

Abstract
Dye-decolorizing peroxidases (DyPs) are a family of H2O2-dependent heme peroxidases, which have shown potential applications in lignin degradation and valorization. However, the DyP kinetic mechanism remains underexplored. Using structural biology and solvent isotope (sKIE) and viscosity effects, many mechanistic characteristics have been uncovered for the B-class ElDyP from Enterobacter lignolyticus. Its structure revealed that a water molecule acts as the sixth axial ligand with two channels at diameters of ~3.0 and 8.0 Å leading to the heme center. A conformational change of ERS* to ERS, which have identical spectral characteristics, was proposed as the final step in DyPs’ bisubstrate Ping-Pong mechanism. This step is also the rate-determining step in ABTS oxidation. The normal KIE of wild-type ElDyP with D2O2 at pH 3.5 suggested that cmpd 0 deprotonation by the distal aspartate is rate-limiting in the formation of cmpd I, which is more reactive under acidic pH than under neutral or alkaline pH. The viscosity effects and other biochemical methods implied that the reducing substrate binds with cmpd I instead of the free enzyme. The significant inverse sKIEs of kcat/KM and kERS* suggested that the aquo release in DyPs is mechanistically important and may explain the enzyme’s adoption of two-electron reduction for cmpd I. The distal aspartate is catalytically more important than the distal arginine and plays key roles in determining DyPs’ acidic pH optimum. The kinetic mechanism of D143H-ElDyP was also briefly studied. The results obtained will pave the way for future protein engineering to improve DyPs’ lignolytic activity.
Keywords: DyP, inverse solvent isotope effect, viscosity, aqo release, conformational change
INTRODUCTION
Dye-decolorizing peroxidases (DyPs) are a new family of heme peroxidases, which use H2O2 to catalyze oxidation of various dye substrates, specifically anthraquinone-derived dyes that have high redox potentials.1–3 DyPs have recently received significant attention due to their potential applications in lignin degradation and valorization.4–7 Lignin is a complex aromatic polymer that represents the second most abundant renewable carbon source on Earth after cellulose, constituting around 30% non-fossil organic carbon.8 However, lignin valorization is challenging because it is recalcitrant to degradation.9–11 More than 60% of lignin is wasted by combustion due to lack of methods to convert it into valuable end products.12–14 Additionally, lignin degradation represents major hindrance for the competitiveness of biofuels vs. fossil-based gasoline.15
While class II plant peroxidases such as lignin peroxidase (LiP), manganese peroxidase (MnP), and versatile peroxidase (VP) can break down lignin efficiently,16–17 they have not been used for industrial applications due to their fungal origins, which create challenges in genetic manipulation and large-scale protein production. Thus, the field is currently undergoing a paradigm shift to pursue lignin biodegradation using bacterial enzymes,4, 18 among which DyPs are promising candidates. Several DyPs have been reported to show activities toward lignin model compounds and wheat straw lignocellulose,19–23 albeit with low efficiency. Therefore, DyPs are considered as a potential functional equivalent of fungal LiP in bacteria.
The characteristics of DyPs, including primary sequence, tertiary structure, catalytic residues, substrate specificity, and pH optimum, differ significantly from those well-known heme peroxidases such as class II plant peroxidases and horseradish peroxidase (HRP).3, 24 In particular, DyP employs a distal aspartate, in place of the distal histidine used by other heme peroxidases, as a catalytic residue. This aspartate is also proposed to be responsible for the acidic optimum of DyP activity.25 While it has been suggested that DyPs follow a similar mechanism as the class II plant peroxidases and HRP,19, 21 their mechanistic details remain underexplored. This has severely limited our ability to engineer them in order to improve their lignolytic activity.
In this work, we have selected B-class ElDyP from Enterobacter lignolyticus as a model to study the DyP molecular mechanism. Selection of ElDyP is based on the fact that the host microbe is capable of growing anaerobically using lignin as the sole carbon source.26 Also, the B-class DyPs appear to have the highest activity towards lignin and its non-phenolic model compounds among bacterial DyPs.19, 22 Consequently, the ElDyP may be directly involved in lignin degradation by E. lignolyticus. Additionally, while it is still in dispute,27 the Escherichia coli YfeX, which has 93% sequence identity with ElDyP, was proposed to be the first heme deferrochelatase.28 Detailed mechanistic insights may facilitate to unraveling the true biological functions of DyPs. Here, we used solvent kinetic isotope effects (sKIEs) and viscosity effects determined under steady-state and transient-state conditions, together with structural biology, to dissect the reactions catalyzed by DyPs and to examine the individual roles of catalytic residues. This study provides the insights into the DyP mechanism and paves the way for the efforts in protein engineering.
MATERIALS AND METHODS
Instruments, biochemicals, and chemicals
All activity assays and steady-state kinetics were performed on a Cary 100 Bio UV-Vis spectrometer equipped with a temperature controller and magnetic stirring. Transient-state kinetics was carried out on an Applied Photophysics SX20 stopped-flow spectrometer equipped with sequential mixing, a PDA detector, and a monochromator. All chemical and biochemical reagents were purchased at the highest grade and used without further purification. Protein concentrations were determined by bicinchoninic acid assays (BCA).29 Stocks of H2O2 and ascorbate were prepared fresh before experiments. Concentrations of H2O2 were determined at 240 nm using ε240 = 43.6 M−1cm−1. Buffers of pH 2.0–6.5, 7.0–8.0, 8.5–9.0, and 9.5–11.5 were prepared using sodium citrate, potassium phosphate, Tris-HCl, and 3-(cyclohexylamino)-1-propanesulfonic acid (CAPS), respectively. All kinetic measurements were performed in triplicate and data were processed by OriginPro 2015.
Cloning, expression, and purification of ElDyP and its mutants
The gene corresponding to ElDyP was synthesized by GenScript and inserted into pET28-MHL (Addgene plasmid #26096) using restriction sites NdeI and HindIII to generate pElDyP, which contains a His6 tag and tobacco etch virus (TEV) cleavage site at the N-terminus. Active-site mutants of ElDyP were generated using QuickChange from Agilent Technologies according to manufacturer’s instructions. DNA sequences of the synthesized wild-type (wt) ElDyP and mutant primers are summarized in Table S1 of the Supporting Information (SI) file.
Proteins of wt and mutant ElDyPs were overexpressed in Escherichia coli BL21(DE3) cells (Lucigen). A 90-ml starter culture was grown overnight from a single colony in LB media in the presence of 50 μg/ml of kanamycin at 37 °C with shaking at 225 rpm and then used to inoculate 4.5 liters of LB medium. When the A600 reached 0.6, isopropyl-1-thio-β-D-galactopyranoside (IPTG) and hemin chloride were added to a final concentration of 0.2 mM and 30 μg/ml, respectively. The cells were grown at 30 °C for an additional 15 h and then harvested by centrifugation at 5,000×g for 20 min at 4 °C. The cell pellets were collected and stored at −80 °C until further use.
All of the following steps were carried out at 4 °C. Purification buffers consisted of buffer A (400 mM NaCl and 50 mM potassium phosphate, pH 7.8) and increasing concentrations of imidazole. Buffers B (lysis), C (wash), and D (elution) contained 10, 30, and 250 mM imidazole, respectively. The cell pellets were re-suspended in buffer B and lysed by sonication (25×30-s pulsed cycle). The cell debris was removed by centrifugation at 15,000×g for 45 min and the supernatant was incubated with 10 mL Ni-NTA resin that had been pre-equilibrated with buffer B for 1h. The resin was washed with 10 column volumes of buffer C and then eluted with buffer D. Fractions containing the DyP were collected, concentrated to ~70 mg/mL, and exchanged into buffer E (100 mM NaCl and 50 mM potassium phosphate, pH 7.8) using Amicon Ultra-15 filters (10K, EMD Millipore). The purified protein was stored in aliquots at −80 °C until further use. Protein purity was assessed by 12% acrylamide SDS-PAGE and protein concentration was determined by BCA using a BSA calibration curve. The Reinheitszahl values (Rz) of purified proteins were measured at 2.0–2.3. Their heme stoichiometric ratios were determined to be 1.1–1.3 using pyridine hemochromogen assays and Δε557−541 = 20.7 mM−1cm−1.30
The protein for crystallization was purified in the following way. Ten mg wt-ElDyP were treated with 40 μg subtilisin A at 37 °C for 1 h before diisopropyl fluorophosphate was added to a final concentration of 100 μM to stop proteolysis. The protein was then exchanged into buffer B using a 10K Amicon Ultra-15 centrifugal filter and subsequently incubated with Ni-NTA resin for an additional hour. The flow-through fractions, which contained ElDyP lacking a His6-tag, were collected, concentrated, and subjected to size-exclusion chromatography (SEC) using a HiLoad 26/600 Superdex 200 pg column (GE Healthcare) equilibrated with 150 mM NaCl in 50 mM potassium phosphate, pH 7.5. Fractions corresponding to dimeric ElDyP were pooled, and buffer exchanged into 60 mM NaCl, 20 mM Tris-HCl, pH 7.9, before crystallization.
Crystallization and structure determination
Crystals were grown by the sitting drop vapor diffusion method in a crystallization solution consisting of 100 mM HEPES (pH 7.3) and 26% (w/v) PEG3350. Crystals of wt-ElDyP (5.5 mg/ml) were grown at 16 °C. They began to appear after 48 h and matured after 72 h. Individual crystals were transferred to a buffer containing the crystallization solution with 10% glycerol (v/v) as a cryoprotectant before flash-cooling in liquid nitrogen. Diffraction data were collected at 100 K using beamline 22-BM of the Advanced Photon Source, Argonne National Laboratory, at a wavelength of 1.0 Å and processed with the HKL2000 package. The structure was solved by molecular replacement using a PHENIX software suite with coordinates of PDB entry 5DE0 as a starting model.31 The final structure was obtained by iterative automated model building and refinement using PHENIX and interactive model modification in Coot.32 Figures were prepared using PyMol (Schrödinger, LLC). The final coordinates have been deposited in the RCSB under accession ID 5VJ0.
