

Abstract
Dye-decolorizing peroxidases (DyPs) are a family of heme peroxidases, in which a catalytic distal aspartate is involved in H2O2 activation to catalyze oxidations in acidic conditions. They have received much attention due to their potential applications in lignin compound degradation and biofuel production from biomass. However, the mode of oxidation in bacterial DyPs remains unknown. We have recently reported that the bacterial TcDyP from Thermomonospora curvata is among the most active DyPs and shows activity toward phenolic lignin model compounds (J. Biol. Chem. 2015, 290, 23447). Based on the X-ray crystal structure solved at 1.75 Å, sigmoidal steady-state kinetics with Reactive Blue 19 (RB19), and formation of compound II-like product in the absence of reducing substrates observed with stopped-flow spectroscopy and electron paramagnetic resonance (EPR), we hypothesized that the TcDyP catalyzes oxidation of large-size substrates via multiple surface-exposed protein radicals. Among 7 tryptophans and 3 tyrosines in TcDyP consisting of 376 residues for the matured protein, W263, W376, and Y332 were identified as surface-exposed protein radicals. Only the W263 was also characterized as one of surface-exposed oxidation sites. SDS-PAGE and size-exclusion chromatography demonstrated that W376 represents an off-pathway destination for electron transfer, resulting in the crosslinking of proteins in the absence of substrates. Mutation of W376 improved compound I stability and overall catalytic efficiency toward RB19. While Y332 is highly conserved across all four classes of DyPs, its catalytic function in A-class TcDyP is minimal possibly due to its extremely small solvent accessible areas. Identification of surface-exposed protein radicals and substrate oxidation sites is important for understanding DyP mechanism and modulating its catalytic functions for improved activity on phenolic lignin.
Keywords: DyP, surface-exposed, protein radical, oxidation site, crystal structure, EPR, crosslink, stopped-flow
INTRODUCTION
Dye-decolorizing peroxidases (DyPs) are a newly discovered family of heme peroxidases, which are widely present in bacterial genomes.1 While the biological function of DyPs remains unknown, they can use H2O2 to oxidize various industrial dyes, particularly anthraquinone derivatives that have high redox potentials and are poor substrates for classical peroxidases.2,3 DyPs have recently received much attention due to their potential application in lignin degradation, acting on the phenolic moieties.2,4 Lignin is the most abundant renewable source of aromatic polymers on earth,5 which encases cellulose and hemicellulose fibres and prevents conversion of plant biomass to bioethanol.6 However, degradation of lignin presents challenges, requiring strong acids and bases at high temperature to break it down.7 Thus, there is a growing interest in lignin degradation using enzymatic approaches.8 Studies including ours have demonstrated that DyPs can depolymerize phenolic lignin model compounds and wheat straw lignocellulose, though the efficiency is low.9–12 Thus, more structural and mechanistic works are needed for DyP in order to develop its applications in biofuel production from lignocellulose feedstock.
DyPs are subdivided into four classes, A through D, based on their primary sequences.13 Except for class D that is fungal origin, the rest are mainly from bacteria.13 It has to be noted that sequence identity between classes is very low (< 15%), especially between fungal and bacterial DyPs.14 Several fungal lignin-degrading enzymes such as lignin peroxidase (LiP) and versatile peroxidase (VP) have been found to oxidize substrates via surface-exposed tryptophanyl and tyrosyl radicals.15–17 Similar mode of action was also suggested for the fungal AauDyP from Auricularia auricula-judae.18–21 While such information is critical for understanding DyP mechanism and modulating its catalytic functions through protein engineering, it remains unknown for bacterial A-, B-, and C-class DyPs.
We have recently reported an A-class DyP from Thermomonospora curvata (TcDyP), which shows high peroxidase and dye-decolorizing activities.12 It is also among few DyPs that have reactivity with phenolic lignin model compounds,9–12 indicating its potential application in lignin degradation processes. The catalytic cycle of TcDyP has been demonstrated to be similar to that of plant peroxidase, which incudes an enzyme resting state, compound I and compound II.12,22,23 Compound I ([Fe4+=O]+•) is formed by oxidation of the enzyme resting state ([Fe3+]) with a two-electron-equivalent oxidant like H2O2. Reduction of Compound I with a one-electron-equivalent reducing substrate generates compound II ([Fe4+=O]), which returns to the enzyme resting state by a second one-electron-equivalent reducing substrate. It is of note that the TcDyP compound I is unstable and quickly decays into compound II-like product (C2LP) in the absence of a reducing substrate.12 While A-class DyPs have a promising future in biotechnological applications,11,12,24 study on this class lags far behind the others, especially on the mode of oxidation of large-size substrates. Here we report our results to identify surface-exposed protein radicals and a substrate oxidation site involving aromatic redox-active amino acids in TcDyP.
MATERIALS AND METHODS
Instruments, Biochemicals, and Chemicals
All activity assays and steady-state kinetics were performed on a Cary 100 Bio UV-Vis spectrometer equipped with temperature controller and magnetic stirring. Transient-state kinetics was carried out on an Applied Photophysics SX20 stopped-flow spectrometer equipped with sequential mixing, a PDA detector, and a monochromator. Analytical SEC was performed on a Waters Breeze 2 system equipped with a PDA detector. EPR spectra were recorded on a Bruker E680 spectrometer equipped with a high sensitivity cavity and an ESR900 helium flow cryostat from Oxford Instruments. All chemical and biochemical reagents were purchased at the highest grade and used without further purification. Protein concentrations were determined by BCA assays.25 Stocks of H2O2 were prepared fresh before experiments and their concentrations were determined at 240 nm using ε240 = 43.6 M−1cm−1.
Protein mutation, expression and purification
Mutations were generated using QuickChange (Agilent Technologies) according to manufacturer’s instructions. Fifteen mutants were generated, which included thirteen single mutants (W158F, W194F, W237F, W244F, W263F/S/A, W376F, W396F, Y330F, Y332F/L, and Y351F), one double mutant (W263F/W376F), and one triple mutant (W158F/W237F/W396F). Oligonucleotide primers used in the construction of these mutants are summarized in Table S1. Protein expression and purification were performed as described previously except that the cells were grown for an additional 4 h after IPTG induction.12 The mutant W263S was grown at 18 °C overnight after IPTG addition in order to reach full heme occupancy. The Reinheitszahl values (Rz) of purified proteins were measured to be 1.8–2.0. Their heme contents were determined to be 0.97–1.20 using pyridine hemochromogen assays and Δε557−541 = 20.7 mM−1cm−1.26 All mutants were analyzed by circular dichrosim to ensure that they were properly folded.
Proteins for crystallization were purified in the following way. Ten mg wild-type (wt) TcDyP were treated with 100 μg subtilisin A at 37 °C for 1 h before diisopropyl fluorophosphate was added to a final concentration of 100 μM to stop proteolysis. Following dialysis against 20 mM ethanolamine-HCl (pH 9.0), the protein was subjected to ion-exchange chromatography using a HiTrap Q column (GE Healthcare) and a linear gradient of 0–1.0 M NaCl in 20 mM ethanolamine-HCl (pH 9.0). The eluted protein was applied to a HiPrep 26/60 Sephacryl S-200 HR column (GE Healthcare) equilibrated in 20 mM ethanolamine-HCl (pH9.0) and 200 mM NaCl. Fractions corresponding to dimeric TcDyP were pooled, buffer exchanged into 5 mM ethanolamine-HCl (pH 9.0), and concentrated to10 mg/ml using Amicon 10 KDa centrifugal filters.
Crystallization and structure determination
Crystals were grown by the sitting drop vapor diffusion method in a solution consisting of 100 mM sodium succinate (pH 7.0) and 15% (w/v) PEG3350. Crystals appeared within 24 h and matured by 48 h. Individual crystals were transferred to 100 mM sodium succinate (pH 7.0) containing 35% (w/v) PEG3350, and allowed to equilibrate for 5 minutes prior to flash freezing in liquid nitrogen. Diffraction data were collected at 100 K using beamline 22-BM of the Advanced Photon Source, Argonne National Laboratory at a wavelength of 1.0 Å and processed with the HKL2000 package. The structure was solved by molecular replacement using a PHENIX software suite with coordinates of PDB entry 4GT2 as a starting model.27 The final structure was obtained by iterative automated model building and refinement using PHENIX and interactive model modification in Coot.28 Figures were prepared using PyMol (Schrödinger, LLC). The final coordinates have been deposited in the RCSB under accession ID 5JXU.
EPR samples and data collection
EPR quartz tubes with an external diameter of 4 mm were used for measurements. Samples consisting of 1.0 mM wt-TcDyP in 100 mM NaCl and 50 mM KPi (pH 7.8) and 0.1 mM wt-TcDyP in 100 mM NaCl and 50 mM sodium citrate (pH 3.0) were used for analysis of the enzyme resting state. Compound I samples were prepared by manually mixing the enzyme and 10-fold excess H2O2 in EPR tubes. The mixing was conducted on ice and then the EPR tube was flash-frozen in liquid nitrogen.
