


Abstract
The reactive transport of uranium (U) and vanadium(V) from abandoned mine wastes collected from the Blue Gap/Tachee Claim-28 mine site in Arizona was investigated by integrating flow-through column experiments with reactive transport modeling, and electron microscopy. The mine wastes were sequentially reacted in flow-through columns at pH 7.9 (10 mM HCO3−) and pH 3.4 (10 mM CH3COOH) to evaluate the effect of environmentally relevant conditions encountered at Blue Gap/Tachee on the release of U and V. The reaction rate constants (km) for the dissolution of uranyl–vanadate (U–V) minerals predominant at Blue Gap/Tachee were obtained from simulations with the reactive transport software, PFLOTRAN. The estimated reaction rate constants were within 1 order of magnitude for pH 7.9 (km = 4.8 × 10−13 mol cm−2 s−1) and pH 3.4 (km = 3.2 × 10−13 mol cm−2 s−1). However, the estimated equilibrium constants (Keq) for U–V bearing minerals were more than 6 orders of magnitude different for reaction at circumneutral pH (Keq = 10−38.65) compared to acidic pH (Keq = 10−44.81). These results coupled with electron microscopy data suggest that the release of U and V is affected by water pH and the crystalline structure of U–V bearing minerals. The findings from this investigation have important implications for risk exposure assessment, remediation, and resource recovery of U and V in locations where U–V-bearing minerals are abundant.
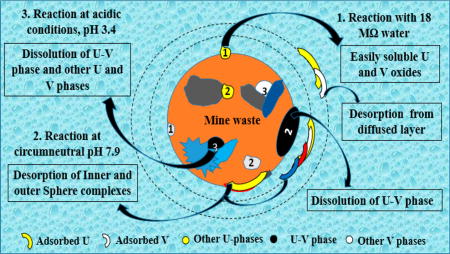
Graphical abstract
INTRODUCTION
The legacy of mill tailings and wastes from mining operations have resulted in the release of elevated concentrations of metals and radionuclides, which pose imminent environmental and human health concerns.1, 2 For instance, the negative health impacts of human exposure to uranium (U) and vanadium(V) through inhalation and ingestion have been well documented.3, 4 Uranium and V co-occur at numerous abandoned mine waste sites on the Navajo Nation territory near the four corners region of the southwestern U.S.5, 6 Uranium and other co-occurring metals from these mine wastes can be released into neighboring water resources affecting their water quality.
The aqueous and solid chemical speciation of U and V at mine waste sites could be affected by a variety of interfacial geochemical processes. These mine waste sites are usually exposed to ambient surface oxidizing conditions in which U(VI) is predominant.6, 7 Aqueous U(VI) can complex with OH− and CO32−, or react with alkaline earth metals and CO32−to form neutral or negatively charged ternary complexes (e.g., U-Ca-CO32−) that affect U mobility in water.8, 9 Vanadium preferentially forms anionic [V10O26(OH)24−, V10O27(OH)5−, V10O286−, VO2(OH)2−, VO3(OH)2−, VO43−, V2O6(OH)3−, V2O74−, V3O93−, and V4O124−], cationic [VO2+], and neutral [VO(OH)3] species of oxyanions and aqueous complexes.10, 11 These oxyanions and aqueous complexes of V and U(VI) can adsorb onto charged surfaces of Al oxides, Fe oxides, and clay minerals.9, 11–18 Uranium and V can also be present in solid form as oxide, phosphate, carbonate, vanadate, and silicate mineral phases.19–22 For instance, their co-occurrence as carnotite [K2(UO2)2V2O8], tyuyamunite [Ca(UO2)2V2O8·(5–8)H2O], metahewettite [CaV6O16·3(H2O)], melanovanadite [CaV4O10·5(H2O)], and V-bearing clays has been observed at the Rough Rock Sandstone of the Cretaceous Mesa Verde Group study region located in southwestern US.23 Uranium oxides such as schoepite [(UO2)8O2(OH)12·12(H2O)] are commonly occurring minerals that also influence uranyl solubility under oxic and suboxic environments between pH 6–9.24, 25 These geochemical processes, which are key to the aqueous and solid speciation of U and other co-occurring metals, should be considered when investigating the reactive transport of metals in abandoned mine wastes.
Reactive transport modeling can be a useful tool to investigate the interfacial mechanisms that affect the surface and subsurface mobilization of U and other metals. Previous studies have used reactive transport modeling to investigate the adsorption behavior of U(VI) complexes onto alluvial aquifer sediments.26, 27 The subsurface desorption kinetics of U(VI) species suggest a nonequilibrium sorption behavior with Fe-oxides, Mn-oxides, and phyllosilicates.7, 28, 29 Uranium transport can also be attributed to dissolution of mineral phases. For example, U transport from contaminated subsurface Hanford sediments was affected by the dissolution of metatorbernite [Cu(UO2)2(PO4)2·8H2O], cuprosklodowskite (Cu[(UO2)-(SiO2OH)]2·6H2O), and Na-boltwoodite [NaUO2(SiO3OH)·1.5H2O].28, 30, 31 The development of similar reactive transport models that represent environmentally relevant oxidizing conditions in abandoned mine waste sites is necessary to better understand the mobility of U, V, and other metals.
Previous laboratory-based studies have shown that U and V can interact through precipitation reactions forming uranyl vanadate minerals with potassium and calcium (e.g., carnotite [K2(UO2)2(VO4)2·3H2O] and tyuyamunite [Ca(UO2)2V2O8· 5–8(H2O)]).21, 32 These studies used batch experiments and thermodynamic predictions to identify precipitation of uranyl vanadates as an effective method to immobilize U and V below the U-maximum contaminant level (MCL, 30 µg L−1) between pH 5.5–7.21, 32 The thermodynamic estimates in these studies were in agreement with those previously calculated by Langmuir.33 However, these theoretically estimated thermodynamic constants are limited to synthetic minerals that have been subjected to specific reactivity conditions which could differ from those of U–V bearing minerals occurring in the environment.
A recent study conducted by our research group at the Blue Gap/Tachee Claim-28 mine site located in Northeastern Arizona reported that the dissolution of U–V bearing minerals affects the mobility of U and V at pH conditions observed in the field, which range from 3.8 to 7.4.6 Results showed that U and V co-occur in mine wastes predominantly as U(VI) and V(V). The Extended X-ray absorption fine structure (EXAFS) fittings suggest that U and V coordination in these mine waste samples is similar to that of minerals such as carnotite or tyuyamunite. Additionally, results from this study suggest dissolution of these U–V bearing minerals as a potential source to the elevated metal concentrations in neighboring surface water resources.6 Although this study represents a valuable first attempt to identify the co-occurrence of U and other metals at this abandoned mine waste site, the mechanisms affecting the reactive transport of U, V, and other metals remain unknown.
