

Abstract
Vast amounts of molecular data are being collected on tumor samples, which provide unique opportunities for discovering trends within and between cancer subtypes. Such cross-cancer analyses require computational methods that enable intuitive and interactive browsing of thousands of samples based on their molecular similarity. We created a portal called TumorMap to assist in exploration and statistical interrogation of high-dimensional complex “omics” data in an interactive and easily interpretable way. In the TumorMap, samples are arranged on a hexagonal grid based on their similarity to one another in the original genomic space and are rendered with Google’s Map technology. While the important feature of this public portal is the ability for the users to build maps from their own data, we pre-built genomic maps from several previously published projects. We demonstrate the utility of this portal by presenting results obtained from The Cancer Genome Atlas project data.
Introduction
Genomic aberrations such as mutations that accumulate in a particular cell’s DNA, together with the tissue microenvironment, contribute to the initiation and progression of malignancies. The Cancer Genome Atlas (TCGA) and similar projects have generated genome-wide molecular characterization information from thousands of tumor samples of various cancer types. These “omic” data, including genomic, transcriptomic, proteomic and epigenomic tumor profiles, enable cross-tumor (“pan-cancer”) analyses to find similarities across cancers originating in different tissues and the identification of clinically and prognostically relevant subtypes that share common molecular driver events and pathway aberrations.
The simultaneous and interactive visualization of multiple genomic datasets can reveal patterns that inspire hypothesis generation (1–4). This is especially true in cancer genomics investigations, where the number of measured features (e.g., 20,000 genes) can be large. Biologists and clinicians need tools to navigate such complex datasets to discover novel clinical and biological insights without additional expertise in computer programming and statistical inference. While several strategies exist to aid in the visualization of samples and to identify subtypes (see related work in Supplementary Methods), no current genomic visualization portals allow users to upload their own data, visualize and interrogate them for statistically significant patterns.
The UCSC TumorMap (https://tumormap.ucsc.edu; Fig. 1A) is a novel interactive visualization and analysis portal to explore patterns among tumor samples arranged relative to one another based on their molecular similarities. In the TumorMap, samples are arranged on the basis of their molecular profile similarity, and attributes associated with samples, such as disease histological subtypes, can be identified easily by eye as contiguous regions as well as discovered using the tool’s Attribute Enrichment Analysis (AEA). Even without the aid of additional computer and programming expertise, users can discover trends using a density analysis and perform statistical enrichment analysis in complex genomics datasets for scientific and therapeutic hypothesis generation. In addition to browsing precomputed maps, the TumorMap allows users to build their own interactive maps from several uploaded high-throughput platforms, as well as integrate multiple platforms into a single landscape (Fig. 1B). Maps can be constructed from any data type comprised of molecular features that encode observations across a set of samples and from which pairwise similarities between samples can be calculated. The use of standardized similarity spaces enables multiple datasets of different feature types to be combined into a single integrated map.
Figure 1.