X- and W-band spectra were collected at the National High Magnetic Field Laboratory (NHMFL, Tallahassee, FL). The X-band (9.4 GHz) measurements were performed on a Bruker E680 spectrometer with a high sensitivity resonator (model No. 4119HS). The X-band spectra were collected at 5 K with a 4000 G scan width using a 100 kHz modulation frequency, 5 G modulation amplitude, and a 1 mW microwave power. The X-band spectra centered at g = 2 at 5–30 K were recorded using a 2 G modulation amplitude, and a 0.2 mW microwave power. The W-band experiments were carried out on a 94 GHz quasi-optical EPR spectrometer (HiPER)29 using an induction-mode cylindrical sample holder. The W-band spectra were collected with a 400 G sweep width, a 5 G modulation amplitude, and an 800 Hz modulation frequency.
Analysis of reactions between TcDyPs and H2O2 by SDS-PAGE and SEC
In a 50 μL solution of 50 mM salt at different pH values (sodium citrate, KPi, and Tris-HCl for pH 3.0–6.0, 7.0–8.0, and 9.0, respectively), 23 μM wt-TcDyP was incubated with 23 μM H2O2 at room temperature for 1 min. An aliquot of 10 μL reaction mixture was withdrawn and the reaction was stopped by addition of equal volume of 2X Laemmli buffer followed by heating at 95 °C for 4 min, which was subsequently used for SDS-PAGE. For the reaction run at pH 8.0, 30 μL reaction mixture was withdrawn and analyzed by SEC using a BioSep SEC-S2000 HPLC column. The column was eluted with 150 mM NaCl in 50 mM KPi (pH 7.5) at a flow rate of 0.5 mL/min. The retention volumes of molecular mass standards were as follows: aprotinin (6.5 kDa, 9.75 ml), horse cytochrome c (12.4 kDa, 9.05 ml), chicken albumin (44.3 kDa, 7.70 ml), bovine albumin (66 kDa, 7.20 ml), yeast alcohol dehydrogenase (150 kDa, 6.85 ml), and horse apoferritin (443 kDa, 6.00 ml).
The same experiment was also carried out with wt-TcDyP at pH 7.8 in the presence of RB19, in which 23 μM enzyme was incubated with 23 μM RB19 for 1 min prior to H2O2 addition. Controls were prepared, in which only RB19, or H2O2, or none of them was added.
Experiments of mutants and H2O2 were performed at pH 7.8 and analyzed by SDS-PAGE and SEC in the same way as described above.
Enzyme assays and steady-state kinetics
The activities of wt and mutant TcDyPs were determined using RB19 as the substrate. Briefly, in a 480 μL solution consisting of 50 mM sodium citrate (pH 3.0), 0.02 mg/mL bovine serum albumin (BSA), 1 mM H2O2, and 100 μM RB19, the purified enzyme was added to a final concentration of 50 nM to initiate the decolorization reaction. Assays were done in triplicates. Enzyme activity was determined by measuring the initial rate of absorbance decrease at 595 nm using ε595nm = 10 mM−1cm−1.
To determine steady-state kinetic parameters, the reactions were performed in the same way as described above except that concentrations of RB19 were varied between 5 to 100 μM in the presence of 1 mM H2O2, and concentrations of H2O2 were varied between 10 μM to 1 mM in the presence of 100 μM RB19. Rates were determined by the slopes of the initial reactions. The data obtained were fitted either to Michaelis-Menten Equation 1 or Hill Equation 2 using SigmaPlot 11. All measurements were done in triplicates.
| (Eq. 1) |
| (Eq. 2) |
Transient-state kinetics of compound I formation
All concentrations listed here were concentrations before mixing. Stopped-flow kinetics was performed in a single mixing mode at 22 °C in KPi (pH 7.8 and 3.0) using 5 μM enzyme and equal volume of H2O2 at various concentrations. All reactions were first investigated using a multiple-wavelength detector (PDA). The PDA data were analyzed using singular value decomposition with the Pro-KIV Global Analysis program provided by Applied PhotoPhysics to obtain the number of reaction intermediates and their corresponding spectra. Reactions were then monitored at a defined wavelength (the Soret band of enzyme resting state) using a monochromator to follow the formation of compound I. The monochromator data were fitted to a single exponential equation to obtain pseudo-first order rate constants (kobs). Second-order rate constants of compound I formation were calculated from plots of kobs vs. H2O2 concentrations. All experiments were performed in triplicate.
Rates of compound I decay (kd) were extracted from PDA data of reactions between 5 μM enzyme and 5 μM H2O2. Spectra extracted at a defined wavelength corresponding to the Soret band of C2LP were fitted to a single exponential equation to obtain the decay rates.
RESULTS AND DISCUSSION
Overall structure of TcDyP and heme access channels
The TcDyP crystal structure was solved by molecular replacement using the DyP model from Streptomyces coelicolor (PDB entry 4GT2) as a search probe and refined to 1.75 Å limiting resolution. Data collection and refinement statistics are listed in Table S2. The structure shows that TcDyP exists as a homodimer, which is consistent with the molecular mass of ~80 KDa estimated from SEC. The structure of a monomer unit is depicted in Figure 1A, for which the dimensions are roughly 49 Å × 46 Å × 39 Å. TcDyP adopts a ferredoxin-like fold consisting of antiparallel β-sheets and peripheral α-helices, which is characteristic of the DyP-type family but distinct from the classical heme peroxidases primarily made of helical proteins.1,14,30 Similar to other heme peroxidases,31 TcDyP has a histidine coordinating to the heme iron as the fifth ligand at a distance of 2.1 Å. However, unlike classical heme peroxidases such as horseradish peroxidase (HRP),30–32 which use the distal histidine to catalyze oxidation, DyPs employ an aspartate in place of the distal histidine to perform the same function.23 Such change is thought to contribute to shifting the optimal pH of enzyme activity from neutral conditions for HRP to acidic conditions for DyPs.23 The distal aspartate is also a part of GXXDG motif, which is recognized as the fingerprint of DyPs involved in heme binding.33
Figure 1.
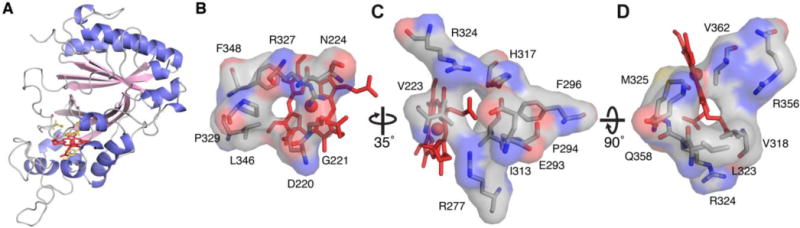
Structures of TcDyP (PDB: 5JXU). (A) α-Helices, β-strands, and loops are colored blue, pink, and grey, respectively. The heme porphyrin and catalytic residues are represented as red and yellow sticks, respectively. The heme iron is shown as a red ball; (B) Access channel 1 leading to the heme distal cavity; (C) Access channel 2 pointing to the propionate group on pyrrole ring C; (D) Access channel 3 pointing to the methyl group on pyrrole ring C.
As shown in Figures 1B–D, three heme access channels are present in TcDyP. Channel 1 has a diameter of ~3.3 Å and leads to the heme distal cavity where H2O2 binds to the heme iron. Thus, channel 1 is proposed as the channel for H2O2 to access the heme. Channel 2 has the largest diameter of ~5.0 Å among all three channels, which points to the propionate group on pyrrole ring C. The heme propionate has been demonstrated to provide a direct electron transfer path from the porphyrin radical to a binding substrate.32 Channel 3 leads to the methyl group on pyrrole ring C, which is much smaller than the channel 2 with a diameter of ~3.5 Å. Both channels 2 and 3 are thought to serve as the tunnels for small-size substrates to access the heme prior to oxidation.
Structural evidence for surface-exposed oxidation sites
Figure 2 shows a superimposition of TcDyP and AauDyP, which reveals that their overall structures are aligned well except for a few flexible loops. Specifically, the heme center and surrounding areas are almost identical. Previousmodeling work with fungal AauDyP has predicted that even the small-sized guaiacol molecule narrowly fits into the distal cavity where the catalytic Asp and Arg residues lie.19 In addition, the widest heme access channel 2 in TcDyP is partially blocked by E293 (Figure 1C). Thus, it cannot accommodate large-size anthraquinone-based dyes such as RB19 as the substrate in the active site. Yet TcDyP has shown high activity toward RB19. We therefore hypothesized that TcDyP catalyzes decolorization reactions via surface-exposed oxidation sites, which requires a long-range electron transfer (LRET) from the porphyrin to redox-active amino acids located at the protein surface to form protein-based radicals.
Figure 2.