The objective of this study is to investigate the reactive transport of U and V from the Blue Gap/Tachee Claim-28 mine waste site through laboratory experiments, reactive transport modeling, and electron microscopy. In the current investigation we seek to determine the equilibrium and reaction rate constants for dissolution of these U–V bearing minerals at circumneutral and acidic pH, by modeling the batch and column experiments performed in this study with PFLO-TRAN.27, 28, 33 This study was developed based on our prior evidence that suggested U–V bearing mineral phases as the potential source to U and V release from Blue Gap/Tachee mine wastes.6 Our integrated approach of using microscopy and reactive transport modeling serves as a foundation to better understand the mechanisms affecting the reactivity of mine wastes and the transport of U and V under environmentally relevant conditions.
MATERIALS AND METHODS
Materials
We collected abandoned mine waste and background sediment samples from the Blue Gap/Tachee Claim-28 mine site in January 2014 to identify the processes and phases that affect the U and V transport into neighboring surface and groundwater resources. Sediment samples from the site included mine waste and baseline reference soil (background soil) previously characterized by Blake et al, 2015.6 The Blake et al. study used powder X-ray diffraction to identify quartz (SiO2; 59%) potassium feldspar (KAlSi3O8; 34%), and kaolinite (Al2Si2O5(OH)4; 7%) as the major mineralogical constituents. Additionally the co-occurrence of U and V, and predominance of U(VI) and V(V) were identified using microscopy, X-ray photoelectron spectroscopy (XPS), and X-ray absorption near edge spectroscopy (XANES) techniques. Furthermore, the coordination numbers obtained from EXAFS fits suggested the structure of the U–V bearing mineral to be similar to that of carnotite or tyuyamunite.6
Batch Experiments
We conducted batch experiments to model the U and V release at circumneutral (8.3) and acidic (3.8) pH, and determined the equilibrium and reaction rate constants for dissolution of U–V bearing mineral phases. The determined equilibrium constant was then used to obtain the reaction rate constants for U–V bearing mineral dissolution during column experiments. Briefly in the batch experiments, we reacted 0.1 g of mine wastes and background soil samples that were sieved to <63 µm size (using U.S. standard sieve series number 230), with 50 mL 10 mM HCO3− (pH 8.3) and 10 mM C6H8O6 (pH 3.8) through gentle shaking in 50 mL centrifuge tubes for 264 h, as described in Blake et al.6 During the experiments we collected 2 mL aliquots 0.5, 1, 2, 6, 24, 48, 96, and 264 h after the experiment began. We diluted these aliquots and analyzed for elemental concentrations using inductively coupled plasma optical emission spectroscopy and mass spectroscopy (ICP-OES/MS). All batch experiments were conducted in triplicate.
Continuous Flow through Column Experiments
We conducted column experiments to investigate the transport of U and V from Blue Gap/Tachee sediments. An initial set of experiments consisted of reacting mine waste and background soil with 18MΩ water at pH 5.4 to evaluate the release of water-soluble U and V. Another set of experiments was performed in parallel for which the mine waste and background soil were sequentially reacted at pH 7.9 (using 10 mM HCO3−) and pH 3.4 (using 10 mM CH3COOH). These pH values and concentrations represent environmentally relevant conditions observed at the study site.6 We prepared samples by crushing and homogenizing sediments with a Spex shatter box and sieving through U.S. standard sieve with series 120-45 to obtain particle sizes between 125 and 355 µm. The homogenized sieved fractions were then packed into cylindrical Pyrex glass columns with an inner diameter of 1 cm and a height of 10 cm using vacuum filter pumps. Solutions of 10 mM HCO3− and 10 mM CH3COOH were prepared using 18MΩ deionized water, NaHCO3 (powder), and 17.4N CH3COOH. The prepared solutions were pumped against gravity through the packed columns at 0.9 mL min−1 using a Masterflex L/S series 8-channel peristaltic pump. The column experiments were conducted on eight columns in parallel (Figure S1). We chose 0.9 mL min−1 based on existing literature7, 28, 34 as a typical groundwater rate to evaluate how solutions at circumneutral and acidic conditions would react with mine wastes. Columns 1, 2, 5, and 6 were packed with mine waste and columns 3, 4, 7, and 8 were packed with background soil. Columns 1–4 were reacted with 18MΩ water for 90 min with a total of 23 (columns 1–2) and 31 (columns 3–4) pore volumes (PV) at the rate of 3.53 and 2.65 cm3 PV−1, respectively. Columns 5–8 were sequentially reacted for 1 week each (10 080 min) with 10 mM HCO3− and 10 mM CH3COOH (Figure S1). The 1 week reaction time resulted in 2570 PV for columns 5 and 6, and 3450 PV for columns 7 and 8. Samples were collected at 10, 20, 30, 40, 50, 60, 70, 80, and 90 min of reaction time from the outlets of columns 1, 2, 3, and 4. Samples were collected from columns 5–8, at 30, 180, 540, 1440, 2880, 4320, 5760, 7,200 and 10 080 min after sequential reaction with 10 mM HCO3− and 10 mM CH3COOH solutions. The sample aliquots were filtered using a 0.45 µm syringe filter and diluted accordingly for analyses. All experiments were performed at room temperature (25 °C) and, consequently, that temperature was set for the reactive transport of modeling.
Aqueous Chemical Analyses
We used a PerkinElmer Optima 5300DV inductively coupled plasma-optical emission spectrometer (ICP-OES) and a PerkinElmer NexION 300D (Dynamic Reaction Cell) inductively coupled plasma-mass spectrometer (ICP-MS) to obtain the trace metal concentrations in experimental aliquots and total acid extracts of mine waste and background soil. The solution pH was measured using a Thermo Scientific Orion Versastar Advanced Electrochemistry pH meter. Additional technical details of these methods are described in the Supporting Information.
Reactive Transport Modeling
We used PFLOTRAN a surface-subsurface multiphase, multicomponent reactive flow and transport model28, 29, 35 to determine the equilibrium constants for U–V bearing mineral dissolution at circumneutral and acidic pH (batch experiments) and identify the processes controlling the reactive transport of U and V from mine waste (column experiments).