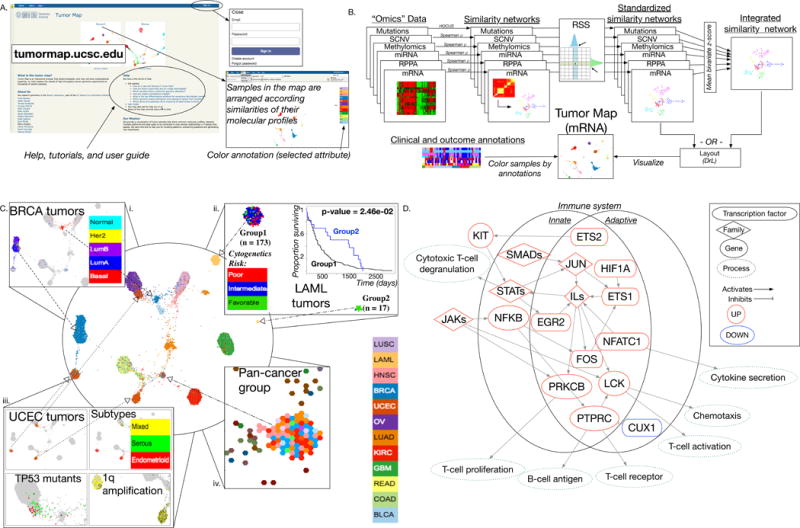
TumorMap framework and application to the analysis of the TCGA Pan-Cancer-12 Dataset. A, The TumorMap is a publicly available web portal. B, Outline of the TumorMap construction workflow. Data from individual molecular platforms (“Omics” Data) are provided as input from which pairwise similarities between samples are calculated to produce “Similarity networks”; these networks are standardized using the Reciprocal Significance of Similarities (RSS; see Supplementary Methods) to create a coherent space of standardized similarity networks. Map layouts are created with the OpenOrd algorithm using coherent sample networks. Integrated multiplatform maps are created from several coherent networks, combined before input to OpenOrd layout procedure. Shown is an mRNA-based gene expression map; colors represent tissue of origin. Attributes such as clinical, molecular, phenotype, or outcome metadata, annotate samples using colors and color gradients based on groupings that can be defined by the user. C, TumorMap rendering of the Pan-Cancer-12 Dataset, an integrated cross-cancer TumorMap based on six molecular data platforms. Several previously reported groups of interest are shown including: (i) BRCA tumors cluster into two major groups, with basal samples grouping with squamous tumors; (ii) LAML tumors separate into two major groups, with one group significantly enriched for favorable cytogenetic risk; (iii) separation of endometrioid UCEC tumors into two major groups, one of which is characterized by a 1q chromosome amplification event. A novel group was also detected; (iv) an integrated pan-cancer cluster, defined by tumors from nine different tissues of origin, exhibits a strong immune signature. D, Pathway diagram of immune signaling-related genes with higher inferred activity among samples belonging to the integrated pan-cancer cluster (shown in C) compared with samples outside of it; networks include genes from both the innate and adaptive immune systems.
Materials and Methods
Overview of the TumorMap
The TumorMap presents tumor samples on a two-dimensional grid and combines the strengths of lattice and landscape visualization approaches. As in other multidimensional scaling approaches, the positioning of samples is guided by feature vector similarities so that nearby samples are “like” one another using a method called OpenOrd; other embedding techniques are also available, such as tSNE and PCA (see Supplementary Methods). Information describing documented and quantified measurements on the samples, such as clinical outcomes or mutation events, are made available as attributes in the display. Up to two attributes can be viewed concurrently. The TumorMap uses the available metadata on samples to perform AEA (see Supplementary Methods) to test for the presence of overrepresented or underrepresented events that distinguish a group of samples from the rest or one group from another.
One of the features that separates the TumorMap portal from other similar tools is the ability to find connections between attributes and their distributions on the map or to each other using a spatial correlation analysis (SCA; see Supplementary Methods). SCA uses the locality of samples on a given map to find associations between attributes that may not be detectable using direct sample overlaps or correlations. The approach takes its inspiration from the field of geospatial analysis to find either cooccurring or mutually exclusive pairs of attributes across regions rather than individual samples. Tumor samples that exhibit similar genomic profiles will tend to reside near one another in a map. Thus, by using a spatially aware correlation analysis, SCA can identify pairs of attributes that are likely to occur in samples with similar molecular state, even though the samples may be distinct. Lee’s L metric (5) is used for this calculation, which was originally introduced to find associations between variables in geographical maps (see Supplementary Methods).
