

Abstract
Influenza viruses cause over 500,000 deaths worldwide1 and are associated with an annual cost of 12 - 14 billion USD in the United States alone considering direct medical and hospitalization expenses and work absenteeism2. Animal models are crucial in Influenza A virus (IAV) studies to evaluate viral pathogenesis, host-pathogen interactions, immune responses, and the efficacy of current and/or novel vaccine approaches as well as antivirals. Mice are an advantageous small animal model because their immune system is evolutionarily similar to that found in humans, they are available from commercial vendors as genetically identical subjects, there are multiple strains that can be exploited to evaluate the genetic basis of infections, and they are relatively inexpensive and easy to manipulate. To recapitulate IAV infection in humans via the airways, mice are first anesthetized prior to intranasal inoculation with infectious IAVs under proper biosafety containment. After infection, the pathogenesis of IAVs is determined by monitoring daily the morbidity (body weight loss) and mortality (survival) rate. In addition, viral pathogenesis can also be evaluated by assessing virus replication in the upper (nasal mucosa) or lower (lungs) respiratory tract of infected mice. Humoral responses upon IAV infection can be rapidly evaluated by non-invasive bleeding and secondary antibody detection assays aimed at detecting the presence of total or neutralizing antibodies. Here, we describe the common methods used to infect mice intranasally (i.n) with IAV and evaluate pathogenesis, humoral immune responses and protection efficacy.
Keywords: Immunology, Issue 127, Influenza A virus, influenza infection, mice, intranasal inoculation, pathogenicity, safety, immunogenicity, protection efficacy, viral challenge, vaccination, humoral responses
Introduction
IAVs are enveloped viruses classified in the Orthomyxoviridae family3. They contain eight single-stranded RNA molecules with negative polarity3. In humans, IAVs cause seasonal epidemics and occasional pandemics of important consequence when novel viruses are introduced in the human population4. Moreover, seasonal IAVs are highly and rapidly transmitted between humans producing an elevated economic loss worldwide every year2,5. IAV symptoms include cough, nasal congestion, fever, malaise, headache, anorexia and myalgia, but the virus can also produce a more severe disease in immunocompromised patients6. In fact, the World Health Organization (WHO) calculates that seasonal influenza viruses cause 300,000 - 500,000 deaths worldwide every year1. There are only two classes of drugs currently approved by the Food and Drug Administration (FDA) for influenza prophylaxis and treatment in humans: neuraminidase (NA) inhibitors (e.g., oseltamivir) and blockers of the M2 ion channel (e.g., amantadine); however, the emergence of drug-resistant virus variants is an increasing concern. Vaccination, therefore, remains the best medical option to protect humans against IAVs infections. To date, three types of influenza vaccines licensed by the FDA for human use are available: recombinant viral hemagglutinin (HA) protein vaccines, inactivated influenza vaccines (IIV), and live-attenuated influenza vaccines (LAIV)5,7. The three vaccines are designed to induce adaptive immune response against the viral HA protein, the major target of neutralizing antibodies against IAVs.
A validated mouse model to study IAV infection in vivo
Animal models have been used to study, among others, IAV pathogenesis8,9,10,11, viral factors that contribute to disease12 and/or viral transmission13,14, and to test the efficacy of new vaccines or antiviral drugs9,10,15. Mice (Mus musculus) are the most extensively used animal model for IAV research for several reasons: 1) the immune system is evolutionarily similar to that present in humans; 2) low cost, including animal purchase, housing and reproduction; 3) small size to easily manipulate and store; 4) minimal host variability to obtain homogeneous responses and results; 5) a large knowledge of mice biology, including genome sequence; 6) many available molecular biology and/or immunology reagents; 7) available knock out (KO) mice to study the contribution of a given host protein on viral infection; and, 8) multiple mouse strains that can be exploited to evaluate the genetic basis of infections.
There are several mouse strains currently available to study IAV in vivo. Age, immune status, sex, genetic background and mouse strain as well as routes of infection, dose and viral strains all influence the outcome of IAV infection in mice. The most common mouse strains used in IAV research are C57BL/6, BALB/C and, more recently, DBA.2 mice since they are more susceptible to IAV disease than the two former strains16,17,18,19,20. Importantly, the immune response also can be different depending on the mouse strain18,19,20. Thus, it is very important to recover all the available information about the mouse and IAV strain to choose the best option for the experiment to be conducted.
Although mouse is a good animal model of infection for in vivo studies with IAV, they have several limitations, which need to be considered in the experimental design. For instance, a major limitation of using mice for in vivo studies is that IAVs do not transmit among mice. Thus, for transmission studies, more accepted animal models (e.g., ferrets or guinea pigs) are used16,17,21. In addition, there are several differences between the manifestations of IAV in mice and humans. Unlike humans, mice do not develop fever upon IAV infection; conversely they present with hypothermia16,17. In mice, IAV replication is concentrated in the lower respiratory tract (lungs) rather than the upper airways. Thus, virulence of IAV in mice is not always correlated to that seen in humans. Altogether, because the advantages outweigh the limited disadvantages, mouse represents the first animal model used to evaluate influenza viral pathogenesis, immunogenicity and protective efficacy in vaccine and antiviral studies. Moreover, it would not be ethically acceptable to conduct studies with IAV using large animal models without previous evidence in a small animal model of IAV infection. In this manuscript, we describe how to infect mice intranasally (i.n.) with IAV, how to monitor the severity and progress of viral infection and how to carry out the experiments required to evaluate humoral immune responses and protection efficacy.
Protocol
All animal protocols described here were approved by the Institutional Animal Care and Use Committee (IACUC) and the Institutional Biosafety Committee (IBC) at the University of Rochester School of Medicine and Dentistry, and comply with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Research Council 22. The facilities and programs of the Vivarium and Division of Laboratory Animal Medicine of the School of Medicine and Dentistry are accredited by AAALAC International and comply with state law, federal statute and National Institutes of Health (NIH) policy. Similar requirements should be applied at each institution to adhere to the animal protocols described in this manuscript.
1. Use of Small Vertebrate Animals
NOTE: The proper Personal Protection Equipment (PPE) is required for working with mice. Minimum requirements include disposable coveralls, shoe covers, head bonnet, mask, and gloves.
