

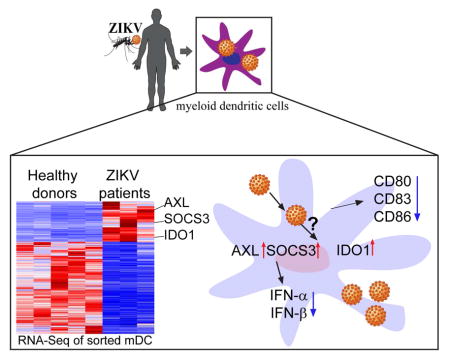
Summary
Although dendritic cells are among the human cell population best equipped for cell-intrinsic antiviral immune defense, they seem highly susceptible to infection with Zika Virus (ZIKV). Using highly-purified myeloid dendritic cells isolated from individuals with naturally-acquired acute infection, we here show that ZIKV induces profound perturbations of transcriptional signatures relative to healthy donors. Interestingly, we noted a remarkable downregulation of antiviral Interferon-stimulated genes and innate immune sensors, suggesting that ZIKV can actively suppress Interferon-dependent immune responses. In contrast, several host factors known to support ZIKV infection were strongly upregulated during natural ZIKV infection; these transcripts included AXL, the main entry receptor for ZIKV, SOCS3, a negative regulator of ISG expression, and IDO-1, a recognized inducer of regulatory T cell responses. Thus, during in vivo infection, ZIKV can transform the transcriptome of dendritic cells in favor of the virus to render these cells highly conducive to ZIKV infection.
Graphical Abstract
Introduction
Zika virus (ZIKV) is an emerging mosquito-borne member of the Flavivirus genus, which includes dengue (DENV), West Nile (WNV), Yellow fever (YFV) and Japanese encephalitis viruses (JEV). Infection with Zika virus causes a transient febrile illness that is typically associated with a skin rash, generalized fatigue and disseminated joint pain (Calvet et al., 2016). In the context of the recent Zika virus epidemic in Asia, Polynesia and the Americas, more complex and severe Zika-associated disease manifestations became apparent, including neonatal birth defects affecting the central nervous system (Cunha et al., 2017; Panchaud et al., 2016), Guillain-Barre syndrome (Nascimento and da Silva, 2017) or profound Thrombocytopenia (Boyer Chammard et al., 2017) in adults. Yet, to a large extent, the mechanisms by which ZIKV causes disease, and the immune responses that humans can generate to restrict ZIKV are unclear. Dendritic cells (DCs) have critical roles in pathogen detection, antigen presentation, and induction of innate and adaptive effector cell immune responses; however, at least in adults, these cells may also represent key target cells for ZIKV infection. This is true for dendritic cells localized in the skin and other mucosal barriers where encounters between host and virus initially occur (Hamel et al., 2015), but may also relate to the myeloid dendritic cells (mDCs) circulating in peripheral blood (Bowen et al., 2017). Although DCs contain a highly complex machinery for detecting viral pathogens and are among the immune cells most effectively equipped for inhibiting viral replication steps through cell-intrinsic immune defense pathways, initial studies suggest that ZIKV has developed successful strategies to escape and counterbalance such antiviral restriction mechanisms. In particular, several pieces of evidence suggest that ZIKV can block type I Interferon-mediated immune responses in dendritic cells by targeting STAT1 and STAT2, critical effectors of the IFN-dependent immune responses, either through proteasomal degradation or through inhibition of STAT protein phosphorylation (Bowen et al., 2017; Grant et al., 2016; Kumar et al., 2016). Simultaneously to inhibiting IFN responses, it is evident that ZIKV is well adjusted to take advantage of the specific subcellular microenvironment of dendritic cells, and can exploit several host factors present in dendritic cells to support individual steps of the viral life cycle. Indeed, recent studies identified a list of 1116 host genes that act as ZIKV or DENV dependency genes and were identified in human cell culture models, but are frequently also expressed in primary dendritic cells (Savidis et al., 2016a). Viral dependence on such host factors represents a specific viral vulnerability and the relative presence or absence of such viral dependency factors may influence the susceptibility to infection among different hosts.
In the present study, we conducted a detailed analysis of host gene expression changes in mDCs, using cell samples from individuals with acute ZIKV infection and in vitro infected primary dendritic cells. These data show that at least during in vivo infection of mDCs, antiviral interferon-stimulated genes and innate immune sensors show a profoundly reduced gene expression intensity, suggesting that type I IFN responses during ZIKV infection are actively suppressed. In combination with existing datasets describing host factors known to support ZIKV infection (Savidis et al., 2016a), we identify a list of viral dependency genes that are consistently upregulated during in vitro and in vivo ZIKV infection, and can support the ZIKV life cycle through a mixture of direct and indirect immune effects. The specific combination of overexpressed viral dependency genes and downregulation of interferon-dependent viral restriction factors reflects a remarkable ability of ZIKV to manipulate the cellular and subcellular milieu of dendritic cells in favor of the virus and offers opportunities for therapeutically targeting viral replicative activity through manipulation of critical host factors.
Results
Viral replication in individuals with acute ZIKV infection
Currently available data suggest that ZIKV can infect a diverse array of human immune cells of both myeloid and lymphoid origin (Hamel et al., 2015; Quicke et al., 2016), but viral preferences for target cell selection in vivo are not well defined. To begin to assess preferred target cells for ZIKV, we analyzed ZIKV replication in circulating cell populations isolated from three ZIKV-infected female adults who travelled to Caribbean destinations during autumn/winter 2015/2016, presented for medical care after developing typical clinical symptoms associated with acute ZIKV infection and were found to have detectable ZIKV plasma RNA, while PCR assays for alternative viruses, in particular Dengue and Chikungunya Virus, were negative as determined by routine clinical and laboratory tests. Interestingly, ZIKV RNA was detected in sorted mDCs from two out of the three study subjects, while sorted B cells, NK cells, CD4 and CD8 T cells and plasmacytoid dendritic cells (pDCs) from any of these patients contained no detectable ZIKV RNA (Table 1); this is consistent with preferential targeting of myeloid DCs by ZIKV in vivo.
Table 1.