Spectroelectrochemical determination of redox potential
In a 3.5-ml cuvette, a 3.0-ml solution consisting of 50mM potassium phosphate (pH 7.0), 100mM NaCl, and a mixture of redox mediators (10 μM each for methyl viologen, anthraquinone-2-sulfonic acid, anthraquinone-2,6-disulfonic acid, 2-hydroxy-1,4-naphthoquinone, 2,5-dihydroxy-1,4-benzoquinone, duroquinone, 1,2-naphthoquinone, and ferricyanide; 3μM each for Safferin and Neutral Red) was purged with water-saturated argon for 1 h. The electrodes (Ag/AgCl, 012167; Pt gauze, 011498; ALS Co, Japan) were connected to a potentiostat. The wt-ElDyP was added to a final concentration of 10μM. The entire mixture was completely reduced to ferrous state with ~5 μl of 100 mM freshly prepared sodium dithionite stock solution. This was then oxidized by stepwise addition of 2–5μl aliquots of argon-purged 2.5 mM K3Fe(CN)6. The reaction mixture was kept under water-saturated argon protection during the whole experiment. After each addition, the reaction was allowed to equilibrate with stirring until the difference in potential readings was less than 1 mV/min. Once the equilibrium was established, the UV-visible spectrum was recorded. The fraction of reduced ElDyP was calculated by the ΔA434 nm and the midpoint reduction potential (E°′) was determined by fitting the data into the following Nernst Eq. 1,
| (Eq. 1) |
where f is the fraction reduced, n is the number of electrons, E° is the potential at each point in mV, and E°′ is the midpoint reduction potential in mV. The determined potential is with respect to a SHE. The experiment was performed again in D2O. All reagents were prepared in 99.9% D2O (Cambridge Isotope Laboratories).
Enzyme assays
Enzyme activities against various substrates were determined using a continuous assay by monitoring the absorbance change at a certain wavelength at 25 °C. Briefly, in a 480 μL solution consisting of 50 mM sodium citrate (pH 3.5), 10 mM H2O2, reducing substrate, and 0.02 mg/mL bovine serum albumin (BSA), the purified wt or mutant ElDyP was added to initiate the reaction. Controls were performed in the absence of enzyme, H2O2, or both. The tested reducing substrates and their concentrations, wt enzyme concentrations, wavelengths monitored, and the corresponding extinction coefficients were summarized in Table 1.
Table 1.
Activities of wt-ElDyP toward various substrates
| Substrate | λ (nm) |
ε (cm−1mM−1) |
[S] (mM) |
[E] (nM) |
SA* (U/mg) |
|---|---|---|---|---|---|
| ABTS | 420 | 36.0 | 5 | 1 | 5560± 60 |
| RB19 | 595 | 10.0 | 0.1 | 50 | 257 ± 7 |
| RB4 | 610 | 4.20 | 0.1 | 50 | 218 ± 4 |
| RB5 | 600 | 8.00 | 0.1 | 50 | 136 ± 3 |
| RBlk5 | 598 | 30.0 | 0.1 | 50 | 22.9 ±0.5 |
| Pyrogallol | 430 | 2.47 | 400 | 200 | 46.3 ± 0.6 |
| HQ | 247 | 21.0 | 5 | 200 | 38.0 ± 1.0 |
| Guaiacol | 465 | 26.6 | 10 | 1000 | 0.34 ± 0.01 |
| VA | 340 | 93.0 | 10 | 1000 | ND* |
| Mn2+ | 270 | 11.6 | 20 | 1000 | 2.08 ± 0.08 |
SA: Specific Activity; ND: Not Detected.
To determine optimal pH, the assays were carried out with 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) and Reactive Blue 19 (RB19) as described above in the pH range of 2.0–8.0 at 25 °C. To determine optimal temperature, the assays were performed in a similar way with ABTS at pH 3.5 at temperatures ranging from 10–60 °C.
Steady-state kinetics
To determine steady-state kinetic parameters (kcat and KM) of the wt and mutant ElDyPs, reactions were performed in the same way as described above except that the concentration of one substrate was varied in the presence of the other substrate at saturation. The data obtained were fitted into either Michaelis-Menten Eq. 2 or Hill Eq. 3.
| (Eq. 2) |
| (Eq. 3) |
Transient-state kinetics
Stopped-flow experiments were performed at 25 °C at pH 2.0–11.5 and investigated at both defined (monochromator) and multiple (PDA) wavelengths. The PDA data were analyzed using singular value decomposition with the Pro-KIV Global Analysis program provided by Applied PhotoPhysics to obtain the number of reaction intermediates and their corresponding spectra. Reactions were then monitored at selected single wavelengths to follow the formation and decay of intermediates. Data were fitted to single exponential expressions to obtain pseudo-first-order rate constants (kobs). Second-order rate constants were calculated from plots of kobs vs. substrate concentrations.
All concentrations mentioned here were concentrations after mixing. Formation of compound (cmpd) I was carried out in a conventional mixing mode with 2.5 μM enzyme in 5 mM phosphate buffer (pH 7.8) containing 10 mM NaCl and equal volume of H2O2 in 50 mM buffer (pH 2.0–11.5) at various concentrations. Formation of cmpd I was monitored at wavelengths corresponding to Soret and Q bands. Regeneration of wt enzyme resting state (ERS*) from cmpd I was performed in a sequential mixing mode, in which 2.5 μM wt enzyme in 5 mM phosphate buffer (pH 7.8) containing 10 mM NaCl was premixed with equal volume of 2.5 μM H2O2 in 50 mM citrate buffer (pH 3.5). After a 2s-delay to reach maximal formation of compound I, ABTS in 50 mM buffer (pH 2.0–11.5) was mixed with cmpd I in the presence of ascorbate at concentrations of 10, 50, and 75 μM for 0.1–1.0, 1.5–5.0, and 7.5 μM ABTS, respectively. Regeneration of ERS* was monitored at 406 nm corresponding to the Soret band of wt-ElDyP.
Solvent kinetic isotope effect (sKIE)
Experiments of steady-state and transient-state kinetics described above were performed again in deuterated buffers, which were prepared in 99.9% D2O (Cambridge Isotope Laboratories). The pD (pH in D2O) was adjusted based on the following relationship: pD = pHobs + 0.38. Reducing substrates and H2O2 were directly dissolved in D2O buffers. Enzymes were highly concentrated (≥ 60 mg/mL) to ensure minimal protium contribution to deuterated buffers and were equilibrated in D2O buffer before analysis.
Viscosity effect
Experiments of steady-state and transient-state kinetics described above were performed at pH 3.5 with wt-ElDyP in sucrose at various concentrations. For stopped-flow experiments, all reagents, including the enzyme, were prepared in a matching sucrose solution.
RESULTS
Protein purification and characterization
Proteins of wt and mutant ElDyPs were purified with an N-His6 tag and a tobacco etch virus (TEV) protease cleavage site. Mutant H215A is not included in this report because the holo-enzyme could not be obtained. This is not surprising, as mutation of the axial ligand in heme proteins (H215 in ElDyP) has been known to result in the failure of heme incorporation.33–34 SDS-PAGE (Figure S1-A in SI) reveals that the molecular weight (MW) of ElDyP is 35 kDa, which is consistent with the calculated value. Since the presence of the His6 tag did not interfere with enzyme activity, it was not removed prior to further biochemical studies. Size-exclusion chromatography (SEC) of wt-ElDyP (Figure 1A) shows that the enzyme mainly exists as a dimer in its native state, which is consistent with the result from its crystal structure. Spectroelectrochemical titrations of wt-ElDyP (Figure 1B) at pH 7.0 gave a Fe3+/Fe2+ midpoint reduction potential (E°′) of −290 ± 0.96 mV vs NHE with Nernstian behavior (n = 1.13 ± 0.05). When the measurement was carried out in D2O at pD 7.0, the E°′ shifted more positively to −263 ± 0.70 mV (n = 0.95 ± 0.02). As shown in Figure 1C and Table S2 in SI, the UV-Vis spectrum of wt-ElDyP displays Soret (406 nm), Q- (507 and 539 nm), and charge-transfer (635 nm) bands, corresponding to the presence of a heme cofactor. The mutants have the same absorbance maxima as the wt except that D143H contains an extra Q-band at 567 nm, suggesting that the heme environment has changed significantly in D143H. The pH dependence of enzyme activity (Figure 1D) demonstrates that the wt and mutant ElDyPs display the highest activities toward ABTS at pH 3.5 and 4.0, respectively. The same pattern was also observed with RB19 (data not shown). It has to be noted that the mutations, especially replacement of D143 with H143, did not result in significant shifts of pH optimum. The temperature-rate profile (Figure S1-B in SI) shows that the highest activity of wt-ElDyP toward ABTS was achieved at 30 °C, while 93% wt activity was observed at 25 °C. Thus, for the sake of simplicity, further studies were carried out at 25 °C (r.t.).
Figure 1.

Biochemical characterization of ElDyP. (A) SEC chromatography of wt-ElDyP (red) and molecular weight standards (black) loaded onto a GE Superdex 200 Increase 10/300 GL column eluted with 50 mM potassium phosphate buffer (pH 7.5) containing 150 mM NaCl. The inset represents a MW calibration curve. (B) Changes in the UV-Vis absorption spectrum showing stepwise oxidation of Fe2+−wt-ElDyP (red) to Fe3+−wt-ElDyP (blue). The inset represents three individual spectroelectrochemical titrations of wt-ElDyP monitored by ΔA434 nm. (C) Normalized UV-Vis absorption spectra of wt and mutant ElDyPs with 450–700 nm region magnified by 4 times. (D) pH-activity profiles of wt and mutant ElDyPs with ABTS.