In accordance with the IACUC protocol, place a maximum of 5 mice per cage. Following IACUC protocol, euthanize mice with two methods of euthanasia (the second must be a physical method) to ensure that the animal is dead. NOTE: In the experiments in which IAV morbidity and mortality are evaluated, mice that lost 20% of their initial body weight were considered to have reached the experimental endpoint, and were euthanized with CO2, and cervical dislocation as the physical secondary method. This percentage of body weight may be different in other institutions. In the procedures where the mice lungs and nasal mucosa are collected after IAV infection, mice are euthanized with a lethal dose of 2,2,2-tribromoethanol (TBE), and by cutting the hepatic vein as the physical secondary method. Female 6-to-8-week-old C57BL/6 mice were purchased and maintained in the animal care facility at the University of Rochester School of Medicine and Dentistry under specific pathogen-free conditions.
2. Biosafety
NOTE: In this report, the IAVs used to infect mice are the common mouse-adapted laboratory strain of influenza A/Puerto Rico/8/34 H1N1 (PR8 WT)22,23 and a temperature sensitive LAIV variant, PR8 LAIV8. Perform all procedures that involve IAV infections (in vitro or in vivo), cell cultures, and biological samples, in a biological safety cabinet under biosafety level (BSL)-2 conditions. Utilize other BSL suits or containment conditions if highly virulent IAV strains are used.
Clean the biosafety cabinet with chlorine dioxide disinfectant before and after performing all the experimental procedures described in this manuscript. Sterilize all dissection material (scissors, scalpel and dissecting forceps) and the Dounce homogenizer before and after their use following proper IBC recommendations. Discard all material produced during the procedures under proper IBC and IACUC guidelines.
3. Intranasal Infection
Place the female 6-to-8-week-old C57BL/6 mice under specific pathogen-free conditions. Organize and label the mouse cages with the virus and dose used. Identify the mice in each cage using an ear punch code or with another approved method such as painting of the tails. Place a maximum of 5 mice per cage.
Prepare the dilution of IAV (Fluorescent Forming Units (FFU)/mouse) under aseptic conditions in a total volume of 30 µL/mouse in sterile 1x phosphate buffer saline (PBS). Maintain the virus inoculum on ice. Calculate the amount of IAV to add in the viral dilution with the formula: ((X FFU per mouse / 30 µL) x final volume) / stock viral titer.
Weigh the mice with a scale and record the weight of each mouse (Day 0). Hold the mouse in a dorsal position by picking it up by the scruff of the neck between the index finger and thumb. Hold the tail against the hand with the pinky finger.
Anesthetize the mouse intraperitoneally (i.p.) with 240 - 250 mg/kg of TBE by inserting the needle in the caudal 2/3 of the right side of the abdomen. Pause briefly before withdrawing the needle. Return the mouse to the cage and wait 5 min.
Apply sterile ophthalmic ointment to the eyes to prevent dryness while the mouse is anesthetized. Check if the mouse is anesthetized by lack of pedal withdrawal reflex (i.e., toe pinch). If mice are not fully anesthetized they will cough up the virus.
When the mouse is fully anesthetized, place the mouse in dorsal recumbency. Put the pipet tip containing 30 µL of the virus inoculum in the nostril and slowly but constantly eject the solution. Be sure the mouse is inhaling the preparation by observing the inoculum drop disappearing.
Check that the mouse is breathing as TBE can depress the temperature and breathing rate. Return the animal to the cage placing it in dorsal recumbency. Monitor the mice for any signs of respiratory distress and if observed, help the mouse to breathe by holding animal vertically and induce impact driven respiration24. Monitor the mouse until it regains consciousness (30 - 45 min after it was anesthetized).
4. Characterization of Viral Pathogenesis (Figure 1)
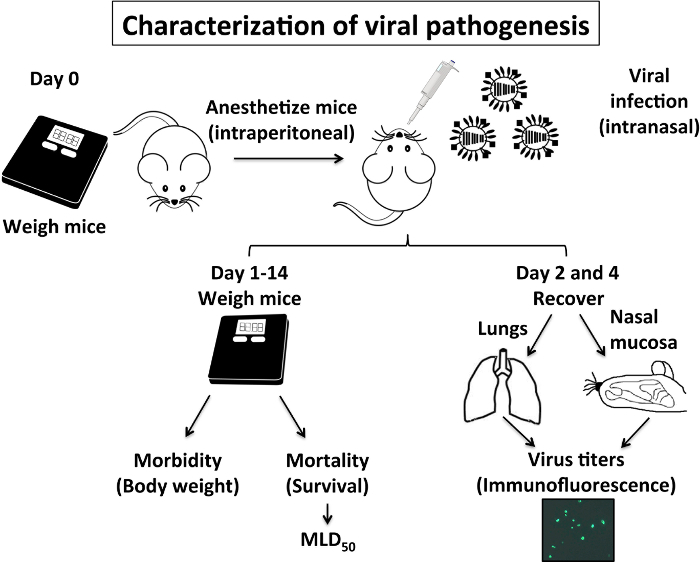
- Evaluation of morbidity and mortality (Figure 1 and Figure 2)
- Infect i.n. 3 - 4 groups of female 6-to-8-week-old C57BL/6 mice (at least 5 mice per group, n = 5) with 10-fold doses (10, 102, 103 and 104 FFU/mouse) of PR8 WT as described in section 3.
- Monitor and weigh the mice with a scale over the next 14 days at approximately the same time to minimize weight variations due to food ingestion. Euthanize mice that lose 20% of their initial body weight as described in section 1.
- After 14 days, euthanize the mice that survive viral infection as described in section 1.
- Calculate the viral 50% mouse lethal dose (MLD50) based on the survival data obtained using the method of Reed and Muench25. First, calculate the proportion distance (PD):
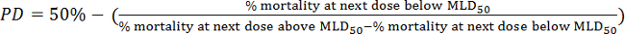
- Next, calculate the MLD50 by the following formula:
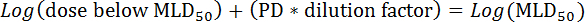
- Evaluation of viral titers in lungs and nasal mucosa (Figure 1 and Figure 2)
- Recovery of lungs and nasal mucosa
- Infect i.n. female 6-to-8-week-old C57BL/6 mice (n = 6) with the viral dose to test 10-104 FFU of PR8 WT as described in section 3.
