

Abstract
Background
The Gene Ontology (GO) is a community-based bioinformatics resource that employs ontologies to represent biological knowledge and describes information about gene and gene product function. GO includes three independent categories: molecular function, biological process and cellular component. For better biological reasoning, identifying the biological relationships between terms in different categories are important.
However, the existing measurements to calculate similarity between terms in different categories are either developed by using the GO data only or only take part of combined gene co-function network information.
Results
We propose an iterative ranking-based method called C r o G O2 to measure the cross-categories GO term similarities by incorporating level information of GO terms with both direct and indirect interactions in the gene co-function network.
Conclusions
The evaluation test shows that C r o G O2 performs better than the existing methods. A genome-specific term association network for yeast is also generated by connecting terms with the high confidence score. The linkages in the term association network could be supported by the literature. Given a gene set, the related terms identified by using the association network have overlap with the related terms identified by GO enrichment analysis.
Keywords: Gene Ontology, Term similarity, Cross categories
Background
The Gene Ontology (GO) is a community-based bioinformatics resource that employs ontologies to represent biological knowledge and describes information about gene and gene product function [1]. It is widely used to infer functional information for gene products, such as gene function enrichment [2], protein function prediction [3, 4], disease association analysis [5–7]. GO contains three key categories: cellular component (CC; where gene products are active), molecular function (MF; the biological function of gene or gene product) and biological process (BP; pathways or larger processes that multiple gene products involved in). Comparing the similarity between GO terms is an important basic for the GO-based application. The methods of measuring term similarities have been extensively studied in last decade [8–19]. However, most of existing methods focus on measuring the similarity in the same GO category and cannot calculate the semantic similarities between GO terms belonging to different GO categories.
Although GO is originally constructed as three independent categories, identifying their biological relationships may be helpful to understand the biological mechanism and infer gene function [20]. Furthermore, identifying relationships between terms in different categories may provide evidence for biological reasoning and hypotheses. For example, anaphase-promoting complex plays an important role in anaphase inhibitory protein degradation and mitotic cyclins, which can be revealed by discovering the relationship between MF term “anaphase-promoting complex binding” and BP term “activation of anaphase-promoting complex activity involved in meiotic cell cycle” [21].
Several methods are proposed to calculate the similarities between terms across GO categories. Let t 1 and t 2 be two terms belonging to two different GO categories. Association rule mining (ASR), which is a well-known data mining algorithm, was used to calculate the similarity of t 1 and t 2, labeled as S i m ASR(t 1,t 2) [22, 23]. By combining the ASR approach and text mining-based method, Myhre et al. generated a ready-for-use cross-category GO structure. The limitation of the ASR-based approach is that “shallow annotation” problem is ignored [24]. Specifically, let t 1 and t 2 be two terms in different categories C 1 and C 2. If both t 1 and t 2 are high-level terms that are near to the root terms of C 1 and C 2, the similarity between t 1 and t 2 may be high no matter whether t 1 and t 2 are biologically related. The reason is that the high-level terms may annotate almost all genes involved in a GO category after propagation [25]. Consequently, term pairs at high levels can have high similarity, which may not reflect the biological relationship between the terms.
To solve the “shallow annotation” problem, a Vector Space Model (VSM)-based approach was developed by Bodenreidar et al.. This method takes the semantic information of genes into account to avoid “shallow annotation” problem. VSM is a classical method, which is widely used to calculate the similarities between documents that can be represented as vectors [23]. Specifically, each term is considered as a vector, which length is the same as all the genes involved in GO. Each element in a vector is a binary value. If there is association between a term and a gene, the binary value is 1, otherwise 0 [26]. The similarity of t 1 and t 2 in different categories can be measured with weighted cosine similarity. The VSM-based approach is based on the interaction of the gene sets annotated by t 1 and t 2. Therefore, the result heavily relies on the quality and coverage of G annotation data. Unfortunately, the gene annotations are far from complete currently [27], which may lead to inaccurate term similarity scores.
To avoid the data availability problem, inspiring from existing integration methods, a novel method CroGO was proposed to calculate the similarity between two GO terms in different categories in our previous work [21]. CroGo incorporate gene co-function network data and gene ontology data to calculate the cross-categories GO term similarities. The experiment result shows that CroGO outperforms the aforementioned methods. However, only part of the information in gene co-function network was used by CroGO, since it only took the direct link in the network into account. Other than the directly connected gene pairs, the indirect gene-gene interactions contained in the gene co-function network should also be considered.
