

Abstract
Ligand-independent activation of EphA2 receptor kinase promotes cancer metastasis and invasion. Activating EphA2 receptor tyrosine kinase with small molecule agonist is a novel strategy to treat EphA2 overexpressing cancer. In this study, we performed a lead optimization of a small molecule Doxazosin that was identified as an EphA2 receptor agonist. 33 new analogs were developed and evaluated; a structure−activity relationship was summarized based on the EphA2 activation of these derivatives. Two new derivative compounds 24 and 27 showed much improved activity compared to Doxazosin. Compound 24 possesses a bulky amide moiety, and compound 27 has a dimeric structure that is very different to the parental compound. Compound 27 with a twelve-carbon linker of the dimer activated the kinase and induced receptor internalization and cell death with the best potency. Another dimer with a six-carbon linker has significantly reduced potency compared to the dimer with a longer linker, suggesting that the length of the linker is critical for the activity of the dimeric agonist. To explore the receptor binding characteristics of the new molecules, we applied a docking study to examine how the small molecule binds to the EphA2 receptor. The results reveal that compounds 24 and 27 form more hydrogen bonds to EphA2 than Doxazosin, suggesting that they may have higher binding affinity to the receptor.
Keywords: EphA2, Doxazosin, small molecular ligand, dimer, internalization
Graphical Abstract
1. Introduction
Erythropoietin-producing hepatocellular receptor 2 (EphA2) is one of the receptor tyrosine kinases that regulate cancer cell migration. It has been well documented that EphA2 is overexpressed in many types of human cancers, and the overexpression is correlated with malignant progression and poor prognosis[1–3]. EphA2 in breast epithelial cells has been reported to induce morphological transformation, while in prostate cancer and glioma cell lines, elevated EphA2 expression causes increased chemotactic cell migration and invasion[4,5]. In contrast to its pro-oncogenic roles, other studies indicate that EphA2 activation by its ligand, ephrin-A1, suppresses tumor progression, including induction of apoptosis, inhibition of cell proliferation, and suppression of cell migration[5–8]. In vivo studies demonstrate that EphA2 activation by systemically administered ephrin-A1 decreases tumorigenicity and invasiveness of carcinoma xenografts[5,8,9]. On the other side, transgenic mice with EphA2 deletion show increased susceptibility to carcinogen-induced skin tumorigenesis[10]. These two opposing observations raise the question about the true role of EphA2 in tumor development and metastasis. In fact, EphA2 plays dual roles in tumorigenesis. Under ligand stimulation, EphA2 inhibits cell migration in keeping with the well-established repulsive roles of Eph receptors in regulating cell motility. However, in the absence of ligand, EphA2 promotes cell migration and invision, which is correlated with its expression level[1–3,11,12]. Unfortunately, EphA2 overexpression is often accompanied by loss of expression or mislocalization of ephrin-A1 in breast cancer, glioma and skin tumors. The reduced ligand-dependent activation of EphA2 coupled with increased EphA2 expression and frequent Akt activation provide a permissive environment to promote ligand-independent pro-invasive Akt-EphA2 crosstalk. This interaction may be in part responsible for EphA2 overexpression during tumor progression and the positive correlation of EphA2 expression and unfavorable prognosis[1,2,13–16].
This hypothesis is also supported by an in vivo study demonstrating that ephrin administration inhibited the progression of tumors with high expression of EphA2[5,8]. However, ephrins cannot cross the blood brain barrier, which makes this endogenous ligand less useful for the treatment of gliomas with over expression of EphA2. This fact led us to hypothesize that small molecule agonists for EphA2 can be exploited as novel cancer therapeutics. In a previous study, we identified a small molecule EphA2 agonist Doxazosin using a combination of structure-based virtual screening and cell-based assays[4] .
In the current study, we lead optimized Doxazosin to activate EphA2 with higher potency and reduce the original targeting effect of Doxazosin on adrenoceptor. Several new analogs of Doxazosin showed significantly increased potency to activate EphA2 and induce subsequent receptor internalization. A detailed structure-activity relationship (SAR) was summarized based on the diversely modified structures and the activities. One particular compound 27 with a dimeric structure showed superior activity within the new compound pool. The novel symmetric structure provides us a new scaffold for further optimization.
2. Results and discussion
2.1. Lead optimization and summarization of the SAR
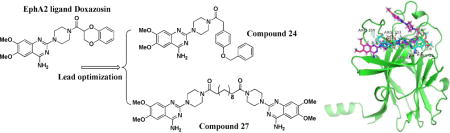
Our previous efforts to develop small molecule EphA2 agonist led to the identification of Doxazosin (2-[4-(2,3-Dihydro-1,4-benzodioxine-2-carbonyl)piperazin-1-yl]-6,7-dimethoxyquinazolin-4-amine), an α1-selective alpha blocker, used to treat high blood pressure and urinary retention associated with benign prostatic hyperplasia (BPH) [17,18]. Doxazosin significantly activated EphA2 at 50μM in vitro, and inhibited prostate tumor metastasis in the mouse model[4]. In the present study, a total of 33 new derivatives based on Doxazosin were synthesized using combinatorial chemistry strategy. First, we kept A and B moieties, and changed C moiety systematically (Figure 1). Subsequently, we synthesized several symmetric molecules with the best C moiety on both sides of the piperazine ring to determine if the whole A and B parts are necessary for the activity, which is adapted from a recent report about a dimeric EphA2 agonist showing promising activity[19]. Based on the symmetric molecular hypothesis, we also designed larger symmetric structures as illustrated by the dimers (Figure 1). We then modified A moiety to ethoxy, B moiety to substituted amines to generate diverse structures and enrich the SAR for the future structure optimization.
Figure 1.
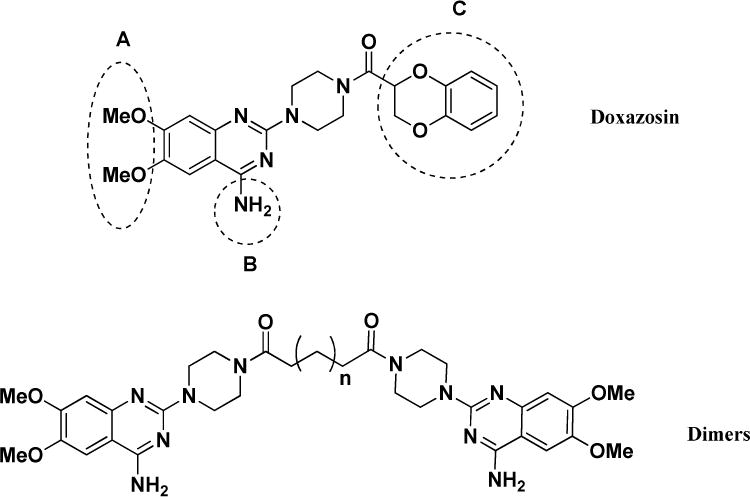
Proposed modification of Doxazosin
In the newly designed derivatives, we explored other different structures at C domain including aromatic rings and alky groups. For the aromatic rings, we also synthesized extended linkers. The synthesis of C moiety substituted derivatives is illustrated in Scheme 1.
Scheme 1.
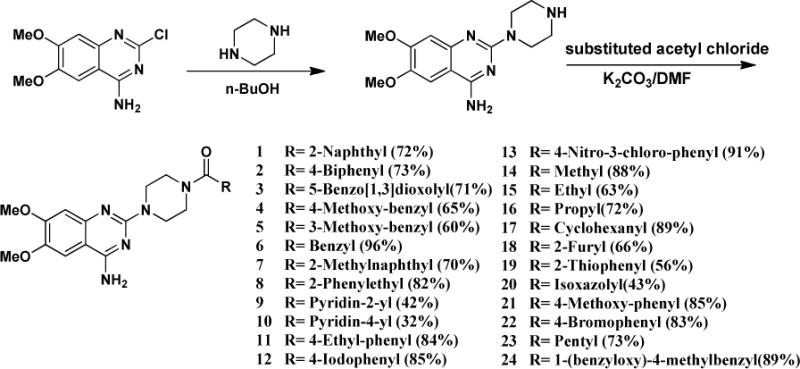
Synthesis of C moiety substituted Doxazosin analogs
As illustrated in Figure 1, we also synthesized dimers with piperazine ring as the core scaffold. A and B whole moiety was replaced by a C moiety structure. The symmetric hypothesis led us to explore the possibility of a bigger dimer based on the whole left domain of the lead compound Doxazosin. Therefore, we synthesized compounds 27 and 28 using different linkers as illustrated in Scheme 3.
Scheme 3.
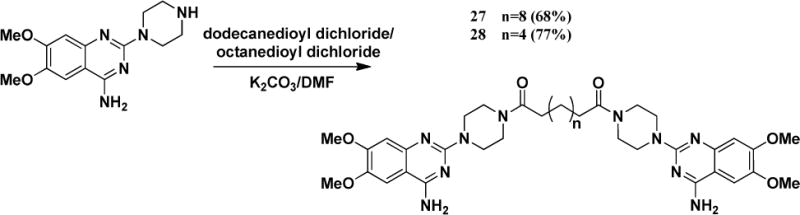
Synthesis of dimer 27 and 28
To investigate if the primary amino group on the pyrimidine ring is needed for the activity, we modified the B moiety into different structures. First, we acetylated Doxazosin and compound 24 to generate compounds 29 and 30, respectively (Scheme 4).
Scheme 4.
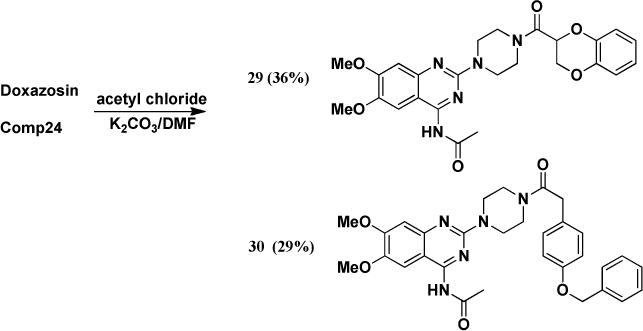
Synthesis of B moiety acetylated analogs
Furthermore, we modified this amino group to alkylated analogs as illustrated in Scheme 5.
Scheme 5.
