying a rat cerebellar culture protocol (Brewer et al., 1993), 7-10 day old Sprague-Dawley rat pups (Harlan) were cryoanesthetized on ice for 2-3 min and rapidly decapitated. The brains were removed and placed in petri dishes containing ice-cold Gays balanced salt solution (GBSS) supplemented with 4.5 mg/ml glucose for dissection. The cerebella were separated and placed into ice cold minimal essential media (MEM) supplemented with 6.5 mg/ml glucose. Meninges were removed and tissue was sectioned into approximately 6 pieces that were transferred to calcium- and magnesium-free Hanks balanced salt solution (CM-HBSS) containing 0.25% trypsin and 0.5 mg/ml DNAase at 37°C. After incubation for 10 min, pieces were transferred into MEM with 10% fetal bovine serum (FBS) and 5% horse serum (HS) to inactivate trypsin. Cells were dispersed by triturating with two fire polished pipettes of decreasing bore size and passed through a 70μm cell strainer. Viable cell density was determined by trypan blue exclusion with a hemocytometer. Cells were diluted to a concentration of 800,000 cells/ml in serum-free Neurobasal-A media supplemented with 2% B27 (Gibco), 500μM L-glutamine and 1% penicillin-streptomycin, and plated on poly-L lysine- coated 12 well plates at 1 ml/well. All of the media was changed after one hour. Half of the media was changed every 3-4 days until DIV10, at which point 100% of the media is changed. This technique permits glia to proliferate to near confluence and produces neurons with prominent processes and intricate neural networks (Mitchell et al., 2009).
4.3. Ethanol or D3T pretreatment and β-amyloid or H2O2 exposure
Culture media were changed to media containing 30mM ethanol (1.8μl ethanol/1ml media) or media alone. Culture plates containing ethanol were placed in airtight containers that had small open dishes of ∼90mM ethanol to minimize evaporative ethanol loss from experimental wells. Control plates were treated similarly, but with water alone. The containers were allowed to equilibrate with incubator atmosphere for 15 min and then sealed. Culture media and solution dishes were changed at day 3. At day 6, 100% of ethanol media or control media was removed and media with or without 25μM β-amyloid [25-35] was added. For D3T treatments, culture media was adjusted to 25μM D3T by changing 50% of media with fresh media containing 50μM D3T. Following 48h D3T treatment, β-amyloid or H2O2 was added at indicated concentrations for 24h and culture media were assayed for LDH activity.
4.4. Lactate dehydrogenase assay for neurotoxicity
Following treatment, media were collected from cultures and spun at 5000g for 5 min. The manufacturer's microplate protocol was used to determine toxicity. Lactate dehydrogenase (LDH) activity correlated linearly (R2 = 0.9992) with the number of live cells lysed in control experiments, over the activity ranges reported herein. Neurotoxicity was reported as LDH activity/100μL culture media.
4.5. Western blot analyses
Protein (5-20μg) from cellular extracts was run on 4-12% polyacrylamide gels. After separation, protein was transferred to polyvinylidene fluoride (PVDF) membranes, that then were blocked with 3% BSA in TBS with 0.1% Tween 20 (TBS-T) for 60 min. Primary antibody was diluted based on manufacturer's recommendation in 3% BSA TBS-T. PVDF membranes were incubated overnight @ 4°C. HRP-conjugated secondary antibodies were incubated for 1h and membranes developed with ECL (Pierce) followed by exposure to x-ray film. Following development, films were scanned and analyzed with NIH ImageJ 1.34s. Unless otherwise stated, actin was used to normalize loading.
4.6. Deconvolution microscopy
Cerebellar cultures were grown on 18mm acid-washed glass coverslips coated with poly-L lysine. At 10 DIV, cultures were treated with 30mM ethanol (MEP) or control (C) media for 6 days and then fixed with a cocktail of 100mM PIPES buffer (pH 6.8) and 4% formaldehyde. Monolayers were then permeabilized+blocked in PBS with 0.1% Triton-X100 and 10% normal goat serum for 3 h. Following permeabilization+blocking, the primary antibodies were diluted to the appropriate concentrations in the same perm/block cocktail, with the exception of addition of 5μM Hoechst33342 (used to visualize nuclei), and incubated overnight. Primary antibody dilutions were as follows: chicken α-β III tubulin, 1:500; mouse α-PSD-95, 1:200; rabbit α-pan NR2, 1:500. Next, coverslips were rinsed 3 times with PBS containing 0.1% Triton-X100 and stained with the appropriate secondary antibodies (Cy2 conjugated α-rabbit, Cy3 α-chicken, and Cy5 α-mouse; all goat at 1:400) for 1h, washed extensively and mounted with fluoro-gel.