- Collect the mouse lungs and nasal mucosa at days 2 (n = 3) and 4 (n = 3).
- Euthanize the mice with a lethal dose (i.p.) of TBE (500 mg/kg). Place the animal in dorsal recumbency and disinfect the fur over the thorax with 70% ethanol. Cut the skin with a scalpel from the sternum to the base of the abdomen. Make 1 cm cuts from the base of the incision to the lateral parts of the mice with the scissors.
- Cut the hepatic vein with scissors to bleed the mouse as the secondary euthanasia method. Place the animal in a ventral position and wait 2 min to allow the blood to drain out. This step is important to reduce blood in the lungs.
- Remove the skin from the entire upper part of the mouse (including the head) to the base of the abdomen where the first incision was made with the aid of dissecting forceps and scissors. Cut the head with the scissors and place it in a sterile dry Petri dish.
- Cut the junctions between the maxilla and the mandible with the scissors and discard the mandible. With the scissors, make two cuts at the ends of the zygomatic arcs and remove them. Remove the eyes with the help of the dissecting forceps.
- Hold the cranium with the dissecting forceps and cut the cranium along the sagittal suture with a scalpel. Lift the two halves of the cranium with the brain to see the nasal mucosa. The nasal mucosas are surrounded by the anterior bones of the skull, including the nasal, maxilla, palatine, zygomatic, and ethmoid bones.
- Using the scalpel, cut the part of the cranium with the brain and discard. Place the other part of the cranium with the nasal mucosa in a sterile tube. Store the tube on ice (4 °C) if the samples are processed the same day, or on dry ice to freeze them quickly if the samples will be processed later.
- To avoid contamination of samples, clean and disinfect the dissecting tools between each tissue recovered and between each animal dissection with chlorine dioxide disinfectant.
- To collect the lungs, place the animal in dorsal recumbency and cut the pleura with the scissors. Open the rib cage by cutting the ribs at 1 cm at both sides of the sternum. Make the cuts from the base of the rib cage to the superior part with the scissors.
- Make a cross section with the scissors between the two incisions in the superior part of the rib cage and remove the chest plate. Hold the lungs with the forceps, cut at the end of the trachea (just before the lungs) with the scissors and put the lungs into a sterile tube. Store the tube on ice (4 °C) if the samples are processed the same day, or on dry ice to freeze them quickly if the samples will be processed later. NOTE: Homogenize the tissue samples on the same day that they are collected, and store at -80 °C to analyze viral titers later. It is important that samples not undergo repeated freeze-thaw cycles before virus titers are determined. Alternatively, store the tissue samples at -80 °C until they will be processed.
- Homogenization of lungs and nasal mucosa
- Place the lungs or nasal mucosa into a sterile Dounce homogenizer and add 1 mL of cold infection media. Homogenize the sample by moving the pestle up and down for approximately 1 min at room temperature (RT) until the lungs are completely disrupted. Put the homogenized sample in a sterile tube and store at 4 °C. Use a new Dounce homogenizer for each sample.
- Centrifuge the samples at 300 x g for 5 - 10 min at RT. Collect the supernatant in a new sterile tube. Store the supernatant at 4 °C if viral titration is performed on the same day and discard the pellet. Alternatively, freeze (-80 °C) the supernatant of the homogenized samples to evaluate viral titers later.
- Virus titration by indirect immunofluorescence
- The day before titration, seed 96-well plates with Madin-Darby Canine Kidney (MDCK) epithelial cells (4 x 104cells/well) to reach ~80 - 90% confluence in tissue culture media. Place the cells in a 37 °C incubator with 5% CO2. The day of viral titration, check the cells under the microscope to confirm a monolayer before starting the viral infection.
- Prepare 1:10 dilutions of the supernatant of the homogenized samples (lung or nasal mucosa) in infection media in a 96-well plate. Perform the viral titration by triplicates to accurately determine viral titers.
- Add 90 µL of infection media to each of the wells in the 96-well plate. Add 10 µL of one of the supernatant of the homogenized samples to wells A1, A2 and A3 to evaluate the viral titer of each sample in triplicate. Add 10 µL of another supernatant of the homogenized samples to wells A4, A5 and A6. Perform the same dilution consecutively with the rest of the samples.
- After the adding the sample supernatant to row A, use a multichannel pipet to mix by pipetting up and down. Transfer 10 µL from row A to row B. Change the tips between dilutions and mix well. Repeat this until reaching the last row (H).
- Remove the tissue culture medium (step 4.2.3.1) using a vacuum aspirator-multichannel adapter from the MDCK cells in the 96-well plates and wash twice with 100 µL/well of 1x PBS.
- Transfer 50 µL/well of the serially diluted supernatant of the homogenized samples to the 96-well plate with MDCK cells. Start transferring the higher dilutions (row H) to the lower dilutions (row A). In this case, it is not necessary to change the tips between different rows.
- Put the 96-well plates on a rocking platform for 1 h at RT to allow viral adsorption. After viral adsorption, remove the inoculum using a vacuum aspirator-multichannel adapter and add 100 µL/well of post-infection media. Incubate infected cells for 8 h in a 33 °C or 37 °C incubator with 5% CO2. Proceed to fixing, staining, imaging, and viral titer calculation as described in steps 4.2.3.6 – 4.2.3.12.
- Remove the tissue culture medium from the 96-well plates using a vacuum aspirator-multichannel adapter. Fix and permeabilize the cells with 100 µL/well of fixation/permeabilization solution for 20 min at RT. CAUTION: Use the fixation/permeabilization solution in a fume hood to prevent formaldehyde exposure.
- Remove the fixation/permeabilization solution using a vacuum aspirator-multichannel adapter, wash with 1x PBS, and incubate the fixed cells with blocking solution for 1 h at RT. Store the cells in blocking solution at 4 °C or proceed to the next step.
- Remove the blocking solution. Add 50 µL/well of 1 µg/mL monoclonal antibody (mAb) anti-NP HB-65 diluted in antibody dilution solution. Incubate for 1 h at 37 °C. Different mAb or polyclonal antibodies (pAb) anti-PR8 can be used.
- Meanwhile, dilute 1:200 the fluorescein isothiocyanate (FITC)-conjugated secondary anti-mouse antibody in antibody dilution solution. Centrifuge the solution at 1,700 x g for 5 - 10 min at RT. Other secondary antibodies conjugated with other fluorophores can be used.