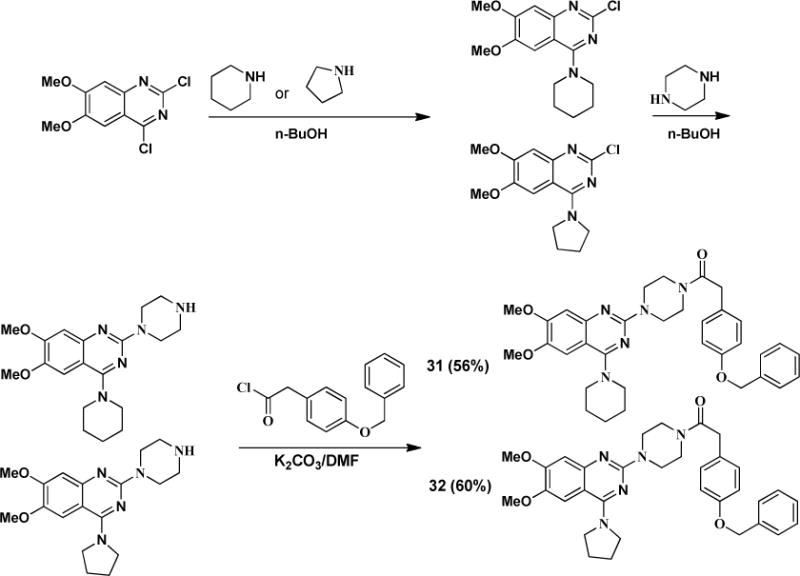
Synthesis of B moiety modified analogs
At last, we modified A moiety to ethoxy group to examine if methoxy is required for the activity. The synthesis is illustrated in Scheme 6.
Scheme 6.
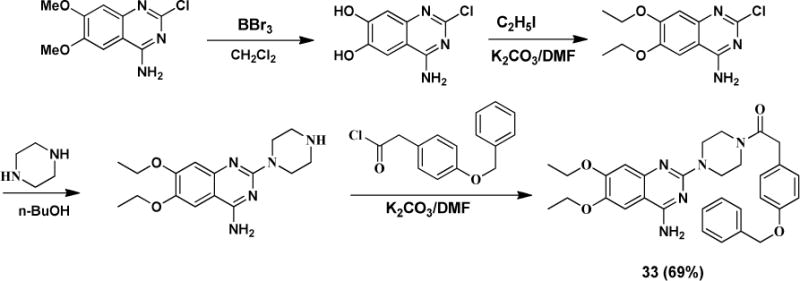
Synthesis of A moiety modified analog
The 33 new analogs show diverse substitution on the three identified structural moieties, and the biological results of the compounds can provide us a detail SAR to elucidate the structure requirement for the activation of EphA2.
To evaluate the compounds, we utilized a MDA-MB-231 breast cancer cell line which overexpresses EphA2 receptor (MDA-MB-231-A2). The ratio of phosphorylated EphA2 protein over total EphA2 protein was measured after treatment with the various compounds as the readout for receptor activation. All the compounds were examined with doses of 2, 10, and 50 μM, and we used 50μM Doxazosin and 0.5% DMSO as the positive and negative control, respectively (Figure 2). EphA2 activation by 50μM Doxazosin was used as the cutoff to identify more potent analogs. Compounds 1–24 all showed different substitution at the C moiety as indicated in Scheme 1. Compounds 1 and 2 with bulky C domain seemed to show higher activity than compound 3. However, 50 μM compounds 1 and 2 did not show a much higher activity than the lead compound Doxazosin at 50 μM. Compounds 4–8 all have one carbon extension at C moiety, and the activity of these compounds are comparable to Doxazosin. Compounds 9, 10, 18, 19 and 20 that have heteroaromatic ring at C moiety showed decreased activity, which rules out other possible substitution with heteroaromatic ring in the future. Compounds 11, 12, 22 and 24 have slightly or very bulky and electron donating substitute group at the aromatic ring of C moiety, and compounds 22 and 24 show similar or significantly increased activity compared to Doxazosin, whereas compound 13 with electron withdrawing substitution group decreases the activity. However, compound 21 shows decreased activity even with an electron donating group at 4′ aromatic ring of C moiety. Compounds 14–17 all have alky substitute groups at C moiety and show decreased activity, suggesting that small non-aromatic substitution at C moiety diminishes the activity and should be avoided in the future modification. Compound 23 has a long alky chain at C moiety and shows slightly decreased activity. Based on the structure of 23, we designed dimer structures as demonstrated by compounds 27 and 28. Compound 27 shows significantly increased activity compared to Doxazosin, whereas compound 28 with short linkage shows decreased activity, suggesting that the length of the linkage of the dimer is critical for the activity. Inspired by the dimer design, we also generated dimer structure based on the piperazine ring as the core as illustrated by compounds 25 and 26. However, both compounds show significantly decreased activity.
Figure 2.
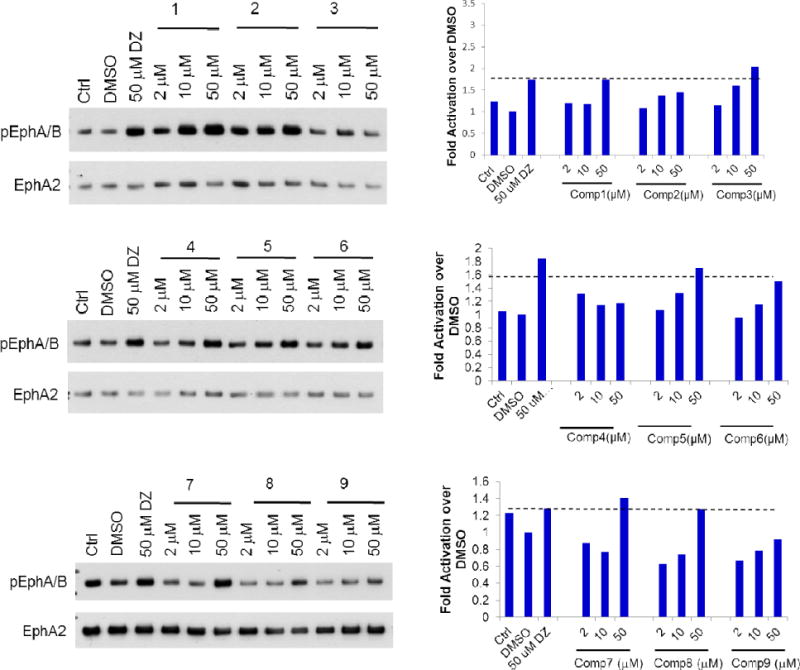
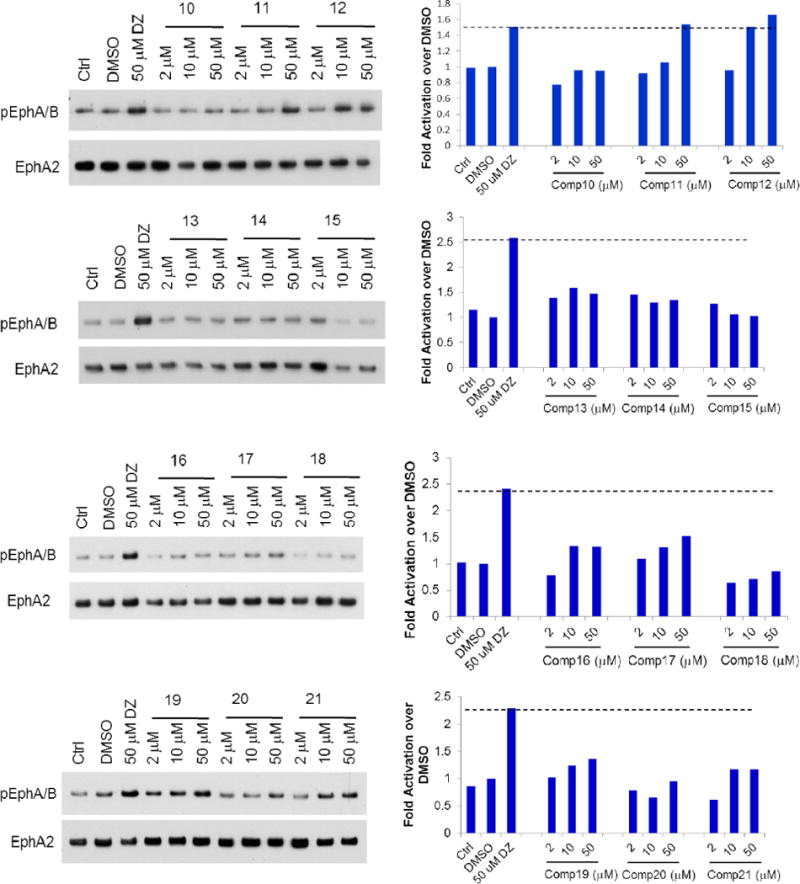
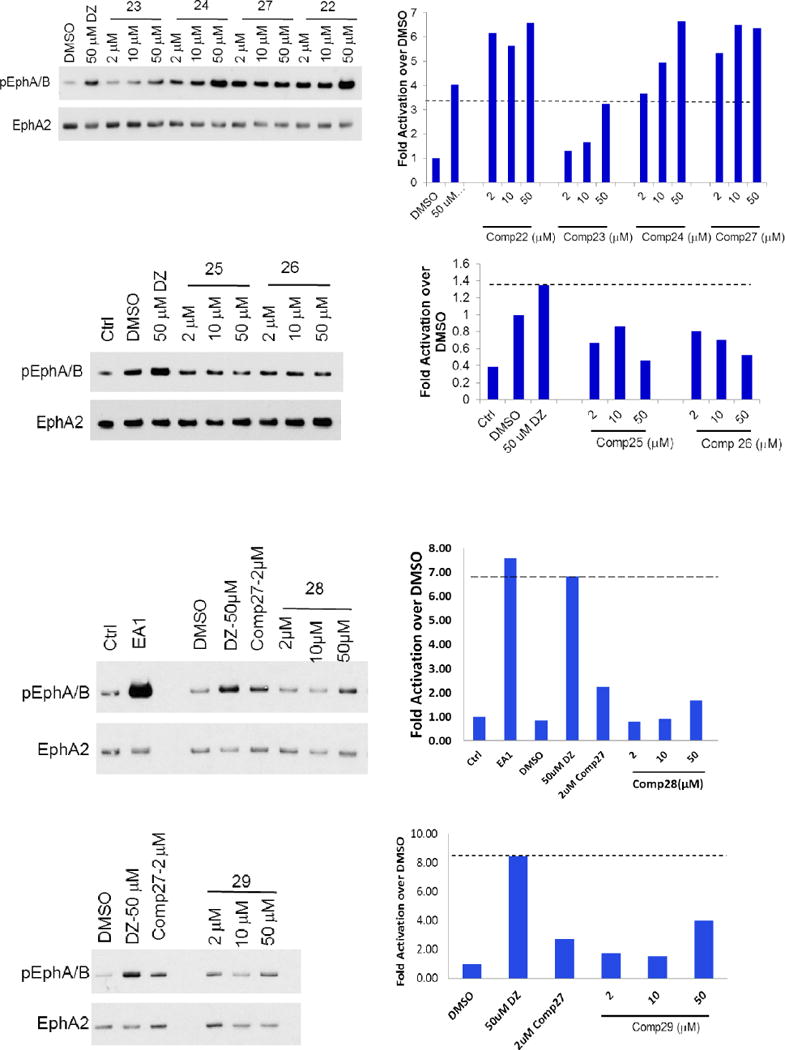
EphA2 activation of the new derivatives. MDA-MB-231-EphA2 cells were treated with the compounds with three doses (in 0.5% DMSO) for 30 minutes and cell lysates were subject to immunoprecipitation and immunoblot for phosphorylated EphA/B kinases (pEphA/B) and total EphA2. Treatment with 1 μg/ml ephrin-A1-Fc (EA1-Fc) or Doxazosin (DZ) 50 μM were served as positive controls. Treatment with 0.5% DMSO was served as vehicle control.