To model the batch and column experiments we used, various U and V bearing aqueous species and mineral, their associated thermodynamic equilibrium constants (Keq) and stoichiometric reaction coefficients (Tables S1 and S2), as inputs for PFLOTRAN. Most of the listed parameters were fixed, except the equilibrium constant of carnotite and reaction rate constants of the U–V bearing minerals, metaschoepite [UO3·2H2O] and rutherfordine [(UO2)CO3] which were varied to fit the experimental data and obtain the equilibrium and reaction rate constants for dissolution of U–V bearing minerals. At circumneutral pH, minerals such as metaschoepite and rutherfordine were considered in the model as they are commonly occurring oxides and carbonates of U that affect U transport.24, 36–38 In addition to metaschoepite and rutherfordine, surface complexation reactions for > SOUO2OH and > SOHUO2CO3 were also considered in order to understand the effect of sorption on the reactive transport of U.39 The equilibrium constants estimated for U–V bearing minerals by modeling the batch experiments at circumneutral and acidic pH were used to estimate their reaction rate constants during column experiments.
Some key assumptions made in the model include the following: (1) U–V bearing minerals are the dominant U phase in the mine waste sample. (2) U–V bearing minerals have similar properties to those of carnotite. (3) U–V bearing mineral particles are spherical in shape. These assumptions were made on the basis of our previously published information on the Blue Gap/Tachee mine site, where the EXAFS fits of U–V bearing minerals (that were dominant in the mine waste) suggested similar coordination as carnotite or tyuyamunite. The spherical shape of the U–V bearing minerals was assumed for calculation simplifications.
Using PFLOTRAN, we modeled the water movement through the column considering fully saturated sediments under oxidizing conditions. The governing mass conservation equation that accounts for the unidirectional change in the total dissolved concentration of the jth primary species is given by (eq 1):
| (1) |
where z [L] is the height of the column (10 cm), t [T] is the time for which column experiments were conducted ψj [ML−3] denotes the total concentration, D [L−2 T−1] denotes hydrodynamic dispersion coefficient, Qj [M L−3 T−1 ] is a source/sink term, Im [M L−3 T−1] is the reaction rate of the mth mineral, vjm[−] is the stoichiometric coefficient for mineral reactions, Sj [ML−3] is the sorbed concentration, q [L T−1] is Darcy’s flux calculated to be approximately 1.15 cm min−1 based on the flow rate used for the column experiments, 0.9 mL min−1 and φ [−] is porosity. Dirichlet boundary conditions were used to model the 1D reactive transport of metals during reaction at pH 7.9 (using 10 mM HCO3−) and pH 3.4 (using 10 mM CH3COOH). Additional information and other supporting equations used in the model are explained in the Supporting Information. (SI eq 2–13)
Solids Characterization
Solid characterization using JEOL 2010 high resolution transmission electron microscopy (HR-TEM) was performed to determine the crystallinity of U–V bearing minerals in the mine waste sample. Synchrotron micro- X-ray fluorescence mapping (µ-SXRF) was conducted in beamline (BL) 10-2 in Stanford Synchrotron Radiation Lightsource (SSRL). Additional technical details of these methods are described in the Supporting Information.
RESULTS AND DISCUSSION
Mine Waste Reactivity in Columns Using 18MΩ Water (pH 5.4)
We investigated the potential release of water-soluble U and V from mine waste solids by reacting columns with 18MΩ water as a function of pore volumes and time (Figure S2). Aqueous concentrations as high as 2.3 × 10−7 M U and 74 × 10−6 M V were measured. For reference, the concentrations of U released from this reaction (~2.3 × 10−7 M) were similar to the concentrations observed in one of the water sources proximate to the site (spring concentrations equal to 2.8 × 10−7 M), as was shown in a previous study conducted by our group. These mobilized concentrations were almost two times higher than the regulated U.S. Environmental Protection Agency maximum contaminant level (MCL) of U, that is,1.3 × 10−7 M (30 µg L−1)40. The U and V concentrations released after the reaction of background soil (control) with 18MΩ water were either below the MCL for U or below the detection limit of the ICP-OES/MS instruments. The presence of U and V in the effluent solution can be attributed to possible contributions from electrostatically bound chemical species in the diffuse layer or dissolution of soluble oxide phases, such as schoepite [(UO2)8O2(OH)12·12(H2O)] and V oxides as observed in other investigations.41–43 The mobility of such labile aqueous species from mine waste is environmentally relevant. Further investigation of the reactive transport of U and V based on co-occurring U–V bearing mineral phases was conducted by sequentially reacting mine waste and background soil with circumneutral and acidic reagents.
Reactive Transport of U and V at Circumneutral pH
Batch Experiments at pH (8.3)
The reactive transport model developed for batch experiments reacting mine waste with 10 mM HCO3− identified dissolution of U–V bearing minerals as the key process affecting the dissolved concentrations of U and V (Figure 1A,B). The equilibrium constant (Keq) for U–V bearing mineral dissolution was estimated to be 10−44.81 assuming the stoichiometry of carnotite (Table 1), which was almost 12 orders of magnitude lower than that reported in the literature for carnotite (Keq = 10−56.38).44 The difference between the Keq for dissolution of U–V bearing minerals and carnotite can be attributed to a difference in reactivity conditions and crystallinity between U–V bearing minerals in the mine wastes and carnotite. In addition, the estimated reaction rate constant (km) for U–V mineral dissolution was 2.13 × 10−14 mol cm−2 s−1 (Table 1). Similarly, the estimated km values for precipitation of metaschoepite and rutherfordine were approximately 1 × 10−15 and 2 × 10−16 mol cm−2 s−1, respectively (Table S3). The nature of a reaction (dissolution or precipitation) can be identified from the sign convention of the reaction rate (Im) appearing in PFLOTRAN, that is, negative for dissolution and positive for precipitation. These results are in agreement with observations previously made at the nearby Colorado Plateau deposits, where precipitation of metaschoepite from aqueous U concentrations between pH 6 and 10 was reported.24 The equilibrium constants estimated by modeling these batch experiments at circumneutral conditions were then used as a foundation for identifying the reaction rate constants for U–V bearing mineral dissolution in the column experiments.
Figure 1.

Measured and simulated effluent concentrations and reactive transport model (PFLOTRAN) of U and V, from mine waste (circle) and background soil (squares) during batch and continuous flow-through column experiments at circumneutral pH (using 10 mM HCO3−). (A) U concentrations from batch experiments versus time; (B) V concentration from batch experiments versus time; (C) U concentrations from column experiments versus pore volumes; (D) V concentration from column experiments versus pore volumes. The curve fitting resulting from the reactive transport model are presented with dashed lines.
Table 1.