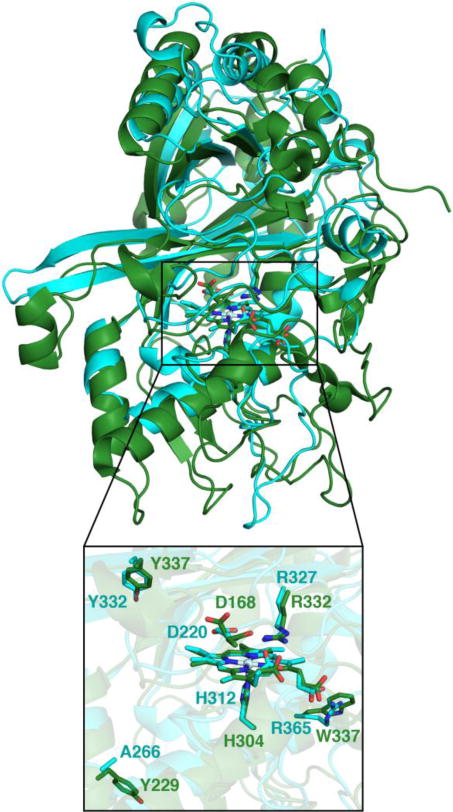
Structural superimposition of TcDyP (cyan) and AauDyP (green, PDB: 4AU9). Catalytic residues of both enzymes are represented as sticks and labeled. Surface-exposed oxidation sites of AauDyP and the corresponding residues in TcDyP are also labeled.
Tyrosines and tryptophans are common redox-active amino acids, which can form radicals under oxidative conditions and have been shown to play important roles in catalysis.34,35 Understanding the exact location and nature of protein radicals and their effect on the structure and function of proteins has been the subject of intense study.36 Such examples include elucidation of roles of tyrosine and tryptophan radicals in myoglobins, cytochrome c peroxidase (CcP), LiP, VP, and more recently fungal DyP.15–21,37,38
DyPs are rich in tryptophans and tyrosines,39 for which their percentage can reach up to 5% of the total residues. On the basis of sequence study of the four classes of DyPs, we have concluded that, while tyrosines predominates in D-class as redox-active amino acids, tryptophans are in the majority for A-class DyPs. For example, while the D-class DyP from Coprinopsis cinerea (Uniprot A8NBN3) contains 23 tyrosines and 8 tryptophans out of 648 residues for the matured protein, A-class TcDyP has 7 tryptophans and 3 tyrosines in a total of 376 amino acid residues. Recent study on D-class AauDyP has shown that, out of 7 tyrosines and 4 trytophans in 448 amino acids, surface-exposed residues including Y229, Y337, and W377 are identified as potential radical sites for dye decolorization.18–20 These results suggest that DyPs may employ multiple protein radicals on the protein surface as a general strategy for oxidizing large-size substrates. Additionally, identification of these radical sites provides a starting point for protein engineering as demonstrated by the creation of a MnCcP variant (W151F/W191F) that has six-fold improved Mn2+ oxidative activity relative to wt-MnCcP.40
Formation of protein radicals in wt-TcDyP as observed by EPR
The spectrum in the top panel of Figure 3A indicates that the wt-TcDyP at pH 7.8 predominantly existed as a rhombically distorted axial resonance at g⊥≈ 6 (gy = 6.20, gx = 5.50, and g‖ = 2.00), corresponding to a high-spin Fe3+. The absolute difference in g values (Δg = gy – gx) at g = 6 was used to determine the percentage of rhombicity, R (= Δg/16 × 100%), which was calculated to be 4.4% at pH 7.8. The resonance between g = 3 and g = 2 suggests the presence of a small proportion of low-spin Fe3+. The signal pattern changed significantly when the EPR was recorded at pH 3.0 (center panel in Figure 3A). A symmetrical signal was observed for high-spin Fe3+ at g⊥= 6.00. The less reactive low-spin Fe3+ species disappeared, which is consistent with the fact that DyPs are most active under acidic conditions.
Figure 3.
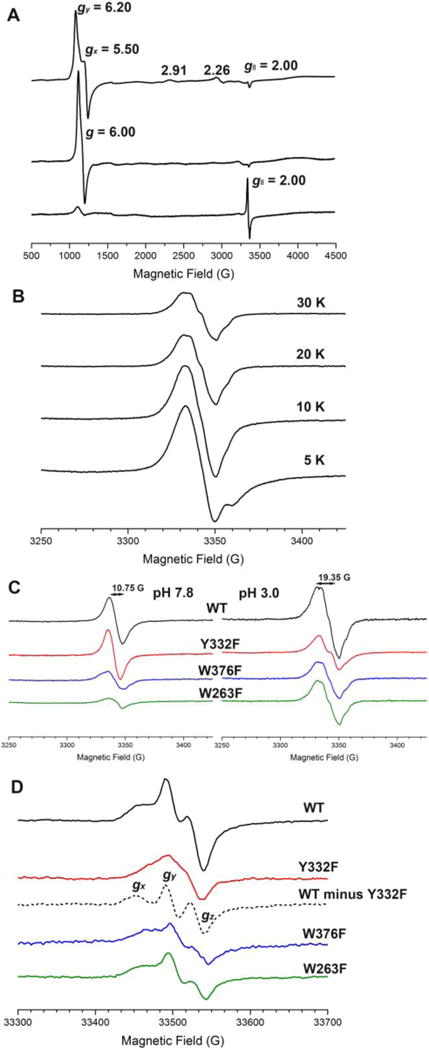
EPR spectra. (A) X-band spectra of 1.0 mM wt-TcDyP at pH 7.8 (top), 0.1 mM wt-TcDyP at pH 3.0 (center), and 0.1 mM wt-TcDyP plus 1.0 mM H2O2 at pH 3.0 (bottom) recorded at 5 K; (B) Temperature-dependent X-band spectra of 0.1 mM wt-TcDyP reacted with 1.0 mM H2O2 at pH 3.0; (C) X-band spectra of 1.0 mM TcDyPs at pH 7.8 (left) and 0.1 mM TcDyPs at pH 3.0 (right) reacted with 10 equiv. H2O2 recorded at 20 K; (D) W-band spectra of 0.2 mM TcDyPs reacted with 10 equiv. H2O2 at pH 3.0 and 30 K.
To explore whether a protein-based radical was generated, the EPR spectrum was recorded for 0.1 mM TcDyP in the presence of 1.0 mM H2O2 at pH 3.0. As shown in the bottom panel of Figure 3A, a radical signal with unresolved hyperfine couplings was observed at g‖ = 2.00, which had an overall width and peak-to-trough width of 90 G and 19 G, respectively. The presence of a small ferric signal was attributed to either nonreactive enzyme or most probably to native enzyme that cycled back. It has been reported that protein-based radicals usually have a signal at g = 2 with a width of ~100 G.41–44 On the other hand, oxoferryl-porphyrin radicals were shown to contain an extremely broad signal starting from g⊥ = 3.45 or 3.27 to g⊥ = 2,45,46 which was not observed in TcDyP. Furthermore, the shape and line width of TcDyP signals obtained at 20 and 30 K (Figure 3B) are similar to the protein radicals of Synechocystis PCC6803 peroxidase-catalase recorded at 60 K and of turnip peroxidase isozymes recorded at 30 K.43,44 Thus, the observed radical signal in the reaction between wt-TcDyP and H2O2 (Figure 3A) is assigned as a protein-based radical, which was further confirmed with the EPR study of mutant TcDyPs at X- (Figure 3C) and W-bands (Figure 3D) described later in this paper.
Since it has been demonstrated in our recent study that the wt-TcDyP compound I is unstable and quickly decays into C2LP in the absence of reducing substrates,12 we proposed that the C2LP corresponds to the observed protein-based radicals. Notably, a temperature-dependence study of wt-TcDyP at pH 3.0 (Figure 3B) illustrates that the shapes of spectral lines varied from 5 K to 20 K, indicating possible interactions between the protein-based radicals and ferry-ion center. This was also confirmed by microwave power saturation experiments, which showed power-dependent line shape changes at 5 K (data not shown). These results suggest that the formed protein-radicals are spatially close to heme, which corresponds to our findings described later in this paper.
Crosslinking of wt-TcDyP
Since many hemoproteins have been reported to undergo protein crosslinking in the presence of H2O2 due to formation of surface-exposed protein radicals,38,47,48 we decided to determine whether the crosslinking also occurred with wt-TcDyP. Thus, reactions of wt-TcDyP with H2O2 were analyzed with SDS-PAGE and SEC.
Figure 4A shows the SDS-PAGE of wt-TcDyP incubated with one equivalent H2O2 under various pH conditions. Except for the 43 KDa band that corresponded to a TcDyP monomer, additional bands at 100 and 250 KDa were also observed, which were assigned as a covalent dimer and oligomer, respectively. While the formation of covalent dimer and oligomer was pH dependent, their trends were different. The covalent dimer and oligomer contents reached their maximum at pH ~8 and ~3, respectively. This indicated that, under an acidic condition and in the absence of a reducing substrate, the rate of formation of covalent oligomers from covalent dimers was much faster than that of covalent dimer formation from monomers. Additionally, when the experiment was carried out in the presence of RB19, the bands corresponding to covalent dimer and oligomer disappeared (lane 4 in Figure 4B), which suggested that substrate binding may prevent the protein crosslinking.