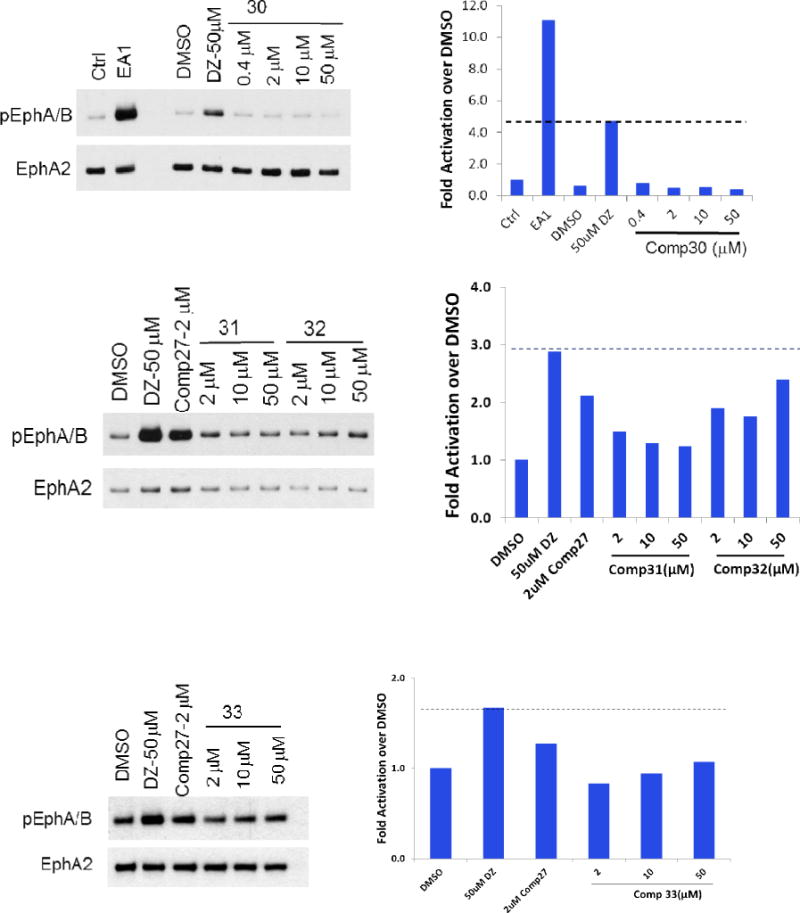
Besides C moiety, we also modified the structures of the A and B moieties. Compounds 29 and 30 were based on compounds Doxazosin and 24, respectively. The amino group was acetylated to generate the new analogs. The results indicate that the amino group seems critical for the activity, and the acetylation significantly diminishes the activity. In compounds 31 and 32, we replaced the amino group with piperidine and pyrrolidine groups, respectively, and all of these changes harm the EphA2 activation. For the A moiety, we changed the dimethoxy of compound 24 to generated compound 33. Unfortunately, this modification diminishes the activity as well. The structure activity relationship demonstrates that A and B moieties of Doxazosin are critical for the activity and C moiety is an open area for further optimization. In addition, the dimer structure of compound 27 provides a new platform to design new core structures for a group of new analogs to activate EphA2.
2.2. EphA2 internalization
EphA2 overexpression has been identified in various different malignant cells[2,6,7,11–14,20,21] Decreased levels of EphA2-ligand binding in cancer cells lead to the aggressive growth of the tumor, which can be overcome in vitro by using either monoclonal antibodies, or small molecule ligand[4,5,8,19,21]. In our previous studies, we have demonstrated that EphA2 ligands negatively regulate tumor cell growth and invasiveness[4]. Ligand binding could even reverse the oncogenic effects of EphA2 overexpression[15,16]. How did EphA2 ligands reverse the oncogenic effects of EphA2 overexpression? Previous studies have demonstrated that ligand binding causes EphA2 to be internalized and degraded[4,22]. Since we have developed small molecule EphA2 ligands based on Doxazosin as the lead compound and they can effectively activate the kinase, it is necessary to determine if the ligand could cause EphA2 internalization.
To directly demonstrate this phenomenon at the cellular level, we used immunofluorescence to monitor localization of EphA2 following the small molecule treatment. This was performed using the MDA-MB-231cell line. This cell line spreads well in culture, thereby facilitating immunofluorescence detection of cell surface and intracellular EphA2 (Figure 3). Ephrin-A1-Fc was used as a positive control, which induced nearly complete internalization of EphA2 within 60 min. Consistent with its agonistic activities, Doxazosin also stimulated significant EphA2 internalization at 50μM. Compound 27, the most active one in the SAR study, was chosen to perform the internalization study. The results showed that EphA2 internalization by compound 27 even happened in very low concentration as 0.4μM, which indicates that compound 27 has much better potency to induce EphA2 internalization than Doxazosin. Together, these data confirm that the new analog indeed triggers EphA2 receptor internalization in keeping with its agonistic activities.
Figure 3. Doxazosin analog compound 27 treatment causes EphA2 receptor internalization.
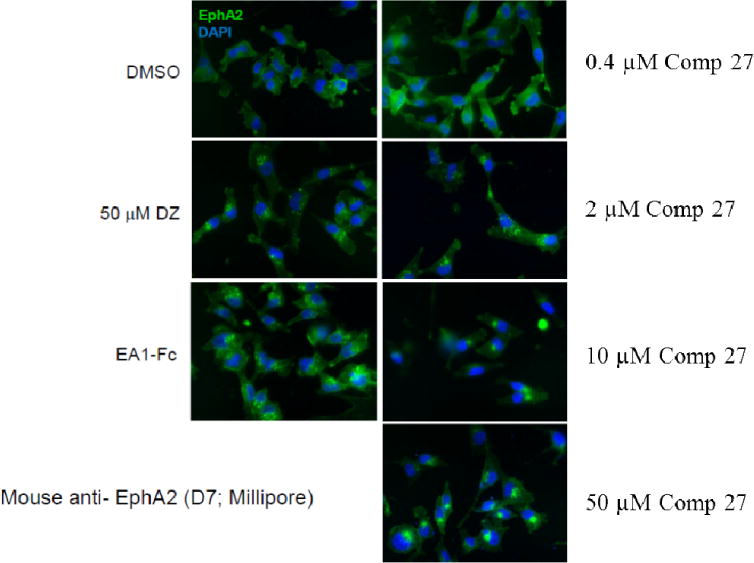
Immunofluorescence staining of MDA-MB-231 cells for EphA2 receptor (green) after treatment for 60 minutes with various concentration of compound 27 in 0.5% DMSO. Treatment with 1 ug/mL ephrin-A1-Fc, 50uM Doxazosin and 0.5% DMSO served as positive and negative controls, respectively. DAPI nuclear staining is shown in blue. Scale bars, 25 um. Cells were seeded on 24-well plates and stimulated after 24–48 hours. The experiment was repeated three times independently, and the representative one is listed.
2.3. Compound 27 inhibits cancer cell colony formation
EphA2 is involved in tumorigenesis, growth and metastasis. Compound 27 can activate EphA2 and induce the internalization of the receptor. Next, we examined if the compound could inhibit tumor growth with a colony formation assay. The same MDA-MB-231 breast cancer cells were treated with compound 27 at various concentrations for two weeks (Figure 4). The results indicated that the compound inhibited colony formation even at nanomolar concentrations. We also determined the activity of compounds 23 and 24. Compound 23 is the monomer of compound 27 and compound 24 showed potent EphA2 activation in the SAR study. The results revealed that compound 23 showed moderate activity to inhibit the colony formation, and compound 24 showed slightly weaker activity compared to compound 27.
Figure 4. Compound 27 inhibits cancer cell colony formation in MDA-MB-231 cells.
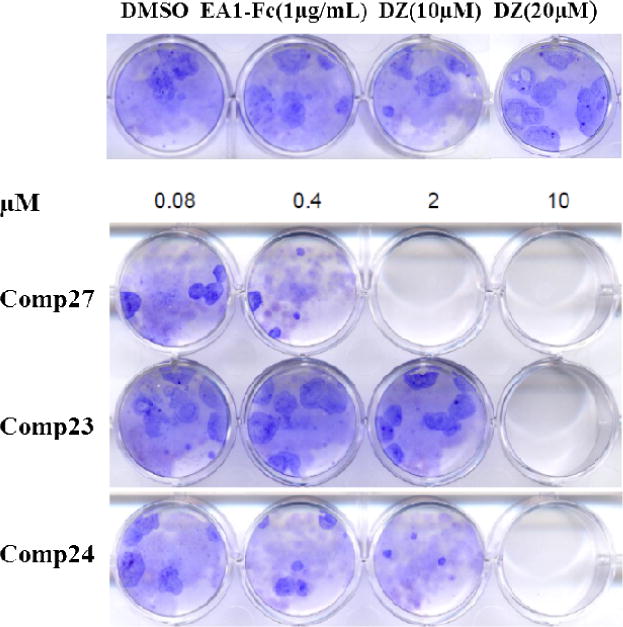
MDA-MB-231 cells (1000 cells/well) were seeded in 12-well plates and cultured for 24 h, followed by treatment with various concentrations of the Doxazosin analogs (0, 0.08, 0.4, 2 and 10 μM) for 14 days. EA1-Fc (1μg/mL) and Doxazosin (10μM and 20μM) were used as positive controls. After washing with PBS, colonies were fixed and stained with crystal violet. The experiment was repeated three times independently, and the representative one is listed.