Parameters of U–V Bearing Minerals Estimated by Modelling the Reactive Transport of U and V during Mine Waste Reaction at Circumneutral and Acidic pHa
| experiments | equilibrium constant (Keq) |
average U–V bearing mineral surface area (cm2 cm−3) |
reaction rate constants (km) (mol cm−2 s−1) |
effective reaction rate constant (mol cm−3 s−1) |
|---|---|---|---|---|
| batch circumneutral (pH 8.3) (<63 µm) | 10−44.81 | 468.8 | 2.13 × 10−14 | 9.99 × 10−12 |
| batch acidic (pH 3.8)(<63 µm) | 10−38.65 | 468.8 | 6.4 × 10−14 | 3 × 10−11 |
| column circumneutral (pH 7.9) (120–355 µm) | 10−44.81 | 62.5 | 4.8 × 10−13 | 3 × 10−11 |
| column acidic (pH 3.4) (120–355 µm) | 10−38.65 | 62.5 | 3.2 × 10−13 | 2 × 10−11 |
The surface area of U–V bearing minerals was estimated using the average U–V bearing mineral diameter (Table S2) and equation 13. The effective reaction rate constant accounts for the effect of grain surface area on the reactive transport of U and V, where k is the reaction rate constant and is the surface area of U–V bearing minerals.
Column Experiments at pH (7.9)
A linear correlation [with a slope of 1.45 and a R2 of 0.993] observed between molar concentrations of U and V (Figure S3A) that were mobilized during mine waste reaction with 10 mM HCO3−, suggested dissolution of U–V bearing mineral phases as a potential source. These results agree with a previous study from our research group in which a linear correlation between U and V was observed (slope= 0.636 and R2 = 0.95) in batch experiments reacting mine wastes with 10 mM HCO3− at pH 8.3.6 The U and V release patterns obtained from these experiments were then simulated using PFLOTRAN to further understand the processes affecting the reactive transport of U and V.
Similar to modeling results from the batch experiments, the reactive transport model for flow-through column experiments on reaction with 10 mM HCO3− suggested dissolution of U–V bearing minerals as the primary mechanism affecting U and V transport. The effluent concentrations of U and V reported as a function of pore volumes (Figure 1C,D) and time (Figure S4), were compared to those obtained from the reactive transport model (PFLOTRAN). PFLOTRAN used Keq = 10−44.81 for U–V bearing mineral dissolution estimated from batch experiments by assuming the stoichiometry of carnotite, to adequately fit the flow-through column experiments data and estimate the km and fitting parameter (nm) for U–V bearing mineral phase dissolution. The fitting parameter nm, to which the nonlinear water–rock reactions are sensitive (SI eq 8), and km were estimated to be 5.2, and 4.8 × 10−13 mol cm−2 s−1, respectively. In addition, the negative reaction rate (Im) indicated the dissolution of U–V bearing minerals. The km estimated for the dissolution of U–V bearing minerals (4.8 × 10−13 mol cm−2 s−1) during column experiments was different from that estimated for the batch experiments that is, km = 2.13 × 10−14 mol cm−2 s−1 (Table 1). The increase in km cannot specifically be attributed to a single process. However, the difference in the effective reaction rate constant (keffective = km × am0) estimated using the average surface area, can be attributed to differences in grain size and reactivity conditions, which include flow and heterogeneity in sample composition (Table 1). Observing the effect of grain size on the reactive transport of U and V is also important45, 46 as the sieved samples may contain nanocrystals of U–V and other co-occurring U and V minerals, bound electrostatically to the surface of coarser particles.47 After the effect of grain size was introduced into PFLOTRAN, by adding U–V bearing minerals of variable sizes and surface areas, their effective reaction rate constants (keffective) were estimated (Table S4). The change in the estimated keffective for various U–V bearing mineral phases was negligible (Table S4), leading to the inference that the effect of grain size on the reactive transport of U and V is minimal (Figure S5).
Other secondary mineral phases of U such as oxides, phosphates, silicates, and vanadates soluble at circumneutral pH that have not been considered in this model could also contribute to the release of U and V from mine waste.32, 41, 48, 49 For instance, a back scatter electron-scanning electron (BSE-SEM) micrograph which shows the association of V with Fe and K was collected (Figure S6). The co-occurrence of U with other elements (e.g., Se and Sr) was also observed using synchrotron µ-XRF mapping (Figure S7). The contribution of these secondary mineral phases can be expected, especially considering (1) the heterogeneity of mine waste, (2) discrepancies between experimental and model U concentrations, and (3) that the early time-behavior of V was higher in the experimental data than the model (see Figure 1D).43 However, after 3–4 h of reaction, the experimental V concentrations agreed well with the model indicating dissolution of U–V bearing minerals. Following the column experiments with 10 mM HCO3− the mine waste and background soil were sequentially reacted under acidic conditions to further understand the reactive transport of U and V.
Reactive Transport of U and V at Acidic pH
Batch Experiments at pH (3.8)
The reactive transport model for batch reactions with mine waste under acidic conditions (pH 3.8) suggests dissolution of the U–V bearing mineral phase as a primary contributor to the reactive transport of U and V. We used PFLOTRAN to fit the time behavior of U and V and obtain the equilibrium (Keq = 10−38.65) and reaction rate constant (km = 6.4 × 10−14 mol cm−2 s−1) for U–V bearing mineral dissolution by assuming the stoichiometry of carnotite (Figure 2A,B). As observed in the column experiments conducted at circumneutral pH, the estimated Keq under acidic conditions was different from that of synthetic carnotite (Keq = 10−56.38).44 Additionally, Keq estimated under acidic conditions (Keq = 10−38.65) was also different from that obtained at circumneutral conditions (Keq = 10−44.81), suggesting the possible dissolution of two different uranyl vanadates. Furthermore, the difference between the Keq of U–V bearing minerals and carnotite at acidic pH can be attributed to difference in reactivity conditions and crystallinity as indicated previously in the reactive transport model at circumneutral pH. We also observed a negative reaction rate (Im) for U–V bearing minerals that represents their dissolution at acidic pH. The Keq for U–V bearing mineral phase dissolution identified by modeling the batch reaction under acidic conditions, was then used to estimate the reaction rate constants for U–V bearing mineral dissolution during column experiments.
Figure 2.

Measured and simulated effluent concentrations and reactive transport model (PFLOTRAN) of U and V, from mine waste (circle) and background soil (squares) during batch and continuous flow-through column experiments at acidic pH (using 10 mM C6H8O6 and CH3COOH). (A) U concentrations from batch experiments versus time; (B) V concentration from batch experiments versus time; (C) U concentrations from column experiments versus pore volumes; (D) V concentration from column experiments versus pore volumes. The curve fittings resulting from the reactive transport model are presented with dashed lines.
