

Abstract
Nanotechnology-mediated antioxidative therapy is emerging as a novel strategy for treating a myriad of important diseases through scavenging excessive reactive oxygen and nitrogen species (RONS), a mechanism critical in disease development and progression. However, similar to antioxidative enzymes, currently studied nano-antioxidants have demonstrated scavenging activity to specific RONS, and sufficient antioxidative effects against multiple RONS generated in diseases remain elusive. Here we propose to develop bioinspired melanin nanoparticles (MeNPs) for more potent and safer antioxidative therapy. While melanin is known to function as a potential radical scavenger, its antioxidative mechanisms are far from clear and its applications for the treatment of RONS-associated diseases have yet to be well explored. In this study, we provide for the first time exhaustive characterization of the activities of MeNPs against multiple RONS including O2•−, H2O2, •OH, •NO, and ONOO−, the main toxic RONS generated in diseases. The potential of MeNPs for antioxidative therapy has also been evaluated in vitro and in a rat model of ischemic stroke. In addition to the broad defense against these RONS, MeNPs can also attenuate the RONS-triggered inflammatory responses through suppressing the expression of inflammatory mediators and cytokines. In vivo results further demonstrate that these unique multi-antioxidative, anti-inflammatory and biocompatible features of MeNPs contribute to their effective protection of ischemic brains with negligible side effects.
Keywords: Melanin, Nanoparticle, Antioxidant, Anti-Inflammation, Ischemic Stroke
Graphical Abstract
INTRODUCTION
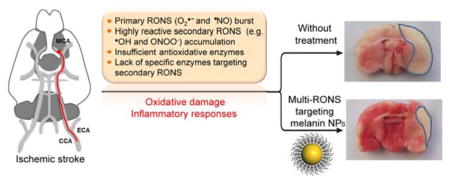
Elevated levels of reactive oxygen and nitrogen species (RONS) have been implicated in the development and progression of a variety of diseases such as cancer, neurologic disorders, and inflammatory diseases.1,2 These active species react with proteins, nucleic acids and lipids, frequently causing irreversible tissue and organ damage. Superoxide anion radical (O2•−) is the main initial RONS, which can be converted into toxic H2O2 by superoxide dismutase (SOD) or self-dismutation.3 To maintain the intracellular redox balance, multiple antioxidative enzymes (e.g., SOD and catalase) work conjointly to protect the human body from oxidative damage.4,5 However, these enzymes are overwhelmed by dramatically elevated O2•−, and their adequate replenishment is generally impossible during disease development. Notably, O2•− can further participate the generation of secondary RONS, in particular hydroxyl radical (•OH) and peroxynitrite (ONOO−), by the reaction with other molecules (e.g., H2O2 and •NO). These secondary RONS are considered more chemically reactive and can dramatically aggravate oxidative injury,6,7 while there are no specific enzymes targeting them. In addition, RONS can trigger severe inflammatory responses by rapid activation of resident immune cells, exerting secondary tissue injury through the production of more RONS and cytokines.8,9 These barriers highlight the need of developing innovative antioxidative strategies to simultaneously target these RONS for effective therapy.
Recent advances in nanotechnology have seen the development of several functional nanomaterials, such as carbon, platinum, ceria, manganese, and vanadium nanoparticles (NPs) with higher antioxidative activity and stability than natural enzymes.10–15 Nevertheless, one significant hurdle to these nano-antioxidants is the high specificity of their antioxidative activity against RONS (mainly H2O2), which may fail to sufficiently prevent oxidative damage, given the multiple RONS generated in diseases. Moreover, the potential deleterious health effects associated with these exogenous nanomaterials, such as the generation of oxidative stress, immune cell activation, mitochondrial respiration, and genotoxicity, remain another intractable problem for in vivo applications.16,17 Thus, we think that an ideal nano-antioxidant needs to possess at least the following features: i) robust scavenging of multiple primary and secondary RONS; ii) highly stable antioxidative activity against oxidative damage; iii) capability to block RONS-triggered inflammatory activation; and iv) good biocompatibility. To the best of our knowledge, it remains challenging for current antioxidative nanomaterials to meet all these criteria.
Naturally occurring biopolymers in living organisms have inspired the generation of biocompatible, bioregenerative, and biodegradable nanomaterials for biomedical applications, with the potential of avoiding the side effects associated with exogenous nanomaterials.18–20 Melanin, a heterogeneous biological polymer presenting in most organisms including human, has attracted increasing attention for molecular imaging and photothermal therapy, due to its intrinsic photoacoustic signal, strong chelating capability to metal ions, and near-infrared absorbance.21–23 Melanin also plays an important role in protecting skin against ultraviolet (UV) irradiation, which is presumably attributable, at least in part, to its ability to intercept and deactivate radical species generated by UV light.24 Notably, melanin contains a population of functional groups such as catechol, amine, and imine, which we hypothesize could detoxify multiple RONS. Although previous studies have shown the potential of melanin in radical scavenging,25,26 its antioxidative mechanisms remain poorly understood, and very few studies have thus far explored their potential for the in vivo treatment of RONS-associated diseases.
Herein, we reveal the detailed mechanisms underlying the antioxidative effects of water-soluble MeNPs, by comprehensive analysis of their scavenging activities against multiple RONS. Our data also provide strong evidence for their roles in suppressing RONS-induced inflammation activation. In vivo studies in a rat model of ischemic stroke clearly demonstrate the feasibility of using melanin as a robust antioxidant to protect the ischemic brain from RONS-induced damage, with negligible side effects. We expect that this study will lay the foundation for further work to elucidate the protective effects of melanin and other similar biomaterials against ischemic brain injury, and yield valuable insight into the development of safe and efficient antioxidants to treat a wide range of RONS-associated diseases.
RESULTS AND DISCUSSION
Preparation of PEG-MeNPs and their O2•− scavenging mechanism
Highly water-dispensable MeNPs were prepared by a straightforward method we recently developed.21 To further improve physiological stability, amine-terminated polyethylene glycol (PEG) was conjugated to the surface of MeNPs. Under basic conditions, the catechol groups in MeNPs can be oxidized into the corresponding quinone, which can then react with the nucleophilic amine groups in the PEG by means of the Schiff base reaction.28–29 Alternatively, the nucleophilic amine groups in the PEG react with dihydroxyindole/indolequinone groups in MeNPs through the Michael addition reaction.22,27–29 The conjugation of PEG to the MeNPs was confirmed by FTIR and 1H NMR analysis (Figure S1). After PEG modification, the MeNPs are stable under the physiological condition without any obvious aggregation, as determined by dynamic light scattering (DLS) (Figure S2). Transmission electron microscopy (TEM) image of PEG-MeNPs showed uniform and monodisperse spherical structures with an average diameter of ~120 nm (Figure 1A). As O2•− is the primary and most toxic ROS, and has also been identified as the main oxidant in various cell types,30–32 we first examined the antioxidative activity of PEG-MeNPs towards O2•− using electron paramagnetic resonance (EPR) spectroscopy. The spin trap agent 5-diethoxyphosphoryl-5-methyl-1-pyrroline N-oxide (DEPMPO) was used to produce a stable paramagnetic adduct DEPMPO-OOH upon reaction with O2•−.33 As shown in Figure 1B, neither water nor PEG molecules had any impact on this reaction, as evidenced by the strong EPR signals of DEPMPO-OOH. In contrast, similar to SOD, both MeNPs and PEG-MeNPs reduced the EPR amplitude of DEPMPO-OOH nearly to background levels, indicating that these NPs were robust scavengers of O2•−.