In this paper, we developed a novel approach, C r o G O2, to measure the cross-categories GO term similarities by incorporating both direct and indirect interactions in the gene co-function network. Comparing with the existing approaches, C r o G O2 has the following advantages:
Comparing with the state-of-art methods, C r o G O2 performs better than existing methods by taking the global interactions in the gene co-functional network into account. It proves that gene co-functional network could be a good complement to GO for cross-categories term similarity calculation.
A novel iterative ranking-based method is developed to measure the relationship between two gene sets based on the gene co-functional network.
A cross-categories term association network was constructed by selecting the term-pairs with high similarity score calculated by C r o G O2. Applying C r o G O2 to identify the highly related terms between BP and MF category has discovered term pairs with solid supports from literature.
Methods
We proposes C r o G O2 to measure the relationships between genes based on the global feature of a gene network and then measure the similarity between GO terms in different categories. To measure the similarity of t 1 and t 2 in different categories, C r o G O2 consists of three steps. First, it measures the interaction between genes based on the gene network. Second, it calculates the similarity between two gene sets annotated by t 1 and t 2 based on gene-gene associations from last step. Third, it combines the network-based gene set similarities and the level information of t 1 and t 2 in GO to calculate the similarity between t 1 and t 2. The diagram of the whole process of C r o G O2 is shown in Fig. 1.
Fig. 1.
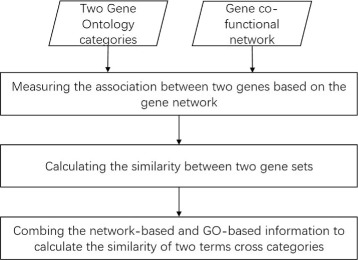
The workflow of C r o G O2
Step 1. measuring the network-based association between two genes
In this step, we use both the direct and indirect interactions between genes in the gene co-functional network to measure the association between two genes. A gene network includes not only the direct interaction between genes but also the global view of associations among genes, which are not connected directly. In this step, we adopted the iterative ranking (IR) [28] algorithm to measure the association between two genes. The basic idea is that the
Figure 2 is an illustration example of our basic idea. Given a gene co-functional network G(V,E), the association score between gene g z and g i is determined by two types of information: the direct link between g z and g i, (g z,g i); the indirect link between g z and g i, {(g z,g j), (g j,g i)},{(g z,g j+1),(g j+1,g i)},{(g z,g j+2),(g j+2,g j+3),(g j+3,g i)}. Mathematically, we calculate the IR score in the following steps.
Fig. 2.
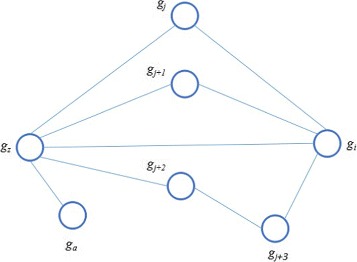
Illustration example for iterative ranking based association score. The nodes and edges represent genes and their interactions respectively
First, a normalized adjacent matrix is generated by using the weighted average of neighbors, labeled as U. Given a gene g i and g j, a normalize association score in U is calculated as follows.
| 1 |
Second, given a gene g z, its association with g i is defined in terms of g j, we update the score iteratively. At each iteration t, the algorithm considers information from neighbors at path length=t (Eq. 2).
| 2 |
where o i represents the original association score between g z and g i, α is a weight parameter between 0 and 1. We can extend the Eq. 2 to calculate the iterative ranking-based association score for the whole network.
| 3 |
where O is the adjacent matrix containing the original gene-gene relations in the input gene co-function network, R t and R t+1 are adjacent matrices saving iterative gene association score in iterative t and t+1. The stopping criterion of the iterative process is defined as follows.
| 4 |
where n is the number of nodes involved in the network. The iteration stops until θ is smaller than a given threshold. The pseudo-code of the algorithm is shown in Algorithm 1.

Step 2. calculating the similarity between two gene sets
Given two terms t 1 and t 2 in different GO categories C 1 and C 2, let G 1 and G 2 be gene set annotated by t 1 and t 2. Based on the global association score between genes calculated in last step, the association score of the two gene sets is calculated in this step. Given an adjacent matrix R, which includes the iterative ranking-based association scores between genes, the network-based similarity between t 1 and t 2 is defined based on their annotation sets as follows.
| 5 |
where G 1 and G 2 represent the gene sets annotated to t 1 and t 2 respectively, |X| is the number of genes in set X, G 1∪G 2 is union of set G 1 and G 2. Noted that we re-defined |G 1−G 2| in our method as follows:
| 6 |
where r ij is association score between genes g i and g j in network R. Particularly, if two gene sets G 1 and G 2 are identical, |G 1−G 2|=0. In summary, the term similarity S i m net(t 1,t 2) represents the association between G 1 and G 2 annotated by t 1 and t 2 based on the gene association in R.
