

Abstract
Metagenomic whole genome sequencing for detection of pathogens in clinical samples is an exciting new area for discovery and clinical testing. A major barrier to this approach is the overwhelming ratio of human to pathogen DNA in samples with low pathogen abundance, which is typical of most clinical specimens. Microbial DNA enrichment methods offer the potential to relieve this limitation by improving this ratio. Two commercially available enrichment kits, the NEBNext Microbiome DNA Enrichment Kit and the Molzym MolYsis Basic kit, were tested for their ability to enrich for microbial DNA from resected arthroplasty component sonicate fluids from prosthetic joint infections or uninfected sonicate fluids spiked with Staphylococcus aureus. Using spiked uninfected sonicate fluid there was a 6-fold enrichment of bacterial DNA with the NEBNext kit and 76-fold enrichment with the MolYsis kit. Metagenomic whole genome sequencing of sonicate fluid revealed 13- to 85-fold enrichment of bacterial DNA using the NEBNext enrichment kit. The MolYsis approach achieved 481- to 9580-fold enrichment, resulting in 7 to 59% of sequencing reads being from the pathogens known to be present in the samples. These results demonstrate the usefulness of these tools when testing clinical samples with low microbial burden using next generation sequencing.
Keywords: Metagenomics, Whole genome sequencing, Enrichment, Clinical samples, Pathogen detection
1. Introduction
Next-generation sequencing is a powerful tool used for a wide range of applications, including environmental and human microbiome analysis, profiling of cancer cells, gene expression analysis, and sequencing of human genomes for variants related to diseases. One exciting and expanding area of investigation is the use of metagenomic whole genome sequencing (WGS) for diagnosing infection. The potential to detect and identify causative organisms that are difficult to find by conventional methods in an unbiased way, as well as provide insight into the characteristics important for clinical management, such as antibiotic resistance and virulence, has garnered great interest in clinical medicine (Goldberg et al., 2015). This was highlighted by the use of this technology in the diagnosis of neuroleptospirosis in a 14 year old boy (Wilson et al., 2014), as well the identification of multiple viruses in cases of encephalitis (Quan et al., 2010; Hoffmann et al., 2015; Naccache et al., 2015) and detection of pathogens associated with diarrhea (Zhou et al., 2015; Moore et al., 2015).
Perhaps the largest barrier to the promise of WGS-based infectious diseases diagnosis is the inability to meaningfully enrich the yield of non-human DNA from human samples. Without the ability to target DNA sequencing, low microbial burden means that the overwhelming majority of DNA sequenced then comes from host cells rather than the pathogen(s). For example, only 0.0046% of reads (475 out of 10,196,620) came from Leptospira species in the above-mentioned neuroleptospirosis case (Wilson et al., 2014), and only 0.0012% of reads (1612 out of 134,068,968 reads) were attributable to Astrovirus in an encephalitis case thought to be caused by this virus (Naccache et al., 2015). While bioinformatic tools exist to help identify and remove human reads (Zhang et al., 2015; Ames et al., 2013), greater sequencing depths are necessary to obtain enough pathogen reads to identify pathogens and extract useful information (e.g., to assess resistance, virulence, strain-type). This increased sequencing depth can quickly escalate the costs of sequencing.
Commercial tools have emerged which are designed to address this problem by enriching for microbial DNA. One method, New England Biolab’s NEBNext Microbiome DNA Enrichment kit, takes advantage of human and other higher order eukaryotic DNA having high CpG methylation rates. By using the methylated CpG-specific binding protein MBD2 fused to a human IgG Fc fragment, human DNA is selectively bound and separated using Protein A-bound magnetic beads (Feehery et al., 2013). An alternative approach, utilized by Molzym’s MolYsis kit, is to selectively lyse human cells using chaotropic reagents and degrade any released DNA with DNase prior to extraction of DNA from microorganisms. Both techniques have been shown to be effective at enriching microbial DNA (Feehery et al., 2013; Zheng et al., 2014; Benitez-Paez et al., 2013; Gebert et al., 2008; Handschur et al., 2009; Hansen et al., 2009; Horz et al., 2008; Loonen et al., 2012; Votintseva et al., 2015), however no direct comparisons between the approaches have been reported. Additionally, previous studies using the MolYsis kit focused on improving techniques such as real-time PCR, whereas use of these methods for WGS, where microbial DNA enrichment could be even more useful, has been less studied (Votintseva et al., 2015). Other methods such as host cell lysis with detergents (Hasan et al., 2016) or ox bile (Zhou & Pollard, 2012) and immunoprecipitation of DNA with inactive methyl-specific restriction endonucleases (Barnes et al., 2014; Liu et al., 2016) have also been reported, but are not available as commercial products.