Protein structure and access channels to heme active site
The crystal structure of wt-ElDyP was solved by molecular replacement using the VcDyP from Vibrio cholerae (PDB entry 5DE0) as a search model and refined to 1.93 Å limiting resolution. Data collection and refinement statistics are given in Table S3 in SI. The final model consists of four copies of the ElDyP monomer, each with a heme bound in its active site. The monomers assemble into symmetric dimers composed of chains A and B, and chains C and D, as judged by buried surface area found at the dimerization interfaces. The ElDyP monomer represented by chain B was used for all structural analyses described in this manuscript. As shown in Figure 2A, the ElDyP displays a ferredoxin-like fold consisting of four-strand, antiparallel β-sheet and peripheral α-helices. The overall structure is similar to B-class DyPB from Rhodococcus jostii RHA1, VcDyP, and YfeX,35–37 but is quite different from the structures of class II plant peroxidases, which contain mainly α-helices.38–40 The heme active site shows that H215 acts as a proximal ligand. The distal site is occupied by D143, R232 and H2O288. The H2O288 acts as a sixth ligand to heme iron and forms hydrogen bonds with both D143 and R232. During enzyme oxidation, H2O2 is expected to displace this H2O288 and initiates the oxidation reaction. R232 also forms a hydrogen bond with the heme 5-propionate group, which should contribute to the noteworthy pKa shift of arginine as described below. Compared with the active site of LiP from Phanerochaete chrysosporium (PDB 1B82),38 a typical distal histidine in class II plant peroxidases (H47 in LiP) and HRP is replaced with a distal aspartate (D143 in ElDyP) that is absolutely conserved across all four classes of DyPs.
Figure 2.
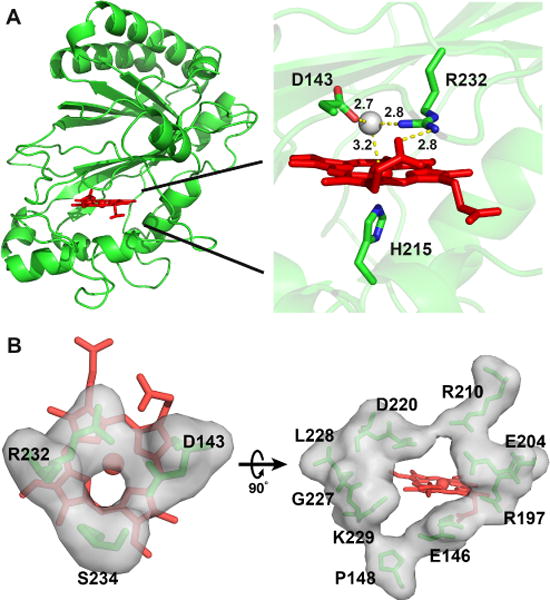
Crystal structure of wt-ElDyP. (A) Overall structure (left) and active site of the enzyme. Catalytic residues, heme, and water 288 are represented in green sticks, red sticks, and a grey ball, respectively. Distances in angstrom are labeled and shown in yellow dashed lines. (B) Surface representations of the small (left, diameter of ~3.0 Å) and large (right, diameter of ~8.0 Å) heme access channels.
Examination of the ElDyP structure revealed two heme access channels. The first of these has a diameter of ~3.0 Å and points to the distal side of the heme (left panel in Figure 2B). This channel is proposed to be the entrance of H2O2 and also exists in DyPB and VcDyP.35–36 The second heme access channel has a diameter of ~8.0 Å and leads to the heme 6-propionate group (right panel in Figure 2B), which is larger than the corresponding channels in DyPB (~6.0 Å) and VcDyP (~7.5 Å).35–36 It has been reported that the propionate group can provide a direct electron transfer path from the porphyrin radical to a bound substrate.41 Charged residues in ElDyP are mainly present in and around these two channels. Other areas are mostly occupied by hydrophobic and polar residues, which may facilitate surface interaction with dyes and lignin, both of which are rich in aromatic moieties.
Enzyme activities
The wt-ElDyP is active toward a wide range of substrates summarized in Table 1. While ABTS and anthraquinone dyes such as reactive blue (RB) could be easily oxidized by ElDyP, low activities were observed with Reactive Black 5 (RBlk5) and small-size molecules like pyrogallol, hydroquinone (HQ), and guaiacol. No activity was detected with veratryl alcohol (VA). Similar to B-class DyPB and C-class DyP2,20, 35 ElDyP was also found to have low MnP activity. Such activity is thought to be important for microbial lignin degradation due to high Mn content of wood that facilitates formation of Mn3+ oxidant.42
To determine the catalytic importance of the distal aspartate and arginine, the corresponding mutants were studied with ABTS and RB19. As shown in Figure 3, mutation of either residue is deleterious, resulting in significant loss of enzyme activity. D143A only retained <2% wt activity toward both tested substrates. D143H displayed 11% and 12% wt activities toward ABTS and RB19, respectively. The higher activity observed with D143H is attributed to the introduction of the distal histidine, which is present in class II plant peroxidases and HRP. R232A retained ~14% wt activity, which is much higher than the activity of D143A, suggesting that the distal aspartate may be catalytically more important than the distal arginine. That said, both residues are required for substrate oxidation.
Figure 3.
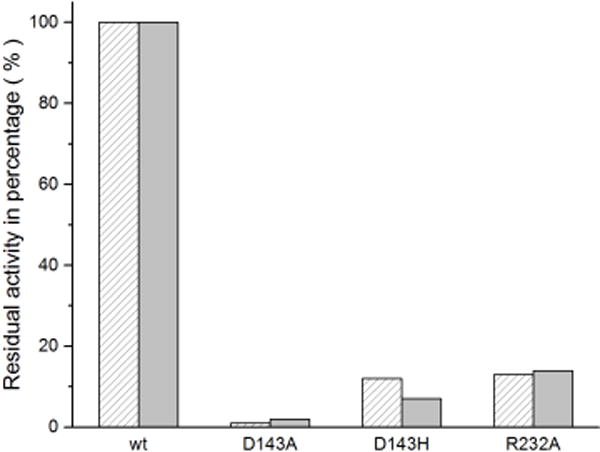
Activity percentages of wt and mutant ElDyPs toward ABTS (sparse bar) and RB19 (solid bar).
sKIE on steady-state kinetics
To characterize wt and mutant ElDyPs, their kinetic parameters were determined with ABTS, RB19, and H2O2. As summarized in Table 2, the turnover numbers (kcat) of wt-ElDyP with the two reducing substrates are among the fastest DyP-catalyzed reactions reported so far.20–21, 35–36, 43 Moreover, the wt enzyme displayed 3- to 9-fold higher affinity (KM) with the reducing substrate than with H2O2. While mutations of distal residues significantly decrease the turnover numbers, the values for D143H and R232A are still comparable with other wt DyPs,35–36 suggesting that ElDyP is a good target to study DyP mechanisms. The affinities of the mutants with reducing substrates and H2O2 are weakened except for D143A, in which the KM values with ABTS and RB19 are similar to those of the wt enzyme.
Table 2.
Steady-state kinetic parameters and sKIEa
| Substrate | Solvent | wt | D143A | D143H | R232A | |
|---|---|---|---|---|---|---|
| ABTS (H2O2) |
H2O | kcat (s−1) | (3.54 ± 0.01) × 103 [(3.73 ± 0.02) × 103] |
(3.45 ± 0.02) × 10 [(4.62 ± 0.01) × 10] |
(4.37 ± 0.05) × 102 [(4.16 ± 0.08 × 102] |
(4.87 ± 0.06) × 102 [(5.25 ± 0.02) × 102] |
| KM (μM) | (6.07 ± 0.01) × 102 [(2.02 ± 0.04) × 103 |
(4.80 ± 0.20) × 102 [(7.50 ± 0.20) × 104] |
(2.50 ± 0.05) × 103 [(4.90 ± 0.30) × 103] |
(1.30 ± 0.02) × 103 [(1.35 ± 0.02) × 104] |
||
| kcat/KM (s−1·M−1) | (5.80 ± 0.10) × 106 [(1.85 ± 0.03) × 106] |
(7.20 ± 0.30) × 104 [(6.10 ± 0.10) × 102] |
(1.75 ± 0.02) × 105 [(8.40 ± 0.40) × 104] |
(3.76 ± 0.03) × 105 [(3.90 ± 0.10) × 104] |
||
| D2O | kcat (s−1) | (5.10 ± 0.20) × 103 | (3.92 ± 0.08) × 102 | |||
| KM (μM) | (1.00 ± 0.10) × 102 | (2.95 ± 0.05) × 102 | ||||
| kcat/KM (s−1·M−1) | (5.10 ± 0.20) × 107 | (1.33 ± 0.05) × 106 | ||||
| sKIE | D(kcat) | 0.69 ± 0.02 | 1.11 ± 0.04 | |||
| D(kcat/KM) | 0.11 ± 0.01 | 0.13 ± 0.01 | ||||
| RB19 (H2O2) |
H2O | kcat (s−1) | (1.67 ± 0.08) × 102 [(1.69 ± 0.01) × 102] |
(2.92 ± 0.03) × 100 [(3.80 ± 0.20) × 100] |
(1.22 ± 0.02) × 10 [(1.77 ± 0.04) × 10] |
(2.30 ± 0.05) × 10 [(3.10 ± 0.10) × 10] |
| KM (μM) | (3.90 ± 0.30) × 10 [(3.70 ± 0.10) × 102] |
(3.93 ± 0.05) × 10 [(1.60 ± 0.20) × 104] |
(7.70 ± 0.20) × 10 [(5.70 ± 0.40) × 102] |
(4.60 ± 0.20) × 10 [(2.20 ± 0.20) × 103] |
||
| kcat/KM (s−1·M−1) | (4.20 ± 0.10) × 106 [(4.60 ± 0.10) × 105] |
(7.40 ± 0.10) × 104 [(2.40 ± 0.20) × 102] |
(1.60 ± 0.30) × 105 [(3.10 ± 0.40) × 104] |
(5.00 ± 0.30) × 105 [(1.40 ± 0.20) × 104] |
Values shown in square brackets are parameters for H2O2.
The effect of D2O on kinetics provides insights into the role of transfer of exchangeable protons during the reaction, thus revealing important mechanistic details. Since RB19 displayed a significant inhibitory effect in D2O, determination of steady-state kinetic parameters in D2O was carried out only with ABTS. As shown in Table 2, inverse sKIEs were observed with ABTS for both the turnover number and catalytic efficiency . Their mechanistic implications will be discussed below in detail.