Step 3. calculating the cross-categories term similarity
In this step, we combine the network-based gene set similarities and the level information in GO to calculate the similarity between t 1 and t 2 in different categories. To overcome the “shallow annotation” problem, we take the level information of t 1 and t 2 in different categories into account.
| 7 |
where and are the number of genes in the category C 1 and C 2. If t x is close to the root of C x, is close to 0; if t x is a specific term (far from the root), is close to 1. Equation (7) shows that the specific term pair are more likely to be identified.
Then, the similarity between t 1 and t 2 is calculated by integrating gene co-functional network, GO structure and gene annotations as:
| 8 |
Our previous work indicated that the relationships between two terms should be directed [21]. Therefore, we applied the term pair assignment method proposed in our previous work to look for the directions of the relationships. First, all similarities of term pairs across categories are computed with Eq. (8). Second, a user defined threshold is applied to filter term relationships with a threshold. Third, given a term t 1 and a term set T 2 that has connection to t 1, the edge direction are deleted from t 1 to t 2 only if there is a term t 3 satisfying that t 3 is a descendant of t 2 (t 2,t 3∈T 2). In the end, we can get the directed relationships between terms in different GO categories.
Results
In our experiment, we used BP and MF category as input to evaluate C r o G O2. To show the significance of C r o G O2, we compare C r o G O2 with CroGO [21], ASR-based [22] and VSM-based [23] methods. All the four methods are applied to a gold-standard set constructed with known pathway-to-reaction associations on yeast, which is also used as the evaluation data set in previous research [20, 21]. Then, we constructed a term association network for yeast between BP category and MF category.
The GO data and gene annotations were downloaded from GO official website in October 2015 [27]. We used yeastNet as the input co-function network, which contains 102,803 edges and 5483 genes [29]. C r o G O2 was implemented with java and JUNG library [30]. In the experiment, parameter α is set as 0.1. To determine the parameter α, we re-ran C r o G O2 by varying the parameter α. C r o G O2 achieve the best performance when α=0.1.
Performance evaluation on gold-standard set
To test the performance of C r o G O2, we generated a “gold-standard” set based on the pathway-to-reaction interactions [20] in yeast. The process includes three parts: 1) a BP term is associated with a pathway based on GO biological process; 2) a metabolic pathway could be associate with several Enzyme Commission (EC) groups based on the enzymes catalysation; and 3) each EC can be linked to a MF term based on the association data from GO database [31–33]. Finally, the gold-standard set includes 334 MF-BP pairs. These 334 MF-BP term pairs are considered as the positive set. We also randomly selected 334 MF-BP term pairs as the random set. Note that similar gold-standard set generation method has been applied in previous research but on different data sources [20, 21]. Similarities of term pairs in both gold-standard set and random set are calculated using all four compared methods. We compared their performance based on receiver operating characteristic (ROC) curve [34] of each approach.
The result showed clearly that C r o G O2 performs better than other three methods. Comparing the AUC score of the four methods showed that C r o G O2 had the highest AUC score (0.87) with the CroGO as the runner-up (Fig. 3). The AUC scores of CroGO, ASR and VSM are 0.82, 0.80 and 0.81 respectively. Table 1 shows that when the false positive threshold is 5%, the true positive rate of C r o G O2 is 66%, while the values of CroGO, ASR and VSM based approaches are 56, 59 and 59% respectively. C r o G O2 also has the highest true positive rate when the false positive rate is equal to 10 and 15%.
Fig. 3.
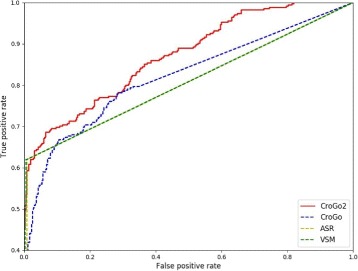
ROC curves for the four methods on the gold-standard sets of yeast. The red, blue, yellow and green lines represent CroGO2 (red), CroGO (blue), and ASR (yellow) and VSM (green) method respectively. Most portion of ROC curves of ASR and VSM are overlapping
Table 1.