Column Experiments at pH (3.4)
Consistent with the results obtained for experiments at circumneutral pH, the dissolution of U–V bearing minerals was identified as the key process affecting the reactive transport of U and V during column experiments under acidic conditions. A linear correlation (slope of 0.862 and a R2 of 0.983) between effluent molar concentrations of U and V was also observed in column experiments under acidic conditions (Figure S3B). However, the rates of dissolution obtained for column experiments at acidic conditions are higher compared to those at circumneutral conditions (Table 1). Given that the pKa of CH3COOH is 4.74, >90% of CH3COOH remains protonated. Therefore, a greater contribution from dissolution by acetic acid (protonated form) over complexation or reduction reactions caused by acetate (nonprotonated form) at pH 3.4 is expected.50 These results agree with observations made in our previous study at Blue Gap/Tachee, in which linear correlations with a slope of 1.08 and a R2 of 0.996 between molar concentrations of U and V were observed.6 Additionally, 99.1% U and 92.8% V of the total elemental content present in the mine waste was extracted through the sequential reaction of solids in column experiments with 10 mM HCO3− and CH3COOH. Note that the acid extractable content of unreacted mine wastes was 1912.12 mg kg−1 U and 858.01 mg kg−1 V (Table S5). The U and V concentrations released during these continuous sequential reactions showed strong agreement with the modeled U and V concentrations, suggesting dissolution of U–V bearing minerals as a primary contributor to the reactive transport of U and V. Different Keq and km values for the dissolution of U–V bearing minerals were obtained for mine waste reaction under circumneutral and acidic conditions during flow-through column experiments (Figure 2C,D and Figure S8). The reactive transport model constructed with a known Keq for metaschoepite (−105.26)42 and Keq for U–V bearing mineral phase (−10−38.65) estimated from a batch reaction under acidic conditions by assuming the stoichiometry of carnotite, was used to identify the km constants for dissolution of U–V bearing minerals. The estimated km for the U–V bearing mineral phase, 3.2 × 10−13 mol cm−2 s−1, was slightly different from those estimated for batch reactions (km = 6.4 × 10−14 mol cm−2 s−1) (Table 1). As discussed previously for column experiments at circumneutral pH (7.9), this decrease in km for U–V bearing mineral dissolution cannot specifically be attributed to a single process. However, the change in the effective reaction rate constant (keffective) can be attributed to differences in grain size and reactivity conditions which include flow and heterogeneity in sample composition (Table 1). The effect of grain size was not particularly tested for the sequential reaction of mine waste with CH3COOH, as the electrostatically bound micrometer sized U–V bearing minerals would already be lost during reaction with 10 mM HCO3−. In addition, the fitting parameter “nm”, necessary to model the nonlinear dissolution of U–V bearing mineral phase was estimated to be 4.5 (SI eqs 8). Our PFLOTRAN simulations also estimated the reaction rate (Im) of U–V bearing mineral phase to be negative suggesting dissolution of the U–V bearing mineral phase. However, the reaction rate for metaschoepite and rutherfordine remained unchanged suggesting minimal contribution from metaschoepite and rutherfordine in the reactive transport of U and V. Although, our experimental and model concentrations agree very well with each other, the early release of lower experimental U and V concentrations can be attributed to (1) sample heterogeneity; (2) restricted access to incorporated U–V bearing mineral phases; and (3) precipitation of an unidentified U and V phase.
The difference in the reactive transport of U and V after reaction under circumneutral and acidic conditions could be attributed to the dissolution of different coexisting U–V bearing mineral phases, possibly amorphous and crystalline, as identified in the unreacted mine waste. The coexistence of amorphous and crystalline U–V bearing minerals was identified through selected area electron diffraction (SAED) and energy dispersive spectroscopy (EDS) analysis performed using a high resolution-transmission electron microscopy (HR-TEM) (Figure 3). For instance, Figure 3A,D represents the back scatter electron image (BSE) of two nanoparticulate grains that were identified as uranyl vanadates co-occurring with other minerals based on the EDS elemental composition spectrum in Figure 3B,E. Diffused rings in the SAED pattern (Figure 3C) suggested the identified U–V bearing mineral phase in Figure 3A to be amorphous. However, unlike Figure 3C, definite patterns in Figure 3F suggested the presence of crystalline U–V bearing mineral that co-occurs with other crystalline mineral phases within a particular grain of the mine waste sample. It is likely that the dissolution of the amorphous U–V bearing mineral phase is dominant at circumneutral conditions (pH 7.9) and the dissolution of crystalline U–V bearing mineral phases is dominant under acidic conditions (pH 3.4). However, further analysis is necessary to better understand the effect of minerology, specific stoichiometry, and crystallinity on the reactive transport of U and V from these mine wastes. The coexistence of crystalline and amorphous U–V bearing minerals in unreacted mine waste samples can be a result of nucleation51, 52 or polymorphism,53 which are common in natural samples, due to their heterogeneity in site and reactivity conditions. These results agree with observations made in other studies that have shown that crystallinity can affect the solubility of mineral phases.54, 55 However, the amorphous characteristics in the U–V bearing mineral phase observed in Figure 3C could also be caused by rapid amorphization due to short-term exposure to the electron beam.56, 57 Therefore, our future work will investigate the crystal chemistry of these U–V bearing minerals.
Figure 3.

Transmission electron microscopy (TEM) images, energy dispersive spectroscopy (EDS) spectra and selected area electron diffraction (SAED) patterns for unreacted mine waste samples indicating the co-occurrence of amorphous and crystalline U–V bearing minerals: (A, D) TEM images of U–V bearing mineral phases; (B, E) EDS spectra identifying the presence of U–V bearing minerals; (C, F) SAED patterns confirming the co-occurrence of amorphous and crystalline U–V bearing mineral phase.
It is possible that other secondary mineral phases such as uranium-bearing oxides, hydrous ferric oxides, and uranium phosphates that have not been considered in the reactive transport model but are soluble at acidic pH, can also affect the reactive transport of U and V.42, 58, 59 For instance, analyses conducted using synchrotron-based micro-XRF mapping on the mine waste sample indicate that U co-occurs with other elements (Figure S7). Despite the chemical complexity of these samples, our reactive transport model can reasonably represent the release of U and V by considering the dissolution of U–V bearing minerals as the key contributing process. However, we acknowledge that contributions from other mineral phases, heterogeneity, and other aqueous processes were not accounted in the reactive transport for U and V, which represents a limitation of this study. Therefore, future research should focus on trying to understand the role of other reaction processes such as the contribution of other secondary mineral/nano-particulate phases of U and V, and the influence of mineral structure of the U–V bearing minerals on the reactive transport U and V from such mine waste sites.