Transient-state kinetics of cmpd I formation
Formation of cmpd I between ElDyPs and H2O2 was investigated using stopped-flow spectroscopy. Representative spectra at acidic and alkaline pH values are shown in Figures 4A–D. The absorbance maxima are summarized in Table S2 of the SI file. While both wt- and R232A-ElDyP displayed similar spectral transition characters under both acidic and alkaline pH values (Figures 4A and 4D), the aspartate mutants gave vastly different transitional spectra under the same conditions (Figures 4B and 4C). In particular, a red shift was observed in the aspartate mutants for the Soret band along with appearance of two peaks in Q-band region (526–556 nm) when buffer pH was increased from 4.0 to 10, suggesting the formation of cmpd II-like intermediates under alkaline and neutral pH in the presence of H2O2 (data not shown).
Figure 4.
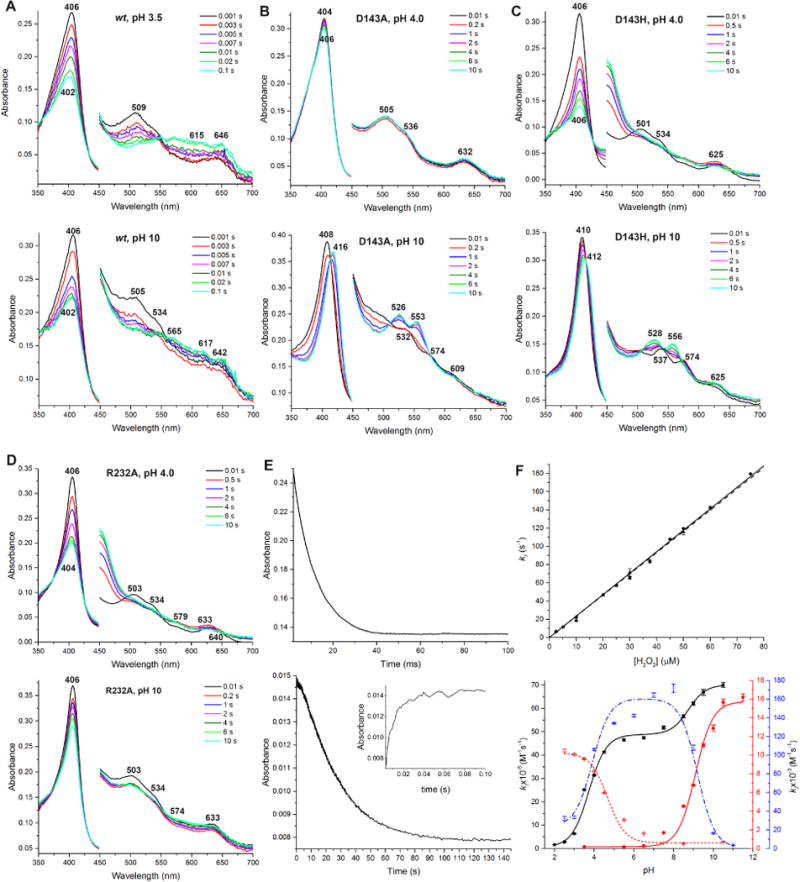
Single mixing of cmpd I formation. Spectra in 450–700 nm are magnified by 4 times. All spectral transitions represent the reactions between 2.5 μM enzymes and 50 μM H2O2. (A) Spectral transition of wt-ElDyP under acidic (top) and alkaline (bottom) conditions. (B) Spectral transition of D143A under acidic (top) and alkaline (bottom) conditions. (C) Spectral transition of D143H under acidic (top) and alkaline (bottom) conditions. (D) Spectral transition of R232A under acidic (top) and alkaline (bottom) conditions. (E) Formation and decay of wt-ElDyP cmpd I monitored at 406 (top) and 615 nm (bottom), respectively. The bottom inset represents cmpd I formation monitored at 615 nm. (F) Second-order rate constants of cmpd I formation. The top panel represents the dependence of k1 monitored at 406 (● and solid line) and 615 nm (▲ and dash line). The bottom panel represents pH-rate profiles of wt (■ and black solid line), D143A (● and red solid line), D143H (✳ and blue dash line), and R232A (+ and red dot line). The color of Y-axes and curves correspond to each other.
To determine second-order rate constants of cmpd I formation (kI), reactions between enzymes and H2O2 were followed via the disappearance of a Soret band (406 nm) corresponding to ERS* (top panel in Figure 4E). For the wt-ElDyP, the reaction was also monitored at 615 nm (bottom panel in Figure 4E), where cmpd I formation and decay were observed. The traces were fitted to single-exponential expressions to obtain kobs, which was then plotted against H2O2 concentrations to derive a kI (top panel in Figure 4F). The results at optimal pH/pD are summarized in Table 3. Both the distal aspartate and arginine are important for cmpd I formation, for which their replacement by alanine resulted in rate decrease by 104 and 102 magnitude, respectively. This is consistent with steady-state kinetic results, in which aspartate was found to be catalytically more important than arginine. Substitution of aspartate with histidine partially recovered enzyme activity in cmpd I formation and was only 22-fold less active than the wt-ElDyP.
Table 3.
Transient-state kinetic parameters, sKIEs, and pKa of ionizable groups in cmpd I formationa
| kI (M−1s−1)b | KIE | pKa | ||
|---|---|---|---|---|
| Asp | Arg | |||
| wt | (2.35 ± 0.02) × 106 [(9.62 ± 0.06) × 105] |
2.44 ± 0.06 | 3.65 ± 0.05 [4.15 ± 0.06] |
8.70 ± 0.20 [8.20 ± 0.20] |
| D143A | (1.97 ± 0.01) × 102 | 9.08 ± 0.08 | ||
| D143H | (1.06 ± 0.01) × 105 [(6.90 ± 0.20) × 104] |
1.54 ± 0.06 | 3.60 ± 0.20c [3.70 ± 0.20]c |
9.30 ± 0.20 [10.10 ± 0.20] |
| R232A | (8.27 ± 0.07) × 103 | 4.80 ± 0.20 | ||
Values shown in square brackets were determined in D2O.
Values were determined at optimal pH/pD of 3.5 and 4.0 for the wt and mutant enzymes, respectively.
For histidine.
pH dependence of cmpd I formation and decay
To study the pH-rate relationships of cmpd I formation, the kI values of wt and mutant ElDyPs were determined at various pH. Plots of kI vs pH are shown in the bottom panel of Figure 4F. Assuming that pH dependence is controlled by protonation/deprotonation of an ionizable group in or close to the active site, the data were fitted to equations 4, 5, 6, and 7 for wt, D143A, D143H, and R232A, respectively. The k1a/k1b and pK1/pK2 represent pH-independent second-order rate constant of cmpd I formation and the pKa of ionizable groups, respectively. The latter is summarized in Table 3.
In contrast to LiP where cmpd I formation is pH independent,44–45 ElDyPs showed a clear dependence on
| (Eq. 4) |
| (Eq. 5) |
| (Eq. 6) |
| (Eq. 7) |
pH. The rate of wt cmpd I formation increases as pH increases. Two ionizable residues with pKa of 3.65 and 8.69 were identified, which were assigned to the distal aspartate and arginine in the heme center, respectively. While the observed pKa of the distal aspartate is exactly the same as the intrinsic value, the arginine pKa shifted drastically from the intrinsic value of 12.48 to 8.70 in the protein. Such a huge shift is not uncommon because the protein microenvironment can change a pKa significantly via hydrogen bonds, electrostatic interactions, and hydrophobicity.46 For example, the pKa of the distal histidine in pristine peroxidase can vary from 5.1 for ERS to 7.8 for cmpd I during H2O2 oxidation.47 The pKa of K115, a catalytic residue in acetoacetate decarboxylase, shifts downward by 4.5 pH units to ~6.0.48 In this regard, the crystal structure of wt-ElDyP has revealed that R232 forms 6 hydrogen bonds (Figure 2A) with neighboring residues (D143 and heme propionate) and solvent molecules (H2O 42 and 288). In addition, there are several hydrophobic residues (I230, V231, and F248) within a distance of 4 Å surrounding R232. These interactions may account for the large pKa shift (3.78 pH units) associated with R232.
To investigate if the distal aspartate and arginine are indeed responsible for these ionizable residues, the pH dependence of their mutants were also studied. It was predicted that mutation of a distal residue to alanine would result in removal of one of the pKa identified in the wt enzyme. Indeed, both D143A and R232A displayed single pKa at 9.08 and 4.81, respectively, confirming assignment of the residues for the observed pKa in the wt enzyme. Moreover, while the rate of cmpd I formation becomes faster with increasing pH for D143A, the corresponding rate in R232A decreases as the pH increases. Their implications will be discussed below. The pH dependence of cmpd I formation for D143H is bell-shaped, which is quite different from the wt-ElDyP and other two mutants. Two pKa values of 3.61 and 9.32 were determined, corresponding to the histidine and arginine in D143H, respectively. The pH dependence of D143H is similar to that of HRP cmpd I formation, in which a bell-shaped curve was obtained with pKa of 4.11 and 10.90.49
The stability of cmpd I was also investigated at different pH values. By monitoring H2O2 oxidation of wt-ElDyP (1:1 ratio) at 615 nm, cmpd I formation followed by decay to ERS* was observed sequentially (bottom panel in Figure 4E). The half-life of cmpd I decayed to ERS* was 0.2, 4.4, and 2.0 min at pH 3.5, 7.5, and 10.0, respectively. Cmpd I would further decay into a new species, possibly cmpd III, in the presence of excess H2O2 over a long time (data not shown). Thus, wt-ElDyP cmpd I has the shortest lifetime and is thus most reactive at acidic pH, suggesting that acidic conditions may facilitate its conversion to other reactive intermediates.