Figure 4.
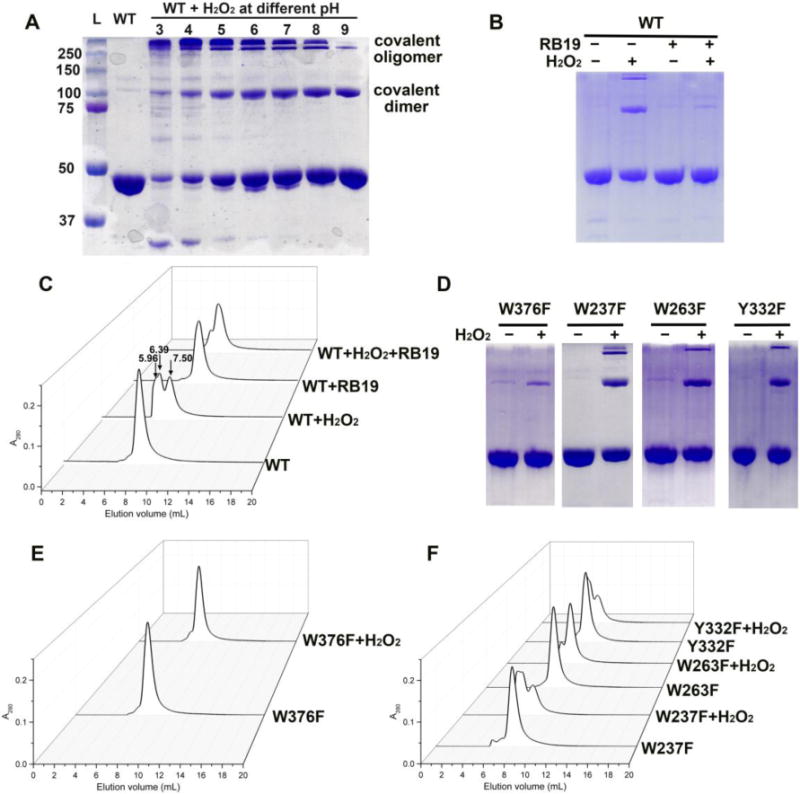
Analysis of reactions by SDS-PAGE and SEC. (A) SDS-PAGE of reactions between wt-TcDyP and H2O2 under various pH conditions; (B) SDS-PAGE of reactions between wt-TcDyP and H2O2 in the presence and absence of RB19 at pH 7.8; (C) SEC of reactions shown in (B); (D) SDS-PAGE of reactions between mutants and H2O2; (E) SEC of reactions between W376F and H2O2; (F) SEC of reactions between mutants and H2O2.
To test whether the crosslinking would affect enzyme activity, the reactions were also analyzed by SEC in the presence and absence of RB19. The native enzyme displayed a peak at 7.50 mL in Figure 4C, corresponding to a TcDyP dimer. When H2O2 was present, two additional peaks, accounting for 54% of the total peak areas, appeared at 6.39 and 5.96 mL, corresponding to a TcDyP tetramer and oligomer, respectively. Peaks were isolatedand subject to enzyme activity assay using RB19. It was found that the peak consisting of tetramer and oligomer lost ~30% activity compared with the dimer peak. The results agree with the gel image shown in Figure 4A, in which ~30% protein exist as covalent dimer and oligomer at pH 8.0. When the H2O2 and RB19 were simultaneously present in the reaction, the tetramer and oligomer peaks only accounted for 6% of total peak areas (Figure 4C), consistent with the results from SDS-PAGE (lane 4 in Figure 4B).
Involvement of W376 in protein crosslinking
Since tryptophan and tyrosine are the most common candidates of radical sites, each of them was mutated to a less oxidizable phenylalanine in TcDyP. It was discovered that, while the other mutants (e.g. W237F, W263F, and Y332F) displayed similar phenomena as the wt when incubated with H2O2 (Figures 4D–F), only the W376F mutant significantly lost its ability to form the protein crosslinks. The bands at 100 and 250 KDa in SDS-PAGE almost disappeared when W376F was oxidized by H2O2 at pH 7.8 (Figure 4D). Accordingly, when the reaction was analyzed by SEC, no peak corresponding to the tetramer and oligomer was observed (Figure 4E).
Since W376 is located on the protein surface with a large solvent accessible area (30.2 Å), it was initially predicted to be a radical site for substrate oxidation. Unexpectedly, activity assay showed that W376F displayed a higher reactivity than the wt with RB19 (116%, Figure 5). Thus, our data are consistent with the idea that W376 was likely to serve as a radical sink and represent an off-pathway destination for electron transfer from the heme center to the protein surface when a dye substrate is absent.
Figure 5.
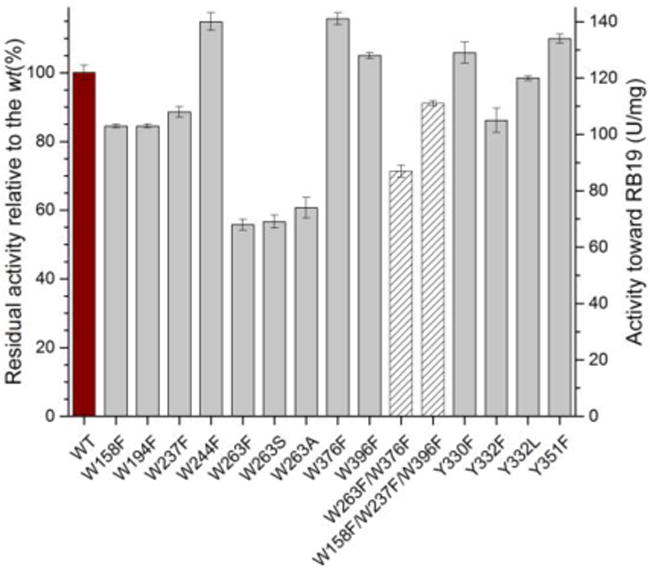
Residual activities of mutant TcDyPs toward RB19. Red, grey, and patterned bars represent wt, single and multiple mutants, respectively.
Surface-exposed protein-based radicals and substrate oxidation sites involving W263 and Y332
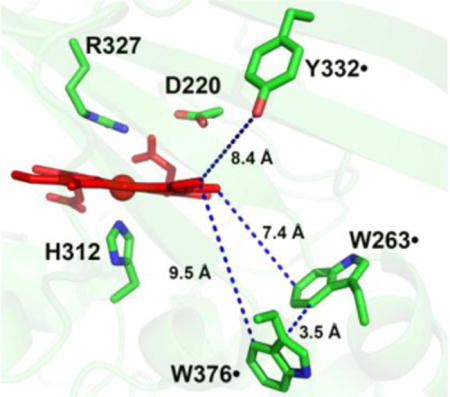
To identify surface-exposed substrate oxidation sites involving tryptophanyl and tyrosyl radicals, the activities of all phenylalanine mutants were determined with RB19 in the presence of 1 mM H2O2. The results are depicted in Figure 5. While all single mutants except W263F retained the wt activity, the W263F mutant lost ~50% its activity. Examination of TcDyP crystal structure reveals that W263 is located on the proximal side of the heme at the dimer interface, and is the closest residue to the heme among all tryptophans and tyrosines at a distance of 7.4 Å (Figure 6). Sequence alignment depicted in Figure S1 shows that W263 is absolutely conserved in A-class DyPs, which suggests that it is functionally important and may serve as one of protein radicals for surface-exposed substrate oxidation.
Figure 6.
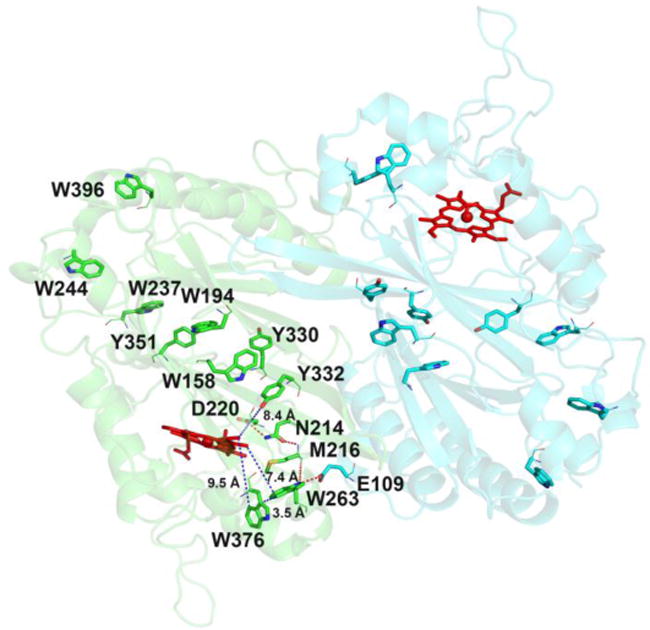
Location of Trp and Tyr residues in wt-TcDyP and the hydrogen bonding network involving W263. Main and side chains of residues are represented as lines and sticks, respectively. Red and blue dots represent hydrogen bonding and distance, respectively. The two monomeric units are colored in green and cyan.