Environmental Implications
This work integrates reactive transport modeling, electron microscopy, and aqueous chemistry methods to evaluate the mechanisms affecting the transport of U and V from mine wastes from a site in northeastern AZ. Outcomes from this investigation include the determination of solubility and reaction rate constants for the dissolution of U–V bearing mineral phases at circumneutral and acidic pH, that can serve as a foundation to better understand their reactivity at relevant field conditions. This information can be useful to better understand the mobility of U and V in neighboring community water sources to assess risks for human exposure. Additionally, the identification of factors affecting the dissolution of U–V bearing minerals under environmentally relevant conditions evaluated in this study is relevant to inform remediation and resource recovery initiatives in sites where these U–V bearing minerals are abundant. For instance, the results from this study have implications for other abandoned uranium mines in the Colorado Plateau, South Dakota (Black Hills), southwest China, southern Jordan, and the calcreted drainages of arid and semiarid western and southern Australia where carnotite, tyuyamunite and other U–V bearing minerals are commonly found.24, 46, 60–65
Spectroscopy and microscopy analyses suggest that U is associated with different elements, which is indicative of the complex mineralogy of these solids. These results are consistent with those reported in the previous study from our research group.6 Despite the chemical and mineralogical complexity of these mine wastes, dissolution of U–V bearing minerals was identified as the key mechanism controlling the reactive transport of U and V under the environmentally relevant conditions selected for this study. In addition, the integrated methodology used for this study is transferable to other mining and milling sites, and abandoned U mine waste sites where a better understanding of the reactive transport of U and its co-occurring elements is necessary.
Supplementary Material
Acknowledgments
The authors would like to specially recognize Aaron Yazzie and the Blue Gap Tachee Chapter for supporting this work. We also acknowledge the contributions of Chris Shuey (sample collection), Michael Spilde (microprobe analyses), Ying-Bing Jiang (TEM analyses), Juan Lezama-Pacheco (µ-SXRF) and Dr. Glenn Hammond (PFLOTRAN). Special thanks to Dr. Lynn Beene, and Dr. Lucia Rodriguez-Freire for their support and thoughtful comments, which contributed to significantly improve this manuscript. Part of this research was carried out at the Stanford Synchrotron Radiation Light source, a national user facility operated by Stanford University on behalf of the US DOE-OBER. Funding for this research was provided by the National Science Foundation (Grants NM EPSCoR #IIA-1301346, CREST 1345169, and CAREER 1652619) and the National Institute of Environmental Health Sciences Superfund Research Program (Award 1 P42 ES025589). Any opinions, findings, and conclusions or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the views of the National Science Foundation or the National Institutes of Health.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.est.7b03823.
Additional materials and methods, five tables (S1 to S5), and eight figures (S1 to S8) (PDF)
Footnotes
AUTHOR INFORMATION
Notes
The authors declare no competing financial interest.
References
- 1.Dawson SE. Navajo Uranium Workers and the Effects of Occupational Illnesses - a Case-Study. Hum. Organ. 1992;51(4):389–397. [Google Scholar]
- 2.Hopenhayn C. Arsenic in drinking water: Impact on human health. Elements. 2006;2(2):103–107. [Google Scholar]
- 3.Byczkowski JZ, Kulkarni AP. Oxidative stress and pro-oxidant biological effects of vanadium. Adv. Environ. Sci. Technol.-New York. 1998;31:235–264. [Google Scholar]
- 4.Hund L, Bedrick EJ, Miller C, Huerta G, Nez T, Ramone S, Shuey C, Cajero M, Lewis J. A Bayesian framework for estimating disease risk due to exposure to uranium mine and mill waste on the Navajo Nation. J. R. Statist. Soc. Ser. A. 2015;178(4):1069–1091. [Google Scholar]
- 5.Chenoweth W. Vanadium mining in the Carrizo Mountains, 1942–1947. San Juan County, New Mexico and Apache County Arizona. Vol. 378. New Mexico Bureau of Mines and Mineral Resources; 1991. p. 33. Open-file Report. [Google Scholar]
- 6.Blake JM, Avasarala S, Artyushkova K, Ali A-MS, Brearley AJ, Shuey C, Robinson WP, Nez C, Bill S, Lewis J. Elevated concentrations of U and co-occurring metals in abandoned mine wastes in a northeastern Arizona Native American community. Environ. Sci. Technol. 2015;49(14):8506–8514. doi: 10.1021/acs.est.5b01408. [DOI] [PubMed] [Google Scholar]
- 7.Qafoku NP, Zachara JM, Liu C, Gassman PL, Qafoku OS, Smith SC. Kinetic desorption and sorption of U(VI) during reactive transport in a contaminated Hanford sediment. Environ. Sci. Technol. 2005;39(9):3157–3165. doi: 10.1021/es048462q. [DOI] [PubMed] [Google Scholar]
- 8.Dong W, Brooks SC. Determination of the Formation Constants of Ternary Complexes of Uranyl and Carbonate with Alkaline Earth Metals (Mg2+, Ca2+, Sr2+, and Ba2+) Using Anion Exchange Method. Environ. Sci. Technol. 2006;40(15):4689–4695. doi: 10.1021/es0606327. [DOI] [PubMed] [Google Scholar]
- 9.Bargar JR, Reitmeyer R, Davis JA. Spectroscopic confirmation of uranium (VI)-carbonato adsorption complexes on hematite. Environ. Sci. Technol. 1999;33(14):2481–2484. [Google Scholar]
- 10.Naeem A, Westerhoff P, Mustafa S. Vanadium removal by metal (hydr)oxide adsorbents. Water Res. 2007;41(7):1596–1602. doi: 10.1016/j.watres.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 11.Burke IT, Mayes WM, Peacock CL, Brown AP, Jarvis AP, Gruiz K. Speciation of arsenic, chromium, and vanadium in red mud samples from the Ajka spill site, Hungary. Environ. Sci. Technol. 2012;46(6):3085–3092. doi: 10.1021/es3003475. [DOI] [PubMed] [Google Scholar]