2.4. Docking of compounds 24 and 27 to EphA2
Doxazosin has been demonstrated as a small molecule ligand of EphA2, and the docking domain has been identified with NMR approach and other biological assays[4,23–25]. Compounds 24 and 27 showed improved EphA2 activation compared to Doxazosin. We would like to see if the new ligand shows any stronger interaction to the binding domain of Doxazosin in EphA2. To further elucidate the binding characteristics of the compounds with EphA2, we performed docking studies with Doxazosin, 24, and 27 with the crystal structure (PDB:3FL7) of the EphA2 extracellular domain[26]. As illustrated in Figure 5 upper panel, the aromatic amino group of Doxazosin forms hydrogen bonds with GLY51, SER68; the oxygen of the Dioxane ring forms hydrogen bond with ARG103. In addition to the polar interaction, the dihydrobenzo-dioxine moiety sits inside a hydrophobic pocket. Compared to Doxazosin, compound 24 forms more hydrogen bonds (Figure 5, middle panel). The methoxy group forms hydrogen bond with GLY51, the aromatic amino group forms hydrogen bonds with GLY51, SER68. The ketone of the amide bond forms hydrogen bond with ARG103. The binding characteristics of compound 24 are quite similar to Doxazosin, and the aromatic group of the amide bond sits inside the same hydrophobic pocket. However, the aromatic group of compound 24 is bulkier than Doxazosin, which pushes the structure a little bit out, and makes the methoxy group closer to GLY51 to form the extra hydrogen bond. The results suggest that compound 24 may have higher binding affinity to EphA2 than Doxazosin, which is consistent with previous EphA2 activation study. However, the most potent compound 27 has very different structure scaffold compared to Doxazosin and compound 24. The binding manner of this ligand looks very different to Doxazosin and compound 24 (Figure 5, low panel). Due to the bulkiness of the structure, it does not fit into the hydrophobic pocket anymore. The molecule adapts itself into two grooves of the protein. Besides the regular hydrophobic interaction, it forms 4 hydrogen bonds as well. One of the nitrogens in the quinazoline ring forms two hydrogen bonds with ARG159, and one of the ketone forms two hydrogen bonds with ARG103. To fit into the two grooves, the length of the linker seems quite critical, and short linker could not provide enough flexibility for the structure to bind to the groove. Compared to compound 27, compound 28 has a shorter multiple carbon linker and does not show any activity to activate EphA2, and could not fit into the two grooves.
Figure 5.
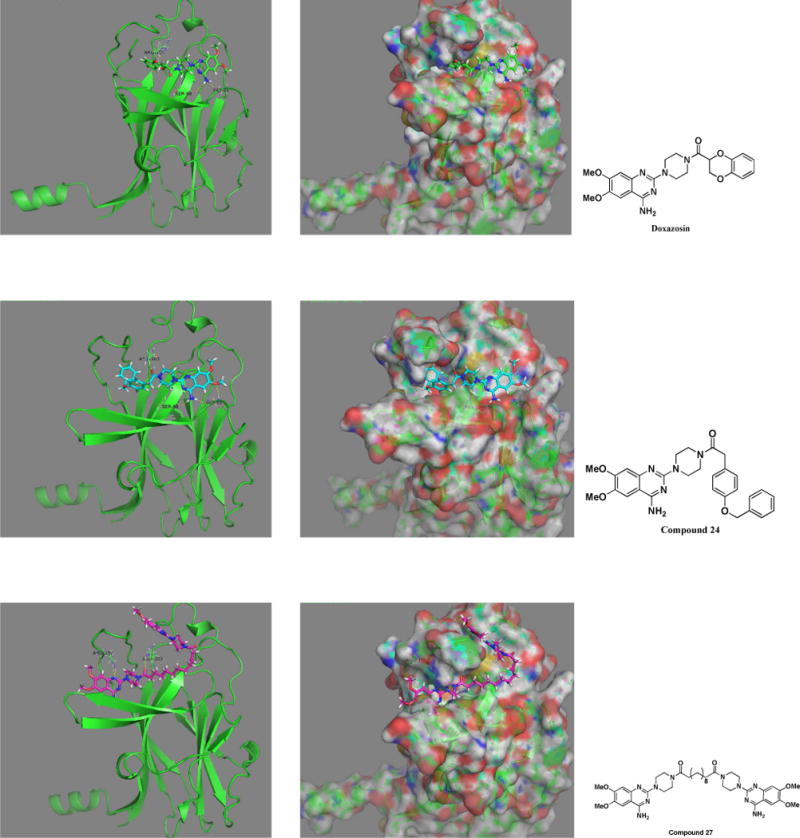
Compounds 27, 24 and Doxazosin docking with EphA2. EphA2 (PDB:3FL7) out membrane part was used to perform the docking study. The original identified Doxazosin binding site was used to fit the compounds. Compound 24 and Doxazosin share the similar binding mode, whereas compound 27 shows a different binding characteristic.
2.5 Adrenoceptor binding activity
Doxazosin is an α-adrenoceptor antagonist, and is well used in clinic for the treatment of hypertension or benign prostate hyperplasia[17,18]. The EphA2 activation of Doxazosin is independent of the α-adrenoceptor activity. During the lead optimization process, we would like to gradually eliminate the α-adrenoceptor binding activity of Doxazosin, and improve the EphA2 activation potency in the new derivatives. To examine if our new analogs still show α-adrenoceptor binding activity, we performed the competition experiments with these ligands. We used Doxazosin as the positive control for the binding assay, and Prazosin as the competitor to the receptor. As shown in Figure 6, Doxazosin shows an IC50 of 0.74 ± 0.30 nM, whereas compounds 24 and 27 show IC50s of 4.28 ± 3.10 nM and 2.50 ± 1.62 nM, respectively. The results suggest that these modifications actually decreased the α-adrenoceptor binding affinity. Although we won’t be able to summarize a structure activity relationship about the α-adrenoceptor binding activity at this stage, the lead optimization process at least showed the feasibility to eliminate the α-adrenoceptor activity of the new derivatives in the future.
Figure 6.
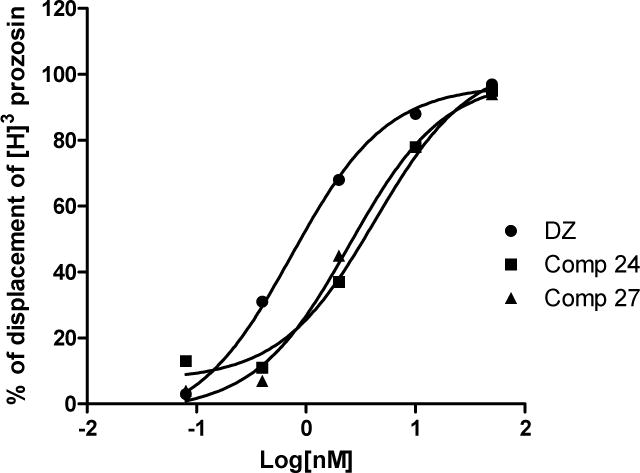
Compounds 27, 24 and Doxazosin binding with α-adrenoceptor. The compounds were tested in triplication. IC50s of the three compounds were listed as mean ± SD
3. Conclusion
We developed a series of EphA2 agonists based on Doxazosin as a lead compound. Several new derivatives showed improved activity to activate EphA2 and induce receptor internalization as well. An SAR was summarized based on the structures of the compounds and their corresponding EphA2 activation. The A and B moieties of Doxazosin are strictly limited to the current structure, and any change in our current optimization leads to decreased or even loss of activity. C moiety seems an open area for further structural optimization in the future. In addition, we developed compound 27 that has the dimer structure, which is different to the main structure scaffold of most analogs. It provides a new platform that can be used to design new dimer core structures as a group of new analogs to activate EphA2.
The two most potent new compounds, 24 and 27, inhibit the colony formation of the cancer cells expressing high level of EphA2. Our molecular docking study revealed that the new compounds may possess stronger interaction to the protein than Doxazosin based on the hydrogen bonds formed between the ligands and EphA2. Finally, the new derivatives showed relatively weaker binding affinity to α-adrenoceptor, suggesting that our lead optimization decreased the original targeting effect of Doxazosin.
4. Experimental section
4.1. Chemistry
Chemicals were commercially available and used as received without further purification unless otherwise noted. Moisture sensitive reactions were carried out under a dry argon atmosphere in flame-dried glassware. Solvents were distilled before use under argon. Thin-layer chromatography was performed on precoated silica gel F254 plates (Whatman). Silica gel column chromatography was performed using silica gel 60Å(Merck, 230–400 Mesh), and hexane/ethyl acetate was used as the elution solvent. Mass spectra were obtained on the electrospray mass spectrometer at Cleveland State University MS facility Center. The molecular weight of the compounds was examined with LC-MS. All the NMR spectra were recorded on a Bruker 400 MHz in either DMSO-d6 or CDCl3. Chemical shifts (δ) for 1H NMR spectra are reported in parts per million to residual solvent protons.
Compounds 1–24, 27 and 28 were prepared as the following procedure
The dimethoxyquinazolin and piperazine were dissolved in n-BuOH with 1 to 5 mole ratio, and the mixture was heated to 100°C for 24 hours, then it was cooled down to room temperature. The precipitated while solid was collected via filtration and washed with n-BuOH. After it was dried, the solid was mixed with various different substituted acetyl chloride and K2CO3 in DMF. It was stirred until the reaction was completed. The reaction was quenched with Na2CO3 aqueous solution and stirred overnight, and the precipitated product was collected via filtration and washed with water and dried to give the corresponding compounds.