Clinical parameters of the ZIKV-infected patients and healthy donors
| Parameters | Patient A | Patient B | Patient C | Healthy donors |
|---|---|---|---|---|
| Age | 35 | 32 | 31 | 28–33 (Median 31,n=5) |
| Gender | Female | Female | Female | Female |
| Pregnancy | no | no | no | no |
| Traveling history | St. Vincent, Cayman | Puerto Rico | Santo Domingo | - |
| Fever | + | + | + | - |
| Rash | + | + | + | - |
| Joint pain | + | + | + | - |
| Other symptom | − | flu-like, cough, diarrhea, fatigue | ankle edema | - |
| Dengue virus PCR test | − | − | − | n.t |
| Chikungunya virus PCR test | − | − | − | n.t |
| ZIKV virus PCR test | + | + | + | n.t |
| Laboratory ZIKV qRT-PCR (plasma) (copies/ml) | 280 | 40 | 190 | n.t |
| Laboratory ZIKV qRT-PCR (mDC) (copies/cell) | − | 6.7 | 7.2 | n.t |
| Laboratory ZIKV qRT-PCR (CD4,CD8,NK,B,monocyte,pDC) | − | − | − | n.t |
n.t.: not tested
To corroborate these findings, we subsequently conducted in vitro infection experiments using PBMCs from adults; given the association between ZIKV infection and neonatal birth defects (Reynolds et al., 2017), cord blood mononuclear cells (CBMCs) were also used. We observed that both PBMCs and CBMCs effectively supported ZIKV infection after in vitro infection, with higher levels of ZIKV transcripts being detectable in cord blood cells (Figure 1A–B). Within both PBMCs and CMBCs, highest levels of ZIKV transcripts were observed in mDCs and lower levels of ZIKV transcripts were also detected in monocytes, NK cells, pDCs and B cells, while CD4+ and CD8+ T cells were almost entirely resistant against ZIKV (Figure 1C and 1D; Figure S1A). Consistent with these observations, we noted that ZIKV RNA levels increased in mDCs over longer durations of in vitro infection, as opposed to ZIKV RNA levels in alternative cell populations that remained stable or declined over time (Figure 1E and Figure S1B); this suggests that mDCs have improved abilities to support productive ZIKV infection. Notably, negative strands of ZIKV RNA, indicative of active viral replication (Bayer et al., 2016; Li et al., 2016), were most frequently and most strongly detected in mDCs, but low levels of negative-stranded ZIKV RNA were also detectable in pDCs, NK cells, B cells and monocytes (Figure 1F and Figure S1C). As with positive-stranded ZIKV RNA, we noted that levels of negative-stranded ZIKV RNA were generally higher in cell subsets from CBMCs compared to their corresponding counterparts from PBMCs (Figure 1F). To further investigate cell susceptibility to viral infection, we used flow cytometry to analyze viral protein expression in sorted, in vitro infected T cells, B cells, NK cells, monocytes and mDCs except pDCs due to the viability issue of in vitro culture. Consistent with our prior results, we found that the ZIKV E protein was most strongly detectable in mDCs, and least in T cells (Figure S1D–E). Notably, AXL and T-cell immunoglobulin and mucin 3 (TIM3), two members of the phosphatidylserine receptor family, were highly expressed in mDCs compared to other cell subsets, consistent with a role as entry receptors for Flaviviruses that increases susceptibility of mDCs to ZIKV (Perera-Lecoin et al., 2013) (Figure S1F and S1G). Together, these data indicate that among human circulating leukocytes, mDCs represent the major target cells for ZIKV infection in vivo and in vitro.
Figure 1. Zika virus replication in in vitro infected cell subsets from adult PBMCs and cord blood.

(A) PBMCs and CBMCs were infected with ZIKV at MOI of 1 and viral replication was measured by qRT-PCR at indicated hours post-infection. Positive-strand viral RNA expression is shown after normalization to beta-actin (n = 10).
(B) Comparison between ZIKV replication in in vivo infected PBMCs (blue) and CBMCs (red). Data show Mean±SD from 10 donors (n=10).
(C) ZIKV replication in sorted cell populations from PBMCs and CBMCs after infection for 24hours with ZIKV at MOI of 1(n = 9).
(D) Comparison between ZIKV replication in B cells (orange), NK cells (pink) and mDCs (purple) from PBMCs and CBMCs (n=9).
(E) Analysis of ZIKV replication in pDCs and mDCs at indicated time points.
(F) Analysis of negative-strand ZIKV RNA in pDCs and mDCs from PBMCs and CBMCs, as determined by qPCR at 24hours p.i.
Horizontal lines reflect the median. * p<0.05; ** p<0.01; *** p < 0.001; **** p< 0.0001
Profound transcriptional changes in ZIKV-infected mDCs in vivo
Flaviviruses can cause considerable perturbations to the physiology and function of human cells, and influence their cellular regulatory circuits at multiple levels (Clarke et al., 2014; Olagnier et al., 2014; Rolfe et al., 2016). To obtain a global perspective of cellular changes induced by ZIKV infection in vivo, we conducted RNA-Seq-based transcriptional profiling experiments to characterize gene expression changes in sorted mDCs from the three study subjects with acute ZIKV infection. We observed that on a global transcriptional level, gene expression signatures differed drastically between mDCs from ZIKV-infected individuals and a reference cohort of gender- and age-matched uninfected individuals, with a total of n=3303 transcripts showing differential gene expression (FDR-adjusted p<0.05 and log2FC>1.5) (Figure 2A–C). Notably, functional annotations of genes differentially expressed in vivo in mDCs from ZIKV-infected patients referred to multiple key functions of dendritic cells, including phagocytosis, immune recognition by innate pattern-recognition receptors, chemotaxis, interferon-dependent immune responses, co-simulation and antigen presentation pathways, as determined by computerized algorithms; most of these functional activities were predicted to be inhibited or negatively enriched (Figure 2D). Consistent with this observation, a computerized canonical pathway analysis showed that key functional networks of dendritic cells were predicted to be inhibited in mDCs from ZIKV patients, such as JAK/STAT signaling, virus-induced NF-κB activation and TLR signaling (Table S2). For comparative purposes, we subsequently performed analogous transcriptional profiling experiments with freshly isolated mDCs that were in vitro infected with ZIKV, and subjected to RNA-Seq assays at selected time points post infection. Although ZIKV RNA was clearly detectable by RNA-Seq at 24h, and more strongly at 48h after infection (Figure 2E), global transcriptional signatures between infected cells and uninfected control cells varied rather modestly, with a maximum of approximately 250 transcripts showing differential gene expression (nominal p<0.05) at any given time point (Figure 2F, Table S3); this indicates that ZIKV-associated global perturbations of gene expression patterns in mDCs were substantially more pronounced in vivo than in our in vitro infection experiments. Several functional pathways, such as antigen presentation and innate immune recognition were also affected by genes found to be differentially-expressed in in vitro infected cells, although predicted effects were statistically less robust than in in vivo infected cells due to lower numbers of genes entered into the analysis (Figure 2G and 2H). Notably, 199 genes were differentially expressed during both in vivo and in vitro infection; these shared transcripts again related to a diverse spectrum of key functional pathways of dendritic cells, including antigen presentation, Toll-like receptor signaling, cell maturation and cytokine production (Figure 2I, Table S4). Together, these results suggest that ZIKV infection of mDCs in vivo is characterized by profound transcriptional changes likely to affect the full spectrum of immune activities of dendritic cells.
Figure 2. Profound changes in the global transcriptional landscape of mDCs infected with ZIKV in vivo.

(A) Principal component analysis (PCA) of RNA-seq data from mDCs isolated from three patients with acute ZIKV infection (red dots) and 5 healthy donors in mDCs (grey dots). The expression values are normalized across the entire data set.