Whole genome amplification (WGA) by multiple displacement amplification (MDA) is one technique used to amplify DNA for methods such as library preparation for next generation sequencing. MDA typically uses the high-fidelity phi29 polymerase combined with random hexamer primers to amplify DNA in a non-PCR based isothermal reaction (Dean et al., 2001). WGA is particularly useful in situations where very little DNA is present, such as single cell sequencing or low biomass environmental samples (Hannemann et al., 2011; Rodrigue et al., 2009; Yilmaz et al., 2010). WGA does, however, have drawbacks, as amplification bias and contaminant DNA in reagents has been observed (Yilmaz et al., 2010; Blainey & Quake, 2011; de Bourcy et al., 2014; Probst et al., 2015).
Prosthetic joint infection (PJI) is a devastating complication of total joint arthroplasty (Tande & Patel, 2014). Targeted treatment requires identification of the causative pathogen(s), which can be challenging (Osmon et al., 2013). Low organism burden, previous antibiotic treatment, polymicrobial infection, and infection by fastidious organisms all complicate the detection and identification of pathogens. WGS as a diagnostic tool has the potential to mitigate many of these factors; however, the low microbial burden in these infections remains a challenge. This situation is not unlike that in other serious infections, such as central nervous system infection, and endocarditis and other endovascular infections.
Herein, microbial DNA enrichment tools were tested for their ability to improve bacterial DNA yields for sequencing, using clinical samples from patients with PJI. Results should inform the ideal use of metagenomics approaches to diagnose bacterial infection.
2. Materials and methods
2.1. Samples
Samples were collected under the Mayo Clinic Institutional Review Board protocol 10–005,574. Sonicate fluid samples were prepared from resected prosthetic hip and knee components in Mayo Clinic’s Clinical Microbiology Laboratory using previously-described vortexing/sonication methods (Trampuz et al., 2007; Piper et al., 2009). Negative sonicate fluid samples were selected which had no clinical, laboratory, pathological, or microbiological findings suggesting infection. A methicillin-resistant Staphylococcus aureus strain (IDRL-6169) previously isolated from a patient with PJI (Vergidis et al., 2011), was used for the spiked sonicate fluid experiments. Three samples meeting the Infectious Diseases Society of America definition of PJI (Osmon et al., 2013), were selected for WGS analysis based on the additional criteria of being monomicrobial, having relatively high bacterial load (> 100 CFUs/10 ml sonicate fluid), and lacking a sinus tract.
2.2. DNA purification and enrichment
DNA isolation was performed using the Mobio BiOstic Bacteremia DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA). Microbial DNA enrichment was performed either before DNA isolation using the MolYsis Basic5 kit (Molzym, Bremen, Germany) or after DNA isolation using the NEBNext Microbiome DNA Enrichment Kit (New England BioLabs, Ipswich, MA), per the manufacturers’ protocols. Agencourt Ampure XP beads (Beckman Coulter, Brea, CA) were used to clean up DNA following the NEBNext Microbiome DNA Enrichment Kit per the manufacturer’s recommendations to remove binding buffer reagents that could interfere with subsequent steps.
2.3. Spiked uninfected sonicate fluid experiments
Sonicate fluid from four clinical samples classified as uninfected were pooled and divided into 500 μl aliquots. S. aureus was suspended in Ringer’s solution and a total of 107 CFUs were spiked into the pool. This inoculum, while larger than expected in clinical samples, was selected to allow for reliable quantification. Unspiked sonicate fluid and S. aureus in Ringer’s solution alone were included as controls. DNA was extracted with or without pretreatment with the MolYsis kit. Aliquots of DNA (400 ng) from spiked sonicate fluid were collected from untreated samples for subsequent enrichment using the NEBNext DNA microbiome kit. Total DNA concentration was measured using a Qubit 2.0 Fluorometer (Thermo Fisher Scientific Inc., Waltham, MA). The percent of bacterial DNA was calculated by measuring the concentration of S. aureus DNA by real-time PCR and dividing by the total measured DNA. Experiments were performed in triplicate. Statistical significance amongst groups was calculated using a Kruskal Wallis test and Wilcoxon rank sum test to directly compare groups.
2.4. Real-time PCR quantification of S. aureus DNA
Real-time PCR was performed using a Roche LightCycler (Roche Diagnostics, Indianapolis IN) with the Roche LightCycler SYBR Green FastStart kit (Roche Applied Science, Penzberg, Germany). Forward primer 5′-TGGAGAGTTTGATCCTGGCTCAG-3′ and reverse primer 5′- TACCGCGGCTGCTGGCAC-3′ targeting the 16S ribosomal RNA gene were used to amplify microbial DNA. Following DNA purification, 2 μl of template DNA was added to 20 μl total volume reactions. LightCycler conditions consisted of a five minute 95 °C preincubation, 40 cycles of amplification at 95 °C for 10 s, 62 °C for 15 s, and 72 °C for 30 s, and a melting curve analysis of 65 °C to 95 °C at 0.10 °C/s. Serial dilutions of S. aureus genomic DNA (IDRL-6169) were used to create standard curves for quantification of S. aureus DNA. Negative controls, consisting of water, were also used.