- Remove the primary antibody and wash 3 times with 100 µL/well of 1x PBS. Add 50 µL/well of the secondary antibody dilution and incubate for 30 - 45 min at 37 °C. Remove the secondary antibody and wash 3 times with 100 µL/well of 1x PBS. Leave the last wash in the plate.
- Observe the cells under a fluorescence microscope to determine the number of positive stained (green) cells. Calculate the viral titer by counting FFU/mL. Calculate the viral titer expressed in FFU/mL from the dilution with 30 - 50 positive stained cells using the formula: ((n1 + n2 + n3) / 3) x 20 x 1/dilution.

5. Evaluation of Humoral Immune Responses (Figure 3)
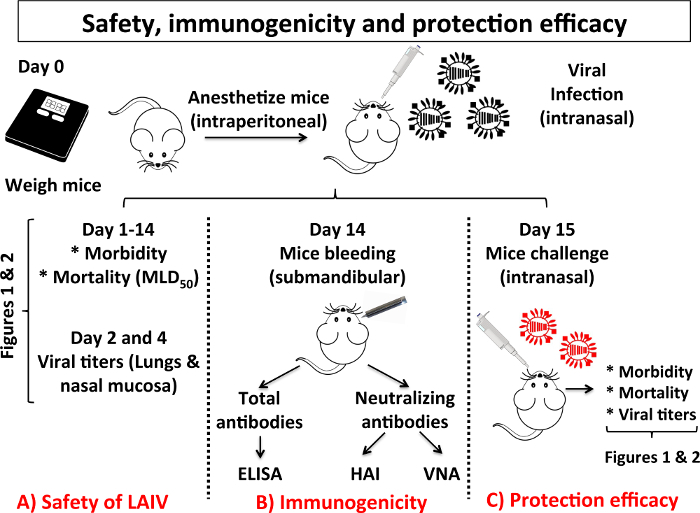
- Infect i.n. 3 groups of female 6-to-8-week-old C57BL/6 mice (n = 5) with the different doses (102, 103, and 104 FFU/mouse) of PR8 LAIV, and mock-infect one group (n = 5) with 1x sterile PBS as a negative control. Perform the i.n mouse infections as described in section 3.
- Mouse bleeding by submandibular puncture
- Fourteen days after immunization, collect the mice blood by submandibular bleeding using a 4 mm lancet26, or using another IACUC approved method.
- Hold the mouse by picking it up by the scruff of the neck between the index finger and thumb. Hold the tail against the hand with the pinky finger to immobilize the mouse.
- Put the mouse in a lateral position to see the cheek. Make the puncture at the juncture of the retro-orbital and submandibular veins with the jugular vein. Sink the lancet (4-mm) and recover the blood in a sterile tube (approximately 0.2 - 0.4 mL/mouse are recovered). Apply pressure on the puncture with a sterile napkin for a few seconds. Place the mouse back into the cage.
- Put the tubes for 1 - 2 h at 37 °C and then centrifuge them at 700 x g for 30 min at RT to separate the serum from the blood. Transfer the upper layer that consists of the serum with a pipet to a new sterile tube. Discard the pellet of red blood cells (RBCs). Store the serum at -20 °C.
- Analysis of total antibodies against viral proteins
- Preparation of infected MDCK cell extracts
- The day before infection, seed one 100-mm dish with 4 - 5 x 106 MDCK cells (to reach ~80 - 90% confluence the next day) in tissue culture media. Place the cells in a 37 °C incubator with 5% CO2. The day of viral infection, check the cells under the microscope to confirm a monolayer before starting the viral infection.
- Prepare a viral dilution with a low (0.001) multiplicity of infection (MOI) of PR8 WT in infection media in 4 mL total final volume. Calculate the amount of PR8 WT with the formula ((X MOI x n°cells) x final volume) / stock viral titer.
- Aspirate the tissue culture medium from the MDCK cell plate and wash twice with 4 mL of 1x PBS. Add the viral inoculum (4 mL) and put the plate on a rocking platform for 1 h at RT to allow viral adsorption. Remove the viral inoculum with vacuum and add 8-10 mL of post-infection media containing 1 µg/mL of TPCK-treated trypsin. Incubate infected cells for 48 - 72 h in a 33 °C or 37 °C incubator with 5% CO2.
- Detach the cells with the aid of a cell scraper and collect the cells and tissue culture medium with a pipet in a 15-mL tube. Centrifuge the tube at 400 x g for 5 - 10 min at RT. Aspirate the supernatant and resuspend the cell pellet in 1 mL of RIPA buffer and put the mix in a 1.5-mL sterile tube. Incubate on ice for 20-30 min.
- Centrifuge the tube at 1,300 x g for 20 - 30 min at 4 °C. Carefully, recover the supernatant. Store the cell extract in 100 µL aliquots at -20 °C.
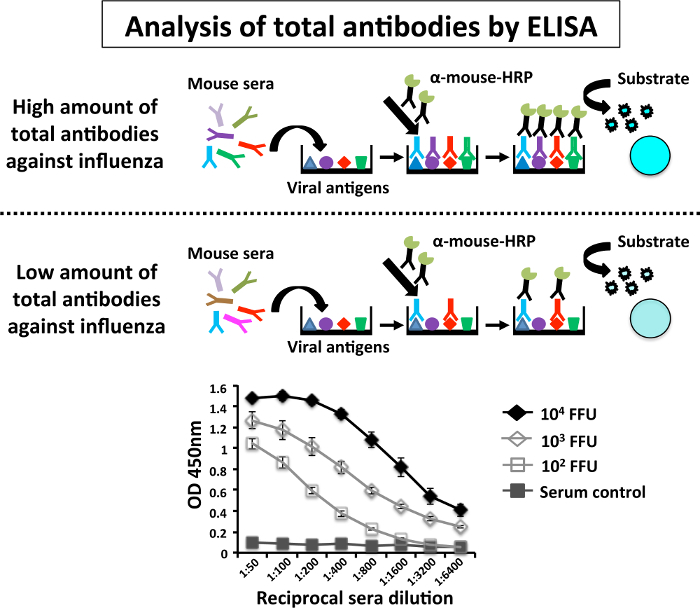
- ELISA (Figure 4)
- Coat polystyrene 96-well plates with the appropriate dilution of infected-MDCK cell extract in 1x PBS (usually between 1:200 - 1:1,000). Coat another plate with the same dilution of mock-infected MDCK cell extract as a negative control. Incubate overnight (O/N) at 4 °C.