(B) Hierarchical clustering and heatmap of genes that are differentially expressed (DEGs) between ZIKV-infected patients and healthy donors. Displayed DEG had log2 FC≥1.5 fold changes in gene expression intensity and FDR-adjusted p-value <0.05, relative to the control cohort.
(C) Volcano plot displaying genes detected by RNA-Seq. Pink dots represent genes which show FDR-adjusted p<0.05; red dots indicate genes with FDR-adjusted p<0.05 and log2FC>1.5.
(D) Functional annotations of differentially-expressed genes, as determined using Ingenuity pathway analysis. Negative z-scores indicate decreased functional activity (blue), grey bars reflect z-scores with unknown functional activity changes.
(E) Zika viral genome coverage detected by RNA-seq in in vitro infected mDCs at indicated hours post-infection. Alignment was performed using Bedtools software.
(F) Waterfall plot representing the total number of up-regulated and down-regulated genes between in vitro infected mDCs and their respective controls at indicated time points. Transcripts with a nominal p-value <0.05 were considered as DEGs.
(G) Predicted functional pathways of indicated DEG, as analyzed by Ingenuity Pathway analysis. z-scores >2 indicate functional pathway activation (red), whereas negative z-scores indicated pathway de-activation (blue). n.s.: not significant n.d: not detected.
(H) Venn diagram indicates shared DEGs between in vivo and in vitro infected mDC samples, heatmap represents expression pattern of 199 genes that were differentially-expressed both in in vivo and in vitro infected mDCs.
(I) Predicted functional pathways of shared 199 DEGs analyzed by Ingenuity Pathway analysis. Directional changes in gene expression intensity were not determined in this analysis.
ZIKV infection inhibits type I IFN responses in vivo
mDCs are among the cell populations most effectively equipped to recognize viral pathogens and initiate cell-intrinsic immune responses able to restrict viral replication, typically via activation of type I interferon responses; this makes dendritic cells an unfavorable target cell population for viral infection, and creates a hostile microenvironment for many viruses known to cause disease in humans. To investigate why mDCs can effectively serve as target cells for ZIKV, we mined the global transcriptional profiling data from dendritic cells of ZIKV-infected individuals for expression levels of Interferon-stimulated genes (ISGs). We noted considerable changes in the expression profile of ISGs, of which n=1035 were differentially expressed (FDR-adjusted p<0.05 and log2FC>1.5) in response to ZIKV infection in vivo (Figure 3A). Notably, most of these ISG (71%) were downregulated relative to uninfected control data (Figure 3B). Consistent with these observations, we observed that a pre-defined list of ISG with known antiviral activity against Flaviviridae showed significantly reduced expression intensity during acute ZIKV infection (Itsui et al., 2006; Jiang et al., 2010; Metz et al., 2012; Schoggins et al., 2011; Zhao et al., 2012)(Figure 3C, Figure S2A and Table S5). Notably, we observed that IFITM1 and IFITM3, which can inhibit viral genome entry into the cytosol and represent key viral restriction factors during the earliest stages of infection (Savidis et al., 2016b), did not show altered gene expression intensity relative to uninfected cells (Figure S3A); the same was true for cytoplasmic sensors of microbial RNA and DNA, including RIG-I and other RIG-like receptors which have an active role in recognizing Flaviviral RNA. Key transcription factors regulating the induction of type I IFN responses were also downregulated during acute ZIKV infection (Figure 3C), as was interferon-α itself, and, to a lesser degree, interferon-β (Figure 3D). In vitro infected mDCs also showed increased downregulation of key effectors of the IFN signature, including IFN-α, and, less profoundly, IFN-β (Figure 3E), although global ISG alterations in in vitro infected mDCs showed a more heterogeneous picture (Figure S2B). Interestingly, ZIKV infection systematically downregulated type I, type II and type III interferon-stimulated genes in infected dendritic cells, contrary to in vitro findings (Bowen et al., 2017); this suggests that ZIKV infection may not only affect intrinsic antiviral immunity but also influence the induction of adaptive immune responses typically regulated by type II IFN (Figure S2C). In line with inhibited type I IFN responses, we observed that costimulatory molecules and cellular activation markers were downregulated during in vitro infection with ZIKV (Figure 3F). Thus, these data suggest that at least during in vivo ZIKV infection, antiviral IFN immune responses and innate immune sensors are actively suppressed to subnormal levels, suggesting that ZIKV infection induces transcriptional changes specifically designed to create an intracellular milieu conducive to supporting ZIKV infection.
Figure 3. Zika virus infection leads to downregulation of Interferon stimulated genes (ISGs) and impaired dendritic cells function.

(A) Heatmap representing interferon stimulated genes (ISGs) that were differentially-expressed between mDCs isolated from ZIKV-infected patients and healthy controls.
(B) Volcano plot reflecting all differentially-expressed ISG (FDR-adjusted p<0.05 and log2FC>1.5, blue dots). Differentially-expressed ISG with known antiviral function are highlighted in orange.
(C) Heatmap reflecting expression intensity of a known list of antiviral ISG during in vivo and in vitro infection of mDCs with ZIKV.
(D–E) Changes in mRNA expression of IFN-α or IFN-β from (D) in vivo infected compared to healthy donors and (E) from in vitro infected normalized to uninfected cells.
(F) Surface expression of indicated dendritic cell immune activation (CD83) and maturation markers (CD80 and CD86) at 0h, 24h and 48h post infection (+). Data from uninfected controls (−) are shown for comparison.
Horizontal lines reflect the median. * p<0.05; ** p<0.01; *** p < 0.001; **** p< 0.0001
Upregulation of ZIKV dependency factors in mDCs in vivo
To further explore how ZIKV can transform the transcriptome of mDCs in favor of the host, we analyzed transcriptional profiling data from in vivo infected mDCs with regard to genes (n=1116) known to support ZIKV replication, as determined by a recent genome-wide siRNA and CRISPR/Cas9 screen using the H1-Hela cell line and the ZIKV MR766 strain (Table S6) (Savidis et al., 2016a). Unexpectedly, we observed that significant proportions of these viral dependency genes (60%) were downregulated during in vivo infection, suggesting that their role for actively supporting and enabling ZIKV infection is less pronounced, at least during in vivo infection of human dendritic cells. 166 of these dependency genes demonstrated no detectable expression at all in mDCs, in neither of the two study cohorts, and 781 transcripts were not differentially expressed between mDCs from infected vs uninfected individuals. Out of n=169 dependency genes that were differentially-expressed in mDCs from ZIKV-infected patients, n=102 transcripts were downregulated during in vivo infection, suggesting that their role for actively supporting and enabling ZIKV infection is less pronounced, at least during in vivo infection of human dendritic cells. In contrast, we found that 67 of these viral dependency genes were significantly upregulated in cells infected with ZIKV in vivo (Figure 4A); these transcripts included several proteins involved in the viral life cycle such as the endoplasmic reticulum membrane complex 2 (EMC2) and 6 (EMC6) possibly contributing to viral endocytosis (Savidis et al., 2016a), or TRAPPC4 and BNIP1 which participate in viral trafficking from the endoplasmic reticulum to the Golgi apparatus (Aoki et al., 2009; Sztul and Lupashin, 2009) (Table S6).