2.5. Metagenomic whole genome sequencing
All DNA for WGS was amplified using the Illustra GenomiPhi V2 whole genome amplification kit (GE Healthcare Bio-Sciences, Pittsburgh, PA) to obtain sufficient amounts of DNA for library preparation. MolYsis pretreatment routinely resulted in insufficient amounts of DNA for library preparation without WGA, even for kits such as Nextera XT which require as little as 1 ng of DNA. Paired-end libraries were prepared using the NEBNext Ultra DNA Library Prep Kit (New England BioLabs) by the Mayo Clinic Medical Genome Facility. Samples were sequenced with the Illumina HiSeq 2500 in rapid run mode with 2 × 250 bp reads. Samples were either run as single samples or multiplexed with 2 or 6 samples per lane, based on the number of samples available to sequence at a time.
2.6. WGS data analysis
Paired-end reads were pre-processed using seqtk (version 1.0-r82, [https://github.com/lh3/seqtk]), Trimmomatic (version 0.35) (Bolger et al., 2014) and the Livermore Metagenomics Analysis Toolkit (LMAT) (Ames et al., 2013) with its pre-processing scripts. BioBloom tools was used to pre-filter human and PhiX reads (Chu et al., 2014). Taxonomy for individual reads was assigned using LMAT with the kML + Human.v4-14.20.g10.db database, which attempts to make a taxonomic assignment to all reads present. Reads assigned to the known pathogen’s genus group (including at the species, strains, and species’ mobile genetic elements) were considered as being from the known pathogen. The percent of reads from the pathogen was calculated by dividing the number of assigned pathogen reads by the total number of reads prior to pre-processing.
3. Results
Pooled sonicate fluid from prostheses resected due to aseptic failure was spiked with S. aureus and used as a tool to compare the ability of the different methods studied to enrich for bacterial DNA. Spiked sonicate fluid underwent DNA extraction alone, with pretreatment with MolYsis prior to DNA extraction, or with the NEBNext Microbiome DNA enrichment kit after DNA extraction. The percent of S. aureus DNA was then determined by measuring the amount of S. aureus DNA by real-time PCR in relation to the total DNA concentration.
DNA extraction of spiked sonicate fluid without enrichment yielded 1.1% of the total DNA being from S. aureus (Fig. 1). Removal of human DNA with the NEBNext microbial DNA enrichment kit improved the relative amount of S. aureus DNA to 6.1%, representing a 5.7-fold enrichment of bacterial DNA. Treatment of samples with the MolYsis kit prior to DNA extraction resulted in a significantly higher proportion of S. aureus DNA concentration at 81%, representing a 76-fold increase in bacterial DNA. The average bacterial DNA concentration in the spiked sonicate samples was 0.27 ng/μl in the unenriched samples and 0.25 ng/μl in the MolYsis treated samples. Unspiked sonicate fluid controls contained a calculated 0.03% bacterial DNA (6.8 pg/μl), indicating minimal influence of any bacterial DNA that may be present. The percent of bacterial DNA was significantly different between the unenriched and both enriched samples (p=0.0495).
Fig. 1.
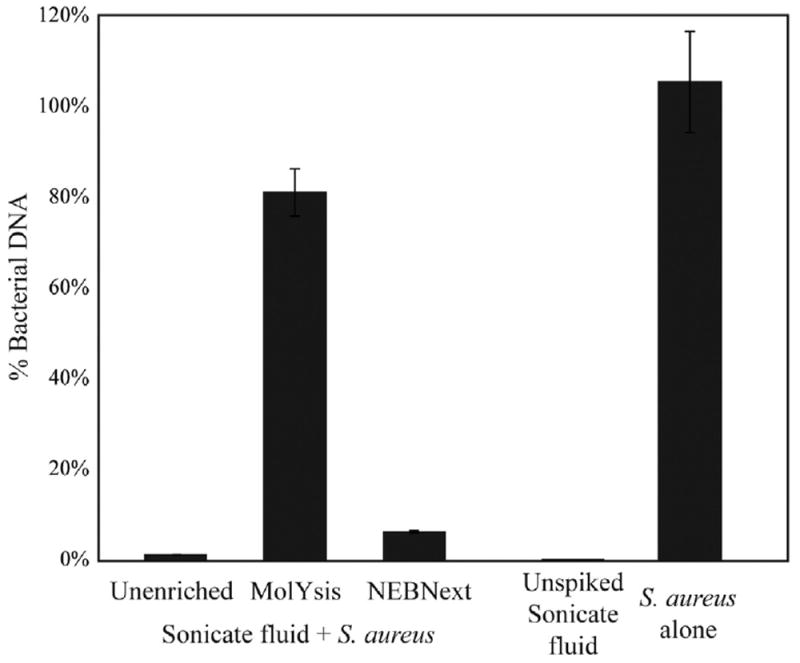
Enrichment of S. aureus DNA in spiked sonicate fluid samples. Culture-negative sonicate fluid was spiked with S. aureus prior to enrichment with MolYsis or NEBNext microbiome DNA enrichment kits and DNA purification. Unspiked sonicate fluid and S. aureus in Ringer’s solution were included as controls. Percent bacterial DNA was calculated by comparing the S. aureus DNA content by real-time PCR results for S. aureus 16S ribosomal RNA gene to total DNA in the sample as measured using a Qubit fluorometer.