To further examine the importance of W263, activities of W263F mutant were tested against a series of substrates commonly used for DyPs, which included ABTS, catechol, guaiacol, hydroquinone, and reactive blue 4 and 5. The W263F mutant retained 40–60% of the wt activity (data not shown), consistent with the result obtained with RB19. Additionally, residues W158, W237, and W396 have large solvent exposed areas as summarized in Table S3. We therefore hypothesized that a triple mutant of these positions would magnify the activity loss if they were involved in substrate oxidation via surface-exposed protein radicals. Activity assays on the W158F/W237F/W396F showed that the triple mutant retained more than 90% wt activity (Figure 5), implying that these positions are not functionally important. Thus, of all the tryptophans in TcDyP, only W263 was identified as the one that participates directly in surface-exposed substrate oxidation. It has to be noted that TcDyP must have multiple substrate oxidation sites on the protein surface because (1) W263 mutation only resulted in 50% loss of the wt activity; (2) W263 mutation resulted in fewer radicals than the wt (Figures 3C and 3D), which will be described in the next section; and (3) steady-state kinetics of TcDyPs with RB19 displayed a sigmoidal curve, which will be discussed in the section after next.
Sequence alignment in Figure S1 has shown that Y332 is highly conserved across all four classes of DyPs. It is located at the TcDyP dimer interface on the distal side of the heme with a distance of 8.4 Å (Figure 6). Additionally, this tyrosine has been proposed as a potential surface-exposed protein radical site in B- and D-class DyPs.18,19,49 As depicted in Figure 2, structural alignment of TcDyP (A-class) and AauDyP (D-class) based on the heme cofactor reveals that the conserved tyrosines in two proteins are overlapped. The same result was obtained when structures of TcDyP and VcDyP from Vibrio cholerae (B-class) were aligned (data not shown). Thus, Y332 was proposed to be one of the surface-exposed protein radical sites in TcDyP, which was supported by the EPR study described in the next section. To test whether Y332 could also serve as a surface-exposed oxidation site, its mutations were performed. Enzyme assays with RB19 showed that the mutants retained more than 90% wt activity (Figure 5). While it is in contrast with VcDyP, in which the tyrosine to leucine mutant abolishes enzyme activity,49 it agrees with AauDyP, in which the mutation also minimally affects enzyme activity.18 The observed weak effects of Y332 are mainly attributed to its small solvent accessible surface area (2.76 Å2) relative to the tyrosine at the same position in VcDyP (12.47 Å2). In addition, the solvent accessible surface areas for Y330 and Y351 are extremely small at 0 and 0.67 Å2, respectively. Their mutations resulted in increased activities toward RB19 as shown in Figure 5, which suggests that Y330 and Y351 do not participate in the formation of protein radicals for surface-exposed substrate oxidation.
EPR study of mutant TcDyPs
Since W263, W376, and Y332 were proposed to be the sites of protein-based radicals, their EPR in the presence of H2O2 were investigated and compared with the wt-TcDyP. As described above, TcDyP compound I is unstable and quickly decays into C2LP, which is hypothesized as protein-based radicals. If the hypothesis is true, the mutants are expected to show radical signals with decreased intensity as well as changes in their hyperfine coupling. Thus, the W263F, W376F, and Y332F mutants were incubated with H2O2 at pH 7.8 and 3.0 for 8 s prior to being flash-frozen in liquid nitrogen. The X-band EPR spectra are shown in Figure 3C. Similar to the wt-TcDyP (Figure 3A), the mutants generated fewer radicals at pH 7.8 (left panel, [TcDyP] = 1.0 mM) than at pH 3.0 (right panel, [TcDyP] = 0.1 mM). Since the EPR samples had the same volume, were prepared in the same manner, and were measured by the same spectrometer using the same settings, the radical intensity should be directly related to the radical yield. To our expectations, all mutants except Y332F at pH 7.8 had lower radical yields than the wt. Compared with the wt signal, small, yet significant changes of hyperfine coupling were also observed for all mutants except Y332F at pH 7.8. The change was much clearer at pH 3.0, which represents an optimal pH condition for TcDyP activity. At pH 7.8, Y332F had the same radical yield and shape as the wt, suggesting that Y332 does not participate in radical formation. This agrees well with the observation that the Y332 mutants and wt had similar compound I decay rates (kd) at pH 7.8 (Table 1), which will be discussed later.
Table 1.
Summary of enzyme steady-state and transient-state kinetics
| RB19 (H2O2 at 1 mM)
|
H2O2 (RB19 at 100 μM)
|
Compound I
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| TcDyPs | kcat (s−1) |
KM (μM) |
kcat/KM (M−1 s−1) |
ha | kcat (s−1) |
KM (μM) |
kcat/KM (M−1 s−1) |
kobsb (M−1 s−1) |
kdc (s−1) |
| wt | 96 ± 2 | 42 ± 1 | 2.3×106 | 2.9 | 96 ± 2 | 188 ± 13 | 5.1×105 | 4.2×106 | 2.10 |
| W263F | 49 ± 1 | 33 ± 1 | 1.5×106 | 3.4 | 60 ± 1 | 219 ± 17 | 2.7×105 | 3.5×106 | 0.71 |
| W263S | 51 ± 1 | 32 ± 1 | 1.6×106 | 2.9 | 47 ± 1 | 178 ± 13 | 2.4×105 | 3.1×106 | 0.62 |
| W263A | 52 ± 1 | 28 ± 1 | 1.9×106 | 3.5 | 63 ± 2 | 303 ± 25 | 2.0 ×105 | 2.6×106 | 0.82 |
| W376F | 119 ± 5 | 49 ± 2 | 2.4×106 | 2.5 | 133 ± 3 | 295 ± 18 | 4.5×105 | 2.9×106 | 0.19 |
| W376F/W263F | 69 ± 1 | 26 ± 1 | 2.7×106 | 2.7 | 90 ± 3 | 283 ± 23 | 3.2×105 | 2.9×106 | 0.17 |
| Y332F | 83 ± 1 | 34 ± 1 | 2.5×106 | 3.4 | 108 ± 4 | 393 ± 32 | 2.7×105 | 3.4×106 | 1.90 |
| Y332L | 97 ± 1 | 37 ± 1 | 2.5×106 | 3.4 | 118 ± 2 | 279 ± 13 | 4.2×105 | 3.2×106 | 1.80 |
Hill constant, h;
Second -order rate constants (kobs) of compound I formation in the presence of H2O2;
Rate constants (kd) of compound I decaying to compound II-like product when the reaction was performed with 5 μM enzyme and 5 μM H2O2 at pH 7.8.
To further characterize the radicals, W-band EPR spectra of 0.2 mM wt and mutant TcDyPs in the presence of 2.0 mM H2O2 were recorded at pH 3.0 and 30 K. The enzymes were incubated with H2O2 for 60 s before being flash-frozen in liquid nitrogen. As shown in Figure 3D, the wt signals consist of at least two types of protein radicals: tyrosyl and tryptophanyl radicals. The Y332F mutant signals are significantly different from the wt signals. Subtraction of Y332F from the wt gave spectral features of gx, gy, and gz values at 2.0078, 2.0049, and 2.0025 (the dotted line in Figure 3D), respectively, which is consistent with the previous observed g values for tyrosyl radicals.50 The broad peaks in Y332F, showing g values ranging from 2.002 to 2.005, were likely resulted from tryptophanyl and other unknown radicals.
Moreover, the broader EPR line shape observed at pH 3.0 than at pH 7.8 implies that additional radical species exist at pH3.0, most likely contributed from Y332 and possibly other residues. Overall, the EPR study of mutants supports the hypothesis that surface-exposed W263, W376, and Y332 generate protein radicals in the presence of H2O2 under acidic conditions.
Steady-state kinetics of W263, W376, and Y332 mutants
To further understand the importance of W263, W376, and Y332, we conducted steady-state kinetic evaluations of a panel of site-directed mutants in the presence of RB19 and H2O2. The results are summarized in Table 1. We have recently reported that small- and large-size substrates display different kinetics with wt-TcDyP,12 which could be attributed to distinct oxidation sites for small- and large-size substrates. The small-size substrates like guaiacol could be oxidized at the heme center via heme access channels 2 and 3 shown in Figure 1. The large-size dye substrates such as RB19 and lignin model compounds are likely to be oxidized on the protein surface via LRET processes as described above. As depicted in Figure 7, unlike the H2O2 that shows the classical Michaelis-Menten kinetics, oxidation of RB19 by the wt and mutant TcDyPs displays a sigmoidal kinetics, suggesting that cooperativity exists between the enzyme and dye substrate. The estimated Hill constant (h) is around three, implying that the TcDyP has multiple substrate binding sites for RB19. Such phenomena were also observed for other hemoproteins. For example, hemoglobin binds four oxygen molecules at four separate sites, displaying a Hill constant of three.51 Thus, single mutations are unlikely to abolish TcDyP activity.