Figure 1.
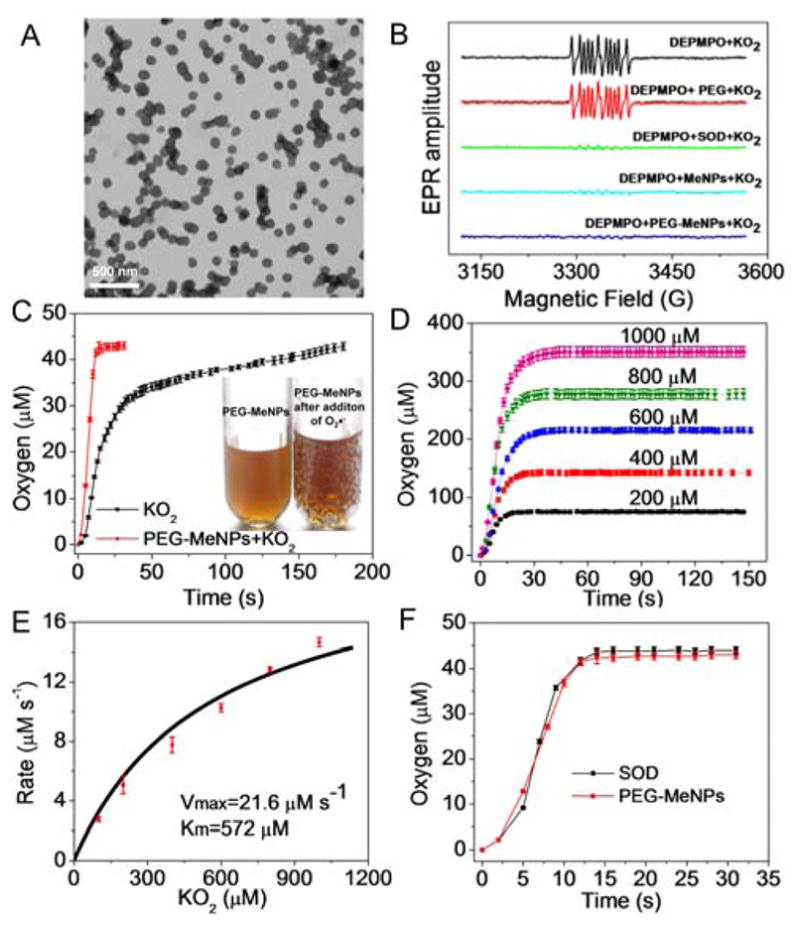
(A) TEM image of PEG-MeNPs. (B) Effect of PEG, SOD, MeNPs, and PEG-MeNPs on EPR signals of DEPMPO-OOH. (C) O2 production from the KO2 solution (100 μM) with vs. without PEG-MeNPs. The insert is the digital picture of the PEG-MeNPs solution before vs. after addition of KO2. (D) PEG-MeNP-catalyzed dismutation of O2•− under different concentrations of KO2. (E) Michaelis-Menten kinetic plot of the initial reaction rate vs. the KO2 concentration for PEG-MeNP-catalyzed dismutation of O2•−. (F) O2 production from the KO2 solution (100 μM) catalyzed by SOD vs. PEG-MeNPs. The concentration of PEG-MeNPs for catalysis is 0.1 nM. Assay was performed in triplicate.
To understand the mechanisms involved in the potent detoxification of O2•− by PEG-MeNPs, we employed X-ray photoelectron spectroscopy (XPS) to monitor the changes of various elements in PEG-MeNPs after reaction with O2•−. XPS analysis revealed only slight increase of relative oxygen level in the PEG-MeNPs (Figure S3), indicating that covalent reaction with O2•− may not be the dominant mechanism of their O2•− scavenging activity. This is consistent with the results of TEM and X-ray diffraction (XRD), in which no apparent difference in the shape, particle size, or structure of PEG-MeNPs was found after addition of O2•− (Figures S4 and S5). On the other hand, a large quantity of bubbles was generated when O2•− was added into the PEG-MeNPs solution (Figure 1C), suggesting that the O2•− scavenging mechanism could probably be attributed to the SOD-mimic catalytic transformation of O2•− to O2. To verify this, a dissolved-oxygen meter was employed to monitor the kinetics of O2 generation. Without the NPs, relatively slow O2 production was observed because of the self-dismutation of O2•−. With PEG-MeNPs, the O2 generation rate was dramatically accelerated, while the total amount of O2 produced remained unaltered (Figure 1C). A further increase in O2•− concentration elicited similar catalytic effects (Figure 1D). After subtraction of self-produced O2 from the control group followed by plotting of the reaction rate to the concentration of O2•−, a typical Michaelis-Menten kinetic curve was obtained (Figure 1E), further confirming the enzymatic behavior of the NPs. The turnover number (kcat) was calculated to be 216,000 s−1. In addition, Figure 1F shows that the PEG-MeNPs exhibited a similar catalytic activity to SOD, a key antioxidative defense in the human body against O2•− radicals. Notably, unlike enzymes which are generally susceptible to pH changes,34 the catalytic activity of PEG-MeNPs remains similar under different pHs or between different batches (Figure S6). Moreover, PEG-MeNPs maintained the high catalytic activity even after one-year storage at 4 °C (Figure S7). All these results indicate the excellent antioxidative stability of PEG-MeNPs.
Considering that melanin is known to possess stable un-paired electrons at the center of the stacked units,27 the efficient and stable catalysis of O2•− dismutation by PEG-MeNPs may be ascribed to the radicals in the polymer matrix, which could act as catalytic centers for electron removal from superoxide.15 EPR analysis further confirmed the strong free radical signal from PEG-MeNPs (Figure 2A). It is worth noting that these free radicals were quite stable under different pHs, in the presence of the radical scavenger glutathione (GSH), or after reaction with O2•− for multiple times.
Figure 2.