The enrichment methods were also tested with sonicate fluids from clinical PJI cases. These infections typically have bacterial loads much lower than those in the spiked sonicate fluids studied. Three monomicrobial PJI sonicate samples were selected, based on their having a high microbial burden of a single pathogen. These samples had previously been evaluated in the Mayo Clinic Clinical Microbiology Laboratory and, by culture, only yielded S. aureus, Staphylococcus epidermidis, and Enterococcus faecalis from the respective samples. The samples were purified as in the spiked experiments with either no enrichment, MolYsis pretreatment, or enrichment with the NEBNext Microbiome kit. Following WGA and metagenomic sequencing, LMAT (Ames et al., 2013) was used to assign taxonomy to each read in order to determine the relative amount of sequences that came from the known PJI pathogen for each infection. All reads that were assigned to the known pathogen’s genus were included in the pathogen read calculations as this tool is unable to reliably classify the majority of reads to the species level for some groups (e.g., Enterococcus species and coagulase-negative Staphylococcus species) and therefore assigns them to the lowest common ancestor, which is at the genus level. WGS analysis revealed a pattern of enrichment similar to that of the spiked sonicate fluids. Without enrichment, 0.0062 to 0.016% of reads were from the known pathogens (Table 1). Enrichment with the NEBNext microbial DNA enrichment kit improved this yield to 0.18 to 0.53%, representing a 13- to 85-fold enrichment of bacterial DNA. As with the spiked samples, MolYsis treatment resulted in higher enrichment, with 7.0 to 59.4% of reads being from the known pathogen, representing a 481- to 9580-fold increase over the unenriched sample. Reads were mapped to the species’ representative genome to confirm uniform coverage of reads (Fig. S1). Notably, other bacterial and viral species were also detected in the samples (Table S1), particularly Pseudomonas, Streptococcus, and Propionibacterium species, a finding consistent with previous reports of common contaminants in metagenomic studies of low biomass samples (Salter et al., 2014; Laurence et al., 2014). Similar types of contamination were also found in negative controls consisting of the WGA kit alone with no template added (Table S1), albeit at higher levels due to the lack of template DNA to compete with contaminant DNA. This suggests that the primary source for these contaminant reads is the WGA kit itself.
Table 1.
Effect of enrichment methods by metagenomic whole genome sequencing. PJI samples underwent enrichment with either MolYsis or NEBNext microbiome kits prior to metagenomic WGS sequencing. “% of reads indicates” the percentage of all assigned reads attributable to the known pathogen genus as assigned by LMAT relative to the total number of reads. Read numbers are reported in parentheses. “Enrichment factor” indicates the fold increase of percent of assigned reads compared to the unenriched sample.
| No enrichment | NEBNext microbiome DNA enrichment | MolYsis enrichment | |
|---|---|---|---|
| S. aureus PJI | |||
| % of reads | 0.016% (4158 of 25,609,460) | 0.21% (350,625 of 169,981,133) | 7.7% (2,286,890 of 29,530,730) |
| Enrichment factor | 13× | 481× | |
| S. epidermidis PJI | |||
| % of reads | 0.0071% (1682 of 23,606,476) | 0.18% (133,680 of 74,544,475) | 7.0% (2,268,087 of 32,184,381) |
| Enrichment factor | 25× | 986× | |
| E. faecalis PJI | |||
| % of reads | 0.0062% (1671 of 26,949,030) | 0.53% (497,206 of 94,522,959) | 59.4% (16,407,878 of 27,643,294) |
| Enrichment factor | 85× | 9580× |
4. Discussion
The very low relative abundance of bacterial to human DNA in many clinical specimens deriving from patients with infection presents a unique challenge when using WGS to detect and identify pathogens. We sought to compare the effectiveness of two commercially available kits in enriching for bacterial DNA. Whether testing uninfected sonicate fluid spiked with S. aureus or PJI samples harboring known pathogens, we observed that both techniques achieved the goal of enriching for bacterial DNA, however the MolYsis method was more effective in achieving this goal, with an enrichment of nearly 500- to 10,000-fold achieved in clinical PJI samples based on metagenomic WGS.
Microbial DNA enrichment methods such as these are powerful tools for the identification and characterization of pathogenic or commensal bacteria. By increasing the relative amount of bacterial DNA, one can go from barely detecting an organism of interest to obtaining enough sequencing reads to assemble nearly complete genomes at sufficient depths to carry out additional characterization such as SNP analysis, antimicrobial resistance or virulence prediction or strain-typing. This has the potential to take WGS approaches beyond pathogen detection to providing information useful in treatment decisions, such as selection of the most appropriate antibiotic (Bradley et al., 2015; Stoesser et al., 2013), and to assessing modes of transmission.