Figure 7.
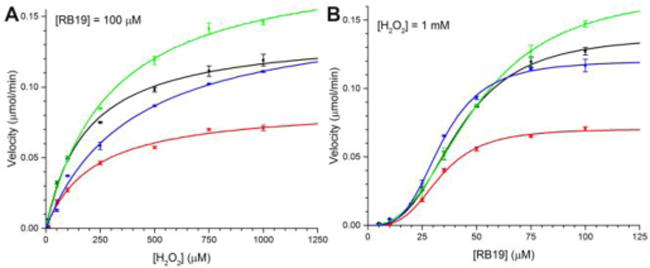
Steady-state kinetics of wt-TcDyP (black), W263F (red), W376F (green), and Y332F (blue). (A) With H2O2; (B) With RB19.
As shown in Figure 6, not only is W263 the closest residue to the heme among all tryptophans and tyrosines, but also it forms a hydrogen bond network to D220 via M216 and N214. The D220 is a catalytic residue located in the heme distal cavity, which is characteristic of DyP enzymes and plays critical roles in compound I formation when the heme Fe3+ is oxidized by H2O2.12 Additionally, W263 forms a hydrogen bond with E109 from another monomer unit, suggesting that W263 could also be involved in dimer formation of the native enzyme. To characterize the role of W263, we mutated this residue to phenylalanine, alanine, and serine. Phenylalanine was selected because it is a conservative substitution of tryptophan, but lacks of the ability to produce a stable radical and form hydrogen bonds. Alanine was chosen because it is a non-polar (hydrophobic) amino acid without aromacity, which could help reveal the importance of indole ring in W263. Serine was selected because it is a preferred substitution for tryptophans in many peroxidases,15,17,18 which may help to address polar interactions involving the indole ring.
While all W263 mutants had lower catalytic efficiency than the wt toward both RB19 and H2O2, the mutations we examined showed a much more pronounced effect on H2O2 consumption than on RB19 oxidation as summarized in Table 1. All W263 mutants displayed similar and increased binding affinities toward RB19 relative to the wt (~30% increase), which suggests that π−π interaction is unlikely important for the binding of dye substrates, as side chains of W263S and W263A lack of aromatic rings. Moreover, their turnover numbers toward RB19 dropped half to ~50 s−1, which is consistent with our expectation as the mutants will not be able to generate a stable radical. While the wt and W263S had almost the same H2O2 affinities, W263F and W263A displayed much lower H2O2 affinities than the wt. In addition, the turnover number of W263S toward H2O2 decreased the most among three W263 mutants. This implies that the hydrogen bond network connecting W263 and D220 is likely to be more involved in H2O2 binding than electron transfer.
While mutation of W376 to F376 had no effect on the binding of RB19, it affected H2O2 binding significantly (~60% decrease in binding affinity). This is not surprising as it is only 9.5 and 3.5 Å away from the heme center and W263, respectively. The close proximity of W376 and W263 within Van der Waals distance (3–6 Å) suggests that an electron transfer could occur between them. The turnover numers of W376F with RB19 and H2O2 were determined to be 1.3- and 1.4-fold faster than those of the wt. Combined with the fact that W376 is responsible for crosslinking in the absence of dye substrates, we concluded that W376 is likely to be involved in off-pathway electron transfer processes. If so, mutation of W376 would be expected to suppress this undesired pathway and thereby increase catalytic efficiency of the enzyme. To test this possibility, a double mutant, W376F/W263F, was constructed and purified. Indeed, the double mutant displayed increased catalytic efficiency toward RB19 and H2O2 by 80% and 20% relative to the W263F, respectively. The 80% improvement of the double mutant with RB19 over the single mutant is significant and demonstrates the importance of W376 mutation in biotechnological development of TcDyP applications.
The Y332 is located at the distal side of the heme with a distance of 8.4 Å. Because it has a small solvent accessible surface area and is close to the distal cavity where Fe3+ in heme is oxidized by H2O2, its mutations are predicted to have minimal effects on RB19 oxidation, but significant impacts on reactions with H2O2. Indeed, the mutants displayed slightly increased binding affinities with RB19, resulting in improved catalytic efficiency relative to the wt. However, the mutations drastically decreased binding affinities with H2O2 while the turnover numbers increased. In fact, the Y332F enzyme had the lowest H2O2 binding affinity among all mutants shown in Table 1.
Transient-state kinetics of W263, W376, and Y332 mutants in compound I formation and decay
Since C2LP was assigned as a protein-based radical involving W263, W376, Y332, and others, the corresponding mutants were studied for their transient-state kinetics of compound I formation and decay using a stopped-flow spectrometer. Therefore, the wt and mutant enzymes were mixed with H2O2 in 1:1 ratio at pH 7.8 and pH 3.0. Their spectral changes are shown in Figures 8A–D. Analysis of these spectra by Pro-KIV gave a two-step reaction A→B→C as the best fit. Based on our recent study,12 A, B, and C were assigned as the enzyme resting state (blue lines), compound I (red lines), and C2LP (green lines), respectively. Their spectral character is summarized in Table 2. It has to be pointed out that, while there are minimal effects on the spectral features overall, the C2LP Soret band undergoes a significant blue shift under acidic conditions resulting in an overlap with the Soret band of the enzyme resting state. This spectral overlap therefore limits the accuracy in determining the kinetic parameters of compound I formation and decay at pH 3.0.
Figure 8.
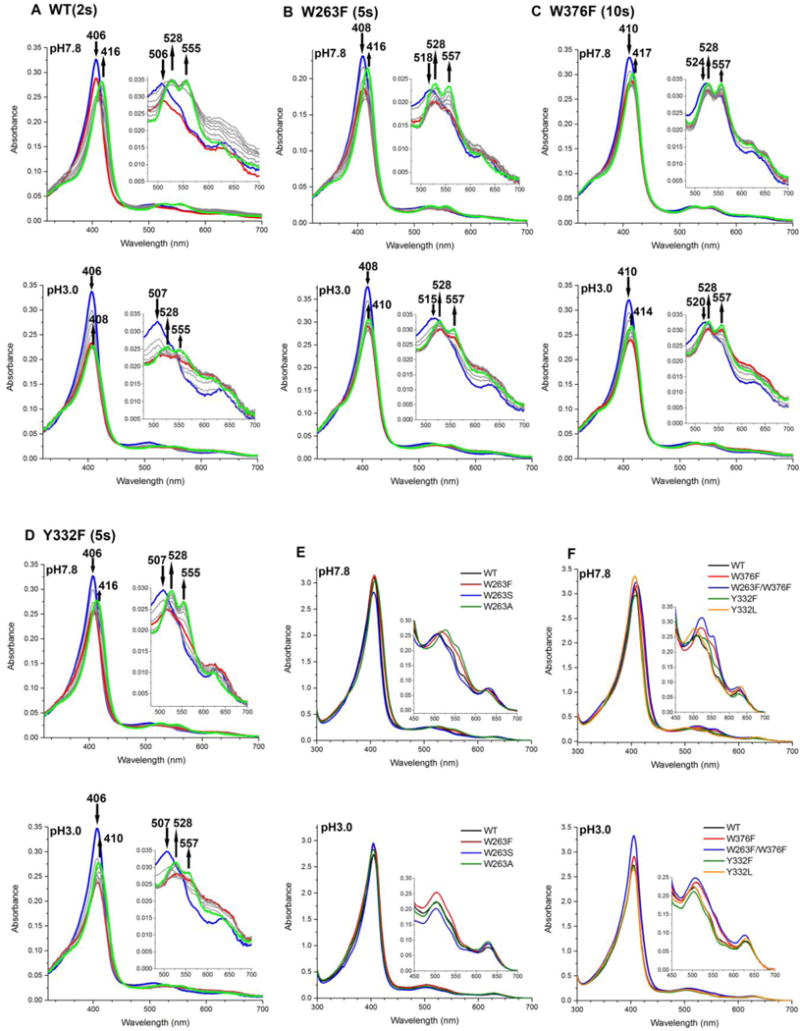
Stopped-flow spectra of reactions between TcDyPs and H2O2 at 1:1 ratio (A–D) and spectral overlay of enzyme resting states (E–F). The blue, red, and green lines in A–D represent initial, intermediate, and final states of the enzymes, respectively. Reactions were performed with 5 μM enzymes at pH 7.8 (top panel) and 3.0 (bottom panel). Arrows indicate changes of absorbance over time.
Table 2.