Figure 4. Upregulation of viral dependency genes during natural ZIKV infection.

(A) Pie chart of the viral dependency genes reported by Savidis et al. (Savidis et al., 2016a). Colors indicate expression intensity of these genes in mDCs from ZIKV-infected patients or healthy donors (left panel). Heatmaps reflect the relative ratio of gene expression intensity between ZIKV-infected patients and controls for 169 viral dependency genes meeting criteria for DEG, and for 502 dependency genes that were expressed in mDCs from ZIKV patients and controls but did not meet criteria for DEGs (right panel).
(B) Venn diagram reflecting overlay between DEGs in in vivo and in vitro infected mDCs, and Zika virus dependency genes reported by Savidis et al. (Savidis et al., 2016a). Numbers of ISGs are listed in parentheses. Gene identities of the 10 transcripts detected in all three gene sets are listed, the green box highlights ISGs.
(C) Heatmap representing gene expression changes of the n=10 viral dependency genes with differential gene expression in both in vitro and in vivo infected mDCs described in (B). Color code reflects the fold-changes in ZIKV-infected samples relative to corresponding controls.
(D) Fold changes of SOCS3 and AXL expression following siRNA-mediated gene silencing relative to control samples (left and middle panels) and intensity of reactive oxidative species (ROS) production following inhibition of IDO-1 activity (right panel).
(E) Monocyte-derived dendritic cells were pretreated with SOCS3 siRNA or AXL siRNA for 48hours, or pretreated with 100μM of the IDO1 inhibitor (NLG919) for 2h. Cells were then infected with Zika virus at MOI of 1. Negative-strand Zika RNA was measured after 24 hours p.i. and normalized to untreated controls.
Horizontal lines reflect the median. * p<0.05; ** p<0.01; *** p < 0.001; **** p< 0.0001.
To identify host transcripts of particular importance for the ZIKV life cycle, we compared viral dependency genes that were differentially-expressed in vivo (n=169) to those differentially-expressed in vitro (n=38) (Figure S5A–D). Overlays of viral dependency genes with transcripts differentially-expressed in vivo and in vitro identified a total of 10 genes that were represented in all three groups of genes, six of which were also classified as interferon-stimulated genes (Figure 4B). Five of these genes, SOCS3, IDO1, DNASE1L3, AXL and HLA-DRB5, were strongly upregulated during acute ZIKV infection in vivo; one transcript, encoding for CXCL16, was downregulated in vivo; in vitro expression pattern of these genes was more heterogeneous and tended to decrease early after infection and increase later on (Figure 4C). To further explore the functions of these genes during ZIKV replication, we generated monocyte-derived DCs (MDDCs) from healthy donors which imitate many characteristics of primary myeloid DCs (Sallusto and Lanzavecchia, 1994; Thurner et al., 1999, 1999), and effectively support ZIKV infection in vitro (Hamel et al., 2015). We confirmed that ZIKV can productively infect MDDCs and results in very similar inhibition of type I interferon, dendritic cell maturation and immune activation as described for primary mDCs (Figure S3A–C). Using siRNA-mediated silencing in MDDCs for DNASE1L3 and HLA-DRB5 or addition of recombinant CXCL16 to the culture medium, we noted no direct impact on viral replicative activity, suggesting that these three genes may modulate host responses to ZIKV through more complex or systemic changes in antiviral immunity that are not reproducible in in vitro infected MDDCs (Figure S4A–S4D). In contrast, siRNA-mediated silencing of AXL and SOCS3 mRNA (Figure 4D and Figure S5A–C) led to significant reductions of negative-stranded ZIKV RNA (Figure 4E), consistent with an important role of these molecules for supporting the efficacy of ZIKV replication. However, addition of a small-molecule inhibitor of IDO1, associated with reduction in reactive oxygen species (Olagnier et al., 2014) (Figure 4D), led to significantly increased expression of negative-stranded ZIKV RNA (Figure 4E). To confirm that manipulation of SOCS3, AXL and IDO-1 impact ZIKV replication in MDDCs, we used the supernatants from infected cells to inoculate the ZIKV-permissive MK2 cell line. Consistent with our prior results, the level of infection in MK2 cells was lower when cells were exposed to supernatants from MDDCs treated with siRNA directed against AXL and SOCS3, and higher when infected with supernatants from MDDCs treated with the IDO-1 inhibitor (Figure S5E and S5F).
AXL, SOCS3 and IDO-1 affect ZIKV replication via alteration of the type I IFN responses
To decipher how silencing of AXL and SOCS3 and, inhibition of IDO-1 activity affected ZIKV replication, we analyzed the alteration of the type I IFN responses. Notably, transcriptional silencing of SOCS3 in MDDCs strongly reduced ZIKV replication and led to increased cellular production of IFN-α (Figure 5A) and IFN-β (Figure S5G), which was inversely associated with levels of ZIKV replication (Figure 5B and Figure S5H). Moreover, silencing of SOCS3 increased expression of several transcripts involved in antiviral immune responses (LCK, PSMB9, BCL2, NMI, CD38 and NFIL3), while reducing expression of FASN, known to facilitate dengue virus replication (Heaton et al., 2010; Tang et al., 2014; Tongluan et al., 2017) (Figure 5C). Collectively, these data suggest that SOCS3 facilitates and supports host cell susceptibility to ZIKV by actively suppressing type I IFN responses, and creating a cellular milieu more conducive to ZIKV replication. While experimental downregulation of AXL, encoding for a known entry receptor for ZIKV, expectedly decreased the replicative activity of ZIKV (Figure 4E), we also noticed a significantly increased expression of IFN-α/β in AXL-deficient cells infected with ZIKV (Figure 5A–B and Figure S5G); this is compatible with a role of AXL for modulating innate immune responses to ZIKV (Meertens et al., 2017). Remarkably, we found that inhibition of IDO1, encoding for a host enzyme that catalyzes Kynurenine production from Tryptophan (Mellor and Munn, 2004), facilitated ZIKV replication in in vitro infected cells, as evidenced by increased rates of ZIKV turnover and decreased expression of IFN-α/β and type I IFN responses following experimental inhibition (Figure 4E,5A–5B and Figure S5H), suggesting IDO1-mediated cell-intrinsic restriction of ZIKV replication. Interestingly, molecular inhibition of IDO1 also resulted in downregulation of transcripts involved in innate immune responses (TRIM14, TLR1, JAK2, STAT6, TRIM22 and TGFB1) and apoptosis genes (CASP8, BCL2 and BAK1) (Figure 5D), consistent with a role in restricting ZIKV replication and influencing target cell survival; these findings are contradictory to the previous characterization of IDO1 as dependency gene for ZIKV in cell lines (Savidis et al., 2016a). Together, these observations show that transcriptional changes in mDCs from ZIKV-infected individuals can influence viral replication dynamics by diverse and possibly opposing mechanisms.
Figure 5. AXL, SOCS3 and IDO-1 alter type I interferon responses during ZIKV infection.