The studied enrichment methods are not without their limitations. There are limited studies regarding the extent to which bacterial DNA is removed during the protocols. Horz et al. tested caries and periodontal samples after MolYsis extraction and reported a wide range of 3.3 to 100% of bacterial DNA remaining after extraction, as measured by16S rRNA gene quantitation (Horz et al., 2010). With the MolYsis approach there are questions as to whether bacteria with weak or absent cell walls (e.g., Mycoplasma, Ureaplasma, or Chlamydia species), or those previously exposed to cell wall-targeting antibiotics, would be lysed and removed by the technique (Horz et al., 2010). While many bacterial and fungal species have been reported as being detectable after MolYsis purification (Gebert et al., 2008; Horz et al., 2008; Votintseva et al., 2015; Meurs et al., 2011; Xu et al., 2012), these studies have been qualitative in nature without any measurement of how much microbial DNA might be lost. Studies examining effects on mixed communities would certainly be beneficial, since biases are likely introduced with this technique. MBD2-Fc proteins utilized by the NEBNext microbiome kit also lack reported data on the impact on microbial DNA recovery. While MBD2 has a high affinity for methylated CpG found in vertebrate DNA, it does still bind non-methylated CpG (Fraga et al., 2003). Bacteria and fungi also methylate cytosines at variable rates, depending on the species and conditions, though typically much less so than vertebrates, particularly at CpG motifs (Fang et al., 2012; Antequera et al., 1984; Lee et al., 2015); this low level of CpG methylation could contribute to decreased specificity. Studies from the manufacturer show that the proportions of bacterial DNA from human saliva or whole black molly fish were largely unaffected by MBD2-Fc enrichment, though some differences were measured, in particular a decrease in Neisseria flavescens reads (Feehery et al., 2013).
The studies presented here do have limitations. We studied sonicate fluid from removed prostheses, which are largely liquid, with variable amounts and sizes of solid tissue pieces. Whether the MolYsis methods work as well for solid tissue samples has not been reported, but it may not be as efficient for these sample types and some degree of tissue disruption/ homogenization may be necessary for maximal effect. In these settings, the MBD2-Fc based approach may be just as, if not more, efficient at microbial DNA enrichment. Because our PJI samples were actual clinical specimens, we cannot know for sure what the true microbial DNA content of the samples was prior to enrichment. Additionally, we used whole genome amplification of all samples prior to sequencing in order to obtain sufficient quantities of DNA for library preparation, a step that could alter proportions of DNA within samples (Probst et al., 2015). WGA has been shown to introduce bias based on GC content (Yilmaz et al., 2010; Probst et al., 2015; Direito et al., 2014), DNA fragment size (Direito, 2014), and relative starting abundance (Raghunathan et al., 2005). While these studies have evaluated the biases introduced in analysis of microbial communities, the impact of WGA on human to bacterial DNA ratios remains unclear. However, the bias against low abundance DNA raises the possibility of exaggerated ratios of microbial DNA before and after enrichment. Despite this, the similar trends observed using spiked sonicate fluid samples, which did not undergo WGA, are reassuring that the WGS results are not simply due to this bias.
Evaluation of the metagenomic WGS results reveals reads from a variety of species beyond the known pathogens identified by culture (Table S1). This can be largely tracked to the WGA kit as no template controls contained the same species considered to be contaminants in PJI samples. Further studies evaluating and comparing WGA kits are needed. This serves as an example of the caution that must be exercised when interpreting metagenomic sequencing data and the importance of controls for these methods, particularly with the increasing interest of these approaches in clinical diagnostics.
Investigators looking at ways to enrich for microbial DNA must take many factors into consideration when choosing the method most appropriate for their studies. The type of sample (e.g., solid tissue versus liquid), most likely pathogen(s) of interest, cost, and required extent of enrichment must all be carefully considered. It should be stressed that the enrichment factor is highly dependent on the starting relative content of bacterial DNA. If the same amount of host DNA is removed, a sample with lower microbial content will have a higher fold-enrichment than the same sample with higher initial microbial DNA, i.e. if microbial DNA made up 1% of the total DNA prior to enrichment, then 500-fold enrichment is not possible.
In conclusion, both methods tested were effective at enriching for microbial DNA, although the MolYsis kit provided for the greater enrichment of the two. We also show that PJI can be diagnosed using a metagenomic strategy.
Supplementary Material
Acknowledgments
We thank the Mayo Clinic Center for Individualized Medicine, Mr. Bruce Eckloff and the Mayo Clinic Medical Genome Facility for assistance with WGS library preparation and sequencing.
Footnotes
Research reported in this publication was supported by the National Institutes of Health under award number R01 AR056647. N.C. is also supported by RO1 CA179243 and R.P. is also supported by R01 AI91594. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.mimet.2016.05.022.