The performance of ASR, VSM, CroGO and CroGO2 measures on yeast gold-standard set
| Organism | Measure | TP rate (when | TP rate (when | TP rate (when |
|---|---|---|---|---|
| FP rate =5%) | FP rate =10%) | FP rate =15%) | ||
| *Yeast | ASR | 59% | / | / |
| VSM | 59% | / | / | |
| CroGO | 56% | 65% | 67% | |
| CroGO2 | 66% | 69% | 71% |
In summary, the evaluation test indicates that C r o G O2 has produced better performance than the other measures.
Robustness test of CroGO2
C r o G O2 combined the co-function network. To test whether varied the co-function network density would affect the performance of C r o G O2, we randomly deleted 50% of edges in the co-function network and used the low-density co-function network as input.
The result shows that there was no significant different between results using two networks with different densities (Fig. 4). The AUC scores using the full network and low-density network are 0.870 and 0.869, which are almost the same.In summary, the experiment result shows that C r o G O2 has high robustness.
Fig. 4.
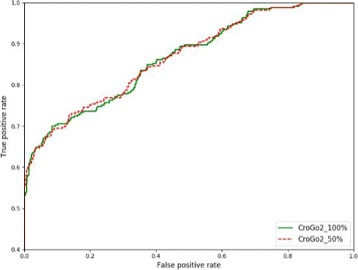
ROC curves for the robustness test of C r o G O2 with different co-function network densities
Discussion
In this section, we linked BP and MF terms to generate a term association network for yeast. The cross-category term association network can provide a convenient way for researchers to use C r o G O2.
A reliable MF-BP association network is generated by calculating pairwise similarities of all MF and BP terms and applying a strict FDR threshold (in this case we use F D R<0.05). Finally, the association network includes 1406 MF terms, 2305 BP terms, and 8531 linkages.
To show the power of the MF-BP association network N, we test whether the result based on association network has an agreement with the result based on GO enrichment. Given a set of genes S with particular function, we can get its enrichment results based on BP category and MF category separately. The enriched term sets of S on BP and MF category are labeled as T BP and T MF respectively. Given T BP and N, we can find out the MF terms, saved as T MF′, connect with terms in T BP based on N. We can check whether overlap terms can be identified between T MF and T MF′. For example, we find a set of genes which are associated with the phenotype “adhesion” from the yeast phenotype ontology [35]. The gene set is {C D C33,C I S3,C W P2,F I G2,F K S3,F L O10,F L O11,F L O5,F L O9,P I R3,S C W4}. Following the aforementioned experiment protocol, the result is shown in Fig. 5. It is shown that three terms (GO:0005199, GO:0030246 and GO:0048029) can be identified by both GO enriched-based and MF-BP association network-based methods.
Fig. 5.
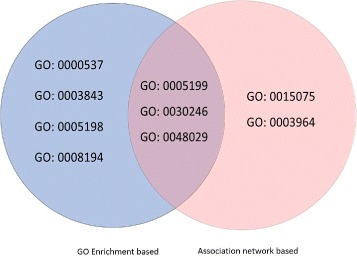
Venn diagram of T MF and T MF′. T MF is the set of enriched MF terms. T MF′ is the set of MF terms associated with the enriched BP terms
Furthermore, the top 20 term associations, which do not have identical annotation set, are shown in Table 2. We found biological evidence from literature or term definition for 15 of them. The rest 5 new conceptual connections may be new knowledge not found in previous study.
Table 2.