Image stacks were collected blindly on a Delta Vision deconvolution microscope (6× objective). Deconvolved image stacks were then rendered in 3D and quantified with Imaris 6.3.1. An algorithm mask was constructed using Imaris and inclusion criteria were set for PSD-95 immunoreactive surfaces, so that all visibly positive surfaces were included. This same algorithm was then used to analyze all subsequent image stacks and data was retrospectively analyzed for statistical significance using either Systat 11.00.01 or GraphPad Prism 5. At least 20 neurons were counted per group.
4.7 Statistics
Deconvolution microscopy and LDH assay data were exported to an Excel spreadsheet post-acquisition and subsequently into GraphPad Prism 5 for analysis of statistical significance. Densitometric data was obtained by scanning x-ray film at 600dpi resolution and analyzed using NIH ImageJ. Statistical tests were either 1-way ANOVA with Bonferroni's posthoc or Student's t-test where appropriate. Data shown are representative of at least 3 independent experiments and are represented as mean ± sem.
Highlights.
Ethanol preconditioning of cerebellar cultures increases synaptic NMDA receptors
Preconditioning also phospho-activates non-receptor tyrosine kinases Src and Pyk2
Peroxiredoxin 2 (Prx2), linked to synaptic NMDA receptor signaling, is increased
NMDAR inhibitor memantine, which abrogates ethanol protection, blocks Prx2 elevations
Cruciferous D3T elevates Prx2 like ethanol and prevents β-amyloid neurotoxicity
Acknowledgments
The research was supported by NIH R01 AA013568 and T32 AA013527, and the Illinois Department of Public Health Alzheimer's Disease Research Fund.
Footnotes
Conflict of interest statement: The authors state that they have no competing interests.
Author contributions: RM: Participated in design, research, data analysis and interpretation, and manuscript preparation.
NT: Participated in research and data analysis. EC: Participated in research and data analysis.
EN: Participated in data analysis and interpretation, and manuscript preparation. MC: Participated in design, data analysis and interpretation, and manuscript preparation.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amadoro G, Ciotti MT, Costanzi M, Cestari V, Calissano P, Canu N. NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc Natl Acad Sci U S A. 2006;103:2892–7. doi: 10.1073/pnas.0511065103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aracava Y, Pereira EF, Maelicke A, Albuquerque EX. Memantine blocks alpha7* nicotinic acetylcholine receptors more potently than n-methyl-D-aspartate receptors in rat hippocampal neurons. J Pharmacol Exp Ther. 2005;312:1195–205. doi: 10.1124/jpet.104.077172. [DOI] [PubMed] [Google Scholar]
- Belmadani A, Kumar S, Schipma M, Collins MA, Neafsey EJ. Inhibition of amyloid-beta-induced neurotoxicity and apoptosis by moderate ethanol preconditioning. Neuroreport. 2004;15:2093–6. doi: 10.1097/00001756-200409150-00019. [DOI] [PubMed] [Google Scholar]
- Bordji K, Becerril-Ortega J, Nicole O, Buisson A. Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid-ss production. J Neurosci. 2010;30:15927–42. doi: 10.1523/JNEUROSCI.3021-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botia B, Seyer D, Ravni A, Benard M, Falluel-Morel A, Cosette P, Jouenne T, Fournier A, Vaudry H, Gonzalez BJ, Vaudry D. Peroxiredoxin 2 is Involved in the Neuroprotective Effects of PACAP in Cultured Cerebellar Granule Neurons. Journal of Molecular Neuroscience. 2008;36:61–72. doi: 10.1007/s12031-008-9075-5. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–76. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Pocernich CB. The glutamatergic system and Alzheimer's disease: therapeutic implications. CNS Drugs. 2003;17:641–52. doi: 10.2165/00023210-200317090-00004. [DOI] [PubMed] [Google Scholar]