Transient-state kinetics of wt cmpd I reduction using ABTS and its pH dependence
The reaction was performed in a sequential mode. Cmpd I was generated by mixing wt-ElDyP with H2O2. After a 2 s delay to achieve maximal cmpd I formation, the mixture was then mixed with ABTS in the presence of ascorbate. Since the absorbance spectrum of the ABTS oxidation product overlaps with that of the wt-ElDyP and interferes with the reaction monitored, ascorbate was used to instantaneously reduce the oxidation product back to ABTS.50 Additionally, only low concentrations of ABTS (<10 μM) were required in order to maintain pseudo-first-order reaction conditions,50 as too high concentrations would make the rate too fast to determine. A typical spectral transition at pH 3.5 is shown in Figure 5A, where the characteristics corresponding to cmpd II are absent.21, 35 Analysis of the PDA data using the Pro-KIV Global Analysis program (Applied PhotoPhysics) did not show significant preference for a two-step reaction over a one-step reaction. Furthermore, when the data were fitted to a two-step reaction, the calculated spectra corresponding to cmpds I and II were almost identical. These results suggest that regeneration of ERS* from cmpd I is likely to proceed via a 2-electron reduction. A one-electron reduction of cmpd I to ERS* should produce cmpd II, which was not observed with our experiments. It has also to be noted that ABTS is a one-electron reductant. Thus, it is proposed that two molecules of ABTS donate their electrons to the protein simultaneously (one electron from each ABTS molecule). This requires the enzyme to have multiple sites for substrate binding, which has been found in several DyPs.51–54
Figure 5.
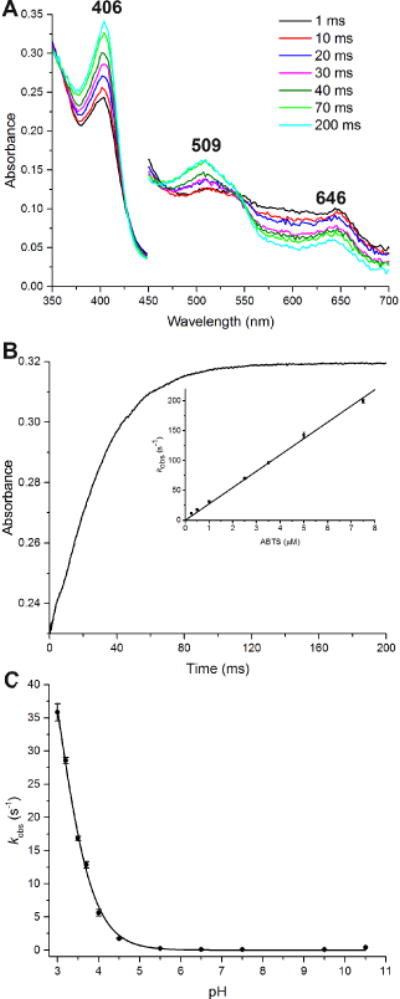
Transient-state kinetics of ERS regeneration from wt-ElDyP cmpd I. (A) Spectral transition of 2.5 μM wt cmpd I reduced by 2.5 μM ABTS at pH 3.5 in the presence of 10 μM ascorbate. The cmpd I was produced by reacting 2.5 μM wt-ElDyP with 2.5 μM H2O2 followed by 2-s delay. (B) Regeneration of ERS from cmpd I monitored at 406 nm. The inset represents determination of second-order rate constant kERS in the presence of ABTS. (C) pH-dependence of 2.5 μM wt cmpd I reacted with 0.5 μM ABTS at various pH.
The reduction of cmpd I was monitored at 406 nm corresponding to ERS* regeneration and a typical absorbance change is shown in Figure 5B. The kobs was obtained by fitting the curve to a single exponential growth equation. The overall second-order rate constants (kERS*) were determined by plotting the kobs vs ABTS concentrations (inset in Figure 5B), giving a value of 2.74 × 107 M−1 s−1. Thus, the rate of ERS*regeneration from wt cmpd I in the presence of ABTS is more than 10-fold faster than that of cmpd I formation (2.35 × 106 M−1s−1). It has to be pointed out that the rate of ERS* regeneration is expected to be substrate-dependent. Consequently, it is possible that a slower rate than the cmpd I formation could be obtained if a less reactive reducing substrate were employed.
The rate of wt cmpd I reduction using ABTS was also determined at pH ranging from 3.0 to 10.5. Conditions lower than pH 3.0 were not studied due to protein instability. As shown in Figure 5C, the overall kobs increases significantly as the pH decreases, demonstrating that cmpd I reduction prefers acidic conditions. When the pH is higher than 5.0, the kobs approaches zero. Additionally, fitting the data to Eq. 7 gave a pKa of 3.00, indicating that an ionizable residue, possibly D143, is involved in this reduction process.
sKIE on transient-state kinetics
D2O2 is a good mechanistic probe for studying compd I formation, which can be easily prepared from H-D exchange of concentrated H2O2 in D2O. Thus, both the enzyme (wt or D143H-ElDyP) and H2O2 at high concentrations were dissolved in D2O before they were mixed together using a stopped-flow spectrometer. Since the D2O2, rather than the H2O2, oxidizes the enzyme to produce cmpd I in D2O, the KIE, instead of sKIE, should be used to describe cmpd I formation. As shown in Figure 6A and summarized in Table 3, the rate of wt cmpd I formation at pD 3.5 was slowed in D2O, resulting in a normal KIE of 2.44. A proton inventory experiment showed a linear relationship between the rates and fraction of D2O, indicating that a single proton was involved in the rate-determining step (RDS) of cmpd I formation. The sKIE of D143H was determined to be 1.54 at pH/pD 4.0, which was much smaller than that of the wt enzyme. The KIE of wt cmpd I formation was also investigated as a function of pH/pD, which increased as the pH/pD decreased, to a maximum of 5.3 at pH/pD 2.0 (black lines in Figure 6B). When the pH/pD was greater than 5.5, the KIE approached to unity. For D143H, a normal KIE was observed when pH/pD£9.0 and the value slightly increased as the pH/pD dropped, to a maximum of 2.1 at pH/pD 3.0. Similarly to HRP,49 an inverse KIE was observed at high alkaline pH/pD (> 9.0), reaching 0.26 at pH/pD 10.0. Additionally, most pKas determined in D2O shift upward (Table 3), consistent with the known effects of D2O on pKa.55
Figure 6.
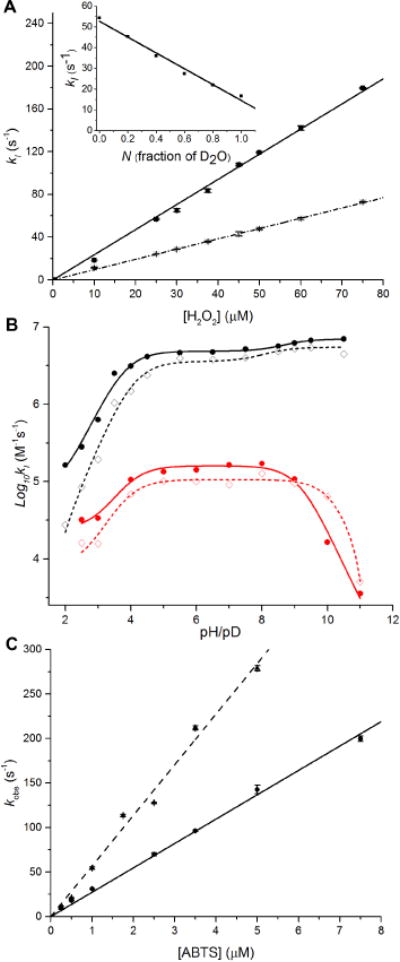
KIE and sKIE of transient-state kinetics. Solid and dash lines represent reactions performed in H2O and D2O, respectively. (A) Rates of wt-ElDyP cmpd I formation at pH/pD 3.5. The inset represents proton inventory plot of rates vs fraction of D2O. (B) pH/pD dependence of wt (black lines) and D143H-ElDyP (red lines) cmpd I formation on a logarithmic scale. (C) Rates of cmpd I reduction using ABTS as the reducing substrate at pH/pD 3.5.
As shown in Figure 6C, the second-order rate constants of cmpd I reduction (kERS) with ABTS in H2O and D2O were determined to be 2.74× 107 and 5.68 × 107 M−1s−1, respectively, at pH/pD 3.5. This resulted in an inverse sKIE [D(kERS)] of 0.48, which suggests that cmpd I reduction to ERS* is likely to involve water elimination from the heme iron. Their mechanistic implications will be discussed in the next section.
Viscosity effects on steady-state and transient-state kinetics
The effects of solvent viscosity upon kcat/KM and kcat were determined to evaluate whether diffusion into and out of the active site plays a significant role in the steady-state kinetic parameters of ElDyP. Additionally, viscosity could cause a small sKIE as D2O is 25% more viscous than H2O at r.t. To probe this possibility, sucrose was used as a viscosogen to modulate the viscosity of reaction medium. The results are summarized in Figure 7. For a completely diffusion-controlled reaction, the slope should equal to 1 as represented by the blue lines. When the slope approaches to zero, the reaction is independent of diffusion. Reactions with slopes between 0 and 1 are partially diffusion-controlled. It was found that kcat/KM of wt-ElDyP with ABTS was 40% diffusion-controlled (black solid line in Figure 7), suggesting that binding of a reducing substrate is partially diffusion-controlled and limits the rate of the overall reaction. The kcat, was observed to be viscosity independent (black dash line in Figure 7), however, implying that product release is not rate-limiting. Similar observations were also made for reactions employing glucose as a viscosogen (data not shown), demonstrating that the viscosity effects are viscosogen independent for steady-state kinetics.
Figure 7.