Top 20 term associations that were identified by C r o G O2
| BP Name | MF Name | Evidence |
|---|---|---|
| butanediol biosynthetic process | (R,R)-butanediol dehydrogenase activity | New |
| glutamine biosynthetic process | glutamate-ammonia ligase activity | [36] |
| putrescine biosynthetic process | ornithine decarboxylase activity | [37, 38] |
| acetyl-CoA biosynthetic process from acetate | acetate-CoA ligase activity | New |
| alanine catabolic process | L-alanine:2-oxoglutarate aminotransferase activity | [39] |
| siroheme biosynthetic process | precorrin-2 dehydrogenase activity | [40] |
| trehalose catabolic process | alpha,alpha-trehalase activity | [41] |
| asparagine catabolic process | asparaginase activity | [42] |
| lysine biosynthetic process | aromatic-amino-acid:2-oxoglutarate aminotransferase activity | [43, 44] |
| glycerol biosynthetic process | glycerol-1-phosphatase activity | New |
| threonine catabolic process | L-threonine ammonia-lyase activity | New |
| peptide alpha-N-acetyltransferase activity | N-terminal protein amino acid acetylation | [45] |
| glutathione catabolic process | gamma-glutamyltransferase activity | [46] |
| alanine biosynthetic process | L-alanine:2-oxoglutarate aminotransferase activity | [47] |
| positive regulation of histone H3-K36 methylation | TFIIF-class binding TF activity | New |
| siroheme biosynthetic process | uroporphyrin-III C-methyltransferase activity | [48] |
| siroheme biosynthetic process | sirohydrochlorin ferrochelatase activity | [40] |
| glutathione biosynthetic process | glutamate-cysteine ligase activity | [49, 50] |
| positive regulation of telomere maintenance via telomerase | Hsp90 protein binding | [51, 52] |
| chorismate biosynthetic process | 3-deoxy-7-phosphoheptulonate synthase activity | [53] |
Conclusions
Identifying the relationships between GO terms in different categories is vital for understanding the biological mechanism and inferring gene function. Recently, researchers have begun to employ gene co-function networks to calculate the similarity between terms in different GO categories. In this article, we proposed a novel approach, called C r o G O2, to measure the cross-categories GO term similarities by incorporating level information in gene ontology with both direct and indirect interactions in the gene co-function network. C r o G O2 has the following advantages: 1) CroGO2 performs better than existing methods by taking the global interactions in the gene co-functional network into account; 2) A novel iterative ranking-based method is developed to measure the relationship between two gene sets; 3) A cross-categories term association network was constructed by selecting the high-quality associations. To demonstrate the advantages of C r o G O2, we compare it with three existing approaches CroGO, ASR and VSM. The experiment on a gold standard set shows that C r o G O2 performs better than other methods. Furthermore, C r o G O2 has the high robustness to the co-function network density. We also generated a genome-specific term association network of yeast. The linkages in the association network can be supported by literature. Given a gene set, the related terms identified by using the association network have overlap with the related terms identified by GO enrichment analysis.
Acknowledgments
This work was supported by National Natural Science Foundation of China (Grant No. 61702421), Natural Science Basic Research Plan in Shaanxi Province of China (Grant No. 2017JQ6047), China Postdoctoral Science Foundation (Grant No. 2017M610651), the Fundamental Research Funds for the Central Universities (Grant No. 3102016QD003), National Natural Science Foundation of China (Grant No. 61602386 and 61332014).
Funding
The publication costs for this article were funded by Northwestern Polytechnical University.
Availability of data and materials
The datasets during and/or analysed during the current study available from the corresponding author on reasonable request.
About this supplement
This article has been published as part of BMC Bioinformatics Volume 18 Supplement 16, 2017: 16th International Conference on Bioinformatics (InCoB 2017): Bioinformatics. The full contents of the supplement are available online at http://dx.doi.org/https://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-18-supplement-16.
Authors’ contributions
JP and XS conceived the project; JP, YW and HW designed the algorithm and experiments; HW and JP wrote this manuscript; JL, WH helped to test the algorithm. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jiajie Peng, Email: jiajiepeng@nwpu.edu.cn.
Honggang Wang, Email: 591981909@qq.com.
Junya Lu, Email: lujunya@mail.nwpu.edu.cn.
Weiwei Hui, Email: wwhui@mail.nwpu.edu.cn.
Yadong Wang, Email: ydwang@hit.edu.cn.
Xuequn Shang, Email: shang@nwpu.edu.cn.