(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)(naphthalen-2-yl)methanone(1)
White solid, melting point 2226–229°C, 72% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 8.016 (4H, m), 7.580 (3H, m), 7.440 (1H, s), 7.167 (2H, br), 6.745 (1H, s), 3.795 (12H, m), 3.490 (2H, br). 13C-NMR (100MHz, DMSO-d6) δ 169.642, 161.632, 158.757, 154.699, 149.117, 145.535, 133.709, 132.754, 130.970, 129.745, 128.525, 127.826, 125.353, 105.645, 104.111, 103.460, 67.487, 56.294, 66.889, 25.592. DUIS-MS calculated for C25H25N5O4 [M + H]+ : 444.1, found : 443.5
[1,1′-biphenyl]-3-yl(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)methanone(2)
White solid, melting point 179–182°C, 73% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.735 (4H, m), 7.498 (6H, m), 7.152 (2H, br), 6.740 (1H, s), 3.794 (12H, m), 3.464 (2H, m). 13C-NMR (100MHz, DMSO-d6) δ 169.475, 161.620, 158.802, 154.687, 149.184, 145.519, 140.765, 139.825, 137.207, 129.509, 128.279, 127.297, 126.522, 125.633, 105.669, 104.085, 103.455, 56.287, 66.879. DUIS-MS calculated for C27H27N5O3 [M + H]+ : 470.1, found : 469.2
(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)(benzo[d][1,3]dioxol-5-yl)methanone (3)
White solid, melting point 267–269°C, 71% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.431 (1H, s), 7.145 (2H, br), 6.981 (3H, m), 6.736 (1H, s), 6.094 (2H, s), 3.791 (10H, m), 3.541 (4H, br). 13C-NMR (100MHz, DMSO-d6) δ 169.133, 161.610, 158.769, 154.682, 149.187, 148.700, 147.605, 145.499, 130.049, 121.990, 108.458, 105.666, 104.094, 103.440, 101.901, 56.503, 56.283, 55.878, 44.074, 19.023. DUIS-MS calculated for C22H23N5O5 [M + H]+ : 438.1, found : 437.5
1-(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)-2-(4-methoxyphenyl)ethanone(4)
White solid, melting point 240–243°C, 65% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.422 (1H, s), 7.168 (4H, m), 6.881 (2H, d, J= 8.8 Hz), 6.734 (1H, s), 3.831 (3H, s), 3.788 (3H, s), 3.729 (3H, s), 3.671 (4H, m), 3.601 (2H, br), 3.532 (4H, br). 13C-NMR (100MHz, DMSO-d6) δ 169.795, 161.584, 158.522, 154.669, 149.169, 145.485, 130.419, 128.163, 114.233, 105.668, 104.061, 103.412, 56.276, 55.662, 45.928, 44.254, 43.886, 41.754. DUIS-MS calculated for C23H27N5O4 [M + H]+ : 438.1, found : 437.5
1-(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)-2-(3-methoxyphenyl)ethanone(5)
White solid, melting point 198–201°C, 60% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.423 (1H, s), 7.231 (1H, m), 7.143 (1H, br), 6.817 (3H, m), 6.737 (1H, s), 3.830 (3H, s), 3.788 (3H, s), 3.738 (5H, m), 3.598 (9H, m). 13C-NMR (100MHz, DMSO-d6) δ 169.382, 161.590, 159.707, 158.745, 154.676, 149.159, 145.495, 137.832, 129.834, 121.631, 115.114, 112.281, 105.662, 104.060, 103.416, 67.488, 56.275, 55. 643, 45.977, 44.237, 43.878, 41.783. DUIS-MS calculated for C23H27N5O4 [M + H]+ : 438.1, found : 437.5
1-(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)-2-phenylethanone(6)
White solid, melting point 221–223°C, 96% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.420 (1H, s), 7.272 (5H, m), 7.140 (2H, br), 6.733 (1H, s), 3.829 (3H, s), 3.786 (3H, s), 3.775 (2H, s), 3.607 (8H, m). 13C-NMR (100MHz, DMSO-d6) δ 169.500, 161.590, 158.759, 154.674, 149.173, 145.492, 136.385, 129.469, 128.807, 126.828, 105.672, 104.064, 103.419, 56.279, 55.877, 45.955, 44.239, 43.883, 41.778. DUIS-MS calculated for C22H25N5O3 [M + H]+ : 408.1, found : 407.2
1-(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)-2-(naphthalen-2-yl)ethanone(7)
White solid, melting point 236–238°C, 70% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.862 (3H, m), 7.774 (1H, s), 7.436 (4H, m), 7.133 (2H, br), 6.725 (1H, s), 3.957 (2H, s), 3.823 (3H, s), 3.782 (3H, s), 3.610 (8H, m). 13C-NMR (100MHz, CDCl3-d6) δ 169.688, 160.521, 158.510, 155.158, 149.759, 145.970, 133.577, 132.459, 128.540, 127.446, 126.517, 125.780, 105.814, 102.901, 101.134, 56.160, 46.256, 43.913, 41.707. DUIS-MS calculated for C26H27N5O3 [M + H]+: 458.1, found : 457.5
1-(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)-2-phenylpropan-1-one(8)
White solid, melting point 207–209°C, 82% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.400 (1H, s), 7.306 (5H, m), 7.112 (2H, br), 6.704 (1H, s), 4.155 (1H, q, J= 6.8 Hz), 3.816 (3H, s), 3.775 (3H, s), 3.680-3.399 (6H, m), 3.007 (1H, m), 1.316 (3H, d, J= 6.8 Hz). 13C-NMR (100MHz, CDCl3-d6) δ 172.254, 160.514, 158.541, 155.142, 149.783, 145.917, 142.085, 128.993, 127.214, 126.849, 105.782, 102.866, 101.177, 56.091, 45.570, 43.777, 42.118, 20.719. DUIS-MS calculated for C22H23N5O5 [M + H]+:422.1, found : 421.5
(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)(pyridin-2-yl)methanone (9)
White solid, melting point 250–252°C, 42% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 8.625 (1H, d, J= 4.4 Hz), 7.952 (1H, t, J= 8 Hz), 7.615 (1H, d, J= 8 Hz), 7.507 (1H, m), 7.432 (1H, s), 7.156 (2H, br), 6.738 (1H, s), 3.831-3.726 (12H, m), 3.469 (2H, m). 13C-NMR (100MHz, DMSO-d6) δ 167.378, 161.623, 158.737, 154.599, 149.175, 148.901, 145.529, 137.864, 125.097, 123.712, 105.685, 104.109, 103.456, 56.299, 55.886, 47.042, 44.409, 43.866, 42.193. DUIS-MS calculated for C20H22N6O3 [M + H]+: 395.1, found : 394.4
(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)(pyridin-4-yl)methanone(10)
White solid, melting point 235–237°C, 32% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 8.697 (2H, d, J= 4.8 Hz), 7.444 (3H, m), 7.163 (2H, br), 6.735 (1H, s), 3.830-3.710 (14H, m) (Note: it seems that two protons are missing but they may overlap with the peak from water). 13C-NMR (100MHz, DMSO-d6) δ 167.422, 161.632, 158.709, 154.711, 150.553, 149.161, 145.559, 143.952, 121.794, 105.669, 104.104, 103.463, 56.301, 55.896, 47.319, 44.215, 43.737, 41.990. DUIS-MS calculated for C20H22N6O3 [M + H]+ : 395.1, found : 394.4
(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)(4-ethylphenyl)methanone(11)
White solid, melting point 240–242°C, 84% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.433 (1H, s), 7.370 (2H, d, J= 8 Hz), 7.302 (2H, d, J= 8 Hz), 7.154 (2H, br), 6.735 (1H, s), 3.831-3.445 (14H, m), 2.660 (2H, q, J= 7.6 Hz), 1.214 (3H, t, J= 7.6 Hz). 13C-NMR (100MHz, DMSO-d6) δ 169.768, 161.614, 158.774, 154.687, 149.181, 145.696, 133.742, 128.206, 127.749, 105.667, 104.088, 103.453, 56.284, 55.877, 28.458, 15.801. DUIS-MS calculated for C23H27N5O3 [M + H]+ : 422.1, found : 421.5
(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)(4-iodophenyl)methanone(12)
White solid, melting point 254–258°C, 85% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.843 (2H, d, J= 8 Hz), 7.423 (1H, s), 7.252 (2H, d, J= 8 Hz), 7.144 (2H, br), 6.735 (1H, s), 3.841-3.716 (14H, m). 13C-NMR (100MHz, DMSO-d6) δ 168.865, 161.618, 158.734, 154.704, 149.161, 145.533, 137.730, 135.857, 129.657, 105.670, 104.110, 103.454, 96.865, 56.301, 55.893. DUIS-MS calculated for C21H22IN5O3 [M + H]+ : 520.0, found : 519.3
(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)(4-chloro-3-nitrophenyl)methanone(13)
White solid, melting point 268–272°C, 91% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 8.179 (1H, s), 7.883 (1H, d, J= 8 Hz), 7.792 (1H, d, J= 8 Hz), 7.434 (1H, s), 7.159 (2H, br), 6.740 (1H, s), 3.833-3.401 (14H, m). 13C-NMR (100MHz, DMSO-d6) δ 166.451, 161.632, 158.722, 154.699, 149.143, 148.029, 145.547, 136.683, 132.589, 126.400, 124.905, 105.664, 104.089, 103.456, 56.293, 55.892. DUIS-MS calculated for C21H21ClN6O5 [M + H]+: 473.0, found : 472.9
1-(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)ethanone(14)
White solid, melting point 244–247°C, 88% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.426 (1H, s), 7.143 (2H, br), 6.745 (1H, s), 3.836-3.684 (10H, m), 3.490 (4H, m), 2.050 (3H, s). 13C-NMR (100MHz, DMSO-d6) δ 168.845, 161.606, 158.768, 154.682, 149.209, 145.469, 105.675, 104.106, 103.410, 56.289, 55.885, 46.158, 44.261, 43.872, 41.361, 21.803. DUIS-MS calculated for C16H21N5O3 [M + H]+: 332.1, found : 331.4
1-(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)propan-1-one(15)
White solid, melting point 269–272°C, 63% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.428 (1H, s), 7.141 (2H, br), 6.746 (1H, s), 3.837-3.686 (10H, m), 3.507 (4H, m), 2.366 (2H, q, J= 7.2 Hz), 1.017 (3H, t, J= 7.2 Hz). 13C-NMR (100MHz, DMSO-d6) δ 171.950, 161.602, 158.795, 154.682, 149.227, 145.466, 105.680, 104.110, 103.413, 56.289, 55.879, 45.251, 44.304, 43.930, 41.538, 26.125, 9.897. DUIS-MS calculated for C17H23N5O3 [M + H]+ : 346.1, found : 345.4
1-(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)butan-1-one(16)
White solid, melting point 259–262°C, 72% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.426 (1H, s), 7.142 (2H, br), 6.743 (1H, s), 3.836 (3H, s), 3.791 (3H, s), 3.735 (2H, m), 3.683 (2H, m), 3.507 (4H, m), 2.337 (2H, t, J=7.2 Hz), 1.555 (2H, m), 0.914 (3H, t, J= 7.6 Hz). 13C-NMR (100MHz, DMSO-d6) δ 171.13, 161.60, 158.78, 154.68, 149.23, 145.47, 105.68, 104.11, 103.41,67.49, 56.29, 55.86, 45.41, 44.37, 43.96, 41.49, 34.76, 25.59, 18.69, 14.31. DUIS-MS calculated for C18H25N5O3 [M + H]+: 360.1, found : 359.4
(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)(cyclohexyl)methanone(17)
White solid, melting point 220–222°C, 89% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.429 (1H, s), 7.138 (2H, br), 6.740 (1H, s), 3.837 (3H, s), 3.792 (3H, s), 3.703 (4H, m), 3.520 (4H, m), 2.627 (1H, m), 1.701 (5H, m), 1.336 (4H, m), 1.174 (1H, m). 13C-NMR (100MHz, DMSO-d6) δ 174.107, 161.596, 158.774, 154.691, 149.220, 145.480, 105.684, 104.138, 103.432, 56.299, 55.879, 45.298, 44.640, 43.992, 41.567, 29.631, 26.081, 25.656, 19.020. DUIS-MS calculated for C21H29N5O3 [M + H]+ : 400.2, found : 399.5