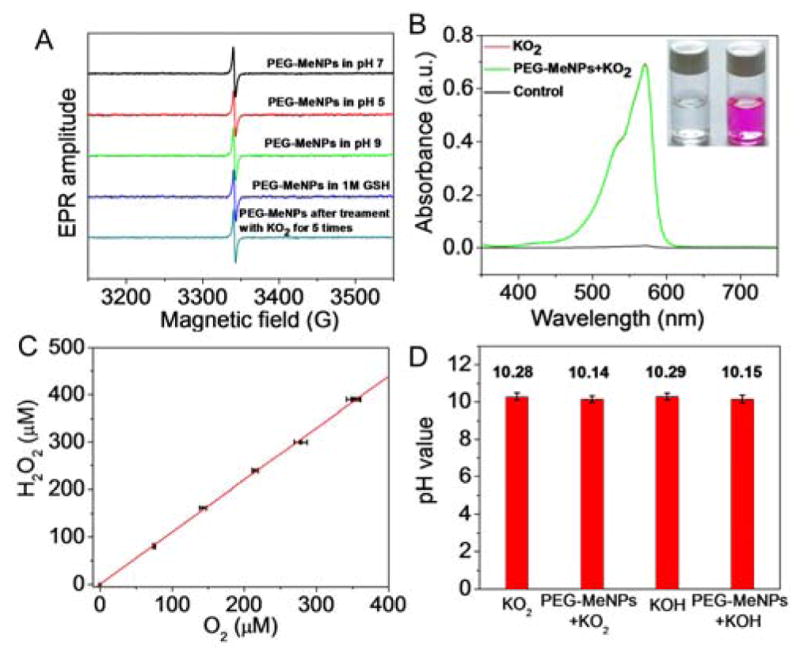
(A) EPR spectra of PEG-MeNPs under different pH values, in the presence of GSH, or after reaction with KO2 for 5 times (5 min each). (B) Absorbance changes in the mixture of Amplex Red and HRP induced by KO2 with or without PEG-MeNPs. The insert is the digital picture of Amplex Red and HRP solution before vs. after addition of KO2 and PEG-MeNPs. (C) The ratio between H2O2 and O2 produced by PEG-MeNP-catalyzed dismutation of KO2. (D) pH values of KO2 and KOH solutions with vs. without PEG-MeNPs. The concentration of PEG-MeNPs is 0.1 nM. Assay was perfomed in triplicate.
Next we quantified the generation of H2O2, another ROS product of the dismutation reaction of O2•−, using Amplex Red as the indicator. Amplex Red is colorless but reacts with H2O2 to form a colored product (resofurin) in the presence of horseradish peroxidase (HRP).35 Upon the addition of O2•− and PEG-MeNPs, the Amplex Red and HRP solution became pink immediately (Figure 2B), indicating the presence of H2O2. As a reference, the O2•− solution was allowed to undergo self-dismutation for 5 min and added into the solution of Amplex Red and HRP, after which the absorbance of the produced resofurin was measured. The spectrum overlap between the two reactions suggested that the total amount of H2O2 generated by PEG-MeNPs catalysis was equal to that by self-dismutation of O2•−. The molar correlation between the generated H2O2 and O2 was also consistently ~1:1 in the PEG-MeNP-catalyzed reactions (Figure 2C). It is noteworthy that when we prolonged the pre-incubation time between PEG-MeNPs and O2•−, the absorbance intensity of produced resofurin was found to be lower than that induced by self-dismutation of O2•− (Figure S8). We postulated that the decreased H2O2 could be the result of the reaction between PEG-MeNPs and H2O2 (Figure S9), as melanin in human hairs is known to be bleached by H2O2.36 This H2O2 attenuation capability makes PEG-MeNPs superior to SOD, which is inert to H2O2 and requires other antioxidative molecules (e.g., catalase, glutathione, or Vitamin E) to scavenge this ROS. It should be noted, however, that these detoxifying molecules are generally overwhelmed by dramatically elevated radical levels and their adequate replenishment is impossible in diseases,37 which may also explain why SOD treatment has shown limited therapeutic efficacy in oxidative stress-associated diseases.38
To further clarify the catalytic mechanism of MeNPs, we also measured OH−, which is stoichiometrically generated in the dismutation reaction. As expected, the pH increase induced by self-dismutation of KO2 was almost the same as that caused by the addition of the same molar KOH (Figure 2D). The addition of PEG-MeNPs to KO2 led to a slightly lower pH value (~10.14) as compared to KOH control (~10.29). This may be attributed to the buffering capacity of the residual catechol groups in PEG-MeNPs,39 as suggested by the pH value (~10.15) of the mixture of PEG-MeNPs and KOH. Considering all these results, we speculated that the SOD-mimic catalysis of O2•− by melanin could be rep-resented by the following half-reactions:
Multi-antioxidative activities of PEG-MeNPs
Scavenging secondary RONS remains a formidable challenge for currently investigated antioxidative materials. •OH and ONOO− are among the most toxic secondary RONS generated in diseases,40 both of which can contribute to lipid peroxidation, protein oxidation, and nucleic acid damage (Figure 3A). In general, •OH is generated via Fenton-type reactions between H2O2 and reduced transition metal ions (e.g., Cu+ and Fe2+) in vivo.41,42 O2•− is essential in this process because it not only produces a large amount of H2O2 through self-dismutation and SOD catalysis, but also serves as an reducing agent for metal ions.43 By EPR analysis, we observed that PEG-MeNPs could also scavenge •OH, as evidenced by the decreased signal of DEPMPO-OH, a paramagnetic adduct arising from the reaction of DEPMPO with •OH (Figure 3B). Notably, when PEG-MeNPs was pre-added into the reaction system containing DEPMPO and Cu+ ions, the subsequent addition of H2O2 did not lead to EPR signal of DEPMPO-OH compared to the control group. We reasoned that PEG-MeNPs could impede the Fenton reaction, presumably due to the strong chelating capability of melanin with transition metal ions,44 thereby blocking the generation of •OH.
Figure 3.