References
- 1.Consortium GO, et al. Gene ontology consortium: going forward. Nucleic Acids Res. 2015;43(D1):D1049–56. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cacchiarelli D, Trapnell C, Ziller MJ, Soumillon M, Cesana M, Karnik R, Donaghey J, Smith ZD, Ratanasirintrawoot S, Zhang X, et al. Integrative analyses of human reprogramming reveal dynamic nature of induced pluripotency. Cell. 2015;162(2):412–24. doi: 10.1016/j.cell.2015.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cho H, Berger B, Peng J. Compact Integration of Multi-Network Topology for Functional Analysis of Genes. Cell Syst. 2016;3(6):540–8. doi: 10.1016/j.cels.2016.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peng J, Wang T, Wang J, Wang Y, Chen J. Extending gene ontology with gene association networks. Bioinformatics. 2015;32(8):1185–94. doi: 10.1093/bioinformatics/btv712. [DOI] [PubMed] [Google Scholar]
- 5.Menche J, Sharma A, Kitsak M, Ghiassian SD, Vidal M, Loscalzo J, Barabási AL. Uncovering disease-disease relationships through the incomplete interactome. Science. 2015;347(6224):1257601. doi: 10.1126/science.1257601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peng J, Lu J, Shang X, Chen J. Identifying consistent disease subnetworks using DNet. Methods. 2017;131:104–10. doi: 10.1016/j.ymeth.2017.07.024. [DOI] [PubMed] [Google Scholar]
- 7.Peng J, Bai K, Shang X, Wang G, Xue H, Jin S, Cheng L, Wang Y, Chen J. Predicting disease-related genes using integrated biomedical networks. BMC Genomics. 2017;18:1043. doi: 10.1186/s12864-016-3263-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peng J, Uygun S, Kim T, Wang Y, Rhee SY, Chen J. Measuring semantic similarities by combining gene ontology annotations and gene co-function networks. BMC Bioinformatics. 2015;16:44. doi: 10.1186/s12859-015-0474-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peng J, Li H, Liu Y, Juan L, Jiang Q, Wang Y, Chen J. InteGO2: a web tool for measuring and visualizing gene semantic similarities using Gene Ontology. BMC Genomics. 2016;17(5):530. doi: 10.1186/s12864-016-2828-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mazandu GK, Chimusa ER, Mulder NJ. Gene ontology semantic similarity tools: survey on features and challenges for biological knowledge discovery. Brief Bioinforma. 2016. p. 1-16. [DOI] [PubMed]
- 11.Teng Z, Guo M, Liu X, et al. Measuring gene functional similarity based on group-wise comparison of GO terms. Bioinformatics. 2013;29(11):1424–32. doi: 10.1093/bioinformatics/btt160. [DOI] [PubMed] [Google Scholar]
- 12.Yu G, Luo W, Fu G, Wang J. Interspecies gene function prediction using semantic similarity. BMC Syst Biol. 2016;10(4):495. doi: 10.1186/s12918-016-0361-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu G, Li F, Qin Y, Bo X, Wu Y, Wang S. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics. 2010;26(7):976–8. doi: 10.1093/bioinformatics/btq064. [DOI] [PubMed] [Google Scholar]
- 14.Peng J, Xue H, Shao Y, Shang X, Wang Y, Chen J. A novel method to measure the semantic similarity of HPO terms. Int J Data Min Bioinforma. 2017;17(2):173–88. doi: 10.1504/IJDMB.2017.084268. [DOI] [Google Scholar]
- 15.Chen G, Zhao J, Cohen T, et al. Using Ontology Fingerprints to disambiguate gene name entities in the biomedical literature. Database J Biol Databases & Curation. 2015;2015(13):bav034. doi: 10.1093/database/bav034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng J, Hui W, Shang X. Measuring phenotype-phenotype similarity through the interactome. BMC Bioinforma. In press. [DOI] [PMC free article] [PubMed]
- 17.Cheng L, Jiang Y, Wang Z, Shi H, Sun J, Yang H, Zhang S, Hu Y, Zhou M. DisSim: an online system for exploring significant similar diseases and exhibiting potential therapeutic drugs. Sci Rep. 2016;6:30024. doi: 10.1038/srep30024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng L, Li J, Ju P, Peng J, Wang Y. SemFunSim: a new method for measuring disease similarity by integrating semantic and gene functional association. PLoS ONE. 2014;9(6):e99415. doi: 10.1371/journal.pone.0099415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peng J, Zhang X, Hui W, Lu J, Li Q, Shang X. Improving the measurement of semantic similarity by combining gene ontology and co-functional network: a random walk based approach. BMC Syst Biol. 2017. In press. [DOI] [PMC free article] [PubMed]
- 20.Myhre S, Tveit H, Mollestad T, Lægreid A. Additional gene ontology structure for improved biological reasoning. Bioinformatics. 2006;22(16):2020–7. doi: 10.1093/bioinformatics/btl334. [DOI] [PubMed] [Google Scholar]
- 21.Peng J, Chen J, Wang Y. Identifying cross-category relations in gene ontology and constructing genome-specific term association networks. BMC Bioinformatics. 2013;14(2):S15. doi: 10.1186/1471-2105-14-S2-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar A, Smith B, Borgelt C. Dependence relationships between gene ontology terms based on TIGR gene product annotations. In: Proceedings of the 3rd International workshop on computational terminology,2004. p. 31–8.