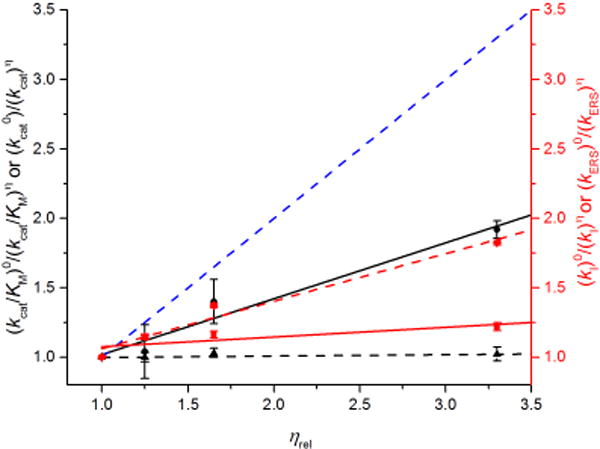
Effects of solvent viscosity on kinetic parameters at pH 3.5. Blue dash lines reflect theoretical limit for a completely diffusion-controlled reaction. Effects on kcat/KM (black solid line, slope = 0.404) and kcat (black dash line, slope = 0.007) with ABTS are scaled by left black Y-axis. Effects on second-order rate constants of cmpd I formation (kI, red solid line, slope = 0.069) and reduction using ABTS (kERS, red dash line, slope = 0.342) are scaled by right red Y-axis.
To characterize the origin of the viscosity effects found in steady-state kinetics, transient-state kinetics of cmpd I formation and reduction were performed in sucrose at various concentrations. It was found that the second-order rate constants of wt cmpd I formation (kI) are insensitive to solvent viscosity (red solid line in Figure 7). However, the reduction of cmpd I in the presence of ABTS and ascorbate was determined to be 34% diffusion-controlled (red dash line in Figure 7), which accounts for the 85% viscosity effect on kcat/KM observed in steady-state kinetics.
DISCUSSION
Based on the results described above, a bisubstrate Ping-Pong mechanism involving a final step of conformational change is proposed in Scheme 1. Several unique features associated with DyPs have been identified and are described below.
Scheme 1.
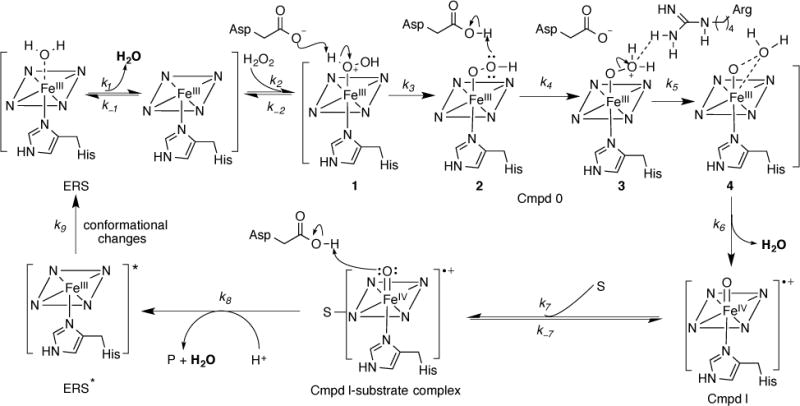
Proposed bisubstrate Ping-Pong mechanism for DyPs involving a final step of conformational change
Cmpd 0 deprotonation is rate-limiting in DyP cmpd I formation
ElDyP activation by H2O2 to form cmpd I was proposed in Scheme 1 based on the Poulos-Kraut mechanism.56 After H2O2 displaces H2O and binds with ERS as the sixth ligand, the conserved distal aspartate acts first as a general base to deprotonate H2O2 to form the iron-peroxide complex named cmpd 0, then as a general acid to protonate the outer oxygen of cmpd 0. Electrostatic interactions between the distal arginine and the outer oxygen atom are proposed to promote heterolysis of the O–O bond to form cmpd I.57 Since the former two steps (k3 and k4) involve formation and breakage of an O–H bond, a primary KIE is expected with D2O2 if any of these steps is rate-limiting. However, no primary KIE should be observed if the O–O bond scission (k5) is rate-limiting. We found that the wt-ElDyP displayed an observed isotope effect of 2.44 in D2O. Because H2O2 can exchange with D2O to produce D2O2 quickly, the observed isotope effect should be a combination of the primary KIE (if it exists) and sKIE. Since two water molecules are released during cmpd I formation (k1 and k6), an inverse sKIE will be expected as described in the next sub-section. Thus, the observed normal isotope effect suggests that a primary KIE larger than 2.44 should be present, as the inverse sKIE will cancel the isotope effect resulting from a normal primary KIE. Proton inventory showed that only one proton is involved in cmpd I formation. Because the viscosity did not contribute to the observed isotope effect, the results suggest that either H2O2 deprotonation or subsequent outer oxygen protonation should be rate-limiting, which could be determined by studying pH-dependence of cmpd I formation.
In sharp contrast with LiP, where the rates of cmpd I formation are pH-independent,44–45 the rates of ElDyP cmpd I formation displayed pH-dependence (Figure 4F, bottom panel). The rate increases as pH increases for both the wt enzyme and D143A-ElDyP. Moreover, cmpd I was formed only at alkaline pH for D143A. These observations indicate that the step of cmpd 0 deprotonation (k3) is likely to be a RDS for ElDyP cmpd I formation. While the distal aspartate is responsible for cmpd 0 deprotonation, alkaline conditions are expected to facilitate this process, resulting in an increased rate. When the aspartate was replaced with an alanine, deprotonation could only occur at alkaline pH due to the assistance of OH−, which resulted in the rate decreased by four orders of magnitude. Furthermore, pH-dependence of wt cmpd I formation in H2O and D2O (black lines in Figure 6B) revealed that the isotope effect becomes less pronounced as the acidity decreases, reaching 1.18 at pH 9.0. Under alkaline conditions, OH− and OD− are expected to participate in cmpd I formation as a specific base and thus, to have a small KIE. This provides additional evidence to support our hypothesis that cmpd 0 deprotonation is a RDS in wt cmpd I formation. The trend of pH-dependence was reversed in R232A, in which cmpd I was formed only at acidic pH and the rate increased as pH decreased. This suggests that O–O bond scission in ElDyP does not necessarily require the assistance of the distal arginine, though loss of it will result in a rate-drop by more than two orders of magnitude. Moreover, unlike the wt and D143A-ElDyPs, the step to protonate the outer oxygen atom (k4) may become rate-limiting in R232A, in which the aspartate acts as a general acid. Acidic pH should facilitate the formation of a protonated aspartate. Together, these results demonstrate that cmpd 0 deprotonation is rate-limiting for wt-ElDyP and that the aspartate is catalytically more important than the arginine.
Conformational change is the final step in the DyP mechanism
Transient-state kinetics of ERS* regeneration revealed that cmpd I reduction (2.74× 107 M−1 s−1) was more than 10 times faster than that of cmpd I formation (2.35 × 106 M−1s−1) when ABTS was used as a reducing substrate. Thus, either cmpd I formation or a step after ERS* regeneration would be rate-limiting in the overall ABTS oxidation catalyzed by wt-ElDyP. This RDS should be described by kcat in steady-state kinetics.
The inverse sKIE is not very common. Its chemical origins have been attributed to catalysis involving cysteine, water dissociation from metal centers, and medium effects.58 Because the closest C249 in ElDyP is 12.6 Å away from the heme iron, it is unlikely that cysteines participate in catalysis. Thus, the observed inverse sKIE should arise from either water release from the heme iron or medium effects during turnover. The fractionation factor (Φ, equilibrium distribution of the two isotopes) for L2O (where L = H or D) is inverse when water is bonded to a metal, resulting in a tendency for D2O to accumulate in bulk solvent.59 It is commonly accepted that the water released from M−OH2↔M + H2O equilibrium has Φ≈0.70.60 Additionally, Φ is reported on a per-bond basis and multiplicative. Thus, the observed sKIE is diagnostic of the number of H2O released from the metal center. For example, 0.49 (Φ2), 0.24 (Φ4), and 0.12 (Φ6) are expected for the dissociation of one, two, and three H2O molecules, respectively. Such a method has recently been employed to reveal kinetic mechanisms of two iron-containing enzymes, FIH and ToMO.59, 61–62 While most inverse sKIE resulting from medium effects are due to viscosity-sensitive conformational changes,58, 63–64 solvation can also cause inverse sKIE as demonstrated by the blue single-copper protein azurin from Pseudomonas aeruginosa.65 The effect of solvation on azurin is reflected as a positive shift of E°′ by 10 mV from H2O to D2O65
The D(kcat) values for wt-ElDyP were determined to be 0.70 with ABTS. Although the value equals Φ of one hydroxyl group dissociated from a metal center, which is included in the step of cmpd I reduction to ERS* (k8), our experimental results with ABTS eliminates this possibility as cmpd I reduction is much faster than cmpd I formation. Thus, a conformational change of ERS* to ERS was proposed as the final rate-limiting step in the presence of ABTS. Since the kcat was found to be viscosity-independent, our results indicate that enzyme motion does not involve movement of large loops on protein surface. The observed inverse sKIE is likely due to protein solvation, which is further supported by a positive shift of E°′ from −290 mV in H2O to −263 mV in D2O for wt-ElDyP.
It has to be pointed out that the two conformers, ERS* and ERS, are spectroscopically identical for ElDyP. Moreover, whether this final step of conformational change is rate-limiting or not will depend on the reducing substrate. With fast substrates such as ABTS and RB19, conformational change (k9) is a RDS. However, with slow substrates such as pyrogallol, guaiacol and HQ, cmpd I reduction (k8) is more likely to be rate-limiting, which has been shown in TcDyP and DyPB.19, 21
Aquo release is mechanistically important
While both the D(kcat) and D(kcat/KM) of wt-ElDyP are inverse with ABTS, the inverse effect is much more pronounced with the latter than with the former. The sKIE of kcat/KM was determined to be 0.11, which is in excellent agreement with the theoretical value of 0.12, corresponding to the release of three water molecules (Φ2× Φ2× Φ2 = 0.12). Since the kcat/KM describes the steps preceding the RDS, examination of the mechanism proposed in Scheme 1 reveals that three waters are released before the RDS in the catalytic cycle, which include (1) one released from Fe (III) in ERS during H2O2 displacement. A water molecule was seen to act as the sixth ligand in wt-ElDyP crystal structure (Figure 2A); (2) one dissociated from cmpd 0 prior to the formation of cmpd I. It was proposed that a water molecule is eliminated from cmpd 0-water complex 4, though additional experimental evidence in support of this concept has yet to be obtained; and (3) one released in the reduction of cmpd I to generate ERS* via a two-electron process. To further demonstrate the importance of aquo release, the sKIE of cmpd I reduction (kERS) was investigated. An inverse sKIE of 0.48 was determined, which is again in excellent agreement with the theoretical prediction of 0.49 (Φ2 = 0.49) as only one water is dissociated from the heme iron during cmpd I reduction. The excellent agreement between theoretical and experimental results suggests that aquo release in DyP is mechanistically important.