References
- Ames SK, Hysom DA, Gardner SN, Lloyd GS, Gokhale MB, Allen JE. Scalable metagenomic taxonomy classification using a reference genome database. Bioinformatics. 2013;29:2253–2260. doi: 10.1093/bioinformatics/btt389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antequera F, Tamame M, Villanueva JR, Santos T. DNA methylation in the fungi. J Biol Chem. 1984;259:8033–8036. [PubMed] [Google Scholar]
- Barnes HE, Liu G, Weston CQ, King P, Pham LK, Waltz S, Helzer KT, Day L, Sphar D, Yamamoto RT, Forsyth RA. Selective microbial genomic DNA isolation using restriction endonucleases. PLoS One. 2014;9:e109061. doi: 10.1371/journal.pone.0109061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benitez-Paez A, Alvarez M, Belda-Ferre P, Rubido S, Mira A, Tomas I. Detection of transient bacteraemia following dental extractions by 16S rDNA pyrosequencing: a pilot study. PLoS One. 2013;8:e57782. doi: 10.1371/journal.pone.0057782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blainey PC, Quake SR. Digital MDA for enumeration of total nucleic acid contamination. Nucleic Acids Res. 2011;39:e19. doi: 10.1093/nar/gkq1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley P, Gordon NC, Walker TM, Dunn L, Heys S, Huang B, Earle S, Pankhurst LJ, Anson L, de Cesare M, Piazza P, Votintseva AA, Golubchik T, Wilson DJ, Wyllie DH, Diel R, Niemann S, Feuerriegel S, Kohl TA, Ismail N, Omar SV, Smith EG, Buck D, McVean G, Walker AS, Peto TE, Crook DW, Iqbal Z. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat Commun. 2015;6:10063. doi: 10.1038/ncomms10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Sadeghi S, Raymond A, Jackman SD, Nip KM, Mar R, Mohamadi H, Butterfield YS, Robertson AG, Birol I. BioBloom tools: fast, accurate and memory-efficient host species sequence screening using bloom filters. Bioinformatics. 2014;30:3402–3404. doi: 10.1093/bioinformatics/btu558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bourcy CF, De Vlaminck I, Kanbar JN, Wang J, Gawad C, Quake SR. A quantitative comparison of single-cell whole genome amplification methods. PLoS One. 2014;9:e105585. doi: 10.1371/journal.pone.0105585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean FB, Nelson JR, Giesler TL, Lasken RS. Rapid amplification of plasmid and phage DNA using phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 2001;11:1095–1099. doi: 10.1101/gr.180501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Direito SO, Zaura E, Little M, Ehrenfreund P, Roling WF. Systematic evaluation of bias in microbial community profiles induced by whole genome amplification. Environ Microbiol. 2014;16:643–657. doi: 10.1111/1462-2920.12365. [DOI] [PubMed] [Google Scholar]
- Fang G, Munera D, Friedman DI, Mandlik A, Chao MC, Banerjee O, Feng Z, Losic B, Mahajan MC, Jabado OJ, Deikus G, Clark TA, Luong K, Murray IA, Davis BM, Keren-Paz A, Chess A, Roberts RJ, Korlach J, Turner SW, Kumar V, Waldor MK, Schadt EE. Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat Biotechnol. 2012;30:1232–1239. doi: 10.1038/nbt.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feehery GR, Yigit E, Oyola SO, Langhorst BW, Schmidt VT, Stewart FJ, Dimalanta ET, Amaral-Zettler LA, Davis T, Quail MA, Pradhan S. A method for selectively enriching microbial DNA from contaminating vertebrate host DNA. PLoS One. 2013;8:e76096. doi: 10.1371/journal.pone.0076096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Montoya G, Taysavang P, Wade PA, Esteller M. The affinity of different MBD proteins for a specific methylated locus depends on their intrinsic binding properties. Nucleic Acids Res. 2003;31:1765–1774. doi: 10.1093/nar/gkg249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebert S, Siegel D, Wellinghausen N. Rapid detection of pathogens in blood culture bottles by real-time PCR in conjunction with the pre-analytic tool MolYsis. J Infect. 2008;57:307–316. doi: 10.1016/j.jinf.2008.07.013. [DOI] [PubMed] [Google Scholar]
- Goldberg B, Sichtig H, Geyer C, Ledeboer N, Weinstock GM. Making the leap from research laboratory to clinic: challenges and opportunities for next-generation sequencing in infectious disease diagnostics. mBio. 2015;6 doi: 10.1128/mBio.01888-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschur M, Karlic H, Hertel C, Pfeilstocker M, Haslberger AG. Preanalytic removal of human DNA eliminates false signals in general 16S rDNA PCR monitoring of bacterial pathogens in blood. Comp Immunol Microbiol Infect Dis. 2009;32:207–219. doi: 10.1016/j.cimid.