(A) Expression of IFN-α after 24 hours post-infection with ZIKV in MDDCs manipulated as described in Figure 4E. Fold-changes normalized to untreated controls are shown.
(B) Correlations between the expression levels of IFN-α and the corresponding ZIKV negative strand RNA levels after silencing SOCS3 (left, n=10), silencing AXL (middle, n=7) or addition of the IDO1 inhibitor NLG99 (right, n=7). Cumulative data were analyzed using generalized estimated equations adjusted for repeated measures.
(C) and (D) Volcano plots reflecting changes in expression of host genes involved in flavivirus pathogenesis following downregulation of SOCS3 (C) or inhibition of IDO1 (D). Differentially-expressed genes (nominal p<0.05) are indicated in red. Heatmaps reflect gene expression changes of the differentially-expressed genes; color coding indicates gene expression intensity.
Horizontal lines reflect the median. * p<0.05; ** p<0.01; *** p < 0.001; **** p< 0.0001.
Discussion
Dendritic cells are highly specialized immune cells with unique functional abilities to sense viral pathogens, present foreign antigens to innate and adaptive effector cells and initiate antiviral immune defense operations to protect the host. In addition, dendritic cells also possess a remarkable cell-intrinsic ability to restrict viral replication steps through a wide spectrum of antiviral effector molecules synthesized and activated in response to secretion of type I Interferons. For these reasons, dendritic cells are frequently regarded as unfavorable target cells for viral infection. Counterintuitively to these observations, several in vitro studies have suggested that dendritic cells can represent preferred host cells for ZIKV infection (Bowen et al., 2017; Hamel et al., 2015), suggesting that ZIKV has developed specific strategies to transform the hostile microenvironment in mDCs into a territory that is supportive and conducive to viral infection. Here, we performed a detailed analysis of dendritic cells infected in vivo with ZIKV in individuals who naturally acquired the disease during recreational travel to Caribbean destinations in autumn/winter 2015/2016. We show that among circulating leukocellular subsets from individuals with naturally-acquired infection, mDCs clearly represent a cell compartment highly susceptible to ZIKV infection, and the only cell type in which cell-associated viral RNA could be detected directly; this strongly supports the notion that mDCs indeed represent a predominant target cell population for ZIKV among circulating leukocytes in vivo. Unfortunately, we had no opportunity to analyze ZIKV infection in ex vivo isolated cell subsets from fetal tissues or pediatric patients, but our in vitro experiments suggest that fetal or neonatal mDCs exhibit even higher susceptibility to ZIKV relative to adult cells, possibly due to the more immature phenotype of these cells during fetal and neonatal development (Hamel et al., 2015; Richter et al., 2014). Notably, recent studies also demonstrated that myeloid cells, including specific monocyte subsets, are highly susceptible to ZIKV during pregnancy and in infants, and highlighted the fact that ZIKV can reprogram their gene expression patterns to limit antiviral immune defense. (Foo et al., 2017; Michlmayr et al., 2017).
To analyze why ZIKV preferentially infects target cells that are among the most immunocompetent and least vulnerable immune cells circulating in human blood and tissues, we conducted genome-wide transcriptional profiling assays from cells collected from individuals with acute ZIKV infection. Remarkably, these data highlighted that upon infection with ZIKV, the cellular transcriptome of mDCs shows profound alterations in favor of the virus, through combined effects that results in downregulation of antiviral effector genes in conjunction with upregulation of a spectrum of host factors known to support ZIKV replication. The inhibition of antiviral interferon-stimulated genes to subnormal levels of expression represents a particularly striking observation, given that a number of prior studies have concluded, based on in vitro infection experiments, that at least at the level of mRNA expression, ZIKV infection induces strong upregulation of IFN itself and several antiviral ISG in dendritic cells (Bowen et al., 2017), in human cell lines (Frumence et al., 2016) and in primary human skin fibroblasts (Hamel et al., 2015). For instance, transcripts encoding for the antiviral factors IFITM1 and IFITM3 were both upregulated in mDCs in in vitro studies, while a downregulation of IFITM3 expression was noted in mDCs in vivo. The reasons for this discrepancy are not clear at present but may relate to altered gene expression kinetics of these two transcripts during naturally-acquired infection. In addition, previous studies have shown that IFITM1 is expressed on the plasma membrane and the early endosomes while IFITM3 can be preferentially detected in late endosomes(Bailey et al., 2014; Gorman et al., 2016; Monel et al., 2017; Savidis et al., 2016b). Nevertheless, there is evidence that ZIKV can antagonize interferon-signaling pathways by targeting key proteins involved in signaling downstream of the IFN receptor. Indeed, ZIKV can inhibit phosphorylation (Bowen et al., 2017), induce degradation of STAT2 (Grant et al., 2016) and block JAK1 function (Wu et al., 2017), all of which leads to inactivation of IFN-dependent immune activity on a protein level. A future analysis of proteomic and phosphoproteomic changes in dendritic cells from ZIKV-infected individuals will be highly informative to investigate these effects in vivo. Interestingly, while type I and type III Interferons can restrict ZIKV infection (Bayer et al., 2016; Chaudhary et al., 2017; Savidis et al., 2016b), type II interferons appeared to activate and promote viral replication in cell lines (Chaudhary et al., 2017); however our data suggest that at least in mDCs, ZIKV infection is associated with non-selective suppression of ISG from any of these three subclasses of interferons.
In addition to downregulation of antiviral ISG during in vivo infection, our work highlights a complementary upregulation of a substantial list of genes known to support ZIKV life cycle through direct and indirect interactions with ZIKV. Our studies show that at least 67 transcripts previously shown to support ZIKV replication in in vitro assays were upregulated during natural infection; these transcripts seem therefore of particular relevance for reinforcing and enabling ZIKV replication in infected humans. Among these genes was AXL, a member of the TAM signaling cascade that acts as a main viral entry receptor previously identified in human neural cells, skin cells and MDDCs (Hamel et al., 2015; Meertens et al., 2017; Richard et al., 2017), suggesting that AXL is an essential host component for supporting ZIKV infection under in vivo infection conditions in DCs. Interestingly, our data also showed that knockdown of AXL in MDDCs can increase the expression levels of IFN-α and IFN-β, indicating that AXL has an additional role for suppressing type I IFN responses in human dendritic cells. Notably, a role for AXL in modulating IFN-I responses and promoting host susceptibility to viral infections has previously been described in in vitro and animal experimental systems (Bhattacharyya et al., 2013; Meertens et al., 2017; Rothlin et al., 2007). Remarkably, we noted that upregulation of AXL coincided with increased expression of SOCS3, consistent with mutually-interdependent interactions between members of the TAM signaling network and the suppressor of cytokine signaling (SOCS) family (Carow and Rottenberg, 2014). Through its known ability to inhibit the JAK/STAT pathway, the main downstream effector pathway for Interferon signaling (Carow and Rottenberg, 2014), SOCS3 is therefore likely to play a key role for the observed downregulation of interferon-stimulated genes in ZIKV infected patients. Simultaneous induction of SOCS3 and AXL during ZIKV infection may occur as an inhibitory feedback mechanism designed to dampen hyperactivated dendritic cells; as such, it is possible that upregulation of SOCS3 and AXL, and the associated inhibition of ISG expression, was preceded by pathogen-induced innate immune activation during the earliest stages of infection.