- 12.Peacock CL, Sherman DM. Vanadium (V) adsorption onto goethite (α-FeOOH) at pH 1.5 to 12: a surface complexation model based on ab initio molecular geometries and EXAFS spectroscopy. Geochim. Cosmochim. Acta. 2004;68(8):1723–1733. [Google Scholar]
- 13.Catana G, Rao RR, Weckhuysen BM, Van Der Voort P, Vansant E, Schoonheydt RA. Supported vanadium oxide catalysts: Quantitative spectroscopy, preferential adsorption of V4+/5+, and Al2O3 coating of zeolite Y. J. Phys. Chem. B. 1998;102(41):8005–8012. [Google Scholar]
- 14.Sylwester E, Hudson E, Allen P. The structure of uranium (VI) sorption complexes on silica, alumina, and montmorillonite. Geochim. Cosmochim. Acta. 2000;64(14):2431–2438. [Google Scholar]
- 15.Stewart BD, Cismasu AC, Williams KH, Peyton BM, Nico PS. Reactivity of Uranium and Ferrous Iron with Natural Iron Oxyhydroxides. Environ. Sci. Technol. 2015;49(17):10357–10365. doi: 10.1021/acs.est.5b02645. [DOI] [PubMed] [Google Scholar]
- 16.Liu C, Shang J, Shan H, Zachara JM. Effect of subgrid heterogeneity on scaling geochemical and biogeochemical reactions: A case of U(VI) desorption. Environ. Sci. Technol. 2014;48(3):1745–1752. doi: 10.1021/es404224j. [DOI] [PubMed] [Google Scholar]
- 17.Kosmulski M. pH-dependent surface charging and points of zero charge: III. Update. J. Colloid Interface Sci. 2006;298(2):730–741. doi: 10.1016/j.jcis.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 18.Schroth BK, Sposito G. Surface charge properties of kaolinite. MRS Online Proc. Libr. 1996;432:87. [Google Scholar]
- 19.Bernier-Latmani R, Veeramani H, Vecchia ED, Junier P, Lezama-Pacheco JS, Suvorova EI, Sharp JO, Wigginton NS, Bargar JR. Non-uraninite products of microbial U (VI) reduction. Environ. Sci. Technol. 2010;44(24):9456–9462. doi: 10.1021/es101675a. [DOI] [PubMed] [Google Scholar]
- 20.Hansley PL, Spirakis CS. Organic matter diagenesis as the key to a unifying theory for the genesis of tabular uranium-vanadium deposits in the Morrison Formation, Colorado Plateau. Econ. Geol. Bull. Soc. Econ. Geol. 1992;87(2):352–365. [Google Scholar]
- 21.Tokunaga TK, Kim Y, Wan J. Potential remediation approach for uranium-contaminated groundwaters through potassium uranyl vanadate precipitation. Environ. Sci. Technol. 2009;43(14):5467–5471. doi: 10.1021/es900619s. [DOI] [PubMed] [Google Scholar]
- 22.Spano TL, Dzik EA, Sharifironizi M, Dustin MK, Turner M, Burns PC. Thermodynamic investigation of uranyl vanadate minerals: Implications for structural stability. Am. Mineral. 2017;102(6):1149–1153. [Google Scholar]
- 23.Franczyk KJ. Stratigraphic revision and depositional environments of the Upper Cretaceous Toreva Formation in the northern Black Mesa area. Navajo and Apache Counties: AZ; Bulletin 1685: U.S. Geological Survey; 1988. [Google Scholar]
- 24.Hostetler P, Garrels R. Transportation and precipitation of uranium and vanadium at low temperatures, with special reference to sandstone-type uranium deposits. Econ. Geol. Bull. Soc. Econ. Geol. 1962;57(2):137–167. [Google Scholar]
- 25.Giammar DE, Hering JG. Time scales for sorption-desorption and surface precipitation of uranyl on goethite. Environ. Sci. Technol. 2001;35(16):3332–3337. doi: 10.1021/es0019981. [DOI] [PubMed] [Google Scholar]
- 26.Davis JA, Meece DE, Kohler M, Curtis GP. Approaches to surface complexation modeling of uranium (VI) adsorption on aquifer sediments. Geochim. Cosmochim. Acta. 2004;68(18):3621–3641. [Google Scholar]
- 27.Curtis GP, Davis JA, Naftz DL. Simulation of reactive transport of uranium (VI) in groundwater with variable chemical conditions. Water Resour. Res. 2006;42(4):W04404. [Google Scholar]
- 28.Hammond GE, Lichtner PC. Field-scale model for the natural attenuation of uranium at the Hanford 300 Area using high-performance computing. Water Resour. Res. 2010;46(9):W09527. [Google Scholar]
- 29.Zachara JM, Chen X, Murray C, Hammond G. River stage influences on uranium transport in a hydrologically dynamic groundwater-surface water transition zone. Water Resour. Res. 2016;52:1568. [Google Scholar]
- 30.Singer DM, Zachara JM, Brown GE., Jr Uranium speciation as a function of depth in contaminated Hanford sediments-A micro-XRF, micro-XRD, and micro-and bulk-XAFS study. Environ. Sci. Technol. 2009;43(3):630–636. doi: 10.1021/es8021045. [DOI] [PubMed] [Google Scholar]
- 31.Stubbs JE, Veblen LA, Elbert DC, Zachara JM, Davis JA, Veblen DR. Newly recognized hosts for uranium in the Hanford Site vadose zone. Geochim. Cosmochim. Acta. 2009;73(6):1563–1576. [Google Scholar]
- 32.Tokunaga TK, Kim Y, Wan J, Yang L. Aqueous uranium (VI) concentrations controlled by calcium uranyl vanadate precipitates. Environ. Sci. Technol. 2012;46(14):7471–7477. doi: 10.1021/es300925u. [DOI] [PubMed] [Google Scholar]
- 33.Langmuir D. Aqueous Environmental Geochemistry. Prentice Hall; 1997. [Google Scholar]
- 34.Liu C, Zachara JM, Qafoku NP, Wang Z. Scale-dependent desorption of uranium from contaminated subsurface sediments. Water Resour. Res. 2008;44(8):W08413. [Google Scholar]
- 35.Steefel CI, DePaolo DJ, Lichtner PC. Reactive transport modeling: An essential tool and a new research approach for the Earth sciences. Earth Planet. Sci. Lett. 2005;240(3):539–558. [Google Scholar]
- 36.Shabansefidan F, Reddy MA. Investigation of Uranium Solubility and its Transport Path in the Razgah Basin’s Groundwater in the Northwest of Iran, Using Saturation Indexes. Intl. J. Comput. Sci. Manage. Studies. 2012;4:39. [Google Scholar]
- 37.Puigdomenech I, Bruno J. Modelling uranium solubilities in aqueous solutions: Validation of a thermodynamic data base for the EQ3/6 geochemical codes. Swedish Nuclear Fuel and Waste Management Company; 1988. [Google Scholar]