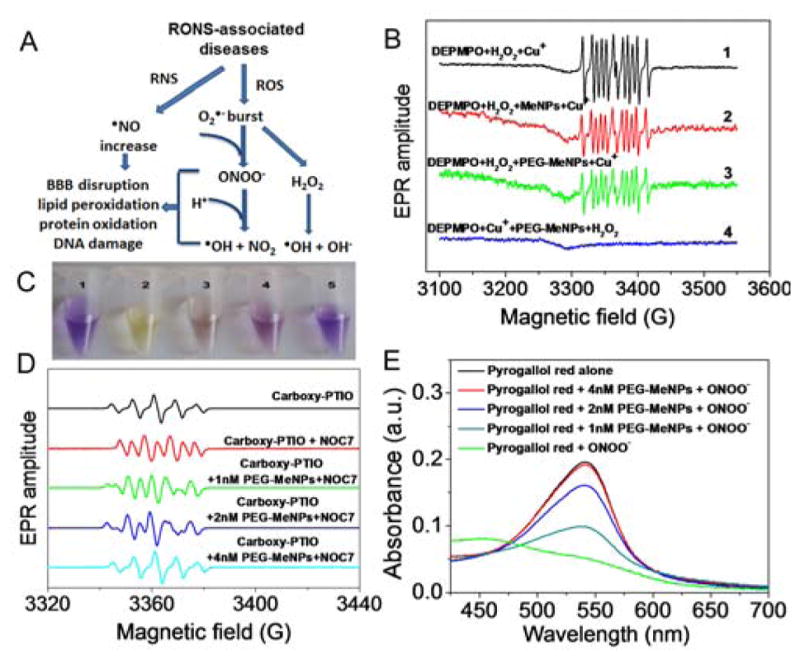
(A) Schematic illustration of RONS metabolism. (B) EPR spectra of DEPMPO-OH obtained by trapping •OH with spin-trap reagent DEPMPO in the absence vs. presence of MeNPs or PEG-MeNPs. The •OH was generated by the Fenton reaction between H2O2 and Cu+ ions. For reaction #2 and #3, MeNPs and PEG-MeNPs were, respectively, added to the mixture of DEPMPO and H2O2, followed by the addition of Cu+ ions. In reaction #4, PEG-MeNPs were pre-incubated with DEPMPO and Cu+, followed by the addition of H2O2. (C) Digital picture of the carboxy-PTIO solution alone (#1) vs. the carboxy-PTIO solutions with •NO-generating NOC7 and PEG-MeNPs (#2–5: 0, 1, 2, and 4 nM). (D) EPR spectra of the five samples from (C). (E) ONOO− scavenging effect of PEG-MeNPs.
Nitric oxide (•NO) plays important physiological roles in normal conditions,45 but is overproduced during diseases and induces oxidative damage.46,47 It can interact with O2•−, leading to the production of peroxynitrite (ONOO−) and •OH. ONOO− has also been implicated as a causative factor in the disruption of the blood-brain barrier (BBB) in some neurologic diseases, due to its much higher penetrating capacity across lipid bilayers compared to O2•− (~400-fold).48 The antioxidative activity of PEG-MeNPs towards •NO was assessed using carboxy-PTIO as the indicator and NOC7 as the •NO donner. In the absence of PEG-MeNPs, carboxy-PTIO reacted with •NO to generate carboxy-PTI, resulting in the color change from purple to yellow and an increase of the EPR signal from 5 to 7 lines (Figure 3C,D). In contrast, the addition of carboxy-PTIO into the mixture of PEG-MeNPs and NOC7 showed a dose-dependent effect in preventing color fading and the formation of carboxy-PTI, suggesting the •NO scavenging by PEG-MeNPs. Next, the ONOO− scavenging activity of PEG-MeNPs was studied using the ONOO−-induced pyrogallol red bleaching assay. Results showed that the shift of absorbance due to the ONOO−-mediated oxidation of pyrogallol red was significantly inhibited in the presence of PEG-MeNPs (Figure 3E). Similarly, PEG-MeNPs also showed high stability in scavenging •NO and ONOO− (Figure S10).
Nitration and nitrosation are two key reactions between phenolic compounds with •NO and ONOO−.49,50 Given the presence of the residual catechol groups in PEG-MeNPs, we hypothesized that one major mechanism contributable to the PEG-MeNP-mediated scavenging of •NO and ONOO− might be the nitration and nitrosation of PEG-MeNPs by •NO and ONOO−. To confirm this, the absorbance changes of PEG-MeNPs were first tested after addition of •NO or ONOO−. PEG-MeNPs showed a time-dependent increase of absorbance in the region of 300–400 nm after addition of •NO (Figure S11A,B), and in the entire visible light region of the spectrum upon reaction with ONOO− (Figure S11C,D). In addition, XPS analysis also showed an in-creased level of nitrogen in the •NO- or ONOO−-treated PEG-MeNPs compared to the non-treated NPs (Figure S12), further confirming the nitration and nitrosation of PEG-MeNPs. Notably, the nitration and nitrosation caused by •NO or ONOO− did not induce obvious size and structural changes of the NPs as determined by TEM and XRD analysis (Figure S13 and S14), neither affected the radicals in PEG-MeNPs (Figure S15). It should be noted that the nitration and nitrosation are complicated for different phenols and the exact structure of melanin remains un-clear, and thus further studies are needed to reveal the reaction mechanisms and possible products of PEG-MeNPs after reaction with •NO or ONOO−.
Neuroprotective and anti-inflammatory activities of PEG-MeNPs
Ischemic stroke, as a result of the blockage of blood supply to the brain, is a major cause of death and disability worldwide.51 Intravenous administration of tissue plasminogen activator and endovascular surgery to break down or remove the blood clots are the only clinically approved methods for preventing ischemia-induced irreversible brain damage.52 Nevertheless, because fewer than 10% of ischemic stroke patients have timely access to these treatments, the vast majority of patients sustain neurological deficits and even death, due to the lack of effective clinical options for persistent occlusion-induced brain injury.53 Oxidative stress and inflammatory activation induced by the excessive production of RONS are critical mechanisms of brain injury in ischemic stroke.54 However, there are no clinically validated RONS-scavenging antioxidants that can safely and effectively protect ischemic brain from oxidative damage.
Given the strong antioxidative potency of PEG-MeNPs, we proceeded to examine their potential for preventing brain damage from ischemic stroke. As neurons are vulnerable to oxidative stress during an ischemic attack, we first used Neuro 2A cells to evaluate the toxicity of PEG-MeNPs and their protective effects against RONS-induced oxidative stress. AlamarBlue and LDH assays showed that PEG-MeNP treatment did not induce obvious cytotoxicity under the experimental conditions (Figure 4A and Figure S16A). In addition, ATP levels in the cells were unaltered by PEG-MeNPs (Figure S16B), indicating that these NPs did not affect mitochondrial function. Next, antimycin A (AMA), an inhibitor of the mitochondrial respiratory complex III,55 was used to stimulate oxidative stress in Neuro 2A cells. Intracellular O2•− was dramatically enhanced upon AMA treatment (Figure 4B,C). As a comparison, when the cells were pre-treated with PEG-MeNPs, they showed decreased levels of O2•− in a dose-dependent manner after AMA stimulation.
Figure 4.