- 23.Bodenreider O, Aubry M, Burgun A. Non-lexical approaches to identifying associative relations in the gene ontology. Pac Symp Biocomput. 2005;10(C9):91. [PMC free article] [PubMed] [Google Scholar]
- 24.Sevilla JL, Segura V, Podhorski A, Guruceaga E, Mato JM, Martinez-Cruz LA, Corrales FJ, Rubio A. Correlation between gene expression and GO semantic similarity. IEEE/ACM Trans Comput Biol Bioinforma (TCBB) 2005;2(4):330–8. doi: 10.1109/TCBB.2005.50. [DOI] [PubMed] [Google Scholar]
- 25.Wang JZ, Du Z, Payattakool R, Philip SY, Chen CF. A new method to measure the semantic similarity of GO terms. Bioinformatics. 2007;23(10):1274–81. doi: 10.1093/bioinformatics/btm087. [DOI] [PubMed] [Google Scholar]
- 26.Baeza-Yates R, Ribeiro-Neto B, et al. Modern information retrieval. 1999; 43(1):26–8.
- 27.Consortium GO, et al. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017;45(D1):D331–8. doi: 10.1093/nar/gkw1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Negahban S, Oh S, Shah D. Iterative ranking from pair-wise comparisons. Advances in neural information processing systems. 2012;3(93):2483–91. [Google Scholar]
- 29.Lee I, Li Z, Marcotte EM. An improved, bias-reduced probabilistic functional gene network of baker’s yeast, Saccharomyces cerevisiae. PloS ONE. 2007;2(10):e988. doi: 10.1371/journal.pone.0000988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.OMadadhain J, Fisher D, Smyth P, White S, Boey YB. Analysis and visualization of network data using JUNG. J Stat Soft. 2005;10(2):1–35. [Google Scholar]
- 31.Hill DP, Davis AP, Richardson JE, Corradi JP, Ringwald M, Eppig JT, Blake JA. Program description: Strategies for biological annotation of mammalian systems: implementing gene ontologies in mouse genome informatics. Genomics. 2001;74:121–8. doi: 10.1006/geno.2001.6513. [DOI] [PubMed] [Google Scholar]
- 32.Camon EB, Barrell DG, Dimmer EC, Lee V, Magrane M, Maslen J, Binns D, Apweiler R. An evaluation of GO annotation retrieval for BioCreAtIvE and GOA. BMC Bioinformatics. 2005;6:S17. doi: 10.1186/1471-2105-6-S1-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caspi R, Foerster H, Fulcher CA, Hopkinson R, Ingraham J, Kaipa P, Krummenacker M, Paley S, Pick J, Rhee SY, et al. MetaCyc: a multiorganism database of metabolic pathways and enzymes. Nucleic Acids Res. 2006;34(suppl 1):D511–6. doi: 10.1093/nar/gkj128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heagerty PJ, Zheng Y. Survival model predictive accuracy and ROC curves. Biometrics. 2005;61:92–105. doi: 10.1111/j.0006-341X.2005.030814.x. [DOI] [PubMed] [Google Scholar]
- 35.Harris MA, Lock A, Bähler J, et al. FYPO: the fission yeast phenotype ontology. Bioinformatics. 2013;29(13):1671. doi: 10.1093/bioinformatics/btt266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gawronski J, Benson DR. Microtiter assay for glutamine synthetase biosynthetic activity using inorganic phosphate detection. Anal Biochem. 2004;327:114–8. doi: 10.1016/j.ab.2003.12.024. [DOI] [PubMed] [Google Scholar]
- 37.Choi H, Kyeong H, Choi JM, Kim H. Rational design of ornithine decarboxylase with high catalytic activity for the production of putrescine. Appl Microbiol Biotechnol. 2014;98(17):7483–90. doi: 10.1007/s00253-014-5669-8. [DOI] [PubMed] [Google Scholar]
- 38.Hanfrey CC, Sommer S, Mayer MJ, Burtin D, Michael AJ. Arabidopsis polyamine biosynthesis: absence of ornithine decarboxylase and the mechanism of arginine decarboxylase activity. Plant J. 2001;27(6):551–60. doi: 10.1046/j.1365-313X.2001.01100.x. [DOI] [PubMed] [Google Scholar]