- 38.Kubatko K-A, Helean KB, Navrotsky A, Burns PC. Thermodynamics of uranyl minerals: Enthalpies of formation of rutherfordine, UO2CO3, andersonite, Na2CaUO2(CO3).3 (H2O)5, and grimselite, K3NaUO2(CO3).3H2O. Am. Mineral. 2005;90(8–9):1284–1290. [Google Scholar]
- 39.Bond DL, Davis JA, Zachara JM. Uranium (VI) release from contaminated vadose zone sediments: Estimation of potential contributions from dissolution and desorption. Developments in Earth and Environmental Sciences. 2007;7:375–416. [Google Scholar]
- 40.USEPA Addressing Uranium Contamination on the Navajo Nation. U.S. Environmental Protection Agency, Pacific Southwest, Region 9; San Francisco, CA: 2015. [Google Scholar]
- 41.Lollar BS. Environmental Geochemistry. Vol. 9 Elsevier; 2005. [Google Scholar]
- 42.Riba O, Walker C, Ragnarsdottir KV. Kinetic studies of synthetic metaschoepite under acidic conditions in batch and flow experiments. Environ. Sci. Technol. 2005;39(20):7915–7920. doi: 10.1021/es050678k. [DOI] [PubMed] [Google Scholar]
- 43.Yoshida S, Iguchi T, Ishida S, Tarama K. Some physico-chemical properties of vanadium oxide supported on silica or γ-alumina. Bull. Chem. Soc. Jpn. 1972;45(2):376–380. [Google Scholar]
- 44.Langmuir D. Uranium solution-mineral equilibria at low temperatures with applications to sedimentary ore deposits. Geochim. Cosmochim. Acta. 1978;42(6):547–569. [Google Scholar]
- 45.Hochella MF, Lower SK, Maurice PA, Penn RL, Sahai N, Sparks DL, Twining BS. Nanominerals, mineral nanoparticles, and earth systems. Science. 2008;319(5870):1631–1635. doi: 10.1126/science.1141134. [DOI] [PubMed] [Google Scholar]
- 46.Kim C, Chi C, Miller S, Rosales R, Sugihara E, Akau J, Rytuba J, Webb S. (Micro) spectroscopic analyses of particle size dependence on arsenic distribution and speciation in mine wastes. Environ. Sci. Technol. 2013;47(15):8164–8171. doi: 10.1021/es4010653. [DOI] [PubMed] [Google Scholar]
- 47.McCarthy JF, Zachara JM. Subsurface transport of contaminants. Environ. Sci. Technol. 1989;23(5):496–502. [Google Scholar]
- 48.Um W, Wang Z, Serne RJ, Williams BD, Brown CF, Dodge CJ, Francis AJ. Uranium phases in contaminated sediments below Hanford’s U tank farm. Environ. Sci. Technol. 2009;43(12):4280–4286. doi: 10.1021/es900203r. [DOI] [PubMed] [Google Scholar]
- 49.Delemos JL, Bostick BC, Quicksall AN, Landis JD, George CC, Slagowski NL, Rock T, Brugge D, Lewis J, Durant JL. Rapid dissolution of soluble uranyl phases in arid, mine-impacted catchments near Church Rock, NM. Environ. Sci. Technol. 2008;42(11):3951–3957. doi: 10.1021/es071738k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goss K-U. The p K a values of PFOA and other highly fluorinated carboxylic acids. Environ. Sci. Technol. 2007;42(2):456–458. doi: 10.1021/es702192c. [DOI] [PubMed] [Google Scholar]
- 51.Kim K-W, Hyun J-T, Lee K-Y, Lee E-H, Lee K-W, Song K-C, Moon J-K. Effects of the different conditions of uranyl and hydrogen peroxide solutions on the behavior of the uranium peroxide precipitation. J. Hazard. Mater. 2011;193:52–58. doi: 10.1016/j.jhazmat.2011.07.032. [DOI] [PubMed] [Google Scholar]
- 52.Schoonen M, Barnes H. Reactions forming pyrite and marcasite from solution: I. Nucleation of FeS2 below 100°C. Geochim. Cosmochim. Acta. 1991;55(6):1495–1504. [Google Scholar]
- 53.Lippmann F. Sedimentary Carbonate Minerals. Vol. 6 Springer Science & Business Media; 2012. [Google Scholar]
- 54.Stefánsson A, Gíslason SR. Chemical weathering of basalts, Southwest Iceland: effect of rock crystallinity and secondary minerals on chemical fluxes to the ocean. Am. J. Sci. 2001;301(6):513–556. [Google Scholar]
- 55.Gorman-Lewis D, Burns PC, Fein JB. Review of uranyl mineral solubility measurements. J. Chem. Thermodyn. 2008;40(3):335–352. [Google Scholar]
- 56.Rey A, Utsunomiya S, Giménez J, Casas I, de Pablo J, Ewing RC. Stability of uranium (VI) peroxide hydrates under ionizing radiation. Am. Mineral. 2009;94(2–3):229–235. [Google Scholar]
- 57.Utsunomiya S, Ewing RC. Radiation-induced decomposition of U (VI) alteration phases of UO2. MRS Online Proc. Libr. 2006;932(73.1) doi: 10.1557/PROC-932-73.1. [DOI] [Google Scholar]
- 58.Smith SC, Douglas M, Moore DA, Kukkadapu RK, Arey BW. Uranium extraction from laboratory-synthesized, uranium-doped hydrous ferric oxides. Environ. Sci. Technol. 2009;43(7):2341–2347. doi: 10.1021/es802621t. [DOI] [PubMed] [Google Scholar]
- 59.Ilton ES, Zachara JM, Moore DA, Mckinley JP, Eckberg AD, Cahill CL, Felmy AR. Dissolution study of metatorbernite: Thermodynamic properties and the effect of pH and phosphate. Environ. Sci. Technol. 2010;44(19):7521–7526. doi: 10.1021/es101619f. [DOI] [PubMed] [Google Scholar]
- 60.Mann A. Chemical Ore Genesis Models for the Precipitation of Carnotite in Calcrete. Commonwealth Scientific and Industrial Research Organisation; 1974. [Google Scholar]
- 61.Hillebrand W. Carnotite and tyuyamunite and their ores in Colorado and Utah. Am. J. Sci. 1924;45:201–216. [Google Scholar]
- 62.Crook T, Blake G. On carnotite and an associated mineral complex from South Australia. Mineral. Mag. 1910;15(71):271–284. [Google Scholar]
- 63.Sharma RK, Putirka KD, Stone JJ. Stream sediment geochemistry of the upper Cheyenne River watershed within the abandoned uranium mining region of the southern Black Hills, South Dakota, USA. Environ. Earth Sci. 2016;75(9):1–12. [Google Scholar]
- 64.Xu J, Zhu S-Y, Luo T-Y, Zhou W, Li Y-L. Uranium mineralization and its radioactive decay-induced carbonization in a black shale-hosted polymetallic sulfide ore layer, southwest China. Econ. Geol. Bull. Soc. Econ. Geol. 2015;110(6):1643–1652. [Google Scholar]
- 65.Abed AM, Saffarini GA, Sadaqah RM. Spatial distribution of uranium and vanadium in the upper phosphorite member in Eshidiyya basin, southern Jordan. Arabian J. Geosci. 2014;7(1):253–271. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.