- Chae HZ, Robison K, Poole LB, Church G, Storz G, Rhee SG. Cloning and sequencing of thiol-specific antioxidant from mammalian brain: alkyl hydroperoxide reductase and thiol-specific antioxidant define a large family of antioxidant enzymes. Proc Natl Acad Sci U S A. 1994;91:7017–21. doi: 10.1073/pnas.91.15.7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MA, Neafsey EJ, Wang K, Achille NJ, Mitchell RM, Sivaswamy S. Moderate ethanol preconditioning of rat brain cultures engenders neuroprotection against dementia-inducing neuroinflammatory proteins: possible signaling mechanisms. Mol Neurobiol. 2010;41:420–5. doi: 10.1007/s12035-010-8138-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumming RC, Dargusch R, Fischer WH, Schubert D. Increase in expression levels and resistance to sulfhydryl oxidation of peroxiredoxin isoforms in amyloid beta-resistant nerve cells. J Biol Chem. 2007;282:30523–34. doi: 10.1074/jbc.M700869200. [DOI] [PubMed] [Google Scholar]
- Freeman D, Cedillos R, Choyke S, Lukic Z, McGuire K, Marvin S, Burrage AM, Sudholt S, Rana A, O'Connor C, Wiethoff CM, Campbell EM. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One. 2013;8:e62143. doi: 10.1371/journal.pone.0062143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Y, Ji X, Hu X, Luo Y, Zhang L, Li P, Liu X, Yan F, Vosler P, Gao Y, Stetler RA, Chen J. Transgenic overexpression of peroxiredoxin-2 attenuates ischemic neuronal injury via suppression of a redox-sensitive pro-death signaling pathway. Antioxid Redox Signal. 2012;17:719–32. doi: 10.1089/ars.2011.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci. 2003;26:81–9. doi: 10.1016/S0166-2236(02)00040-1. [DOI] [PubMed] [Google Scholar]
- Hardingham GE. Pro-survival signalling from the NMDA receptor. Biochem Soc Trans. 2006;34:936–8. doi: 10.1042/BST0340936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Lu W, Ali DW, Pelkey KA, Pitcher GM, Lu YM, Aoto H, Roder JC, Sasaki T, Salter MW, MacDonald JF. CAKbeta/Pyk2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus. Neuron. 2001;29:485–96. doi: 10.1016/s0896-6273(01)00220-3. [DOI] [PubMed] [Google Scholar]
- Kato S, Kato M, Abe Y, Matsumura T, Nishino T, Aoki M, Itoyama Y, Asayama K, Awaya A, Hirano A, Ohama E. Redox system expression in the motor neurons in amyotrophic lateral sclerosis (ALS): immunohistochemical studies on sporadic ALS, superoxide dismutase 1 (SOD1)-mutated familial ALS, and SOD1-mutated ALS animal models. Acta Neuropathol. 2005;110:101–12. doi: 10.1007/s00401-005-1019-3. [DOI] [PubMed] [Google Scholar]
- Kornhuber J, Quack G. Cerebrospinal fluid and serum concentrations of the N-methyl-D-aspartate (NMDA) receptor antagonist memantine in man. Neurosci Lett. 1995;195:137–9. doi: 10.1016/0304-3940(95)11785-u. [DOI] [PubMed] [Google Scholar]
- Krapfenbauer K, Yoo BC, Fountoulakis M, Mitrova E, Lubec G. Expression patterns of antioxidant proteins in brains of patients with sporadic Creutzfeldt-Jacob disease. Electrophoresis. 2002;23:2541–7. doi: 10.1002/1522-2683(200208)23:15<2541::AID-ELPS2541>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Krapfenbauer K, Engidawork E, Cairns N, Fountoulakis M, Lubec G. Aberrant expression of peroxiredoxin subtypes in neurodegenerative disorders. Brain Res. 2003;967:152–60. doi: 10.1016/s0006-8993(02)04243-9. [DOI] [PubMed] [Google Scholar]
- Lin CH, Huang YJ, Lin CJ, Lane HY, Tsai GE. NMDA neurotransmission dysfunction in mild cognitive impairment and Alzheimer's disease. Curr Pharm Des. 2014;20:5169–79. doi: 10.2174/1381612819666140110115603. [DOI] [PubMed] [Google Scholar]
- Lipton SA. The molecular basis of memantine action in Alzheimer's disease and other neurologic disorders: low-affinity, uncompetitive antagonism. Curr Alzheimer Res. 2005;2:155–65. doi: 10.2174/1567205053585846. [DOI] [PubMed] [Google Scholar]