- Remove the supernatant using a vacuum aspirator-multichannel adapter and wash once with 100 µL/well of 1x PBS with a multichannel pipet. Block non-specific binding by adding 100 µL/well of blocking solution for 1 h at RT.
- Meanwhile, make 2-fold serial dilutions (starting dilution 1:50) of the sera from each mouse infected in step 5.1 in antibody dilution solution in a 96-well plate.
- Remove the blocking solution from the polystyrene 96-well plates using a vacuum aspirator-multichannel adapter. Add 50 µL of each mouse serum dilution in the appropriate well and incubate 1 h at 37 °C.
- Remove the mice sera with a vacuum aspirator-multichannel adapter. Wash the wells 3 times with distilled water using a multichannel pipet. Add 50 µL/well of an anti-mouse secondary antibody conjugated with Horseradish Peroxidase (HRP) diluted 1:2,000 in antibody dilution solution. Incubate 1 h at 37 °C.
- Remove the secondary antibody with a vacuum aspirator-multichannel adapter. Wash the wells 3 times with distilled water using a multichannel pipet. Prepare the tetramethylbenzidine (TMB) substrate solution by mixing 1:1 solution A and B. Add 100 µL/well of substrate and incubate for 5 - 10 min at RT in the dark.
- Stop the reaction by adding 100 µL/well of 0.2 N H2SO4. Read the plates at 450 nm on an ELISA plate reader.
- Subtract the value obtained in each dilution of the MDCK mock-infected 96-well plate from the value obtained in the MDCK-infected 96-well plate. Calculate the average values for each dilution with the different sera and represent them in a graph showing standard deviations (SD).
- Analysis of neutralizing antibodies
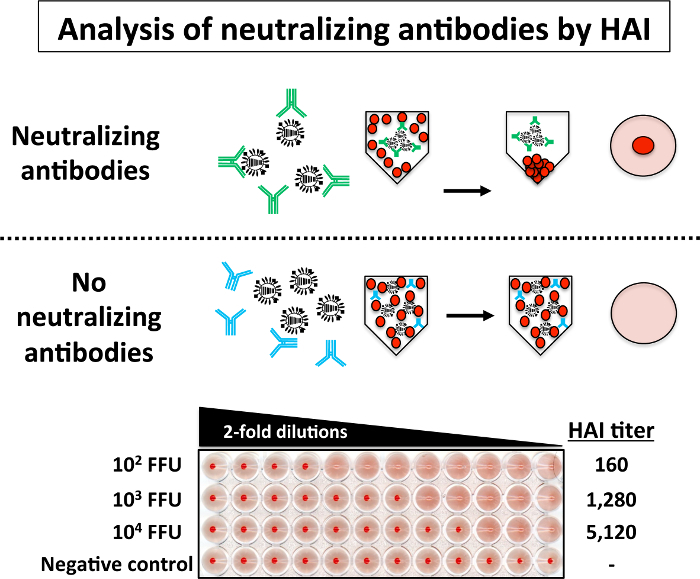
- Hemagglutination Inhibition (HAI) assay (Figure 5)
- Before performing the HAI assay, treat the mouse sera with receptor destroying enzyme (RDE) to eliminate all non-specific inhibitors present in the serum. Add 1 volume of mouse serum to 4 volumes of RDE working dilution (serum dilution is now 1:5). Incubate O/N (12 - 18 h) in a 37 °C water bath.
- Heat the serum samples at 56 °C for 30 min to inactivate the RDE.
- Pre-determine the hemagglutination activity (HAU) of PR8 WT by standard HA assay28. Once the viral HAU is determined, dilute the viral stock to 8 HAU per 50 µL in 1x PBS.
- Prepare 2-fold serial dilutions (12 dilutions) of RDE-treated sera in a 96-well V-bottom plate. Use one row for each serum sample (e.g., A1 to A12).
- Add 25 µL of 1x PBS from A1 to A12 wells. Test each of the serum samples in one row. Add 25 µL of RDE-treated sera (1:5) to 25 µL of 1x PBS in the first well of the column (A1). The first serum dilution is 1:10.
- Once all RDE-treated sera are added to the first column, (e.g., A1, B1, C1), use a multichannel pipet to mix the sera and 1x PBS by pipetting up and down. Transfer 25 µL of the diluted sera from the first column to the second column (e.g., from A1 to A2). Change the tips between dilutions and mix.
- Repeat steps 5.3.1.4.2. until reaching the last column (e.g., A12, B12, C12). Discard the 25 µL from the last column, keeping the volume consistent in all the wells (25 µL). Include one row of wells containing only 25 µL of 1x PBS without any sera as a negative control.
- Add 25 µL of the IAV diluted to 8 HAU/50 µL to each of the wells with sera in the 96-well V-bottom plate; the final volume in each well is 50 µL, containing 4 HAU of virus. Mix the contents by repeated pipetting. Incubate for 1 h at RT.
- Add 50 µL of 0.5% turkey RBCs to each of the wells. Mix the contents by repeated pipetting. Incubate the plate for 30 - 45 min on ice. Read the HAI assay visually when the RBCs in the control wells (1x PBS) form a red precipitate (i.e., pellet) at the bottom of the wells. NOTE: RBCs from other species such as chicken, guinea pig or horse can also be used.
- Calculate the HAI titers by identifying the last well in which the RBCs form a red pellet and hemagglutination does not occur. NOTE: The reciprocal value of this dilution is the HAI titer. The reciprocal value is multiplied by a factor of 20 to correct for the 1:20 dilution of the serum in the first row.
- Virus microneutralization assay (VNA) (Figure 6)
- The day before VNA, prepare MDCK cells in a 96-well plate to reach 80-90% confluence the next day as described in step 5.2.1.1. Prepare a dilution of PR8 WT in infection media to have 200 FFU/25 µL.
- Inactivate the mouse sera at 56 °C for 1 h. Make 2-fold serial dilutions (12 dilution) per mouse sera in triplicate using a 96-well plate.