- 39.Sookoian S, Pirola CJ. Alanine and aspartate aminotransferase and glutamine-cycling pathway: Their roles in pathogenesis of metabolic syndrome. World J Gastroenterol. 2012;18(29):3775–81. doi: 10.3748/wjg.v18.i29.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bali S, Rollauer S, Roversi P, Rauxdeery E, Lea SM, Warren MJ, Ferguson SJ. Identification and characterization of the missing terminal enzyme for siroheme biosynthesis in proteobacteria. Mol Microbiol. 2014;92:153–63. doi: 10.1111/mmi.12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Streeter JG. Accumulation of alpha,alpha-trehalose by Rhizobium bacteria and bacteroids. J Bacteriol. 1985;164:78–84. doi: 10.1128/jb.164.1.78-84.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sieciechowicz KA, Joy KW, Ireland RJ. The metabolism of asparagine in plants. Phytochemistry. 1988;27(3):663–71. doi: 10.1016/0031-9422(88)84071-8. [DOI] [Google Scholar]
- 43.Quezada H, Marinhernandez A, Arreguinespinosa R, Rumjanek FD, Morenosanchez R, Saavedra E. The 2-oxoglutarate supply exerts significant control on the lysine synthesis flux in Saccharomyces cerevisiae. FEBS J. 2013;280(22):5737–49. doi: 10.1111/febs.12490. [DOI] [PubMed] [Google Scholar]
- 44.Wulandari AP, Miyazaki J, Kobashi N, Nishiyama M, Hoshino T, Yamane H. Characterization of bacterial homocitrate synthase involved in lysine biosynthesis. FEBS Lett. 2002;522:35–40. doi: 10.1016/S0014-5793(02)02877-6. [DOI] [PubMed] [Google Scholar]
- 45.Arnesen T. Protein N-terminal acetylation: NAT 2007–2008 Symposia. BMC Proc. 2009;3(6):1–3. doi: 10.1186/1753-6561-3-s6-s1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whitfield JB. Gamma glutamyl transferase. Crit Rev Clin Lab Sci. 2008;38(4):263–355. doi: 10.1080/20014091084227. [DOI] [PubMed] [Google Scholar]
- 47.Kim K, Park C, An J, Ham B, Lee B, Paek K. CaAlaAT1 catalyzes the alanine: 2-oxoglutarate aminotransferase reaction during the resistance response against Tobacco mosaic virus in hot pepper. Planta. 2005;221(6):857–67. doi: 10.1007/s00425-005-1500-1. [DOI] [PubMed] [Google Scholar]
- 48.Leustek T, Smith M, Murillo M, Singh DP, Smith AG, Woodcock SC, Awan SJ, Warren MJ. Siroheme biosynthesis in higher plants analysis of an S-Adenosyl-L-Methionine-Dependent uroporphyrinogen III Methyltransferase from Arabidopsis Thaliana. J Biol Chem. 1997;272(5):2744–52. doi: 10.1074/jbc.272.5.2744. [DOI] [PubMed] [Google Scholar]
- 49.Musgrave W, Yi H, Kline D, Cameron J, Wignes JA, Dey S, Pakrasi HB, Jez JM. Probing the origins of glutathione biosynthesis through biochemical analysis of glutamate-cysteine ligase and glutathione synthetase from a model photosynthetic prokaryote. Biochem J. 2013;450:63–72. doi: 10.1042/BJ20121332. [DOI] [PubMed] [Google Scholar]
- 50.Orr WC, Radyuk SN, Prabhudesai L, Toroser D, Benes J, Luchak JM, Mockett RJ, Rebrin I, Hubbard JG, Sohal RS. Overexpression of glutamate-cysteine ligase extends life span in Drosophila melanogaster. J Biol Chem. 2005;280(45):37331–8. doi: 10.1074/jbc.M508272200. [DOI] [PubMed] [Google Scholar]
- 51.Lee JH, Khadka P, Baek SH, Chung IK. CHIP promotes human telomerase reverse transcriptase degradation and negatively regulates telomerase activity. 285. 2010;53:42033–45. doi: 10.1074/jbc.M110.149831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holt SE, Aisner D, Baur JA, Tesmer VM, Dy M, Ouellette MM, Trager JB, Morin GB, Toft DO, Shay JW, et al. Functional requirement of p23 and Hsp90 in telomerase complexes. Genes Dev. 1999;13(7):817–26. doi: 10.1101/gad.13.7.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nijkamp K, Van Luijk N, De Bont JAM, Wery J. The solvent-tolerant Pseudomonas putida S12 as host for the production of cinnamic acid from glucose. 69. 2005;2:170–7. doi: 10.1007/s00253-005-1973-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets during and/or analysed during the current study available from the corresponding author on reasonable request.