- Mitchell RM, Neafsey EJ, Collins MA. Essential involvement of the NMDA receptor in ethanol preconditioning-dependent neuroprotection from amyloid-beta in vitro. J Neurochem. 2009;111:580–8. doi: 10.1111/j.1471-4159.2009.06351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munday R, Munday CM. Induction of phase II enzymes by 3H-1,2-dithiole-3-thione: dose-response study in rats. Carcinogenesis. 2004;25:1721–5. doi: 10.1093/carcin/bgh162. [DOI] [PubMed] [Google Scholar]
- Nabavi S, Fox R, Proulx CD, Lin JY, Tsien RY, Malinow R. Engineering a memory with LTD and LTP. Nature. 2014;511:348–52. doi: 10.1038/nature13294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neafsey EJ, Collins MA. Moderate alcohol consumption and cognitive risk. Neuropsychiatr Dis Treat. 2011;7:465–84. doi: 10.2147/NDT.S23159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares D, Deshpande VK, Shi Y, Lahiri DK, Greig NH, Rogers JT, Huang X. N-methyl D-aspartate (NMDA) receptor antagonists and memantine treatment for Alzheimer's disease, vascular dementia and Parkinson's disease. Curr Alzheimer Res. 2012;9:746–58. doi: 10.2174/156720512801322564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S, Soriano FX, Leveille F, Martel MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska V, McKenzie G, Craigon M, Corriveau R, Ghazal P, Horsburgh K, Yankner BA, Wyllie DJ, Ikonomidou C, Hardingham GE. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci. 2008;11:476–87. doi: 10.1038/nn2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papouin T, Oliet SH. Organization, control and function of extrasynaptic NMDA receptors. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130601. doi: 10.1098/rstb.2013.0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosini M, Simoni E, Bartolini M, Cavalli A, Ceccarini L, Pascu N, McClymont DW, Tarozzi A, Bolognesi ML, Minarini A, Tumiatti V, Andrisano V, Mellor IR, Melchiorre C. Inhibition of acetylcholinesterase, beta-amyloid aggregation, and NMDA receptors in Alzheimer's disease: a promising direction for the multi-target-directed ligands gold rush. J Med Chem. 2008;51:4381–4. doi: 10.1021/jm800577j. [DOI] [PubMed] [Google Scholar]
- Severino PC, Muller Gdo A, Vandresen-Filho S, Tasca CI. Cell signaling in NMDA preconditioning and neuroprotection in convulsions induced by quinolinic acid. Life Sci. 2011;89:570–6. doi: 10.1016/j.lfs.2011.05.014. [DOI] [PubMed] [Google Scholar]
- Sivaswamy S, Neafsey EJ, Collins MA. Neuroprotective preconditioning of rat brain cultures with ethanol: potential transduction by PKC isoforms and focal adhesion kinase upstream of increases in effector heat shock proteins. Eur J Neurosci. 2010;32:1800–12. doi: 10.1111/j.1460-9568.2010.07451.x. [DOI] [PubMed] [Google Scholar]
- Sugimoto H. The new approach in development of anti-Alzheimer's disease drugs via the cholinergic hypothesis. Chem Biol Interact. 2008;175:204–8. doi: 10.1016/j.cbi.2008.05.031. [DOI] [PubMed] [Google Scholar]
- Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. 2009;7:65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Taylor M, Davey F, Ren Y, Aiton J, Coote P, Fang F, Chen JX, Yan SD, Gunn-Moore FJ. Interaction of amyloid binding alcohol dehydrogenase/Abeta mediates up-regulation of peroxiredoxin II in the brains of Alzheimer's disease patients and a transgenic Alzheimer's disease mouse model. Mol Cell Neurosci. 2007;35:377–82. doi: 10.1016/j.mcn.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Yu XM, Askalan R, Keil GJ, 2nd, Salter MW. NMDA channel regulation by channel-associated protein tyrosine kinase. Src Science. 1997;275:674–8. doi: 10.1126/science.275.5300.674. [DOI] [PubMed] [Google Scholar]