Notably, genes found to be upregulated in mDCs from patients with ZIKV infection included IDO1, which encodes for indoleamine 2,3-dioxygenase and can facilitate viral infection through IDO1-induced Tryptophan depletion, specifically in the context of herpes simplex virus type 2 (HSV-2), measles virus, and vaccinia virus infection (Carow and Rottenberg, 2014). Contrary to prior experiments in cell lines, we observed that in vitro infected primary MDDCs exhibited improved abilities to support ZIKV infection after experimental inhibition of IDO1, which suggests an inhibitory role of IDO1 within the ZIKV life cycle in these cells. Moreover, we showed that inhibition of IDO1 resulted in downregulation of type I IFN responses, coupled with a reduced expression of cell apoptosis genes; together, these observations suggest that IDO1 can protect the host against ZIKV through activation of innate immune responses and induction of cellular death programs in infected target cells. Nevertheless, IDO1 expression in dendritic cells is also known to dampen T cell-mediated immune responses and antagonize host antiviral immunity through the induction of regulatory T cells (Mellor and Munn, 2004); therefore, cell-intrinsic restriction of ZIKV by IDO1 may be counterbalanced by Treg-mediated inhibition of innate and adaptive effector cell responses favoring viral replication. Together, these results suggest that transcripts classified as viral dependency genes may have discrete, and partially contradictory effects on viral replicative activity and antiviral immune defense.
Collectively, this study, we describe immune changes in individuals with naturally-acquired ZIKV infection, highlights several distinct features of the innate immune response in ZIKV-infected individuals, and illustrates the remarkable ability of ZIKV to transform mDCs into a cellular microenvironment able to effectively support viral infection. A continued investigation of immune characteristics occurring in vivo in individuals with ZIKV-induced disease may help to better understand the pathogenesis and human host response to ZIKV.
Experimental procedures
Study participants
Zika virus infected study participants were recruited from the Massachusetts General Hospital and the Brigham and Women’s Hospital (Boston, MA). PBMC samples were used under IRB protocols. Clinical and demographical characteristics of study subjects are summarized in Table 1. Study subjects gave written informed consent to participate in accordance with the Declaration of Helsinki. Cord blood samples were obtained from the New York Blood Center (New York), under an IRB-approved protocol.
Flow cytometry and cell sorting
PBMCs or CBMCs were stained with antibodies against surface markers and subsequently analyzed by a FACS LSR Fortessa or sorted using a FACS Aria cell sorter (BD Biosciences). In addition, Expression of CD80, CD83, CD86 in mDC or MDDCs were also analyzed (see the supplemental experimental procedures for details). All flow data was analyzed by FlowJo V10. Total ROS production was measured using 1μM of the CM-H2DCFDA probe (Thermo Fisher Scientific) according to the product manual. Phorbol 12-myristate 13-acetate (PMA) was served as positive control and DMSO only as negative control.
Generation of Monocyte-derived dendritic cells
Monocytes were cultured using R10 medium supplemented with 100 ng/ml of GM-CSF (PeproTech) and 50 ng/ml IL-4 (PeproTech). MDDCs were harvested at day 5 (Nair et al., 2012), see the supplemental experimental procedures for details.
Transcriptional profiling of primary mDCs
Total RNA was extracted from sorted cell populations using a PicoPure RNA Isolation Kit. RNA-Seq libraries for isolated mDCs from each group were generated as previously described (Trombetta et al., 2014), see the supplemental experimental procedures for details.
Cells
LLC-MK2 and C6/36 cells were obtained from ATCC and maintained in D10 medium, see the supplemental experimental procedures for details.
Zika virus production and titration
ZIKV Strain PRVABC59 was used in this study (Lanciotti et al., 2016). The virus was propagated by C6/36 cell and titrated by flow cytometry, see the supplemental experimental procedures for details.
ZIKV infection of cells
PBMCs and CBMCs cells were infected with ZIKV at a multiplicity of infection (MOI) of 1. The cells were incubated for 2 h at 37°C. Next, the viral inoculum was removed and the cells were washed four times with PBS, see the supplemental experimental procedures for details.
Negative-stranded ZIKV mRNA
A standard RT-PCR was carried out by using primers containing the T7 promoter sequence as listed in Table S1 and then ZIKV RNA fragments were generated by in vitro transcription using a MAXIscript kit. Copies of viral RNA were quantified by normalizing each sample’s threshold cycle (CT) value to a ZIKV RNA standard curve obtained as described before (Hamel et al., 2015).
ZIKV qRT-PCR and qPCR
Total RNA was isolated and qRT-PCR was performed for quantifying total viral RNA. In addition, cDNA was generated with random primers or ZIKV-F primers. the qPCR was used to quantifying the expression level of candidates and negative ZIKV strand, see the supplemental experimental procedures for details.
siRNA-mediated gene knockdown
Silencing of AXL, SOCS3, HLA-DRB5, DNASE1L3 or scramble control using siRNA pools (On-TARGET plus SMARTpool, Dharmacon) were performed by nucleofection of MDDCs. The efficiency of knockdown was determined at the mRNA level by qPCR for all candidates and confirmed at the protein level only for AXL and SOCS3, see the supplemental experimental procedures for details.
Statistics
Significance differences between the different subsets were assessed using Mann-Whitney U tests or Wilcoxon matched-pairs signed-rank test. When appropriate, statistical analysis was corrected for multiple comparisons using a Friedman test with post-hoc Dunn’s test. Correlations between type I interferon and negative-stranded ZIKV RNA were analyzed using generalized estimated equations adjusted for repeated measures.
Accession number
The RNA-seq data reported in this paper have been deposited to the NCBI GEO and are available under accession number GSE101878.
Supplementary Material
Normalized data for DEGs and ISGs identified in in vivo infected DC, as determined by Ingenuity Pathways Analysis of canonical pathways and disease & functions. Relevant pathways and disease functions for mDCs are highlighted in light green.
Normalized data for DEGs from in vitro infected DC, as determined by Ingenuity Pathways Analysis of canonical pathways for each indicated time point after infection.
Ingenuity Pathways Analysis of canonical pathways of the 199 genes shared between DEGs from in vitro and in vivo infected DC. Relevant pathways for mDCs are highlighted in light green
Table S5, related to Figure 3. Functional categories of the 69 pre-defined antiviral ISGs that were differentially expressed in in vivo infected mDCs.
List of the DENV or ZIKV viral dependency genes overlapping with DEGs detected in in vitro and in vivo infected DC.
Acknowledgments
X.G.Y is supported by NIH (grants HL134539, AI089339, HL121890, HL126554, AI116228, and AI087452) and by the Ragon Institute of MGH, MIT and Harvard.