- Add 40 µL of serum to 160 µL of infection media in the first well of the first row, (e.g., A1) (serum dilution 1:5). Mix by pipetting. Add 100 µL of infection media to the other wells (A2 to H12).
- Transfer 100 µL from the first row to the second row to make 1:2 dilutions (e.g., from A1 to A2). Change tips between dilutions and mix by pipetting. Repeat this until the last row (e.g., A12). Discard 100 µL from the last row, keeping the volume consistent in all the wells (100 µL).
- Add 100 µL of IAV dilution to all wells. Incubate 1 h at RT. Meanwhile, remove the tissue culture medium from the MDCK cells in the 96-well plate using a vacuum aspirator-multichannel adapter and wash twice with 100 µL/well of 1x PBS.
- In each 96-well plate of MDCK cells, leave two rows for the controls. One row is the negative control (cells without virus and sera), and the other row is the positive control of infection (cells with virus in the absence of sera or sera from mock-infected mice).
- Transfer 50 µL/well from the virus-antibody plate to the 96-well plates of MDCK cells. Put the plates on a rocking platform for 1 h at RT to allow viral adsorption.
- Remove the virus-antibody inoculum using a vacuum aspirator-multichannel adapter and add 100 µL/well of post-infection media containing 1 µg/mL of TPCK-treated trypsin.
- Incubate the cells for ~3 - 4 days in a 33 °C or 37 °C incubator with 5% CO2 until cytopathic effect (CPE) is observed in the positive control (infected cells in the absence of sera or with control serum).
- Remove the tissue culture medium using a vacuum aspirator-multichannel adapter and add 100 µL/well of 0.1% crystal violet. Incubate for 1 h at RT. Remove the crystal violet solution using a vacuum aspirator-multichannel adapter. Wash the wells 3 times with distilled water. Dry the plates at RT.
- Calculate the VNA titers by identifying the highest dilution that retains a confluent cell monolayer of MDCK cells. NOTE: The reciprocal value of this corresponding dilution is the neutralizing antibody titer. The reciprocal value is multiplied by a factor of 10 to correct the 1:10 dilution of the sera in the first well of the row.

6. Evaluation of Protection Efficacy Vaccines (Figure 3)
Vaccinate i.n. a group of female 6-to-8-week-old C57BL/6 mice (n = 11) with the PR8 LAIV dose to test (FFU/mouse) and mock-vaccinate another group of mice (n = 11) with 1x sterile PBS. Perform the mouse i.n. immunizations as previously described in section 3.
Fourteen days post-vaccination, bleed the mice and collect the sera as described in section 3. Analyze the presence of total and neutralizing antibodies against PR8 WT as described in section 5.1.1.
Fifteen days post-vaccination, measure and record the mouse body weight (Day 0). Challenge mice i.n. with a lethal dose of the PR8 WT (homologous challenge) as previously described in section 3.
Measure the body weight and survival (n = 5 / group) during 14 days after challenge to evaluate the morbidity and mortality produced by the PR8 WT challenge in vaccinated mice (section 4.1).
Recover mouse lungs and nasal mucosa (n = 3/day) at day 2 and day 4 post-challenge with PR8 WT. Homogenize and analyze the lungs and nasal mucosa as described in section 4.2 to assesses the viral replication of PR8 WT in vaccinated mice.
Representative Results
Characterization of viral pathogenesis in mice
The pathogenesis of IAV is related to the morbidity and mortality caused by its infection. These two parameters can be evaluated in mice easily: IAV morbidity is associated with body weight loss in infected mice and the percentage of survival will indicate the mortality rate (Figure 1). The body weight and survival in IAV-infected mice are usually monitored daily for at least two weeks after infection8,9,10,29. The survival data obtained can determine the MLD50 that was calculated using the method of Reed and Muench25. Another indicator of IAV pathogenesis is related to viral replication in the lower (lungs) and in the upper (nasal mucosa) respiratory tract (Figure 1). The information obtained with the viral titer in these organs corresponds to the morbidity and mortality values, and taken together provide a good indication of viral pathogenesis. It is known that IAV replication in mouse lungs peak between days 2 and 4 post-infection (p.i.), and so we recommended recovering these organs at days 2 and 4 p.i.
Figure 1: Characterization of IAV Pathogenesis in Mice: IAV pathogenesis in mice is related to its morbidity (body weight loss) and mortality (% survival), and also to the ability of IAV to replicate in the upper (nasal mucosa) and lower (lungs) respiratory tract. Briefly, on Day 0 mice are weighed and anesthetized intraperitoneally with 2,2,2-tribromoethanol (TBE) before they are infected intranasally with IAV. From day 1 to day 14, mice are weighed daily to evaluate body weight loss (morbidity) and % survival (mortality) to calculate the mouse lethal dose 50 (MLD50). On days 2 and day 4 post-infection, mice lungs and nasal mucosa are recovered and homogenized to evaluate viral replication. Mice experiments to characterize IAV pathogenesis using PR8 WT are performed under BSL-2 conditions. Please click here to view a larger version of this figure.
In Figure 2, the data obtained in the evaluation of PR8 WT pathogenesis is represented. Four groups of 6-to-8-week-old C57BL/6 mice (n = 11) were infected i.n. with the indicated doses (10, 102, 103 and 104 FFU/mouse) of PR8 WT. The body weight and survival of 5 mice per group were measured by 14 days p.i. (Figure 2A-B). All mice immunized with 104 FFU of PR8 WT lost weight quickly (Figure 2A) and all of them died at days 5 and 6 p.i. (Figure 2B). Mice immunized with 103 FFU of PR8 WT lost body weight at later times p.i. (Figure 2A) and they all died by days 7 and 8 p.i. (Figure 2B). All mice immunized with 102 FFU of PR8 WT lost body weight (Figure 2A) but only 2 of them died at day 9 p.i. (Figure 2B). Finally, mice immunized with 10 FFU of PR8 WT did not lose weight (Figure 2A) and all of them survived infection (Figure 2B). The MLD50 of PR8 WT in this experiment calculated with the Reed and Muench method25 was 1.5 x 102 FFU (Figure 2C).