Results
We applied the TumorMap to the analysis of the TCGA Pan-Cancer-12 cohort (6). This cohort contains over five thousand tumor samples, spanning 12 different tumor types. These tumors were characterized using six different platforms, including mRNA-Seq, miRNA-Seq, reverse-phase protein arrays, DNA methylation, somatic copy-number alterations, and somatic single-nucleotide variants. Separate maps were created for each of the six individual platforms after computing all pairwise sample similarities and transforming the similarities using a new reciprocal significance of similarity method we developed that produces coherent and cross-platform intercompatible spaces (Supplementary Fig. S1A–S1C). In addition, several maps were calculated from data- and pathway-integration strategies (Supplementary Fig. S1D and S1E). Annotations for the tumor samples and patients were loaded into the map that included 4,164 attributes, which collectively describe curated phenotypic and outcome-related information (e.g., tissue of origin, tumor stage, histology, etc.; see Supplementary Methods). To benchmark the metrics and layout engine used to derive the maps, we examined relationships between tumor types on the TumorMap created using single data types. Consistent with previous TCGA analyses (7), the organization of the tumors on the maps mirrored their tissue-of-origin and histological type (Supplementary Methods; Supplementary Fig. S2A–S2G). Therefore, we judged the maps to be biologically relevant.
To evaluate the utility of the SCA to detect molecular associations, we asked whether the method could identify known molecular events associated with the loss of function of the RB1 tumor suppressor based on the mRNA-Seq gene expression map. SCA revealed an association between RB1 mutation and CDKN2A deletion as well as between RB1 and PTEN mutations (Supplementary Fig. S2H). Even though patients with RB1 mutations are, for the most part, distinct from those with CDKN2A deletions, the analysis detects that these two mutually exclusive events occur in patients that are near one another on the map, consistent with the fact that RB1 and CDKN2A aberrations affect the same pathway. In addition to CDKN2A, SCA revealed an association between mutations in RB1 and PTEN (Supplementary Fig. S2I). The two events occurred in patients that clustered together in the TumorMap, consistent with several previous analyses (see ref. 8 for an example in GBM). Thus, SCA is able to detect associations between molecular attributes computed across individual samples, as well as across regions representing sets of transcriptionally similar samples. Many more such associations between molecular attributes have been detected by SCA and can be explored using the online interface.
Associations within and between tumor types revealed by an integrated map
Next, we explored whether integrating multiple types of molecular data can reveal novel information not apparent from the analysis of single data types. We investigated sample groupings revealed by an integration of six TCGA omics platforms. We developed a Reciprocal Significance of Similarity transformation of data (see Supplementary Methods) to create an integrated map from an equal contribution of the six different data platforms, each representing a distinct feature type: mRNA transcription, miRNA transcription, protein expression, methylation levels, somatic copy number changes, and somatic single nucleotide variants (Supplementary Fig. S1E, iv; Supplementary Fig. S3A and S3B). Several known connections between the tumor types are visible on the resulting map. For example, squamous-like characteristics of basal breast carcinoma (BRCA) tumors, reported by TCGA (9) and others, are clearly portrayed in the integrated map (Fig. 1C, i; Supplementary Fig. S1E, iv) but are not apparent in the individual platform maps (Supplementary Fig. S1C i–vi). Most of the basal BRCA tumors cluster separately from the rest of the BRCA samples and are near the majority of squamous tumors, such as head and neck squamous cell carcinoma and lung squamous cell carcinoma. This suggests that platform integration pulls out valuable biological insights that are not revealed by the analysis of a single platform.
The integrated map separates acute myelogenous leukemia patients into distinct cytogenetic subgroups, which are characterized by differential survival outcomes with statistical significance (Fig. 1C, ii). This map also revealed that the uterine corpus endometrial carcinoma (UCEC) tumors separate into three major molecular subtypes (Fig. 1C, iii). Although some of the individual UCEC tumors are scattered around the map, 126 cluster near luminal BRCA tumors, 177 near COAD tumors, and another 171 near ovarian serous cystadenocarcinoma (OV) tumors. The majority of the luminal BRCA-like and COAD-like UCEC samples are endometrioid tumors, whereas most of the OV-like UCEC tumors are serous. The OV-like tumors are further characterized by mutations in TP53. When comparing the two endometrioid clusters, the most significant distinguishing feature is an amplification of the long arm of chromosome 1, a known poor prognostic marker in some endometrial tumors (10) and some other cancers (11). These two major endometrioid subtypes were not found by previous Pan-Cancer-12 analyses (7).