It has been postulated that cmpd I undergoes different redox pathways depending on whether the coproduced water is bound with the intermediate or not.66 If the water is absent (dry form), the cmpd I will proceed through a two-electron equivalent reduction. However, only one-electron equivalent processes are accessible in the presence of water (wet form). Thus, the coproduced water may act as a gate to control enzyme function and activity.66 In ElDyP cmpd I reduction, only one water was found released based on the sKIE of 0.48. If the coproduced water is bound with cmpd I before its reduction, two water molecules should be expected, resulting in a sKIE of 0.49×0.49=0.24. Therefore, it was deduced that the wt-ElDyP cmpd I is in a dry form and may undergo a two-electron reduction, which agrees with our experimental results: absence of cmpd II species in stopped-flow spectroscopy during cmpd I reduction.
Reducing substrates bind with cmpd I only
Viscosity experiments performed under steady-state kinetic conditions showed that, while kcat was unchanged, the kcat/KM with reducing substrates decreased as the viscosity increased. The overall reaction of ABTS oxidation (kcat/KM) is 40% diffusion-controlled, suggesting that binding of reducing substrate is important. Additionally, reduction of cmpd I (kERS) using ABTS accounts for most of the overall viscosity observed in steady-state kinetics. Thus, it is proposed that the reducing substrate only binds with cmpd I. This is further supported by isothermal titration calorimetry (ITC) and surface plasmon resonance (SPR) experiments (data not shown), where ABTS did not show bindings to free enzyme except for non-specific bindings under very high substrate concentrations. Similar results were also obtained in our study of TcDyP with lignin model compounds.23 Therefore, unlike some heme peroxidases, in which the reducing substrate binds randomly with free enzyme and cmpd I,67–69 sequential binding of H2O2 with free enzyme followed by reducing substrate with cmpd I is proposed for DyPs.
DyPs’ acidic optimum results from distal aspartate and other acidic residues
It has been claimed that the acidic optimum of DyP activity is due to the presence of the distal aspartate.1–3, 25 While our results provided the first experimental evidence to support this claim, they also demonstrated that the distal aspartate is not the only determinant. Other acidic residues must have contributed to the enzyme’s acidic optimum, as mutations of D143 shifted the pH optimum only by 0.5 pH unit to 4.0 (Figure 1D). Should the distal aspartate be the sole origin of the acidic optimum, the mutant D143H would be expected to have the highest activity under neutral conditions due to the pKa of histidine. This was clearly not the case. Consequently, in addition to the distal aspartate, other acidic residues around the heme center must also be responsible for the DyPs’ acidic optimum, which are yet to be identified. Moreover, the pH dependence of ERS* regeneration showed that the acidic optimum is essential for cmpd I reduction (Figure 5C), which explains why the ElDyP is inactivated above pH 5.0 (Figure 1D) though cmpd I formation is actually enhanced at higher pH values (bottom panel in Figure 4F). The distal aspartate is likely to be the ionizable group and act as a general acid to protonate cmpd I in order to eliminate the oxygen atom attached to Fe(IV) as a water. These results provide convincing evidence that the distal aspartate plays key roles in catalysis and leads to acidic optimum of DyPs.
D143H-ElDyP shows mechanistic difference from the wt enzyme
D143H-ElDyP was selected for study because class II plant peroxidases and HRP contain a distal histidine. Elucidation of its properties will provide the basis to understand mechanistic difference between the DyPs and fungal lignolytic enzymes. The sKIE of cmpd I formation was determined to be much smaller for D143H than for the wt at the same pH/pD (Table 3 and Figure 6B). Its pH-independent, second-order rate constants of cmpd I formation were determined to be 1.56 × 105 and 1.06 × 105 M−1s−1 in H2O and D2O, respectively (red lines in Figure 6B), yielding a pH-independent sKIE of 1.47. This is close to the pH-independent sKIE of 1.60 for HRP.49 The decreased sKIE relative to the wt enzyme at the same pH suggests that deprotonation and protonation of cmpd 0 are less important for cmpd I formation in D143H than in the wt enzyme. When the pH was higher than 9.0, an inverse sKIE was observed, which could be ascribed to restrictions on torsional motions of exchangeable protons as reported for β–lactam synthetase.64
The D(kcat) of D143H with ABTS was close to unity, implying that the rate-limiting conformational changes are insensitive to the solvent. This is in contrast to wt-ElDyP and indicates that the distal aspartate is important in determining the heme microenvironment, which is also reflected in their UV-absorbance spectra (Figure 1C). An extra Q-band was observed at 567 nm when the distal aspartate was replaced with a histidine. The D(kcat/KM) of D143H was nearly identical to that of the wt (Table 2), displaying a large inverse sKIE. Similar to wt-ElDyP, the large inverse sKIE was attributed to water dissociation from the heme iron, which should result in an expected D(kcat/KM) of 0.12. Thus, the predicted value is in excellent agreement with the observed one at 0.13.
CONCLUSION
A bisubstrate Ping-Pong mechanism involving a final step of conformational change was proposed as a possible DyP mechanism based on the sKIEs and viscosity effects on steady-state and transient-state kinetics. The conformational change is also rate-limiting in ABTS oxidation. Spectra of ERS* and ERS present in the conformational change were indistinguishable. The normal KIE and pH-dependence of cmpd I formation demonstrated that cmpd 0 deprotonation is rate-limiting for wt-ElDyP, which is less important for D143H. Cmpd I reduction to ERS* is likely a two-electron process in the presence of ABTS, in which the viscosity effects observed accounted for almost all of the viscosity effects found in kcat/KM. Together with the results of ITC and SPR (data not shown), this suggested that H2O2 and reducing substrates bind with free enzyme and cmpd I, respectively. The significant inverse sKIEs of kcat/KM and kERS* were ascribed to water dissociation from the heme iron, which are in excellent agreement with the predicted values. This demonstrates the unique mechanistic importance of aquo release in DyP-catalyzed reactions. Steady-state and transient-state kinetics showed that the distal aspartate is catalytically more important than the distal arginine. The drastic pKa shift of 3.79 pH units for the distal arginine is likely caused by its extensive networks of hydrogen bonds. While the distal aspartate plays key roles in determining DyPs’ acidic optimum, other acidic residues in the heme center also contribute to this unique property. Thus, the results reported here provide a detailed picture of DyP mechanism at the molecular level, which will pave the way for future protein engineering to improve DyPs’ lignolytic activity.
Supplementary Material
Acknowledgments
This work was supported by the awards from Johnson Cancer Research Center and NIH P30GM110761 pilot project to P.L. and NIH GM121511 to B.V.G. G.H. is supported by an NIH K-INBRE postdoctoral fellowship under grant number P20GM103418. Crystal X-ray data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-BM beamline at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. W-31-109-Eng-38.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
DNA sequences, data collection and refinement statistics of the crystal structure, absorbance maxima of enzyme resting state and cmpd I, and biochemical characterizations (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Singh R, Eltis LD. Arch Biochem Biophys. 2015;574:56–65. doi: 10.1016/j.abb.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 2.Strittmatter E, Plattner DA, Piontek K. Encyclopedia of Inorganic and Bioinorganic Chemistry. John Wiley and Sons, Ltd; New York: 2014. pp. 1–13. [Google Scholar]
- 3.Sugano Y. Cell Mol Life Sci. 2009;66:1387–1403. doi: 10.1007/s00018-008-8651-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown ME, Chang MCY. Curr Opin Chem Biol. 2014;19:1–7. doi: 10.1016/j.cbpa.2013.11.015. [DOI] [PubMed] [Google Scholar]
- 5.de Gonzalo G, Colpa DI, Habib MHM, Fraaije MW. J Biotechnol. 2016;236:110–119. doi: 10.1016/j.jbiotec.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 6.Bugg TD, Ahmad M, Hardiman EM, Singh R. Curr Opin Biotech. 2011;22:394–400. doi: 10.1016/j.copbio.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 7.Linger JG, Vardon DR, Guarnieri MT, Karp EM, Hunsinger GB, Franden MA, Johnson CW, Chupka G, Strathmann TJ, Pienkos PT, Beckham GT. Proc Natl Acad Sci USA. 2014;111:12013–12018. doi: 10.1073/pnas.1410657111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ralph J, Brunow G, Boerjan W. Lignins In eLS. John Wiley & Sons Ltd; Chichester: 2007. No. a0020104. [Google Scholar]