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Hannemann J, Meyer-Staeckling S, Kemming D, Alpers I, Joosse SA, Pospisil H, Kurtz S, Gorndt J, Puschel K, Riethdorf S, Pantel K, Brandt B. Quantitative high-resolution genomic analysis of single cancer cells. PLoS One. 2011;6:e26362. doi: 10.1371/journal.pone.0026362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen WL, Bruggeman CA, Wolffs PF. Evaluation of new preanalysis sample treatment tools and DNA isolation protocols to improve bacterial pathogen detection in whole blood. J Clin Microbiol. 2009;47:2629–2631. doi: 10.1128/JCM.00821-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan MR, Rawat A, Tang P, Jithesh PV, Thomas E, Tan R, Tilley P. Depletion of human DNA in spiked clinical specimens to improve the sensitivity of pathogen detection by next generation sequencing. J Clin Microbiol. 2016 doi: 10.1128/JCM.03050-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann B, Tappe D, Hoper D, Herden C, Boldt A, Mawrin C, Niederstrasser O, Muller T, Jenckel M, van der Grinten E, Lutter C, Abendroth B, Teifke JP, Cadar D, Schmidt-Chanasit J, Ulrich RG, Beer M. A variegated squirrel Bornavirus associated with fatal human encephalitis. N Engl J Med. 2015;373:154–162. doi: 10.1056/NEJMoa1415627. [DOI] [PubMed] [Google Scholar]
- Horz HP, Scheer S, Huenger F, Vianna ME, Conrads G. Selective isolation of bacterial DNA from human clinical specimens. J Microbiol Methods. 2008;72:98–102. doi: 10.1016/j.mimet.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Horz HP, Scheer S, Vianna ME, Conrads G. New methods for selective isolation of bacterial DNA from human clinical specimens. Anaerobe. 2010;16:47–53. doi: 10.1016/j.anaerobe.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Laurence M, Hatzis C, Brash DE. Common contaminants in next-generation sequencing that hinder discovery of low-abundance microbes. PLoS One. 2014;9:e97876. doi: 10.1371/journal.pone.0097876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WC, Anton BP, Wang S, Baybayan P, Singh S, Ashby M, Chua EG, Tay CY, Thirriot F, Loke MF, Goh KL, Marshall BJ, Roberts RJ, Vadivelu J. The complete methylome of Helicobacter pylori UM032. BMC Genomics. 2015;16:424. doi: 10.1186/s12864-015-1585-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Weston CQ, Pham LK, Waltz S, Barnes H, King P, Sphar D, Yamamoto RT, Forsyth RA. Epigenetic segregation of microbial genomes from complex samples using restriction endonucleases HpaII and McrB. PLoS One. 2016;11:e0146064. doi: 10.1371/journal.pone.0146064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loonen AJ, Jansz AR, Stalpers J, Wolffs PF, van den Brule AJ. An evaluation of three processing methods and the effect of reduced culture times for faster direct identification of pathogens from BacT/ALERT blood cultures by MALDI-TOF MS. European journal of clinical microbiology & infectious diseases]–>Eur J Clin Microbiol Infect Dis. 2012;31:1575–1583. doi: 10.1007/s10096-011-1480-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meurs KM, Heaney AM, Atkins CE, DeFrancesco TC, Fox PR, Keene BW, Kellihan HB, Miller MW, Oyama MA, Oaks JL. Comparison of polymerase chain reaction with bacterial 16S primers to blood culture to identify bacteremia in dogs with suspected bacterial endocarditis. J Vet Intern Med/American College of Veterinary Internal Medicine. 2011;25:959–962. doi: 10.1111/j.1939-1676.2011.0742.x. [DOI] [PubMed] [Google Scholar]
- Moore NE, Wang J, Hewitt J, Croucher D, Williamson DA, Paine S, Yen S, Greening GE, Hall RJ. Metagenomic analysis of viruses in feces from unsolved outbreaks of gastroenteritis in humans. J Clin Microbiol. 2015;53:15–21. doi: 10.1128/JCM.02029-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naccache SN, Peggs KS, Mattes FM, Phadke R, Garson JA, Grant P, Samayoa E, Federman S, Miller S, Lunn MP, Gant V, Chiu CY. Diagnosis of neuroinvasive Astrovirus infection in an immunocompromised adult with encephalitis by unbiased next-generation sequencing. Clin Infect Dis. 2015;60:919–923. doi: 10.1093/cid/ciu912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmon DR, Berbari EF, Berendt AR, Lew D, Zimmerli W, Steckelberg JM, Rao N, Hanssen A, Wilson WR. Diagnosis and management of prosthetic joint infection: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2013;56:e1–e25. doi: 10.1093/cid/cis803. [DOI] [PubMed] [Google Scholar]