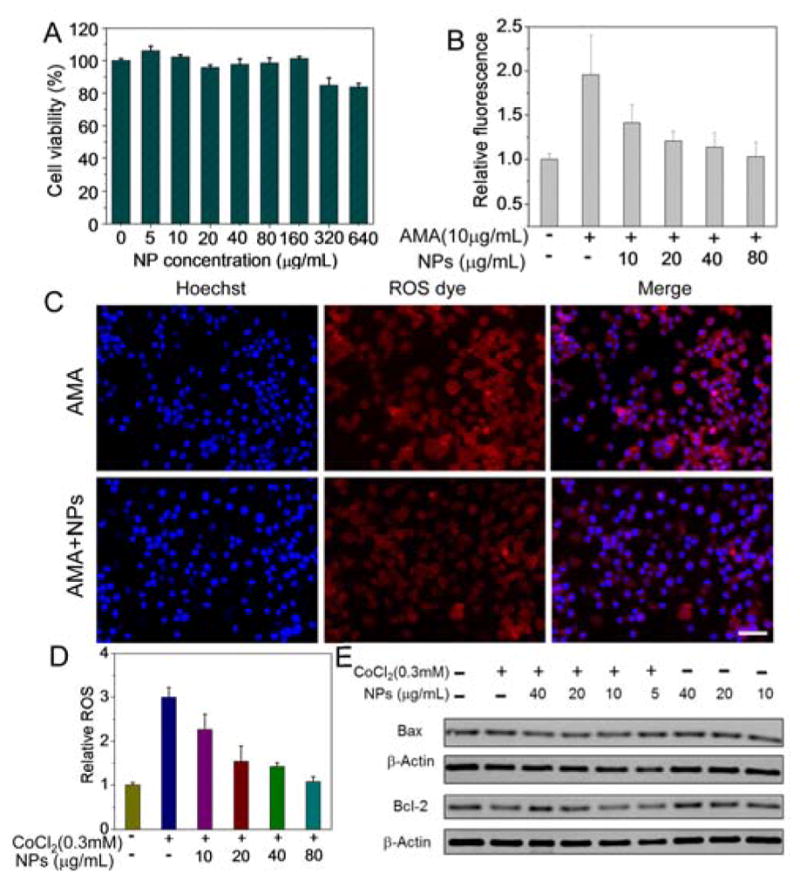
(A) Cytotoxicity of PEG-MeNPs. (B) Intracellular O2•− scavenging by PEG-MeNPs in AMA-treated Neuro 2A cells. (C) Confocal fluorescence images of O2•− levels in the AMA-treated Neuro 2A cells. Scale bar is 50 μm. (D) ROS levels in non-treated and PEG-MeNP-treated Neuro 2A cells under CoCl2-induced hypoxic conditions. (E) Western blot analysis of the expression of Bax and Bcl-2 in the CoCl2-stimualted Neuro 2A cells with vs. without PEG-MeNPs, as well as in the cells only treated with PEG-MeNPs.
To directly assess the neuroprotective effect of PEG-MeNPs in ischemia, we next mimicked ischemic injury in vitro by incubating Neuro 2A cells with cobalt chloride (CoCl2). CoCl2 has been used to induce hypoxic microenvironments and sub-sequent generation of excess ROS in different cell lines, a frequent in vitro model for the mechanistic study of antioxidant treatments for ischemia-associated neuronal disorders.56 CoCl2 dramatically increased the ROS level in Neuro 2A cells and subsequently caused cell death as a result of oxidative stress, whereas PEG-MeNPs clearly reduced both responses (Figure 4D and Figure S17A). It has been revealed that the upregulation of proapoptotic Bax and downregulation of antiapoptotic Bcl-2 are involved in CoCl2-induced cell death in neurons.57 In fact, stable expression of Bcl-2 is crucial for suppressing the overproduction of O2•− in cells, and the overexpression of Bcl-2 is beneficial for decreasing the infarct area following ischemia.58 We therefore investigated whether the neuroprotection of PEG-MeNPs was correlated with the regulation of both proteins. Western blot analysis showed that PEG-MeNPs did not alter the expression of either Bax or Bcl-2 in Neuro 2A, but explicitly prevented Bax upregulation and Bcl-2 downregulation when the cells were also treated with CoCl2 (Figure 4E and Figure S17B). Conversely, enhanced Bax expression and reduced Bcl-2 expression were detected in cells treated with CoCl2 alone.
Non-neuronal cells such as microglia, neutrophils, and macrophages can also be rapidly activated by RONS during ischemia, resulting in the secretion of many detrimental inflammatory mediators (Figure 5A). This can further aggravate neuronal injury in the cerebral penumbra and extend the injury to surrounding tissues.59 The anti-inflammatory effect of PEG-MeNPs was assessed using lipopolysaccharide (LPS)-stimulated macrophages. We first tested the expression of cyclooxygenase 2 (COX-2), an inflammatory mediator activated by LPS stimulation that also participates in the overproduction of ROS (in particular O2•−),60 in the absence vs. presence of PEG-MeNPs by western blot and immunofluorescence analysis. PEG-MeNPs can be efficiently taken up by macrophages (Figure S18) and alleviate the activation of this inflammatory mediator (Figure 5B,C), leading to significant reduction of ROS levels upon LPS stimulation (Figure 5D). The expression of inducible nitric oxide synthase (iNOS), one major source of toxic •NO,61 was also downregulated in LPS-stimulated macrophages with treatment of PEG-MeNPs (Figure S19). In addition, PEG-MeNPs effectively inhibited the overproduction of the total intracellular RNS in LPS-stimulated macrophages (Figure 5E). Similarly, the expression of cytokines including tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) at the protein level was remarkably decreased (Figure S19).
Figure 5.