- 9.Sanderson K. Nature. 2011;474:S12–S14. doi: 10.1038/474S012a. [DOI] [PubMed] [Google Scholar]
- 10.Zakzeski J, Bruijnincx PCA, Jongerius AL, Weckhuysen BM. Chem Rev. 2010;110:3552–3599. doi: 10.1021/cr900354u. [DOI] [PubMed] [Google Scholar]
- 11.Ragauskas AJ, Beckham GT, Biddy MJ, Chandra R, Chen F, Davis MF, Davison BH, Dixon RA, Gilna P, Keller M, Langan P, Naskar AK, Saddler JN, Tschaplinski TJ, Tuskan GA, Wyman CE. Science. 2014;344:709–720. doi: 10.1126/science.1246843. [DOI] [PubMed] [Google Scholar]
- 12.Rahimi A, Ulbrich A, Coon JJ, Stahl SS. Nature. 2014;515:249–252. doi: 10.1038/nature13867. [DOI] [PubMed] [Google Scholar]
- 13.Boerjan W, Ralph J, Baucher M. Annu Rev Plant Biol. 2003;54:519–546. doi: 10.1146/annurev.arplant.54.031902.134938. [DOI] [PubMed] [Google Scholar]
- 14.Sannigrahi P, Ragauskas AJ. J Biobased Mater Bio. 2011;5:514–519. [Google Scholar]
- 15.Vishtal A, Kraslawski A. Bioresources. 2011;6:3547–3568. [Google Scholar]
- 16.Wong DWS. Appl Biochem Biotech. 2009;157:174–209. doi: 10.1007/s12010-008-8279-z. [DOI] [PubMed] [Google Scholar]
- 17.Pollegioni L, Tonin F, Rosini E. FEBS J. 2015;282:1190–1213. doi: 10.1111/febs.13224. [DOI] [PubMed] [Google Scholar]
- 18.Bugg TDH, Ahmad M, Hardiman EM, Rahmanpour R. Nat Prod Rep. 2011;28:1883–1896. doi: 10.1039/c1np00042j. [DOI] [PubMed] [Google Scholar]
- 19.Ahmad M, Roberts JN, Hardiman EM, Singh R, Eltis LD, Bugg TDH. Biochemistry. 2011;50:5096–5107. doi: 10.1021/bi101892z. [DOI] [PubMed] [Google Scholar]
- 20.Brown ME, Barros T, Chang MCY. ACS Chem Biol. 2012;7:2074–2081. doi: 10.1021/cb300383y. [DOI] [PubMed] [Google Scholar]
- 21.Chen C, Shrestha R, Jia K, Gao PF, Geisbrecht BV, Bossmann SH, Shi JS, Li P. J Biol Chem. 2015;290:23447–23463. doi: 10.1074/jbc.M115.658807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Min K, Gong G, Woo HM, Kim Y, Um Y. Sci Rep. 2015;5(8245) doi: 10.1038/srep08245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang GC, Shrestha R, Jia KM, Geisbrecht BV, Li P. Org Lett. 2017;19:1820–1823. doi: 10.1021/acs.orglett.7b00587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshida T, Sugano Y. Arch Biochem Biophys. 2015;574:49–55. doi: 10.1016/j.abb.2015.01.022. [DOI] [PubMed] [Google Scholar]
- 25.Colpa DI, Fraaije MW, van Bloois E. J Ind Microbiol Biot. 2014;41:1–7. doi: 10.1007/s10295-013-1371-6. [DOI] [PubMed] [Google Scholar]
- 26.DeAngelis KM, D’Haeseleer P, Chivian D, Fortney JL, Khudyakov J, Simmons B, Woo H, Arkin AP, Davenport KW, Goodwin L, Chen A, Ivanova N, Kyrpides NC, Mavromatis K, Woyke T, Hazen TC. Stand Genomic Sci. 2011;5:69–85. doi: 10.4056/sigs.2104875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dailey HA, Septer AN, Daugherty L, Thames D, Gerdes S, Stabb EV, Dunn AK, Dailey TA, Phillips JD. Mbio. 2011;2:e00248–11. doi: 10.1128/mBio.00248-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Letoffe S, Heuck G, Delepelaire P, Lange N, Wandersman C. Proc Natl Acad Sci USA. 2009;106:11719–11724. doi: 10.1073/pnas.0903842106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 30.Berry EA, Trumpower BL. Anal Biochem. 1987;161:1–15. doi: 10.1016/0003-2697(87)90643-9. [DOI] [PubMed] [Google Scholar]
- 31.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Crystallogr D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emsley P, Lohkamp B, Scott WG, Cowtan K. Acta Crystallogr D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reyes C, Qian F, Zhang A, Bondarev S, Welch A, Thelen MP, Saltikov CW. J Bacteriol. 2012;194:5840–5847. doi: 10.1128/JB.00890-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilks A, Sun J, Loehr TM, Demontellano PRO. J Am Chem Soc. 1995;117:2925–2926. [Google Scholar]
- 35.Roberts JN, Singh R, Grigg JC, Murphy MEP. Biochemistry. 2011;50:5108–5119. doi: 10.1021/bi200427h. [DOI] [PubMed] [Google Scholar]
- 36.Uchida T, Sasaki M, Tanaka Y, Ishimorit K. Biochemistry. 2015;54:6610–6621. doi: 10.1021/acs.biochem.5b00952. [DOI] [PubMed] [Google Scholar]
- 37.Liu XH, Yuan ZL, Wang JX, Cui YQ, Liu S, Ma YL, Gu LC, Xu SJ. Biochem Bioph Res Co. 2017;484:40–44. doi: 10.1016/j.bbrc.2017.01.081. [DOI] [PubMed] [Google Scholar]
- 38.Blodig W, Smith AT, Doyle WA, Piontek K. J Mol Biol. 2001;305:851–861. doi: 10.1006/jmbi.2000.4346. [DOI] [PubMed] [Google Scholar]
- 39.Sundaramoorthy M, Gold MH, Poulos TL. J Inorg Biochem. 2010;104:683–690. doi: 10.1016/j.jinorgbio.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saez-Jimenez V, Acebes S, Garcia-Ruiz E, Romero A, Guallar V, Alcalde M, Medrano FJ, Martinez AT, Ruiz-Duenas FJ. Biochem J. 2016;473:1917–1928. doi: 10.1042/BCJ20160248. [DOI] [PubMed] [Google Scholar]
- 41.Poulos TL. Chem Rev. 2014;114:3919–3962. doi: 10.1021/cr400415k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.ten Have R, Teunissen PJM. Chem Rev. 2001;101:3397–3413. doi: 10.1021/cr000115l. [DOI] [PubMed] [Google Scholar]
- 43.van Bloois E, Pazmino DET, Winter RT, Fraaije MW. Appl Microbiol Biot. 2010;86:1419–1430. doi: 10.1007/s00253-009-2369-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marquez L, Wariishi H, Dunford HB, Gold MH. J Biol Chem. 1988;263:10549–10552. [PubMed] [Google Scholar]
- 45.Andrawis A, Johnson KA, Tien M. J Biol Chem. 1988;263:1195–1198. [PubMed] [Google Scholar]
- 46.Pace CN, Grimsley GR, Scholtz JM. J Biol Chem. 2009;284:13285–13289. doi: 10.1074/jbc.R800080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chreifi G, Baxter EL, Doukov T, Cohen AE, McPhillips SE, Song JH, Meharenna YT, Soltis SM, Poulos TL. Proc Natl Acad Sci USA. 2016;113:1226–1231. doi: 10.1073/pnas.1521664113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Highbarger LA, Gerlt JA, Kenyon GL. Biochemistry. 1996;35:41–46. doi: 10.1021/bi9518306. [DOI] [PubMed] [Google Scholar]
- 49.Dunford HB, Hewson WD, Steiner H. Can J Chem. 1978;56:2844–2852. [Google Scholar]
- 50.Goodwin DC, Yamazaki I, Aust SD, Grover TA. Anal Biochem. 1995;231:333–338. doi: 10.1006/abio.1995.0059. [DOI] [PubMed] [Google Scholar]
- 51.Linde D, Pogni R, Canellas M, Lucas F, Guallar V, Baratto MC, Sinicropi A, Saez-Jimenez V, Coscolin C, Romero A, Medrano FJ, Ruiz-Duenas FJ, Martinez AT. Biochem J. 2015;466:253–262. doi: 10.1042/BJ20141211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shrestha R, Chen XJ, Ramyar KX, Hayati Z, Carlson EA, Bossmann SH, Song LK, Geisbrecht BV, Li P. ACS Catal. 2016;6:8036–8047. doi: 10.1021/acscatal.6b01952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Strittmatter E, Serrer K, Liers C, Ullrich R, Hofrichter M, Piontek K, Schleicher E, Plattner DA. Arch Biochem Biophys. 2015;574:75–85. doi: 10.1016/j.abb.2014.12.016. [DOI] [PubMed] [Google Scholar]
- 54.Yoshida T, Tsuge H, Hisabori T, Sugano Y. FEBS Lett. 2012;586:4351–4356. doi: 10.1016/j.febslet.2012.10.049. [DOI] [PubMed] [Google Scholar]
- 55.Krezel A, Bal W. J Inorg Biochem. 2004;98:161–166. doi: 10.1016/j.jinorgbio.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 56.Poulos TL, Kraut J. J Biol Chem. 1980;255:8199–8205. [PubMed] [Google Scholar]
- 57.Derat E, Shaik S. J Phys Chem B. 2006;110:10526–10533. doi: 10.1021/jp055412e. [DOI] [PubMed] [Google Scholar]
- 58.Karsten WE, Lai CJ, Cook PF. J Am Chem Soc. 1995;117:5914–5918. [Google Scholar]
- 59.Hangasky JA, Saban E, Knapp MJ. Biochemistry. 2013;52:1594–1602. doi: 10.1021/bi3015482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cook PF. Kinetic and regulatory mechanisms of enzymes from isotope effects. In: Cook PF, editor. Enzyme mechanism from isotope effects. CRC Press; Boca Raton: 1991. pp. 203–231. [Google Scholar]
- 61.Liang AD, Wrobel AT, Lippard S. J Biochemistry. 2014;53:3585–3592. doi: 10.1021/bi500387y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Flagg SC, Giri N, Pektas S, Maroney MJ, Knapp MJ. Biochemistry. 2012;51:6654–6666. doi: 10.1021/bi300229y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grissom CB, Cleland WW. Biochemistry. 1988;27:2927–2934. doi: 10.1021/bi00408a039. [DOI] [PubMed] [Google Scholar]
- 64.Raber ML, Freeman MF, Townsend CA. J Biol Chem. 2009;284:207–217. doi: 10.1074/jbc.M805390200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Farver O, Zhang JD, Chi QJ, Pecht I, Ulstrup J. Proc Natl Acad Sci USA. 2001;98:4426–4430. doi: 10.1073/pnas.071043798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jones P. J Biol Chem. 2001;276:13791–13796. doi: 10.1074/jbc.M011413200. [DOI] [PubMed] [Google Scholar]
- 67.Childs RE, Bardsley WG. Biochem J. 1975;145:93–103. doi: 10.1042/bj1450093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang WC, Noel S, Desmadril M, Gueguen J, Michon T. Biochem J. 1999;340:329–336. [PMC free article] [PubMed] [Google Scholar]
- 69.Koop DR, Hollenberg PF. J Biol Chem. 1980;255:9685–9692. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.