To evaluate the PR8 replication in the upper and lower respiratory tract of mice infected i.n. with the indicated doses (10, 102,103, and 104 FFU/mouse), the lungs and nasal mucosa were recovered at days 2 and 4 p.i. (n = 3). After lung homogenization, the amount of virus present in this organ was analyzed by immunofluorescence assay (Figure 2D). The viral titer detected in the lungs of the different mice groups was related with the dose used to immunize them: higher viral titers were detected in lungs of mice immunized with 104 FFU of PR8 WT at days 2 and 4 p.i., while lower viral titers were detected in the lungs of mice immunized with 10 FFU of PR8 WT.
Figure 2: Representation of the Data Obtained in the Evaluation of Viral Pathogenesis: To analyze morbidity and mortality of PR8 WT, 6-to-8-week-old female C57BL/6 mice (n = 5) were infected i.n. with the indicated number of fluorescent-forming units (FFU) of PR8 WT and then monitored daily for 2 weeks for (A) body-weight loss (morbidity) and (B) survival (mortality). Data represent the means and standard deviations of results determined for individual mice (n = 5). (C) Values of % survival evaluated over 2 weeks are used to calculate the MLD50 using the Reed and Muench method25. (D) To evaluate viral replication, 6-to-8-week-old female C57BL/6 mice (n = 6) were infected i.n. with the indicated number of FFU. Viral replication in the lungs or nasal mucosa of infected mice is usually evaluated at days 2 and 4 p.i. using an immunofocus assay. Viral titers are recorded as FFU/mL. Data represent the means and SDs of results obtained in each mouse. Dashed lines are included to indicate the limit of detection (200 FFU/mL) of the assay. Mice and tissue culture experiments to assess IAV morbidity, mortality and viral titers using PR8 were performed under BSL-2 conditions. Please click here to view a larger version of this figure.
Evaluation of the humoral responses induced after vaccination
Humoral responses induced against IAV infection play an essential role in controlling viral infection. For this reason, it is very important to analyze the humoral responses elicited by IAV WT infections or by new IAV vaccines (Figure 3). In the mouse model, 14 days after immunization, mice are bled to analyze the presence of antibodies against the total viral proteins by ELISA (Figure 4), and the presence of neutralizing antibodies targeting the viral HA protein, which is usually analyzed by common serologic approaches, such as a HAI assay (Figure 5) and/or a VNA (Figure 6).
Figure 3: Scheme of the Different Steps in the Characterization of Safety, Immunogenicity and Protection Efficacy of a LAIV: When a new LAIV is developed, the mouse model of IAV infection is usually used to test safety, immunogenicity and protection efficacy. In this scheme, the experiments to assess safety (A), immunogenicity (B) and protection efficacy (C) of a candidate LAIV are indicated. (A) To evaluate the safety of a LAIV it is necessary to assay the same parameters (morbidity, mortality and viral replication in the upper and lower respiratory tract) described in Figure 1. To evaluate immunogenicity, mice are immunized and 14 days after vaccination they are bled by submandibular puncture. Humoral responses are analyzed by assessing the presence of total antibodies against IAV proteins by enzyme-linked immunosorbent assay (ELISA) and by evaluating the presence of neutralizing antibodies by hemagglutination inhibition assay (HAI) and/or virus microneutralization assay (VNA). (C) To evaluate protection efficacy, 15 days after vaccination, mice are challenged with IAV WT. Protection efficacy is evaluated by the measurement of mice morbidity, mortality and viral titers in the lungs of challenged mice as described in Figure 1. Mice experiments to characterize safety, immunogenicity and protection efficacy of a LAIV candidate are performed under BSL-2 conditions. Other BSL suits or containment conditions may be required depending on the IAV strain used for the challenge of vaccinated mice. Please click here to view a larger version of this figure.
To assess immunogenicity of PR8 LAIV, three groups of 6-to-8-week-old C57BL/6 mice (n = 5) were immunized with different doses (102,103, and 104 FFU/mouse) of PR8 LAIV or mock vaccinated with 1x PBS as a negative control. Fourteen days after vaccination, mice sera were collected by submandibular bleeding26 (Figure 3). Antibody responses against total viral proteins were evaluated by ELISA using a 1:200 dilution of cell extract of PR8-infected MDCK cells (Figure 4). Each serum was analyzed individually. The results indicate that the sera from mice immunized with the higher dose (104 FFU) of PR8 LAIV had higher antibody titers against total PR8 proteins than the sera of mice immunized with the lower dose (102 FFU). Sera control (mock-infected mice) did not show reactivity against PR8 antigens.
Figure 4: Schematic Representation of the Enzyme-linked Immunosorbent Assay (ELISA) to Evaluate Humoral Antibody Responses: Levels of PR8-specific antibodies present in infected mice are determined by ELISA in 96-well polystyrene plates coated with cell extracts from mock (control) or PR8-infected MDCK cells. Briefly, at 14 days p.i. (Figure 3), mice immunized with the indicated doses of PR8 LAIV were bled by submandibular puncture and sera were collected. Each serum was evaluated individually by ELISA for IgG antibodies against total PR8 proteins. Graph data represent the means and standard deviations of the results obtained from individual mice sera (n = 6). O.D, optical density. ELISA assays were performed in a BSL-2 cabinet. Please click here to view a larger version of this figure.
To analyze the presence of neutralizing antibodies in the mouse serum using the HAI assay, serial 2-fold dilutions of sera (previously inactivated at 56 °C) of each group of mice immunized with different doses (102, 103, and 104 FFU) of PR8 LAIV were incubated with 4 HAU of PR8 WT for 1 h at RT (Figure 5). Then, 0.5 % of turkey RBCs were added to the PR8-sera mixtures. The upper part of Figure 5 is a schematic representation of the possible HAI results: when the serum contains neutralizing antibodies, RBCs precipitation is observed in the bottom of the well because the antibodies bound to the viral HA protein prevent the hemagglutination of the RBCs. On the other hand, in the absence of neutralizing antibodies, hemagglutination of the RBCs is observed. Then, the HAI titer is calculated as the reciprocal of the dilution of serum in the last well lacking hemagglutination. The HAI results showed in the lower part of Figure 5 indicate that the serum from mouse vaccinated with the greatest amount of PR8 LAIV (104 FFU) have the higher titer of neutralizing antibodies while the serum from a mouse immunized with the lower dose (102 FFU) have the lowest titer of neutralizing antibodies against PR8. No neutralizing antibodies were present in serum control (mock-infected mice).