Footnotes
Author Contributions
X.S., S.H., X.G.Y. and M.L. developed the research idea and study concept, designed the study, and wrote the manuscript. X.G.Y. supervised the study. X.S. and S.H. designed and conducted experiments. K.E. and S.T. provided technical help. H-R.C. and Z.O. conducted the statistical and bioinformatics analysis for RNA-Seq data. M.L., K.A., A.C., S.Y., P.S. and E.S.R. provided samples from ZIKV-infected patients. All authors critically reviewed the manuscript.
Competing Interests
The authors declare that no competing interest exists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bailey CC, Zhong G, Huang I-C, Farzan M. IFITM-Family Proteins: The Cell’s First Line of Antiviral Defense. Annu Rev Virol. 2014;1:261–283. doi: 10.1146/annurev-virology-031413-085537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer A, Lennemann NJ, Ouyang Y, Bramley JC, Morosky S, Marques ETDA, Cherry S, Sadovsky Y, Coyne CB. Type III Interferons Produced by Human Placental Trophoblasts Confer Protection against Zika Virus Infection. Cell Host Microbe. 2016;19:705–712. doi: 10.1016/j.chom.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Zagórska A, Lew ED, Shrestha B, Rothlin CV, Naughton J, Diamond MS, Lemke G, Young JAT. Enveloped viruses disable innate immune responses in dendritic cells by direct activation of TAM receptors. Cell Host Microbe. 2013;14:136–147. doi: 10.1016/j.chom.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen JR, Quicke KM, Maddur MS, O’Neal JT, McDonald CE, Fedorova NB, Puri V, Shabman RS, Pulendran B, Suthar MS. Zika Virus Antagonizes Type I Interferon Responses during Infection of Human Dendritic Cells. PLoS Pathog. 2017;13:e1006164. doi: 10.1371/journal.ppat.1006164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer Chammard T, Schepers K, Breurec S, Messiaen T, Destrem A-L, Mahevas M, Soulillou A, Janaud L, Curlier E, Herrmann-Storck C, et al. Severe Thrombocytopenia after Zika Virus Infection, Guadeloupe, 2016. Emerg Infect Dis. 2017;23:696–698. doi: 10.3201/eid2304.161967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvet GA, Santos FBD, Sequeira PC. Zika virus infection: epidemiology, clinical manifestations and diagnosis. Curr Opin Infect Dis. 2016;29:459–466. doi: 10.1097/QCO.0000000000000301. [DOI] [PubMed] [Google Scholar]
- Carow B, Rottenberg ME. SOCS3, a Major Regulator of Infection and Inflammation. Front Immunol. 2014:5. doi: 10.3389/fimmu.2014.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary V, Yuen K-S, Chan JF-W, Chan C-P, Wang P-H, Cai J-P, Zhang S, Liang M, Kok K-H, Chan C-P, et al. Selective Activation of Type II Interferon Signaling by Zika Virus NS5 Protein. J Virol. 2017;91:e00163–17. doi: 10.1128/JVI.00163-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke P, Leser JS, Bowen RA, Tyler KL. Virus-induced transcriptional changes in the brain include the differential expression of genes associated with interferon, apoptosis, interleukin 17 receptor A, and glutamate signaling as well as flavivirus-specific upregulation of tRNA synthetases. mBio. 2014;5:e00902–00914. doi: 10.1128/mBio.00902-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha AJLA, da de Magalhães-Barbosa MC, Lima-Setta F, de Medronho RA, Prata-Barbosa A. Microcephaly Case Fatality Rate Associated with Zika Virus Infection in Brazil: Current Estimates. Pediatr Infect Dis J. 2017;36:528–530. doi: 10.1097/INF.0000000000001486. [DOI] [PubMed] [Google Scholar]
- Foo S-S, Chen W, Chan Y, Bowman JW, Chang L-C, Choi Y, Yoo JS, Ge J, Cheng G, Bonnin A, et al. Asian Zika virus strains target CD14 + blood monocytes and induce M2-skewed immunosuppression during pregnancy. Nat Microbiol. 2017:1. doi: 10.1038/s41564-017-0016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frumence E, Roche M, Krejbich-Trotot P, El-Kalamouni C, Nativel B, Rondeau P, Missé D, Gadea G, Viranaicken W, Desprès P. The South Pacific epidemic strain of Zika virus replicates efficiently in human epithelial A549 cells leading to IFN-β production and apoptosis induction. Virology. 2016;493:217–226. doi: 10.1016/j.virol.2016.03.006. [DOI] [PubMed] [Google Scholar]
- Gorman MJ, Poddar S, Farzan M, Diamond MS. The Interferon-Stimulated Gene Ifitm3 Restricts West Nile Virus Infection and Pathogenesis. J Virol. 2016;90:8212–8225. doi: 10.1128/JVI.00581-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, Schwarz MC, Sánchez-Seco MP, Evans MJ, Best SM, et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe. 2016;19:882–890. doi: 10.1016/j.chom.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, Perera-Lecoin M, Surasombatpattana P, Talignani L, Thomas F, et al. Biology of Zika Virus Infection in Human Skin Cells. J Virol. 2015;89:8880–8896. doi: 10.1128/JVI.00354-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itsui Y, Sakamoto N, Kurosaki M, Kanazawa N, Tanabe Y, Koyama T, Takeda Y, Nakagawa M, Kakinuma S, Sekine Y, et al. Expressional screening of interferon-stimulated genes for antiviral activity against hepatitis C virus replication. J Viral Hepat. 2006;13:690–700. doi: 10.1111/j.1365-2893.2006.00732.x. [DOI] [PubMed] [Google Scholar]
- Jiang D, Weidner JM, Qing M, Pan X-B, Guo H, Xu C, Zhang X, Birk A, Chang J, Shi P-Y, et al. Identification of five interferon-induced cellular proteins that inhibit west nile virus and dengue virus infections. J Virol. 2010;84:8332–8341. doi: 10.1128/JVI.02199-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Hou S, Airo AM, Limonta D, Mancinelli V, Branton W, Power C, Hobman TC. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 2016;17:1766–1775. doi: 10.15252/embr.201642627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanciotti RS, Lambert AJ, Holodniy M, Saavedra S, Signor LDCC. Phylogeny of Zika Virus in Western Hemisphere, 2015. Emerg Infect Dis. 2016;22:933–935. doi: 10.3201/eid2205.160065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XF, Dong HL, Huang XY, Qiu YF, Wang HJ, Deng YQ, Zhang NN, Ye Q, Zhao H, Liu ZY, et al. Characterization of a 2016 Clinical Isolate of Zika Virus in Non-human Primates. E Bio Medicine. 2016;12:170–177. doi: 10.1016/j.ebiom.2016.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meertens L, Labeau A, Dejarnac O, Cipriani S, Sinigaglia L, Bonnet-Madin L, Le Charpentier T, Hafirassou ML, Zamborlini A, Cao-Lormeau V-M, et al. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell Rep. 2017;18:324–333. doi: 10.1016/j.celrep.2016.12.045. [DOI] [PubMed] [Google Scholar]