(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)(furan-2-yl)methanone(18)
White solid, melting point 230–233°C, 66% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.869 (1H, d, J= 0.8 Hz), 7.438 (1H, s), 7.162 (2H, br), 7.037 (1H, d, J= 3.2 Hz), 6.752 (1H, s), 6.652 (1H, dd, J= 0.8, 3.2 Hz), 3.795 (14H, m). 13C-NMR (100MHz, DMSO-d6) δ 161.633, 158.862, 154.712, 149.213, 147.474, 145.521, 145.205, 116.020, 111.775, 105.700, 104.137, 103.463, 67.487, 56.302, 55.886, 44.153, 25.591. DUIS-MS calculated for C19H21N5O4 [M + H]+: 384.1, found : 383.4
(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)(thiophen-2-yl)methanone(19)
White solid, melting point 222–224°C, 56% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.784 (1H, d, J= 4.8 Hz), 7.475 (1H, d, J= 3.2 Hz), 7.438 (1H, s), 7.161 (3H, m), 6.750 (1H, s), 3.811(10H, m), 3.761 (4H, br). 13C-NMR (100MHz, DMSO-d6) δ 162.930, 161.637, 158, 732, 154.715, 149.215, 145.523, 137.835, 130.013, 129.650, 127.626, 105.698, 104.143, 103.465, 56.308, 55.892, 44.090. DUIS-MS calculated for C19H21N5O3S [M + H]+: 400.1, found : 399.5
(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)(isoxazol-5-yl)methanone (20)
White solid, melting point 249–251°C, 43% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 8.782 (1H, s), 7.442 (1H, s), 7.198 (2H, br), 6.984 (1H, s), 6.758 (1H, s), 3.839-3.611 (14H, m). 13C-NMR (100MHz, DMSO-d6) δ 161.661, 157.432, 151.435, 145.615, 106.739, 104.129, 103.462, 56.310, 55.916. DUIS-MS calculated for C18H20N6O4 [M + H]+ : 385.1, found : 384.4
(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)(4-methoxyphenyl)methanone (21)
White solid, melting point 229–232°C, 85% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.420 (3H, m), 7.148 (2H, br), 7.007 (2H, d, J= 8.4 Hz), 6.738 (1H, s), 3.813 (13H, m), 3.557 (4H, m). 13C-NMR (100MHz, DMSO-d6) δ 169.598, 161.623, 160.689, 158.790, 154.705, 149.211, 145.517, 129.628, 128.371, 114.131, 105.691, 104.134, 103.458, 56.302, 55.803, 44.140. DUIS-MS calculated for C22H25N5O4 [M + H]+ : 424.1, found : 423.5
(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)(4-bromophenyl)methanone (22)
White solid, melting point 128–130°C, 83% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.665 (2H, d, J= 8.4 Hz), 7.440 (1H, s), 7.407 (2H, d, J= 8.4 Hz), 7.164 (2H, br), 6.744 (1H, s), 3.832 (3H, s), 3.793 (3H, s), 3.725 (6H, m), 3.388 (2H, br). 13C-NMR (100MHz, DMSO-d6) δ 168.663, 161.624, 158.747, 154.710, 149.178, 145.538, 136.589, 131.933, 129.770, 123.409, 105.679, 104.115, 103.4584, 56.303, 55.895. DUIS-MS calculated for C21H22N5O3Br [M + H]+ : 474.0, found : 472.5
1-(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)hexan-1-one (23)
White solid, melting point 170–172°C, 73% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.427 (1H, s), 7.146 (2H, br), 6.743 (1H, s), 3.834 (3H, s), 3.790 (3H, s), 3.731 (2H, m), 3.679 (2H, m), 3.500 (4H, m), 2.335 (2H, t, J= 7.2 Hz), 1.516 (2H, m), 1.287 (4H, m), 0.872 (3H, t, J= 6.4 Hz). 13C-NMR (100MHz, DMSO-d6) δ 171.287, 161.603, 158.789, 154.690, 149.226, 145.478, 105.679, 104.123, 103.424, 56.294, 55.878, 45.433, 44.380, 43.959, 41.508, 32.789, 31.497, 24.984, 22.452, 14.356. DUIS-MS calculated for C20H29N5O3 [M + H]+ : 388.1, found : 387.5
1-(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)-2-(4-(benzyloxy)phenyl)ethanone (24)
White solid, melting point 230–234°C, 89% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.381 (6H, m), 7.169 (4H, m), 6.958 (2H, d, J= 8.4 Hz), 6.740 (1H, s), 5.071 (2H, s), 3.830 (3H, s), 3.789 (3H, s), 3.693-3.535 (10H, m). 13C-NMR (100MHz, DMSO-d6) δ 169.755, 161.602, 158.781, 157.409, 154.701, 149.206, 145.510, 137.636, 130.455, 128.410, 115.151, 105.703, 104.125, 103.441, 69.643, 67.493, 56.302, 55.879, 45.937, 44.274, 43.903, 41.764, 25.596. DUIS-MS calculated for C29H31N5O4 [M + H]+: 514.2, found : 513.6
1,12-bis(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)dodecane-1,12-dione (27)
White solid, melting point 218–225°C, 68% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.425 (2H, s), 7.145 (4H, br), 6.740 (2H, s), 3.831 (6H, s), 3.787 (6H, s), 3.700 (8H, m), 3.495 (8H, br), 2.331 (4H, t, J= 7.2 Hz), 1.504 (4H, br), 1.267 (12H, br). 13C-NMR (100MHz, DMSO-d6) δ 171.277, 161.596, 158.786, 154.682, 149.221, 145.467, 105.680, 104.119, 103.417, 56.293, 55.880, 45.427, 44.380, 43.958, 41.504, 32.834, 29. 358, 25.306. DUIS-MS calculated for C40H56N10O6 [M + H]+ : 774.0, found : 772.9
1,6-bis(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)piperazin-1-yl)hexane-1,6-dione (28)
White solid, melting point 279–280°C, 77% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.424 (2H, s), 7.138 (4H, br), 6.743 (2H, s), 2.846-2.507 (28H, m), 2.393 (4H, br), 1.567 (4H, br). 13C-NMR (100MHz, DMSO-d6) δ 171.217, 161.602, 158.789, 154.683, 149.218, 145.471, 105.679, 104.105, 103.413, 56.293, 55.882, 45.433, 44.386, 43.962, 41.521, 32.759, 25.050. DUIS-MS calculated for C34H44N10O6 [M + H]+ : 689.1, found : 688.8
Compounds 25 and 26 were prepared as the following procedure
The substituted acetyl chloride and piperazine were dissolved in DMF and K2CO3 with 2 to 1 mole ratio, and the mixture was stirred at room temperature until the reaction was completed. The reaction was quenched with Na2CO3 aqueous solution and stirred overnight, and the precipitated product was collected via filtration and washed with water and dried to give the corresponding compounds.
piperazine-1,4-diylbis((4-methoxyphenyl)methanone) (25)
White solid, melting point 215°C decomposed, 86% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.403 (4H, d, J= 8.4 Hz), 6.994 (4H, d, J= 8.4 Hz), 3.799 (6H, s), 3.542 (8H, br). 13C-NMR (100MHz, DMSO-d6) δ 169.643, 160.793, 129.640, 127.996, 114.152, 55.746. DUIS-MS calculated for C20H22N2O4 [M + H]+ : 355.1, found : 354.4
piperazine-1,4-diylbis((4-(methylthio)phenyl)methanone) (26)
White solid, melting point 220°C decomposed, 91% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.378 (4H, d, J= 8.4 Hz), 7.315 (4H, d, J= 8.4 Hz), 3.355 (6H, s), 3.533 (8H, br). 13C-NMR (100MHz, DMSO-d6) δ 169.394, 141.119, 132.045, 128.339, 125.694, 14.715. DUIS-MS calculated for C20H22N2O2S2 [M + H] : 387.1, found : 386.5
Compounds 29 and 30 were prepared as the following procedure
Doxazosin or compound 24 were dissolved in DMF and K2CO3, and then acetyl chloride was added to the three reactions respectively with 1 to 5 mole ratio to the two compounds. The mixture was stirred until the reaction was completed. The reaction was quenched with Na2CO3 aqueous solution and stirred overnight, and the precipitated product was collected via filtration and washed with water and dried to give the corresponding compounds.
N-(2-(4-(2,3-dihydrobenzo[b][1,4]dioxine-2-carbonyl)piperazin-1-yl)-6,7-dimethoxyquinazolin-4-yl) acetamide (29)
White solid, melting point 269–271°C, 36% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 10.464 (1H, s), 7.477 (1H, s), 6.874 (5H, m), 5.296 (1H, dd, J= 2.4, 6.4Hz), 4.427 (1H, dd, J= 2.4, 11.6 Hz), 4.220 (1H, dd, J=6.8, 12 Hz), 3.899 (3H, s), 3.846 (3H, s), 3.713 (8H, m), 2.411 (3H, s). 13C-NMR (100MHz, DMSO-d6) δ 171.532, 165.464, 157.725, 156.160, 151.018, 146.535, 143.432, 121.912, 117.443, 105.629, 103.741, 69.985, 65.205, 56.267, 45.373, 44.592, 43.964, 41.858, 25.802. DUIS-MS calculated for C25H27N5O6 [M + H]+ : 494.1, found : 493.5
N-(2-(4-(2-(4-(benzyloxy)phenyl)acetyl)piperazin-1-yl)-6,7-dimethoxyquinazolin-4-yl)acetamide (30)
White solid, melting point 205–209°C, 29% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 10.436 (1H, s), 7.382 (6H, m), 7.181 (2H, d, J= 8.4 Hz), 6.958 (2H, d, J= 8.4 Hz), 6.897 (1H, s), 5.076 (2H, s), 3.889 (3H, s), 3.838 (3H, s), 3.704 (6H, m), 3.583 (4H, br), 2.394 (3H, s). 13C-NMR (100MHz, CDCl3-d6) δ 170.048, 157.680, 156.151, 154.624, 151.293, 147.091, 136.950, 129.637, 128.582, 127.556, 115.237, 105.657, 100.764, 70.084, 56.270, 45.968, 44.102, 41.715, 40.284, 25.679. DUIS-MS calculated for C31H33N5O5 [M + H]+ : 556.2, found : 555.6
Compounds 31 and 32 were prepared as the following procedure
The dichloro-dimethoxyquinazolin and piperidine or pyrrolidine were dissolved in n-BuOH with 2 to 1 mole ratio, and the mixture was heated to 70°C for 24 hours, then it was cooled down to room temperature. The precipitated while solid was collected via filtration and washed with n-BuOH to get the corresponding intermediates. After it was dried, the intermediates and piperazine were dissolved in n-BuOH with 1 to 5 mole ratio, respectively. The two reactions were all heated to 100°C for 24 hours, then it was cooled down to room temperature. The precipitated while solid was collected via filtration and washed with n-BuOH to get two new intermediates, 6,7-dimethoxy-2-(piperazin-1-yl)-4-(piperidin-1-yl)quinazoline, 6,7-dimethoxy-2-(piperazin-1-yl)-4-(pyrrolidin-1-yl)quinazoline. Last, the two intermediates were mixed with 2-(4-(benzyloxy)phenyl)acetyl chloride (1:1 mole ratio) and K2CO3 in DMF, respectively. The reactions were stirred until completed, and were quenched with Na2CO3 aqueous solution and stirred overnight. The precipitated products were collected via filtration and washed with water and dried to give the corresponding compounds 31, 32.