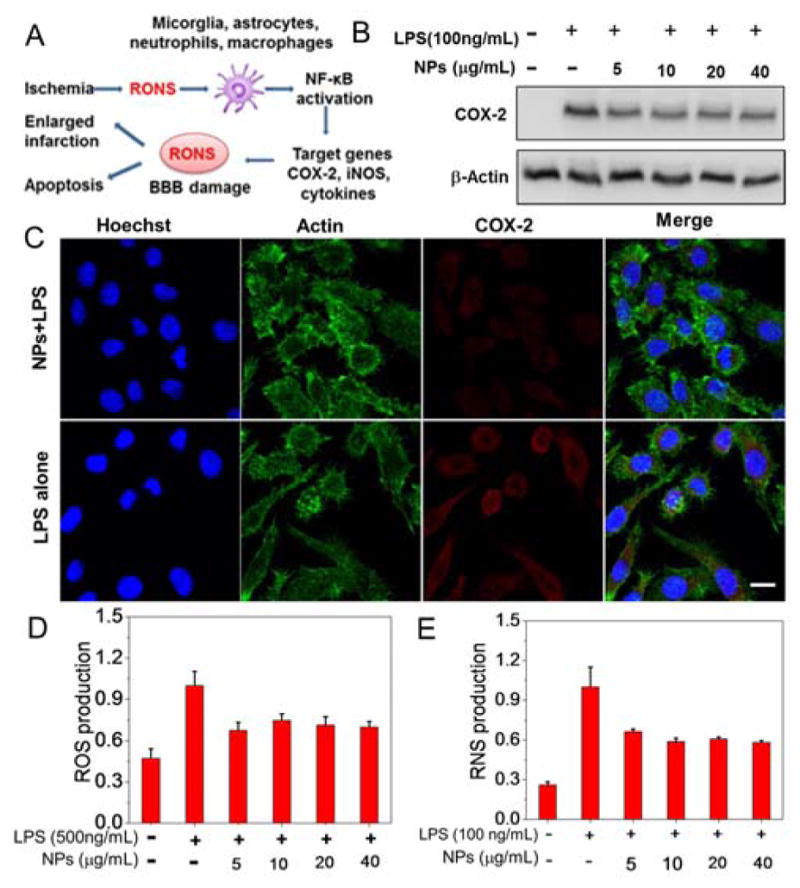
(A) Schematic illustration of inflammatory mechanism triggered by the elevated RONS in non-neuronal cells. (B) Western blot analysis of COX-2 expression in LPS-stimulated macrophages. (C) Immunofluorescence images of the expression of COX-2 in LPS-stimulated macrophages with vs. without pretreatment of PEG-MeNPs. Scale bar is 10 μm. Levels of (D) ROS and (E) RNS in LPS-stimulated macrophages with different concentrations of PEG-MeNPs.
Brain protection by PEG-MeNPs in a rat model of ischemic stroke
With strong evidence of multi-antioxidative and anti-inflammatory activities of PEG-MeNPs in vitro, we then set out to evaluate their in vivo efficacy in ischemic brains by pre-injection into lateral ventricles. Note that intraventricular injection has been widely used in the clinical settings and has several benefits as compared to intravascular delivery, including (i) more precise control of local dosing; (ii) minimization of adverse effects involved in systemic therapy to normal tissues; (iii) circumvention of the BBB; and (iv) in the case of ischemic stroke, circumvention of occluded vessels that may prevent delivery to the ischemic region. After injection of saline or PEG-MeNPs, rats were subjected to 90 min of occlusion of the middle cerebral artery (MCA) (Figure 6A). The brains were then removed, sliced, and stained with 2,3,5-triphenyltetrazolium chloride (TTC), an indicator of cellular respiration widely used for histopathologic staining to identify infarcted brain tissues. A large infarct area (~32%) was detected in the saline control group (Figure 6B,C). In comparison, rats pretreated with PEG-MeNPs were significantly less vulnerable to ischemia (~14% infarct area). Further comparison of the O2•− levels in brains in the saline, NP, and sham groups demonstrated that pretreatment with PEG-MeNPs efficiently suppressed the generation of O2•− (Figure 6D). These results suggest the potential of PEG-MeNPs for the treatment of ischemic brain damage.
Figure 6.
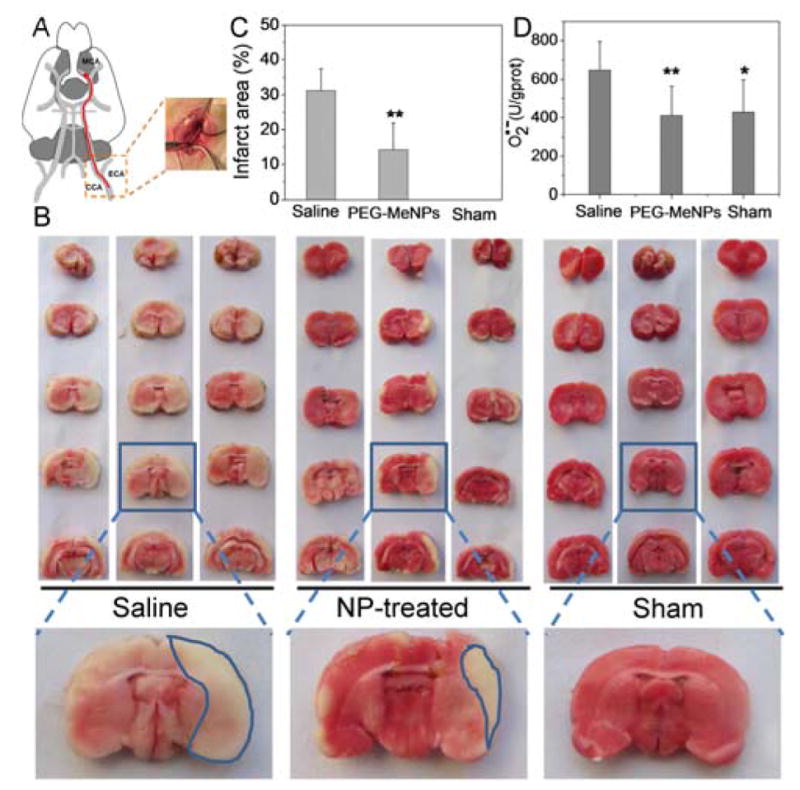
(A) Schematic representation of the ischemic stroke model. (B) Representative images of TTC-stained brain slices from different groups. The corresponding (C) infarct areas and (D) O2•− levels in brain tissues of the three groups (*p<0.05 and **p<0.01 vs. saline control).
In vivo toxicity of PEG-MeNPs
The in vivo safety of PEG-MeNPs was also assessed. After a single intravenous injection of PEG-MeNPs into immunocompetent mice through the tail vein, blood was collected for evaluation of immunostimulatory effects. Results showed that PEG-MeNPs did not induce systemic cytokine responses in mice at either 6 h or 24 h post NP injection (Figure S20). Moreover, PEG-MeNPs exhibited excellent blood compatibility, and did not alter serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALKP), blood urine nitrogen (BUN), total protein (TP), or albumin (Alb) (Figures S21 and S22). Hematoxylin and eosin (H&E) histology analysis of different organs showed no morphological changes or signs of inflammation after NP treatment (Figure S23).