- Mellor AL, Munn DH. Ido expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4:762–774. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- Metz P, Dazert E, Ruggieri A, Mazur J, Kaderali L, Kaul A, Zeuge U, Windisch MP, Trippler M, Lohmann V, et al. Identification of type I and type II interferon-induced effectors controlling hepatitis C virus replication. Hepatol Baltim Md. 2012;56:2082–2093. doi: 10.1002/hep.25908. [DOI] [PubMed] [Google Scholar]
- Michlmayr D, Andrade P, Gonzalez K, Balmaseda A, Harris E. CD14 + CD16 + monocytes are the main target of Zika virus infection in peripheral blood mononuclear cells in a paediatric study in Nicaragua. Nat Microbiol. 2017:1. doi: 10.1038/s41564-017-0035-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monel B, Compton AA, Bruel T, Amraoui S, Burlaud-Gaillard J, Roy N, Guivel-Benhassine F, Porrot F, Génin P, Meertens L, et al. Zika virus induces massive cytoplasmic vacuolization and paraptosis-like death in infected cells. EMBO J. 2017;36:1653–1668. doi: 10.15252/embj.201695597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair S, Archer GE, Tedder TF. ISOLATION AND GENERATION OF HUMAN DENDRITIC CELLS. In: Coligan Al John E., editor. Curr Protoc Immunol. Unit7.32. 0 7. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimento OJM, da Silva IRF. Guillain-Barré syndrome and Zika virus outbreaks. Curr Opin Neurol. 2017 doi: 10.1097/WCO.0000000000000471. [DOI] [PubMed] [Google Scholar]
- Olagnier D, Peri S, Steel C, van Montfoort N, Chiang C, Beljanski V, Slifker M, He Z, Nichols CN, Lin R, et al. Cellular oxidative stress response controls the antiviral and apoptotic programs in dengue virus-infected dendritic cells. PLoS Pathog. 2014;10:e1004566. doi: 10.1371/journal.ppat.1004566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchaud A, Stojanov M, Ammerdorffer A, Vouga M, Baud D. Emerging Role of Zika Virus in Adverse Fetal and Neonatal Outcomes. Clin Microbiol Rev. 2016;29:659–694. doi: 10.1128/CMR.00014-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera-Lecoin M, Meertens L, Carnec X, Amara A. Flavivirus Entry Receptors: An Update. Viruses. 2013;6:69–88. doi: 10.3390/v6010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quicke KM, Bowen JR, Johnson EL, McDonald CE, Ma H, O’Neal JT, Rajakumar A, Wrammert J, Rimawi BH, Pulendran B, et al. Zika Virus Infects Human Placental Macrophages. Cell Host Microbe. 2016;20:83–90. doi: 10.1016/j.chom.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds MR, Jones AM, Petersen EE, Lee EH, Rice ME, Bingham A, Ellington SR, Evert N, Reagan-Steiner S, Oduyebo T, et al. Vital Signs: Update on Zika Virus-Associated Birth Defects and Evaluation of All U.S. Infants with Congenital Zika Virus Exposure - U.S. Zika Pregnancy Registry, 2016. MMWR Morb Mortal Wkly Rep. 2017;66:366–373. doi: 10.15585/mmwr.mm6613e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard AS, Shim B-S, Kwon Y-C, Zhang R, Otsuka Y, Schmitt K, Berri F, Diamond MS, Choe H. AXL-dependent infection of human fetal endothelial cells distinguishes Zika virus from other pathogenic flaviviruses. Proc Natl Acad Sci U S A. 2017;114:2024–2029. doi: 10.1073/pnas.1620558114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter MKS, da Voorham JMS, Pedraza ST, Hoornweg TE, van de Pol DPI, Rodenhuis-Zybert IA, Wilschut J, Smit JM. Immature Dengue Virus Is Infectious in Human Immature Dendritic Cells via Interaction with the Receptor Molecule DC-SIGN. PLOS ONE. 2014;9:e98785. doi: 10.1371/journal.pone.0098785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolfe AJ, Bosco DB, Wang J, Nowakowski RS, Fan J, Ren Y. Bioinformatic analysis reveals the expression of unique transcriptomic signatures in Zika virus infected human neural stem cells. Cell Biosci. 2016;6:42. doi: 10.1186/s13578-016-0110-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothlin CV, Ghosh S, Zuniga EI, Oldstone MBA, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131:1124–1136. doi: 10.1016/j.cell.2007.10.034. [DOI] [PubMed] [Google Scholar]
- Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. 1994;179:1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savidis G, McDougall WM, Meraner P, Perreira JM, Portmann JM, Trincucci G, John SP, Aker AM, Renzette N, Robbins DR, et al. Identification of Zika Virus and Dengue Virus Dependency Factors using Functional Genomics. Cell Rep. 2016a;16:232–246. doi: 10.1016/j.celrep.2016.06.028. [DOI] [PubMed] [Google Scholar]
- Savidis G, Perreira JM, Portmann JM, Meraner P, Guo Z, Green S, Brass AL. The IFITMs Inhibit Zika Virus Replication. Cell Rep. 2016b;15:2323–2330. doi: 10.1016/j.celrep.2016.05.074. [DOI] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurner B, Röder C, Dieckmann D, Heuer M, Kruse M, Glaser A, Keikavoussi P, Kämpgen E, Bender A, Schuler G. Generation of large numbers of fully mature and stable dendritic cells from leukapheresis products for clinical application. J Immunol Methods. 1999;223:1–15. doi: 10.1016/s0022-1759(98)00208-7. [DOI] [PubMed] [Google Scholar]
- Trombetta JJ, Gennert D, Lu D, Satija R, Shalek AK, Regev A. Preparation of Single-Cell RNA-Seq Libraries for Next Generation Sequencing. Curr Protoc Mol Biol. 2014;107:4.22.1–17. doi: 10.1002/0471142727.mb0422s107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Lin W, Kumthip K, Cheng D, Fusco DN, Hofmann O, Jilg N, Tai AW, Goto K, Zhang L, et al. A functional genomic screen reveals novel host genes that mediate interferon-alpha’s effects against hepatitis C virus. J Hepatol. 2012;56:326–333. doi: 10.1016/j.jhep.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Normalized data for DEGs and ISGs identified in in vivo infected DC, as determined by Ingenuity Pathways Analysis of canonical pathways and disease & functions. Relevant pathways and disease functions for mDCs are highlighted in light green.
Normalized data for DEGs from in vitro infected DC, as determined by Ingenuity Pathways Analysis of canonical pathways for each indicated time point after infection.
Ingenuity Pathways Analysis of canonical pathways of the 199 genes shared between DEGs from in vitro and in vivo infected DC. Relevant pathways for mDCs are highlighted in light green
Table S5, related to Figure 3. Functional categories of the 69 pre-defined antiviral ISGs that were differentially expressed in in vivo infected mDCs.
List of the DENV or ZIKV viral dependency genes overlapping with DEGs detected in in vitro and in vivo infected DC.