2-(4-(benzyloxy)phenyl)-1-(4-(6,7-dimethoxy-4-(piperidin-1-yl)quinazolin-2-yl)piperazin-1-yl)ethan one (31)
White solid, melting point 160°C decomposed, 56% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.383 (5H, m), 7.182 (2H, d, J= 8.8 Hz), 6.958 (2H, d, J= 7.2 Hz), 6.848 (1H, s), 5.077 (2H, s), 3.861 (3H, s), 3.813 (3H, s), 3.561 (15H, m), 1.687 (6H, br). 13C-NMR (100MHz, DMSO-d6) δ 169.778, 165.057, 157.608, 154.765, 151.037, 145.215, 137.534, 130.440, 128.408, 115.157, 106.105, 105.426, 104.824, 69.631, 56.021, 55.906, 50.828, 45.820, 44.325, 43.913, 41.647, 25.832, 24.832. DUIS-MS calculated for C34H39N5O4 [M + H]+ : 582.2, found : 581.7
2-(4-(benzyloxy)phenyl)-1-(4-(6,7-dimethoxy-4-(pyrrolidin-1-yl)quinazolin-2-yl)piperazin-1-yl)etha none (32)
White solid, melting point 137°C decomposed, 60% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.382 (6H, m), 7.179 (2H, d, J= 8.4 Hz), 6.958 (2H, d, J= 8.4 Hz), 6.793 (1H, s), 5.075 (2H, s), 3.815 (10H, m), 3.623 (10H, m), 1.939 (4H, m). 13C-NMR (100MHz, DMSO-d6) δ 169.772, 159.752, 157.638, 154.027, 150.966, 144.298, 137.636, 130.425, 128.517, 115.166, 106.435, 105.506, 69.638, 56.086, 55.829, 50.454, 25.716. DUIS-MS calculated for C33H37N5O4 [M + H]+ : 568.2, found : 567.7
Compound 33 was prepared as the following procedure
A solution of 4-amino-2-chloro-6,7-dimethoxyquinazoline in CH2Cl2 was cooled to −70 °C under argon, and boron tribromide (1:1.2 mole ratio) was added. [27] The mixture was allowed to warm to room temperature over a period of 4h and was then cooled to −70 °C; methanol was added to quench the reaction, and the solution was concentrated. The solid residue was washed with ethyl acetate to obtain the intermediate 4-amino-2-chloro-6,7-dihydroxyquinazoline. A mixture of the intermediate, ethyl iodine and K2CO3 in DMF was stirred overnight and purified by silica gel chromatography to afford 4-amino-2-chloro-6,7-diethoxyquinazoline. The diethoxyquinazoline and piperazine were dissolved in n-BuOH with 1 to 5 mole ratio, and the mixture was heated to 100°C for 24 hours, then it was cooled down to room temperature. The precipitated while solid was collected via filtration and washed with n-BuOH. After it was dried, the solid was mixed with 2-(4-(benzyloxy)phenyl)acetyl chloride (1:1 mole ratio) and K2CO3 in DMF. It was stirred until the reaction was completed. The reaction was quenched with Na2CO3 aqueous solution and stirred overnight, and the precipitated product was collected via filtration and washed with water and dried to give compound 33.
1-(4-(4-amino-6,7-diethoxyquinazolin-2-yl)piperazin-1-yl)-2-(4-(benzyloxy)phenyl)ethanone (33)
White solid, melting point 248–254°C, 69% yield for the last step; 1H-NMR (400MHz, DMSO-d6) δ 7.383 (6H, m), 7.177 (2H, d, J= 8.4 Hz), 7.112 (2H, br), 6.958 (2H, d, J= 8.4 Hz), 6.714 (1H, s), 5.075 (2H, s), 4.066 (4H, m), 3.590 (6H, m), 1.369 (6H, m). 13C-NMR (100MHz, CDCl3-d6) δ 170.017, 136.987, 129.646, 128.580, 127.739, 115.192, 103.529, 70.075, 65.149, 46.164, 41.904, 40.295, 29.705, 14.666. DUIS-MS calculated for C31H31N5O4 [M + H]+ : 542.2, found : 541.6
4.2. Biological studies
4.2.1. Cell Culture and Antibodies
MDA-MB-231 cells were obtained from ATCC (Rockville, MD). The generation of overexpression EphA2 protein in the cells was reported in our previous study. [4] The cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 10 mg/ml L-Glutamine, 100 U/mL penicillin-streptomycin, and 0.1 mg/ml streptomycin. EphA2 overexpression cells were grown in the presence of 1 μg/ml puromycin. Cell cultures were grown at 37 °C, in a humidified atmosphere of 5% CO2 in a Thermo CO2 incubator (Grand Island, NY).
Antibodies used included rabbit polyclonal antibodies against pEphA/B (synthesized as described previously) [28] and EphA2 (Santa Cruz, Santa Cruz, CA), as well as mouse monoclonal antibody to EphA2 (clone D7, Millipore, Billerica, MA). Secondary antibodies used were goat anti-rabbit conjugated to HRP (Santa Cruz) and donkey anti-mouse conjugated to Alexa Fluor 488 (Thermo Fisher, Grand Island, NY).
4.2.2. EphA2 activation
MDA-MB-231 EphA2-overexpressing cells were plated in 12-well dishes at a density of 100,000 cells/well and grown for 24 hours prior to stimulation with appropriate compounds and ephrins for 30 min and 10 min, respectively. Compounds were prepared in DMSO at 200 times the final concentrations and 0.5% DMSO was used as vehicle control. Following treatment, cells were washed and lysed in modified RIPA Buffer (20 mM Tris-HCl pH 7.4, 20 mM NaF, 150 mM NaCl, 10% glycerol, 0.1% SDS, 0.5% DCA, 2 mM EDTA, 1% Triton X-100, 2 mM Na3VO4, and protease inhibitors, including 1 mM phenylmethylsulphonyl fluoride, and 2 μg/ml each of aprotinin and leupeptin) for 20 min, followed by immunoprecipitation. For immunoprecipitation, lysates were combined with 10 μg of ephrin-A1-Fc and 10-15 μl of Gamma-Bind Protein G beads (GE Healthcare Bioscience, Pittsburgh, PA) and rotated for 3 hours at 4 °C. 10 μg of human unconjugated Fc was used as a negative control. Beads were pelleted at 9,000 rpm for 2 min at 4 °C, and washed twice with 1 ml of IP Wash Buffer (20 mM Tris, pH 7.4, 10% glycerol, 50 mM NaCl, 0.2% Triton X-100, 0.5 mM Na3VO4, 0.5 mM PMSF). All wash buffer was removed and beads were resuspended in 25 μl of 4× LDS Loading Buffer (Thermo Fisher, Grand Island, NY) with 4 μl of 10X Bolt Sample Reducing Agent (Thermo Fisher), followed by Western Blotting. Samples were boiled 5 min and run on 4–12% Bis-Tris Plus gels (Thermo Fisher), followed by transfer to Immobilon-P PVDF membranes (Millipore, Billerica, MA). Membranes were blocked 1 hr at room temperature in 3% BSA in TBS containing 0.05% Tween-20 (TBS-T) followed by overnight incubation with pEphA/B (1:500) and EphA2 (Santa Cruz) (1:1000) primary antibodies. Membranes were washed in TBS-T and incubated with goat anti-rabbit-HRP (1:5000) secondary antibody 1 hr at room temperature, followed by washing and developing with Luminol Reagent (Santa Cruz). Band intensities were quantified using Quantity One software (Bio-Rad, Hercules, CA).
4.2.3. EphA2 internalization
MDA-MB-231 cells were plated in 24-well plates on 10 mm square coverslips coated with 10 μg/ml fibronectin and incubated 24–48 hrs before treatment with 50 μM Doxazosin and various doses of compound 27 (0.4, 2, 10, 50 μM) in 0.5% DMSO for 1 hr. As another positive control, cells were treated with 2 μg/ml ephrin-A1-Fc. Cells were then washed with PBS and fixed with 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) on ice for 10 min, followed by washing and permeabilization with 0.4% NP-40 for 10 min on ice. Non-specific binding sites were blocked with 50 mM NH4Cl on ice for 10 min, followed by washing with PBS and 0.1% BSA in PBS. Cells were then incubated in primary mouse EphA2 antibody (D7, 1:100) in 0.1% BSA/PBS for 1 hr at room temperature, washed in PBS and BSA/PBS, followed by incubation in secondary donkey anti-mouse Alexa Fluor 488 antibody (1:300) in BSA/PBS for 30 min at room temperature. Cells were washed in PBS and BSA/PBS, followed by mounting in DAPI Mounting Medium (Electron Microscopy Sciences). Images were taken with a Leica microscope.
4.2.4. Clonogenic assay
MDA-MB-231 cells (1000 cells/well) were seeded in 12-well plates and cultured for 24 h, followed by treatment with various concentrations of the Doxazosin analogs (0, 0.08, 0.4, 2 and 10 μM) in 0.5% DMSO for 14 days. After washing with PBS, colonies were fixed in 4% paraformaldehyde in PBS, washed in 20% methanol, and stained with 0.5% crystal violet.