A unique pan-cancer cluster revealed by an integrated map
Finally, the analysis of the integrated map revealed a novel cluster of samples (n = 82) spanning nine different types of tissues (Fig. 1C, iv; Supplementary Fig. S3C and S3D). We examined what distinguishes these samples from other samples in the cohort using several correlative methods. These samples are characterized by elevated T-cell and B-cell immune gene programs based on differential expression analysis, followed by gene set enrichment analysis (see Supplementary Methods; Supplementary Table S1A and S1B; Supplementary Fig. S4A); enriched for ESTIMATE (12) immune signaling scores (Supplementary Methods; Supplementary Fig. S4B; Supplementary Table S1C); differentially expressed genes enriched for the T-cell receptor and interferon pathways (Supplementary Methods; Supplementary Fig. S4C; Supplementary Table S1B); and transcription factors implicated by the MARINa (13) algorithm involved in T-cell, B-cell, and interferon signaling (Supplementary Fig. S4E–S4D; see Supplementary Methods). This group of samples has fewer somatic copy number alterations, both arm-level and focal, than other tumors (Supplementary Fig. S4F and S4G), possibly indicating a lower detection rate of these events due to lower tumor purity, consistent with a DNA methylation purity analysis (Supplementary Fig. S4H; Supplementary Table S1D). A “quiet” CNV and SNV profile alone is unlikely to lead to the definition of this cluster as other examples in the map, such as the “normal-like” BRCA samples, have quiet CNV and SNV profiles, yet cluster with other samples from their tissue type. The other platforms likely contribute to the shared signature of this pan-cancer cluster. The gene network in Fig. 1D summarizes some of the regulatory pathways involving the immune response that characterize this group of samples; an overview of analyses (Supplementary Fig. S5) and additional results regarding the pan-cancer cluster are available (Supplementary Methods).
Discussion
The TumorMap is a new type of integrated genomics portal that uses Google Maps (14) for visualization and exploratory analysis, which biologists and bioinformaticians can use to interrogate rich cancer genomics data. The intuitive and interactive layout facilitates the identification of cancer subtypes based on common molecular activities among a set of tumor samples. A toolbox of statistical tests, including novel Spatial Correlation Analysis, allows researchers to find associations between sample groupings and clinical, phenotypic, molecular, and outcome annotations (see Materials and Methods). We illustrate the power of the approach in its ability to detect known and novel tumor subtypes using single and integrated data platforms. A pan-cancer subtype was identified in the integrated map that may implicate immunotherapy options.
The portal is available at https://tumormap.ucsc.edu/ and a video is available for first time users (Supplementary Video S1). Additional datasets used for this study are also available (Supplementary Table S1E–S1I). The current version allows registered users to both create their own maps and project new samples into publicly available datasets as a backdrop. The TumorMap is easily extendable to applications beyond the comparison of cancers, such as navigation through the landscape of stem and progenitor cells. To facilitate its wider applications and extensions, the code repository is available at https://github.com/ucscHexmap/hexagram.git.
Supplementary Material
Acknowledgments
We thank Rosalyn Huffman for help with article preparation and administrative support. We thank Andrea Preble for feedback on early drafts.
Grant Support
This work was supported by National Cancer Institute grants U24-CA143858 (to D. Haussler and J.M. Stuart) and 1R01CA180778 (to J.M. Stuart), National Human Genome Research Institute (5U54HG006097 to J.M. Stuart), National Institute for General Medical Sciences (5R01GM109031 to J.M. Stuart), National Science Foundation Office of Cyber infrastructure CAREER (0845783 to J.M. Stuart), and Prostate Cancer Foundation (to J.M. Stuart). J.M. Stuart is supported by a Stand Up To Cancer – Prostate Cancer Foundation Prostate Dream Team Translational Research Grant (SU2C-AACR-DT0812). This research Grant is made possible by the generous support of the Movember Foundation. Stand Up To Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C. This work was supported by the St. Baldricks Foundation Treehouse Childhood Cancer Project under award number 427053, the University of California California Precision Medicine Initiative: California Kids Cancer Comparison under award number OPR014109, and the Alex’s Lemonade Stand Foundation Innovation Award. The content is solely the responsibility of the authors and does not necessarily represent the official views of our sponsors.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
A.M. Novak is a consultant at DNAnexus. No potential conflicts of interest were disclosed by the other authors.