- Piper KE, Jacobson MJ, Cofield RH, Sperling JW, Sanchez-Sotelo J, Osmon DR, McDowell A, Patrick S, Steckelberg JM, Mandrekar JN, Fernandez Sampedro M, Patel R. Microbiologic diagnosis of prosthetic shoulder infection by use of implant sonication. J Clin Microbiol. 2009;47:1878–1884. doi: 10.1128/JCM.01686-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst AJ, Weinmaier T, DeSantis TZ, Santo Domingo JW, Ashbolt N. New perspectives on microbial community distortion after whole-genome amplification. PLoS One. 2015;10:e0124158. doi: 10.1371/journal.pone.0124158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan PL, Wagner TA, Briese T, Torgerson TR, Hornig M, Tashmukhamedova A, Firth C, Palacios G, Baisre-De-Leon A, Paddock CD, Hutchison SK, Egholm M, Zaki SR, Goldman JE, Ochs HD, Lipkin WI. Astrovirus encephalitis in boy with X-linked agammaglobulinemia. Emerg Infect Dis. 2010;16:918–925. doi: 10.3201/eid1606.091536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghunathan A, Ferguson HR, Jr, Bornarth CJ, Song W, Driscoll M, Lasken RS. Genomic DNA amplification from a single bacterium. Appl Environ Microbiol. 2005;71:3342–3347. doi: 10.1128/AEM.71.6.3342-3347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigue S, Malmstrom RR, Berlin AM, Birren BW, Henn MR, Chisholm SW. Whole genome amplification and de novo assembly of single bacterial cells. PLoS One. 2009;4:e6864. doi: 10.1371/journal.pone.0006864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, Turner P, Parkhill J, Loman NJ, Walker AW. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12:87. doi: 10.1186/s12915-014-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoesser N, Batty EM, Eyre DW, Morgan M, Wyllie DH, Del Ojo EC, Johnson JR, Walker AS, Peto TE, Crook DW. Predicting antimicrobial susceptibilities for Escherichia coli and Klebsiella pneumoniae isolates using whole genomic sequence data. J Antimicrob Chemother. 2013;68:2234–2244. doi: 10.1093/jac/dkt180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tande AJ, Patel R. Prosthetic joint infection. Clin Microbiol Rev. 2014;27:302–345. doi: 10.1128/CMR.00111-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trampuz A, Piper KE, Jacobson MJ, Hanssen AD, Unni KK, Osmon DR, Mandrekar JN, Cockerill FR, Steckelberg JM, Greenleaf JF, Patel R. Sonication of removed hip and knee prostheses for diagnosis of infection. N Engl J Med. 2007;357:654–663. doi: 10.1056/NEJMoa061588. [DOI] [PubMed] [Google Scholar]
- Vergidis P, Rouse MS, Euba G, Karau MJ, Schmidt SM, Mandrekar JN, Steckelberg JM, Patel R. Treatment with linezolid or vancomycin in combination with rifampin is effective in an animal model of methicillin-resistant Staphylococcus aureus foreign body osteomyelitis. Antimicrob Agents Chemother. 2011;55:1182–1186. doi: 10.1128/AAC.00740-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Votintseva AA, Pankhurst LJ, Anson LW, Morgan MR, Gascoyne-Binzi D, Walker TM, Quan TP, Wyllie DH, Del Ojo EC, Wilcox M, Walker AS, Peto TE, Crook DW. Mycobacterial DNA extraction for whole-genome sequencing from early positive liquid (MGIT) cultures. J Clin Microbiol. 2015;53:1137–1143. doi: 10.1128/JCM.03073-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MR, Naccache SN, Samayoa E, Biagtan M, Bashir H, Yu G, Salamat SM, Somasekar S, Federman S, Miller S, Sokolic R, Garabedian E, Candotti F, Buckley RH, Reed KD, Meyer TL, Seroogy CM, Galloway R, Henderson SL, Gern JE, DeRisi JL, Chiu CY. Actionable diagnosis of neuroleptospirosis by next-generation sequencing. N Engl J Med. 2014;370:2408–2417. doi: 10.1056/NEJMoa1401268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Rudkjobing VB, Simonsen O, Pedersen C, Lorenzen J, Schonheyder HC, Nielsen PH, Thomsen TR. Bacterial diversity in suspected prosthetic joint infections: an exploratory study using 16S rRNA gene analysis. FEMS Immunol Med Microbiol. 2012;65:291–304. doi: 10.1111/j.1574-695X.2012.00949.x. [DOI] [PubMed] [Google Scholar]
- Yilmaz S, Allgaier M, Hugenholtz P. Multiple displacement amplification compromises quantitative analysis of metagenomes. Nat Methods. 2010;7:943–944. doi: 10.1038/nmeth1210-943. [DOI] [PubMed] [Google Scholar]
- Zhang C, Cleveland K, Schnoll-Sussman F, McClure B, Bigg M, Thakkar P, Schultz N, Shah MA, Betel D. Identification of low abundance microbiome in clinical samples using whole genome sequencing. Genome Biol. 2015;16:265. doi: 10.1186/s13059-015-0821-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Deng X, Chen J. Whole-genome sequence of “Candidatus Liberibacter asiaticus” from Guangdong, China. Genome Announcements. 2014;2 doi: 10.1128/genomeA.00273-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Pollard AJ. A novel method of selective removal of human DNA improves PCR sensitivity for detection of Salmonella Typhi in blood samples. BMC Infect Dis. 2012;12:164. doi: 10.1186/1471-2334-12-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wylie K, Ei Feghaly RE, Mihindukulasuriya K, Elward A, Haslam DB, Storch G, Weinstock GM. Metagenomic approach for identification of the pathogens associated with diarrhea in stool specimens. J Clin Microbiol. 2015 doi: 10.1128/JCM.01965-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.