4.2.5. Docking investigation
Docking studies were done to gain insight into the possible mode of interaction between the compounds and EphA2. The structures were drawn with Marvin Sketch (www.ChemAxon.com) and energy minimized in MOE 2010 (www.chemcomp.com) using the MMFF94s force field. The database of compounds was then used for the docking studies. The protein crystal structure of 3FL7.pdb [26] was prepared for docking by adjusting the pH of the system to a pH of 7.4. The binding site in EphA2 was delineated by Doxazosin. After docking, only the top docked pose of each compound was retained for analysis.
4.2.6. Adrenoceptor binding activity
Analysis of Doxazosin and analog binding to rat α1a adrenergic receptor was performed by Eurofins Panlabs Taiwan, Ltd. (Taiwan). [3H]-prazosin at 0.25 nM was used as the radioligand. Compounds 24, 27, and Doxazosin at 5 different doses ranging from 0.08 nM-3 μM in 0.1 % DMSO were incubated with [3H]-prazosin in a final volume of 1 ml of assay buffer (50 mM Tris-HCl, pH 7.4, 0.5 mM EDTA) for 1 h at 25°C. Non-specific binding was defined by use of 10 μM phentolamine. Incubations were terminated by vacuum filtration over 0.1% PEI pretreated glass fiber filters and filters were washed for 10s with ice cold 0.1M NaCl and radioactivity retained on the filters was determined by liquid scintillation spectrometry. IC50 values were determined by a non-linear regression analysis using GraphPad Prism v.5 (GraphPad).
Supplementary Material
Scheme 2.

Synthesis of symmetric structure based on piperazine ring as the core structure
-
➢
Identification of a potent small molecule EphA2 agonist with a dimeric structure based on lead compound Doxazosin.
-
➢
The compound triggers EphA2 receptor internalization at sub micro mole concentrations.
-
➢
The compound forms more hydrogen bonds to EphA2 receptor compared to Doxazosin based on the docking result.
-
➢
The compound shows less binding affinity to adrenoceptor compared to Doxazosin.
Acknowledgments
This work was supported by grants from National Institutes of Health to BW (1R01NS096956-01) and National Science Foundation Major Research Instrumentation Grants (CHE-0923398 and CHE-1126384).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Li X, Wang L, Gu JW, Li B, Liu WP, Wang YG, Zhang X, Zhen HN, Fei Z. Up-regulation of EphA2 and down-regulation of EphrinA1 are associated with the aggressive phenotype and poor prognosis of malignant glioma. Tumour Biol. 2010;31:477–488. doi: 10.1007/s13277-010-0060-6. [DOI] [PubMed] [Google Scholar]
- 2.McCarron JK, Stringer BW, Day BW, Boyd AW. Ephrin expression and function in cancer. Future Oncol. 2010;6:165–176. doi: 10.2217/fon.09.146. [DOI] [PubMed] [Google Scholar]
- 3.Miao H, Li DQ, Mukherjee A, Guo H, Petty A, Cutter J, Basilion JP, Sedor J, Wu J, Danielpour D, Sloan AE, Cohen ML, Wang B. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell. 2009;16:9–20. doi: 10.1016/j.ccr.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petty A, Myshkin E, Qin H, Guo H, Miao H, Tochtrop GP, Hsieh JT, Page P, Liu L, Lindner DJ, Acharya C, MacKerell AD, Jr, Ficker E, Song J, Wang B. A small molecule agonist of EphA2 receptor tyrosine kinase inhibits tumor cell migration in vitro and prostate cancer metastasis in vivo. PLoS One. 2012;7:e42120. doi: 10.1371/journal.pone.0042120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noblitt LW, Bangari DS, Shukla S, Knapp DW, Mohammed S, Kinch MS, Mittal SK. Decreased tumorigenic potential of EphA2-overexpressing breast cancer cells following treatment with adenoviral vectors that express EphrinA1. Cancer Gene Ther. 2004;11:757–766. doi: 10.1038/sj.cgt.7700761. [DOI] [PubMed] [Google Scholar]
- 6.Carles-Kinch K, Kilpatrick KE, Stewart JC, Kinch MS. Antibody targeting of the EphA2 tyrosine kinase inhibits malignant cell behavior. Cancer Res. 2002;62:2840–2847. [PubMed] [Google Scholar]
- 7.Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. Ligation of EphA2 by Ephrin A1-Fc inhibits pancreatic adenocarcinoma cellular invasiveness. Biochem Biophys Res Commun. 2004;320:1096–1102. doi: 10.1016/j.bbrc.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 8.Jackson D, Gooya J, Mao S, Kinneer K, Xu L, Camara M, Fazenbaker C, Fleming R, Swamynathan S, Meyer D, Senter PD, Gao C, Wu H, Kinch M, Coats S, Kiener PA, Tice DA. A human antibody-drug conjugate targeting EphA2 inhibits tumor growth in vivo. Cancer Res. 2008;68:9367–9374. doi: 10.1158/0008-5472.CAN-08-1933. [DOI] [PubMed] [Google Scholar]
- 9.Wang B. Cancer cells exploit the Eph-ephrin system to promote invasion and metastasis: tales of unwitting partners. Sci Signal. 2011;4:pe28. doi: 10.1126/scisignal.2002153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo H, Miao H, Gerber L, Singh J, Denning MF, Gilliam AC, Wang B. Disruption of EphA2 receptor tyrosine kinase leads to increased susceptibility to carcinogenesis in mouse skin. Cancer Res. 2006;66:7050–7058. doi: 10.1158/0008-5472.CAN-06-0004. [DOI] [PubMed] [Google Scholar]
- 11.Astin JW, Batson J, Kadir S, Charlet J, Persad RA, Gillatt D, Oxley JD, Nobes CD. Competition amongst Eph receptors regulates contact inhibition of locomotion and invasiveness in prostate cancer cells. Nat Cell Biol. 2010;12:1194–1204. doi: 10.1038/ncb2122. [DOI] [PubMed] [Google Scholar]
- 12.Biao-xue R, Xi-guang C, Shuan-ying Y, Wei L, Zong-juan M. EphA2-dependent molecular targeting therapy for malignant tumors. Curr Cancer Drug Targets. 2011;11:1082–1097. doi: 10.2174/156800911798073050. [DOI] [PubMed] [Google Scholar]
- 13.Miao H, Wang B. EphA receptor signaling–complexity and emerging themes. Semin Cell Dev Biol. 2012;23:16–25. doi: 10.1016/j.semcdb.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nasreen N, Khodayari N, Mohammed KA. Advances in malignant pleural mesothelioma therapy: targeting EphA2 a novel approach. Am J Cancer Res. 2012;2:222–234. [PMC free article] [PubMed] [Google Scholar]
- 15.Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer. 2010;10:165–180. doi: 10.1038/nrc2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell. 2008;133:38–52. doi: 10.1016/j.cell.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 17.Abolyosr A, Elsagheer GA, Abdel-Kader MS, Hassan AM, Abou-Zeid AM. Evaluation of the effect of sildenafil and/or doxazosin on Benign prostatic hyperplasia-related lower urinary tract symptoms and erectile dysfunction. Urol Ann. 2013;5:237–240. doi: 10.4103/0974-7796.120293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee HN, Lee KS, Lee SY, Shim BS, Lee YS, Hong JH, Lim BH, Lee HM. Effects of Doxazosin on Alpha 1-Adrenergic Receptors in Prostates with Benign Prostatic Hyperplasia. Low Urin Tract Symptoms. 2013;5:82–89. doi: 10.1111/j.1757-5672.2012.00168.x. [DOI] [PubMed] [Google Scholar]
- 19.Duggineni S, Mitra S, Lamberto I, Han X, Xu Y, An J, Pasquale EB, Huang Z. Design and Synthesis of Potent Bivalent Peptide Agonists Targeting the EphA2 Receptor. ACS Med Chem Lett. 2013;4:344–348. doi: 10.1021/ml3004523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han DC, Shen TL, Miao H, Wang B, Guan JL. EphB1 associates with Grb7 and regulates cell migration. J Biol Chem. 2002;277:45655–45661. doi: 10.1074/jbc.M203165200. [DOI] [PubMed] [Google Scholar]
- 21.Lee JW, Han HD, Shahzad MM, Kim SW, Mangala LS, Nick AM, Lu C, Langley RR, Schmandt R, Kim HS, Mao S, Gooya J, Fazenbaker C, Jackson D, Tice DA, Landen CN, Coleman RL, Sood AK. EphA2 immunoconjugate as molecularly targeted chemotherapy for ovarian carcinoma. J Natl Cancer Inst. 2009;101:1193–1205. doi: 10.1093/jnci/djp231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walker-Daniels J, Riese DJ, 2nd, Kinch MS. c-Cbl-dependent EphA2 protein degradation is induced by ligand binding. Mol Cancer Res. 2002;1:79–87. [PubMed] [Google Scholar]
- 23.Himanen JP, Goldgur Y, Miao H, Myshkin E, Guo H, Buck M, Nguyen M, Rajashankar KR, Wang B, Nikolov DB. Ligand recognition by A-class Eph receptors: crystal structures of the EphA2 ligand-binding domain and the EphA2/ephrin-A1 complex. EMBO Rep. 2009;10:722–728. doi: 10.1038/embor.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qin H, Shi J, Noberini R, Pasquale EB, Song J. Crystal structure and NMR binding reveal that two small molecule antagonists target the high affinity ephrin-binding channel of the EphA4 receptor. J Biol Chem. 2008;283:29473–29484. doi: 10.1074/jbc.M804114200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seiradake E, Harlos K, Sutton G, Aricescu AR, Jones EY. An extracellular steric seeding mechanism for Eph-ephrin signaling platform assembly. Nat Struct Mol Biol. 2010;17:398–402. doi: 10.1038/nsmb.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Himanen JP, Yermekbayeva L, Janes PW, Walker JR, Xu K, Atapattu L, Rajashankar KR, Mensinga A, Lackmann M, Nikolov DB, Dhe-Paganon S. Architecture of Eph receptor clusters. Proc Natl Acad Sci U S A. 2010;107:10860–10865. doi: 10.1073/pnas.1004148107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw YJ, Yang YT, Garrison JB, Kyprianou N, Chen CS. Pharmacological exploitation of the alpha1-adrenoreceptor antagonist doxazosin to develop a novel class of antitumor agents that block intracellular protein kinase B/Akt activation. J Med Chem. 2004;47:4453–4462. doi: 10.1021/jm049752k. [DOI] [PubMed] [Google Scholar]
- 28.Miao H, Burnett E, Kinch M, Simon E, Wang B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat Cell Biol. 2000;2:62–69. doi: 10.1038/35000008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.