Authors’ Contributions
Conception and design: Y. Newton, A.M. Novak, S. Chopra, R. Baertsch, S.R. Salama, T.C. Goldstein, D. Haussler, J.M. Stuart
Development of methodology: Y. Newton, A.M. Novak, T. Swatloski, D.C. McColl, S. Chopra, K. Graim, T.C. Goldstein, D. Haussler, J.M. Stuart Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): Y. Newton, T.C. Goldstein, D. Haussler Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): A.M. Novak, T. Swatloski, D.C. McColl, K. Graim, A.S. Weinstein, M. Chopra, T.C. Goldstein, D. Haussler, O. Morozova, J.M. Stuart
Writing, review, and/or revision of the manuscript: Y. Newton, T. Swatloski, D.C. McColl, K. Graim, A.S. Weinstein, S.R. Salama, T.C. Goldstein, O. Morozova, J.M. Stuart
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): Y. Newton, A.M. Novak, T. Swatloski, D.C. McColl, S. Chopra, K. Ellrott, M. Chopra, T.C. Goldstein, J.M. Stuart
Study supervision: T.C. Goldstein, J.M. Stuart
Other (initial software prototyping): A.M. Novak
References
- 1.Nielsen CB, Cantor M, Dubchak I, Gordon D, Wang T. Visualizing genomes: techniques and challenges. Nat Methods. 2010;7:S5–15. doi: 10.1038/nmeth.1422. [DOI] [PubMed] [Google Scholar]
- 2.Kim SK. A gene expression map for Caenorhabditis elegans. Science. 2001;293:2087–92. doi: 10.1126/science.1061603. [DOI] [PubMed] [Google Scholar]
- 3.MacArthur BD, Lachmann A, Lemischka IR, Ma’ayan A. GATE: software for the analysis and visualization of high-dimensional time series expression data. Bioinformatics. 2010;26:143–4. doi: 10.1093/bioinformatics/btp628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolouri H, Zhao LP, Holland EC. Big data visualization identifies the multidimensional molecular landscape of human gliomas. Proc Natl Acad Sci U S A. 2016;113:5394–9. doi: 10.1073/pnas.1601591113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee S-I. A generalized significance testing method for global measures of spatial association: an extension of the Mantel test. Environ Plan A. 2004;36:1687–703. [Google Scholar]
- 6.Cancer Genome Atlas Research Network. Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113–20. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158:929–44. doi: 10.1016/j.cell.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Melamed RD, Wang J, Iavarone A, Rabadan R. An information theoretic method to identify combinations of genomic alterations that promote glioblastoma. J Mol Cell Biol. 2015;7:203–13. doi: 10.1093/jmcb/mjv026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prat A, Adamo B, Fan C, Peg V, Vidal M, Galván P, et al. Genomic analyses across six cancer types identify basal-like breast cancer as a unique molecular entity. Sci Rep. 2013;3:3544. doi: 10.1038/srep03544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knuutila S, Björkqvist AM, Autio K, Tarkkanen M, Wolf M, Monni O, et al. DNA copy number amplifications in human neoplasms: review of comparative genomic hybridization studies. Am J Pathol. 1998;152:1107–23. [PMC free article] [PubMed] [Google Scholar]
- 11.An G, Xu Y, Shi L, Shizhen Z, Deng S, Xie Z, et al. Chromosome 1q21 gains confer inferior outcomes in multiple myeloma treated with bortezomib but copy number variation and percentage of plasma cells involved have no additional prognostic value. Haematologica. 2014;99:353–9. doi: 10.3324/haematol.2013.088211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aytes A, Mitrofanova A, Lefebvre C, Alvarez MJ, Castillo-Martin M, Zheng T, et al. Cross-species regulatory network analysis identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer malignancy. Cancer Cell. 2014;25:638–51. doi: 10.1016/j.ccr.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Google Maps JavaScript API V3 Reference. Google Developers.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.