CONCLUSION
In summary, we reported the evoluation of bioinspired PEG-MeNPs as a novel nano-antioxidant for antioxidative therapy. Comprehensive characterization was carried out in this work to improve our fundamental understanding of the antioxidative mechanisms of PEG-MeNPs. Our findings reveal that PEG-MeNPs have a SOD mimetic catalytic mechanism towards O2•−. Superior to both natural antioxidant enzymes (e.g., SOD) and currently studied nano-antioxidants that target specific RONS, PEG-MeNPs exhibit broad antioxidative activities against multiple toxic RONS including O2•−, H2O2, •OH, ONOO−, and •NO, highlighting their potential as a robust RONS scavenger. In vitro results further demonstrate the neuroprotective and anti-inflammatory activities of PEG-MeNPs. Using a rat model of ischemic stroke, we also showed that pre-injection of PEG-MeNPs can significantly decrease the infarct area of the ischemic brain. While our results support the potential of PEG-MeNPs in protecting ischemic brains from oxidative damage, further investigations will still be needed, such as continuous optimization of the PEG-MeNP platform and the administration timing/dosage, and deeper under-standing of the in vivo protective mechanisms. In addition, despite that our preliminary in vitro and in vivo side effect studies showed no obvious toxicity of PEG-MeNPs, more studies will be needed to examine the long-term toxicity of PEG-MeNPs to normal cells and tissues, their interactions with biological systems, and the potential toxicity from the degradation byproducts of PEG-MeNPs. Of more clinical relevance to this strategy is whether the brain protection effect seen in this study can be achieved when PEG-MeNPs are given in a delayed fashion. This could significantly improve the clinical outcomes and quality of life for ischemic stroke patients who are not eligible for endovascular surgery. As RONS and subsequent inflammatory activation are associated with the etiology of aging and many diseases such as cancer, coronary heart disease, Alzheimer’s disease, Parkinson’s disease, and neurodegenerative disorders, this study may pave the way for the development of safe and effective antioxidants for wide-spread biomedical applications.
Supplementary Material
Acknowledgments
Financial support by NIH R00CA160350 and CA200900, NSFC (21635007,21605137) and National Key Research and Development Program of China (2016YFA0203200), the Youth Innovation Promotion Association of Chinese Academy of Sciences (No. 2014201).
Footnotes
Notes
The authors declare no competing financial interest.
Experimental details and supporting results for XPS, TEM, and XRD characterization of PEG-MeNPs before and after reaction with O2•−, •NO, or ONOO-; catalytic and scavenging stabilities of PEG-MeNPs; reaction with H2O2; in vitro studies and in vivo toxicity tests of PEG-MeNPs; and western blot analysis of anti-inflammatory activities of PEG-MeNPs. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Nathan C, Cunningham-Bussel A. Nat Rev Immunol. 2013;13:349. doi: 10.1038/nri3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnham KJ, Masters CL, Bush AI. Nat Rev. 2004;3:205. doi: 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- 3.Dickinson BC, Chang CJ. Nat Chem Biol. 2011;7:504. doi: 10.1038/nchembio.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mikkelsen RB, Wardman P. Oncogene. 2003;22:5734. doi: 10.1038/sj.onc.1206663. [DOI] [PubMed] [Google Scholar]
- 5.Weidinger A, Kozlov AV. Biomolecules. 2015;5:472. doi: 10.3390/biom5020472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winterbourn CC. Nat Chem Biol. 2008;4:278. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 7.Marla SS, Lee J, Groves JT. Proc Natl Acad Sci USA. 1997;94:14243. doi: 10.1073/pnas.94.26.14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Antioxid Redox Signal. 2014;20:1126. doi: 10.1089/ars.2012.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leavy O. Nat Rev Immunol. 2014;14:357. doi: 10.1038/nri3685. [DOI] [PubMed] [Google Scholar]
- 10.Lucente-Schultz RM, Moore VC, Leonard AD, Price BK, Kosynkin DV, Lu M, Partha R, Conyers JL, Tour JM. J Am Chem Soc. 2009;131:3934. doi: 10.1021/ja805721p. [DOI] [PubMed] [Google Scholar]
- 11.Kim CK, Kim T, Choi IY, Soh M, Kim D, Kim YJ, Jang H, Yang HS, Kim JY, Park HK, Park SP, Park S, Yu T, Yoon BW, Lee SH, Hyeon T. Angew Chem Int Ed. 2012;51:11039. doi: 10.1002/anie.201203780. [DOI] [PubMed] [Google Scholar]
- 12.Lee HJ, Park J, Yoon OJ, Kim HW, Lee DY, Kim do H, Lee WB, Lee NE, Bonventre JV, Kim SS. Nat Nanotechnol. 2011;6:121. doi: 10.1038/nnano.2010.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prasad P, Gordijo CR, Abbasi AZ, Maeda A, Ip A, Rauth AM, DaCosta RS, Wu XY. ACS nano. 2014;8:3202. doi: 10.1021/nn405773r. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe A, Kajita M, Kim J, Kanayama A, Takahashi K, Mashino T, Miyamoto Y. Nanotechnology. 2009;20:455105. doi: 10.1088/0957-4484/20/45/455105. [DOI] [PubMed] [Google Scholar]
- 15.Samuel ELG, Marcano DC, Berka V, Bitner BR, Wu G, Potter A, Fabian RH, Pautler RG, Kent TA, Tsai AL, Tour JM. Proc Natl Acad Sci USA. 2015;112:2343. doi: 10.1073/pnas.1417047112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharifi S, Behzadi S, Laurent S, Forrest ML, Stroeve P, Mahmoudi M. Chem Soc Rev. 2012;41:2323. doi: 10.1039/c1cs15188f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elsaesser A, Howard CV. Adv Drug Deliv Rev. 2012;64:129. doi: 10.1016/j.addr.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 18.Ige OO, Umoru LE, Aribo S. ISRN Mater Sci. 2012;2012:20. [Google Scholar]
- 19.Lee Y, Kim H, Kang S, Lee J, Park J, Jon S. Angew Chem Int Ed. 2016;55:7460. doi: 10.1002/anie.201602525. [DOI] [PubMed] [Google Scholar]