Absorption maxima of resting state and intermediates of TcDyPsa
| TcDyPs | Resting State | Compound I | C2LP |
|---|---|---|---|
| wt | 406, 506, 545, 569 (sh), 626 | 408, 524, 549, 620, 650 (sh) | 416, 528, 555, 619 |
| 406, 507, 545, 569 (sh), 627 | 406, 511, 544, 570, 617, 649 (sh) | 408, 526, 555, 615 (sh), 642 | |
| W263F | 408, 518, 553, 627 | 412, 530, 555, 619, 648 (sh) | 416, 528, 557, 621 (sh) |
| 408, 519, 558, 628 | 410, 530, 558, 616, 650 (sh) | 410, 528, 557, 622 (sh) | |
| W263S | 407, 505, 543, 632 | 406, 528, 614, 644 | 416, 528, 557, 622 |
| 406, 506, 537, 635 | 405, 536, 610, 646 | 408, 526, 555, 621 | |
| W263A | 408, 518, 554, 628 | 410, 530, 555, 617, 646 | 418, 528, 557, 621 |
| 408, 516, 554, 625 | 408, 530, 556, 614, 648 (sh) | 410, 528, 556, 620 | |
| W376F | 410, 524, 553, 623 (sh) | 414, 528, 555, 620, 650 (sh) | 417, 528, 557, 623, 649 (sh) |
| 410, 522, 557, 625 | 412, 529, 557, 618, 649 (sh) | 414, 528, 557, 619, 656 (sh) | |
| W376F/W263F | 410, 524, 553, 621 (sh) | 416, 528, 557, 620, 652 (sh) | 418, 528, 557, 619 |
| 408, 517, 552, 628 | 412, 528, 555, 615, 649 (sh) | 412, 528, 555, 619 | |
| Y332F | 406, 507, 540, 628 | 408, 523, 620, 646 | 416, 528, 555, 622 |
| 406, 507, 542, 573 (sh), 628 | 407, 543, 615 (sh), 649 | 410, 526, 557, 625 | |
| Y332L | 406, 505, 538, 629 | 406, 525, 616, 644 | 412, 526, 555, 618 |
| 406, 506, 535, 583, 634 | 406, 533, 613, 644 (sh) | 408, 522, 555, 630 |
Absorption maxima at pH 7.8 and 3.0 are colored as black and blue, respectively.
To determine the effect of site-directed mutation on compound I formation, the second-order rate constants (kobs) of compound I formation were measured with wt and various mutants at pH 7.8. The results are summarized in Table 1. While all mutations resulted in the decrease of kobs, the W263A, W376F, and W376F/W263F mutants were diminished the most. This is not surprising because (1) W263 is spatially close to the heme and its mutation to A263 represented a significant change in terms of amino acid structure and property; (2) spectra of the resting states of W376 mutants indicated significant perturbation of the heme microenvironment relative to the other mutants (Figures 8E–F). In addition, the second-order rate constant of compound I formation and overall catalytic efficiency toward RB19 for wt-TcDyP were determined to be 4.2 ×106 and 2.3 ×106 M−1 s−1, respectively, which implied that the formation of compound I was not rate-limiting in the catalytic cycle. This is further supported by the steady-state and transient-state kinetics of W376F. While the rate of compound I formation was slower for this mutant than that of the wt, its turnover number toward RB19 was 1.2-fold faster.
The rates of compound I decaying into C2LP (kd) were also determined for reactions between 5 μM enzyme and 5 μM H2O2. As described above, C2LP was assigned as protein-based radicals. Since W263, W376, and Y332 were demonstrated to be the radical sites in the presence of H2O2, mutation of these residues was expected to result in the increase of compound I stability and reduction of decay rates. Indeed, the W263, W376, and Y332 mutants had slower decay rates than the wt. As summarized in Table 1, the decay rates of W263 mutants were 40–50% of the wt, which agrees well with their steady-state kinetics (~50% decrease of kcat from the wt to mutants).
While multiple residues are involved in the formation of protein-based radicals, W376 should act as the destination of off-pathway electron transfer because it promotes the formation of crosslinking in the absence of dye substrates (Figure 4A). Thus, W376 mutation is predicted to have the largest impact on decay rates among all single mutants. Accordingly, the decay rates of W376 mutants were found to be only 8% of the wt. We found that the kd of Y332 mutants was smaller than expected (from 2.10 s−1 for the wt to 1.80–1.90 s−1 for the Y332 mutants), which is attributed to the neutral pH of the reaction condition. As discussed above, Y332 only formed radicals when it was oxidized by H2O2 under acidic conditions (Figure 3C). Thus, mutation of Y332 would minimally affect the compound I stability and its decay rate at pH 7.8.
CONCLUSIONS
The TcDyP structure solved at 1.75 Å contains two ferredoxin-like folds, which is characteristic of proteins from the DyP family. Catalytic residues D220 and R327 are located at the heme distal side and H312 acts as a proximal ligand to the heme. Although bacterial DyPs display activities toward large-size anthraquinone-based dyes and phenolic lignin model compounds, their modes of oxidation remain unknown. Based on the TcDyP structure, we predicted that TcDyP uses surface-exposed protein-based radicals for oxidation of large-size substrates. Thus, efforts were focused on aromatic redox-active amino acids that are rich in DyP enzymes. Among 7 tryptophans and 3 tyrosines that are present in the TcDyP, W263, W376, and Y332 are the closest residues to the heme and were identified as the sites of surface-exposed protein radicals, which correspond to a compound II-like product (C2LP) generated from the decay of compound I. In addition, the absolutely conserved W263 in A-class DyPs was also characterized as a surface-exposed substrate oxidation site, which was supported by ~50% loss of enzyme activity for the mutants and sigmoidal kinetics of TcDyPs toward RB19. W376 was characterized as a major radical sink for the off-pathway electron transfer in the absence of dye substrates, resulting in the formation of protein crosslinks demonstrated by SDS-PAGE, SEC, and significantly slowed decay rates of compound I. Mutation of W376 is beneficial and improves enzyme catalytic efficiency. Sequence alignment revealed that Y332 is highly conserved across all four classes of DyPs. While Y332 was found to function as a surface-exposed protein radical site in TcDyP under acidic conditions, it could not serve as a substrate oxidation site due to its extremely small solvent accessible area. Identification of surface-exposed protein radicals and substrate oxidation sites is important for modulating TcDyP functions and efficiency for improved activity on phenolic lignin.
Supplementary Material
Acknowledgments
This work is supported by the grants from NIH P30GM110761 pilot project and Johnson Cancer Research Center to P.L., NIH R21AI111203 and R21AI113552 to B.V.G., and NIH U19AI091693 and R03AI122860 and NHMFL UCGP grant 5080 to L.S. Crystal X-ray data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-BM beamline at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. W-31-109-Eng-38. EPR data were collected at NHMFL supported by the NSF DMR-1157490 and the State of Florida. The authors would like to thank Drs. Chao Chen and David Meekins for helping construct mutant plasmids and prepare Pymol figures, respectively.