- 20.Lee Y, Lee S, Lee DY, Yu B, Miao W, Jon S. Angew Chem Int Ed. 2016;55:10676. doi: 10.1002/anie.201604858. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y, Ai K, Liu J, Deng M, He Y, Lu L. Adv Mater. 2013;25:1353. doi: 10.1002/adma.201204683. [DOI] [PubMed] [Google Scholar]
- 22.Fan Q, Cheng K, Hu X, Ma X, Zhang R, Yang M, Lu X, Xing L, Huang W, Gambhir SS, Cheng Z. J Am Chem Soc. 2014;136:15185. doi: 10.1021/ja505412p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang R, Fan Q, Yang M, Cheng K, Lu X, Zhang L, Huang W, Cheng Z. Adv Mater. 2015;27:5063. doi: 10.1002/adma.201502201. [DOI] [PubMed] [Google Scholar]
- 24.Brenner M, Hearing V. J Photochem Photobiol. 2008;84:539. doi: 10.1111/j.1751-1097.2007.00226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Panzella L, Gentile G, D’Errico G, Della Vecchia NF, Errico ME, Napolitano A, Carfagna C, d’Ischia M. Angew Chem Int Ed. 2013;52:12684. doi: 10.1002/anie.201305747. [DOI] [PubMed] [Google Scholar]
- 26.Ju KY, Lee Y, Lee S, Park SB, Lee JK. Biomacromolecules. 2011;12:625. doi: 10.1021/bm101281b. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Ai K, Lu L. Chem Rev. 2014;114:5057. doi: 10.1021/cr400407a. [DOI] [PubMed] [Google Scholar]
- 28.Yang J, Cohen Stuart MA, Kamperman M. Chem Soc Rev. 2014;43:8271. doi: 10.1039/c4cs00185k. [DOI] [PubMed] [Google Scholar]
- 29.Kutyrev AA. Tetrahedron. 1991;47:8043. [Google Scholar]
- 30.Kontos HA. Stroke. 2001;32:2712. doi: 10.1161/hs1101.098653. [DOI] [PubMed] [Google Scholar]
- 31.Bergendi L, Beneš L, Ďuračková Z, Ferenčik M. Life Sci. 1999;65:1865. doi: 10.1016/s0024-3205(99)00439-7. [DOI] [PubMed] [Google Scholar]
- 32.Pervaiz S, Clement MV. Int J Biochem Cell Biol. 2007;39:1297. doi: 10.1016/j.biocel.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 33.Zhao H, Joseph J, Zhang H, Karoui H, Kalyanaraman B. Free Radical Bio Med. 2001;31:599. doi: 10.1016/s0891-5849(01)00619-0. [DOI] [PubMed] [Google Scholar]
- 34.Yamakura F, Kobayashi K, Ue H, Konno M. Eur J Biochem. 1995;227:700. doi: 10.1111/j.1432-1033.1995.tb20191.x. [DOI] [PubMed] [Google Scholar]
- 35.Zhou M, Diwu Z, Panchuk-Voloshina N, Haugland RP. Anal Biochem. 1997;253:162. doi: 10.1006/abio.1997.2391. [DOI] [PubMed] [Google Scholar]
- 36.Korytowski W, Sarna T. J Bio Chem. 1990;265:12410. [PubMed] [Google Scholar]
- 37.Schaller B, Graf R. J Cereb Blood Flow Metab. 2004;24:351. doi: 10.1097/00004647-200404000-00001. [DOI] [PubMed] [Google Scholar]
- 38.Veronese FM, Caliceti P, Schiavon O, Sergi M. Adv Drug Deliv Rev. 2002;54:587. doi: 10.1016/s0169-409x(02)00029-7. [DOI] [PubMed] [Google Scholar]
- 39.Della Vecchia NF, Avolio R, Alfè M, Errico ME, Napolitano A, d’Ischia M. Adv Funct Mater. 2013;23:1331. [Google Scholar]
- 40.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Proc Natl Acad Sci USA. 1990;87:1620. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koppenol WH. Free Radical Bio Med. 1993;15:645. doi: 10.1016/0891-5849(93)90168-t. [DOI] [PubMed] [Google Scholar]
- 42.Allen CL, Bayraktutan U. Inter J Stroke. 2009;4:461. doi: 10.1111/j.1747-4949.2009.00387.x. [DOI] [PubMed] [Google Scholar]
- 43.Reddy PH. J Neurochem. 2006;96:1. doi: 10.1111/j.1471-4159.2005.03530.x. [DOI] [PubMed] [Google Scholar]
- 44.Hong L, Simon JD. J Phys Chem B. 2007;111:7938. doi: 10.1021/jp071439h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fang FC. Nat Rev Microbiol. 2004;2:820. doi: 10.1038/nrmicro1004. [DOI] [PubMed] [Google Scholar]
- 46.Samdani AF, Dawson TM, Dawson VL. Stroke. 1997;28:1283. doi: 10.1161/01.str.28.6.1283. [DOI] [PubMed] [Google Scholar]
- 47.Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Giuffrida Stella AM. Nat Rev Neurosci. 2007;8:766. doi: 10.1038/nrn2214. [DOI] [PubMed] [Google Scholar]
- 48.Marla SS, Lee J, Groves JT. Proc Natl Acad Sci USA. 1997;94:14243. doi: 10.1073/pnas.94.26.14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yenes S, Messeguer A. Tetrahedron. 1999;55:14111. [Google Scholar]
- 50.Uppu RM, Lemercier JN, Squadrito GL, Zhang H, Bolzan RM, Pryor WA. Arch Biochem Biophys. 1998;358:1. doi: 10.1006/abbi.1998.0825. [DOI] [PubMed] [Google Scholar]
- 51.Markus HS, Bevan S. Nat Rev Neurol. 2014;10:723. doi: 10.1038/nrneurol.2014.196. [DOI] [PubMed] [Google Scholar]
- 52.Powers WJ, Derdeyn CP, Biller J, Coffey CS, Hoh BL, Jauch EC, Johnston KC, Johnston SC, Khalessi AA, Kidwell CS, Meschia JF, Ovbiagele B, Yavagal DR. Stroke. 2015;46:3020. doi: 10.1161/STR.0000000000000074. [DOI] [PubMed] [Google Scholar]
- 53.Investigators, C. A. S. P. R. Neurology. 2005;64:654. [Google Scholar]
- 54.Eltzschig HK, Eckle T. Nat Med. 2011;17:1391. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ohsawa I, Ishikawa M, Takahashi K, Watanabe M, Nishimaki K, Yamagata K, Katsura K-i, Katayama Y, Asoh S, Ohta S. Nat Med. 2007;13:688. doi: 10.1038/nm1577. [DOI] [PubMed] [Google Scholar]
- 56.Guillemin K, Krasnow MA. Cell. 1997;89:9. doi: 10.1016/s0092-8674(00)80176-2. [DOI] [PubMed] [Google Scholar]
- 57.Torii S, Goto Y, Ishizawa T, Hoshi H, Goryo K, Yasumoto K, Fukumura H, Sogawa K. Cell Death Differ. 2011;18:1711. doi: 10.1038/cdd.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C, Huarte J. Neuron. 1994;13:1017. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 59.Hagberg H, Mallard C, Ferriero DM, Vannucci SJ, Levison SW, Vexler ZS, Gressens P. Nat Rev Neurol. 2015;11:192. doi: 10.1038/nrneurol.2015.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nogawa S, Zhang F, Ross ME, Iadecola C. J Neurosci. 1997;17:2746. doi: 10.1523/JNEUROSCI.17-08-02746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nogawa S, Forster C, Zhang F, Nagayama M, Ross ME, Iadecola C. Proc Natl Acad Sci USA. 1998;95:10966. doi: 10.1073/pnas.95.18.10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.