Footnotes
Supporting Information
A list of primers for mutant plasmid construction, X-ray diffraction data collection and refinement statistics, sequence alignment, and values of solvent accessible areas for all tryptophans and tyrosines in TcDyP are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.Singh R, Eltis LD. Arch Biochem Biophys. 2015;574:56–65. doi: 10.1016/j.abb.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 2.Colpa DI, Fraaije MW, van Bloois E. J Ind l Microbiol Biot. 2014;41:1–7. doi: 10.1007/s10295-013-1371-6. [DOI] [PubMed] [Google Scholar]
- 3.Kim SJ, Shoda M. Appl Environ Microb. 1999;65:1029–1035. doi: 10.1128/aem.65.3.1029-1035.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdel-Hamid AM, Solbiati JO, Cann IKO. Adv Appl Microbiol. 2013;82:1–28. doi: 10.1016/B978-0-12-407679-2.00001-6. [DOI] [PubMed] [Google Scholar]
- 5.Pollegioni L, Tonin F, Rosini E. FEBS J. 2015;282:1190–1213. doi: 10.1111/febs.13224. [DOI] [PubMed] [Google Scholar]
- 6.Mukherjee A, Mandal T, Ganguly A, Chatterjee PK. Chembioeng Rev. 2016;3:86–96. [Google Scholar]
- 7.Rabemanolontsoa H, Saka S. Bioresource Technol. 2016;199:83–91. doi: 10.1016/j.biortech.2015.08.029. [DOI] [PubMed] [Google Scholar]
- 8.Bugg TDH, Rahmanpour R. Curr Opin Chem Biol. 2015;29:10–17. doi: 10.1016/j.cbpa.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 9.Ahmad M, Roberts JN, Hardiman EM, Singh R, Eltis LD, Bugg TDH. Biochemistry. 2011;50:5096–5107. doi: 10.1021/bi101892z. [DOI] [PubMed] [Google Scholar]
- 10.Brown ME, Barros T, Chang MCY. ACS Chem Biol. 2012;7:2074. doi: 10.1021/cb300383y. [DOI] [PubMed] [Google Scholar]
- 11.Min K, Gong G, Woo HM, Kim Y, Um Y. Sci Rep. 2015;5:8245. doi: 10.1038/srep08245. Article number. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen C, Shrestha R, Jia K, Gao PF, Geisbrecht BV, Bossmann SH, Shi JS, Li P. J Biol Chem. 2015;290:23447–23463. doi: 10.1074/jbc.M115.658807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koua D, Cerutti L, Falquet L, Sigrist CJA, Theiler G, Hulo N, Dunand C. Nucleic Acids Res. 2009;37:D261–D266. doi: 10.1093/nar/gkn680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshida T, Sugano Y. Arch Biochem Biophys. 2015;574:49–55. doi: 10.1016/j.abb.2015.01.022. [DOI] [PubMed] [Google Scholar]
- 15.Doyle WA, Blodig W, Veitch NC, Piontek K, Smith AT. Biochemistry. 1998;37:15097–15105. doi: 10.1021/bi981633h. [DOI] [PubMed] [Google Scholar]
- 16.Floudas D, Binder M, Riley R, Barry K, Blanchette RA, Henrissat B, Martinez AT, Otillar R, Spatafora JW, Yadav JS, Aerts A, Benoit I, Boyd A, Carlson A, Copeland A, Coutinho PM, de Vries RP, Ferreira P, Findley K, Foster B, Gaskell J, Glotzer D, Gorecki P, Heitman J, Hesse C, Hori C, Igarashi K, Jurgens JA, Kallen N, Kersten P, Kohler A, Kues U, Kumar TKA, Kuo A, LaButti K, Larrondo LF, Lindquist E, Ling A, Lombard V, Lucas S, Lundell T, Martin R, McLaughlin DJ, Morgenstern I, Morin E, Murat C, Nagy LG, Nolan M, Ohm RA, Patyshakuliyeva A, Rokas A, Ruiz-Duenas FJ, Sabat G, Salamov A, Samejima M, Schmutz J, Slot JC, John FS, Stenlid J, Sun H, Sun S, Syed K, Tsang A, Wiebenga A, Young D, Pisabarro A, Eastwood DC, Martin F, Cullen D, Grigoriev IV, Hibbett DS. Science. 2012;336:1715–1719. doi: 10.1126/science.1221748. [DOI] [PubMed] [Google Scholar]
- 17.Perez-Boada M, Ruiz-Duenas FJ, Pogni R, Basosi R, Choinowski T, Martinez MJ, Piontek K, Martinez AT. J Mol Biol. 2005;354:385–402. doi: 10.1016/j.jmb.2005.09.047. [DOI] [PubMed] [Google Scholar]
- 18.Linde D, Pogni R, Canellas M, Lucas F, Guallar V, Baratto MC, Sinicropi A, Saez-Jimenez V, Coscolin C, Romero A, Medrano FJ, Ruiz-Duenas FJ, Martinez AT. Biochem J. 2015;466:253–262. doi: 10.1042/BJ20141211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strittmatter E, Liers C, Ullrich R, Wachter S, Hofrichter M, Plattner DA, Piontek K. J Biol Chem. 2013;288:4095–4102. doi: 10.1074/jbc.M112.400176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strittmatter E, Serrer K, Liers C, Ullrich R, Hofrichter M, Piontek K, Schleicher E, Plattner DA. Arch Biochem Biophys. 2015;574:75–85. doi: 10.1016/j.abb.2014.12.016. [DOI] [PubMed] [Google Scholar]
- 21.Strittmatter E, Wachter S, Liers C, Ullrich R, Hofrichter M, Plattner DA, Piontek K. Arch Biochem Biophys. 2013;537:161. doi: 10.1016/j.abb.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 22.Sugano Y, Matsushima Y, Tsuchiya K, Aoki H, Hirai M, Shoda M. Biodegradation. 2009;20:433–440. doi: 10.1007/s10532-008-9234-y. [DOI] [PubMed] [Google Scholar]
- 23.Sugano Y, Muramatsu R, Ichiyanagi A, Sato T, Shoda M. J Biol Chem. 2007;282:36652–36658. doi: 10.1074/jbc.M706996200. [DOI] [PubMed] [Google Scholar]
- 24.van Bloois E, Pazmino DET, Winter RT, Fraaije MW. Appl Microbiol Biot. 2010;86:1419–1430. doi: 10.1007/s00253-009-2369-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 26.Berry EA, Trumpower BL. Anal Biochem. 1987;161:1. doi: 10.1016/0003-2697(87)90643-9. [DOI] [PubMed] [Google Scholar]
- 27.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Crystallogr D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emsley P, Lohkamp B, Scott WG, Cowtan K. Acta Crystallogr D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cruickshank PAS, Bolton DR, Robertson DA, Hunter RI, Wylde RJ, Smith GM. Rev Sci Instrum. 2009;80:103102. doi: 10.1063/1.3239402. Article number. [DOI] [PubMed] [Google Scholar]
- 30.Poulos TL. Arch Biochem Biophys. 2010;500:3–12. doi: 10.1016/j.abb.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Veitch NC. Phytochemistry. 2004;65:249–259. doi: 10.1016/j.phytochem.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 32.Poulos TL. Chem Rev. 2014;114:3919–3962. doi: 10.1021/cr400415k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sugano Y. Cell Mol Life Sci. 2009;66:1387–1403. doi: 10.1007/s00018-008-8651-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonini MG, Consolaro MEL, Hart PC, Mao M, de Abreu ALP, Master AM. IUBMB Life. 2014;66:167–181. doi: 10.1002/iub.1258. [DOI] [PubMed] [Google Scholar]
- 35.Gray HB, Winkler JR. Proc Natl Acad Sci USA. 2015;112:10920–10925. doi: 10.1073/pnas.1512704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warren JJ, Ener ME, Vlcek A, Winkler JR, Gray HB. Coordin Chem Rev. 2012;256:2478–2487. doi: 10.1016/j.ccr.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raven EL, Mauk AG. Adv Inorg Chem. 2001;51:1–49. [Google Scholar]
- 38.Pfister TD, Gengenbach AJ, Syn S, Lu Y. Biochemistry. 2001;40:14942–14951. doi: 10.1021/bi011400h. [DOI] [PubMed] [Google Scholar]
- 39.Linde D, Ruiz-Duenas FJ, Fernandez-Fueyo E, Guallar V, Hammel KE, Pogni R, Martinez AT. Arch Biochem Biophys. 2015;574:66–74. doi: 10.1016/j.abb.2015.01.018. [DOI] [PubMed] [Google Scholar]
- 40.Gengenbach A, Syn S, Wang XT, Lu Y. Biochemistry. 1999;38:11425–11432. doi: 10.1021/bi990666+. [DOI] [PubMed] [Google Scholar]
- 41.Houseman ALP, Doan PE, Goodin DB, Hoffman BM. Biochemistry. 1993;32:4430–4443. doi: 10.1021/bi00067a036. [DOI] [PubMed] [Google Scholar]
- 42.Huyett JE, Doan PE, Gurbiel R, Houseman ALP, Sivaraja M, Goodin DB, Hoffman BM. J Am Chem Soc. 1995;117:9033–9041. [Google Scholar]
- 43.Ivancich A, Jakopitsch C, Auer M, Un S, Obinger C. J Am Chem Soc. 2003;125:14093–14102. doi: 10.1021/ja035582+. [DOI] [PubMed] [Google Scholar]
- 44.Ivancich A, Mazza G, Desbois A. Biochemistry. 2001;40:6860–6866. doi: 10.1021/bi002826j. [DOI] [PubMed] [Google Scholar]
- 45.Patterson WR, Poulos TL, Goodin DB. Biochemistry. 1995;34:4342–4345. doi: 10.1021/bi00013a024. [DOI] [PubMed] [Google Scholar]
- 46.Benecky MJ, Frew JE, Scowen N, Jones P, Hoffman BM. Biochemistry. 1993;32:11929–11933. doi: 10.1021/bi00095a024. [DOI] [PubMed] [Google Scholar]
- 47.Rice RH, Lee YM, Brown WD. Arch Biochem Biophys. 1983;221:417–427. doi: 10.1016/0003-9861(83)90160-1. [DOI] [PubMed] [Google Scholar]
- 48.Tew D, Demontellano PRO. J Biol Chem. 1988;263:17880–17886. [PubMed] [Google Scholar]
- 49.Uchida T, Sasaki M, Tanaka Y, Ishimorit K. Biochemistry. 2015;54:6610–6621. doi: 10.1021/acs.biochem.5b00952. [DOI] [PubMed] [Google Scholar]
- 50.Baratto MC, Sinicropi A, Linde D, Saez-Jimenez V, Sorace L, Ruiz-Duenas FJ, Martinez AT, Basosi R, Pogni R. J Phys Chem B. 2015;119:13583–13592. doi: 10.1021/acs.jpcb.5b02961. [DOI] [PubMed] [Google Scholar]
- 51.Ilgenfri G, Schuster TM. J Biol Chem. 1974;249:2959–2973. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.