Figure 5: Hemagglutination Inhibition (HAI) Assay: Levels of neutralizing antibodies against PR8 WT in infected mice can be easily evaluated by HAI assay. Briefly, 2-fold serial dilutions (starting dilution 1:10) of sera from mice immunized with the indicated doses of PR8 LAIV were mixed (1:1) with 4 HAU of PR8 WT for 1 h at RT in a 96-well V-bottom plate. After 1 h incubation, 0.5% RBCs were added to the virus-serum mixtures for 30 - 60 min on ice. HAI titers are determined by identifying the last well in which the RBCs form a red button and hemagglutination does not occur (top). HAI assays with PR8 WT were performed in a BSL-2 cabinet. Please click here to view a larger version of this figure.
In Figure 6, the data obtained in the evaluation of neutralizing antibodies in mouse sera by VNA is represented. 200 FFU of PR8 WT were mixed with serial 2-fold dilutions of sera (previously inactivated at 56 °C) from mice immunized with different doses (102, 103, and 104 FFU) of PR8 LAIV for 1 h at RT. Each serum sample was assessed in triplicate. The virus-sera mixtures were used to infect monolayers of MDCK cells in a 96-well plate. The upper part of Figure 6 is a schematic representation of the possible VNA results: absence of neutralizing antibodies in the serum results in CPE at approximately 3 - 4 days p.i. In contrast, when neutralizing antibodies are present (intact cell monolayer), they bind to the viral HA and prevent viral infection, and there is no CPE. Presence of CPE is usually visualized by staining infected cells with crystal violet, and viral neutralization titers are calculated as the reciprocal of the last dilution at which infection is completely blocked. The results of the VNA are represented in the lower part of Figure 6. The serum from mice immunized with the high amount of PR8 LAIV (104 FFU) have the greatest titer of neutralizing antibodies while sera from mice immunized with the lower dose or PR8 LAIV (102 FFU) have the lowest titer of neutralizing antibodies, similar to results obtained with the HAI assay. No neutralizing antibodies were present in the serum from mock-infected mice.
Figure 6: Virus Microneutralization Assay (VNA): An alternative to the HAI assay, the VNA assay can evaluate the presence of PR8 WT neutralizing antibodies. Briefly, sera from mice vaccinated with the indicated doses of PR8 LAIV were serially 2-fold diluted (start dilution 1:5) in 96-well plates and mixed 1:1 with 200 FFU of PR8 WT for 1 h at RT. After 1 h, the virus-serum mixtures were used to infect 96-well plates of MDCK cells. Infected cells were incubated for 3 - 4 days until complete cytopathic effect. The 96-well plates were then stained with 0.1% crystal violet. VNA titers are determined by identifying the highest dilution to retain a confluent cell monolayer. VNA with PR8 WT were performed under BSL-2 conditions. Please click here to view a larger version of this figure.
Evaluation of protection efficacy of IAV vaccines in mice
When a new LAIV is developed, its safety, immunogenicity and protection efficacy must be tested in vivo, and the mouse model is usually the first animal model chosen to analyze these parameters. Figure 3 is a scheme of the different steps needed to characterize a new LAIV in mice. To evaluate safety of a LAIV (Figure 3A), mice are infected i.n. using a similar protocol to the experimental procedures described in the pathogenesis section (section 4), and the body weight and survival monitoring will provide information related to the morbidity and mortality of the LAIV (Figures 2A-2B), including the MLD50. In addition, upon inoculation with different vaccine doses, the replication of LAIV in the lower (lungs) and in the upper (nasal mucosa) respiratory tract should be assessed8,9,10,11,29 (Figure 2D). To test immunogenicity, sera from mice vaccinated with the LAIV are collected before challenge with PR8 WT (homologous challenge), using similar protocols to those described in the immunogenicity section (section 5) (Figure 3B). The presence of total and neutralizing antibodies in the sera can be detected by ELISA and HAI and/or VNA, respectively.
Lastly, to test protection efficacy, LAIV-vaccinated mice are challenged with PR8 WT and the protection efficacy is characterized by monitoring morbidity, mortality and the presence of challenge PR8 WT in the lungs, as previously described in section 4 (Figure 3C)8,9,10,11. If the LAIV induces protection against PR8 WT challenge, mice will not lose body weight, and they will survive the lethal challenge with IAV WT. Moreover, IAV WT viral titers in the lungs will not be detected, or will be significantly reduced as compared to mock (PBS) vaccinated mice8,9,10,11.
| Tissue culture media and solutions | Composition | Storage | Use | Comments |
| Tissue culture media: Dulbecco’s modified Eagle’s medium (DMEM), 10 % Fetal Bovine Serum (FBS), 1% Penicillin-Streptomycin-L-glutamine (PSG) (DMEM 10 % FBS 1% PSG) | 445 ml DMEM, 50 ml of FBS and 5 ml of 100x 1% Penicillin (100 units/ml)-Streptomycin (100 µg/ml)-L-glutamine (2 mM) (PSG) | Store at 4°C | This media is used for maintenance of Madin-Darby Canine Kidney (MDCK) epithelial cells | |
| Post-infection media: DMEM 0.3 % Bovine Albumin (BA), 1% PSG (DMEM 0.3 % BA 1% PSG) | 491 ml DMEM, 4.2 ml of 35 % BA and 5 ml of 100x PSG | Store at 4°C | This media is used for maintenance of MDCK cells after infection with Influenza A virus (IAV) | |
| 10x Phosphate buffered saline (10x PBS) | 80 g of NaCl, 2 g of KCl, 11.5 g of Na2HPO4.7H2O, 2 g of KH2PO4. Add ddH2O up to 1 L. Adjust pH to 7.3 | Store at room temperature (RT) | Sterilize by autoclave | |
| 1x PBS | Dilute 10x PBS 1:10 with ddH2O | Store at RT | Sterilize by autoclave | |
| Infection media: 1x PBS, 0.3% BA, 1% Penicillin-Streptomycin (PS) (PBS/BA/PS) | 487 ml 1x PBS sterile, 4.2 ml of 35% BA and 5 ml of 100x 1% Penicillin (100 units/ml)-Streptomycin (100 µg/ml) (PS) | Store at 4 °C | This media is used for IAV infections | |
| Fixation/permeabilization solution: 4% formaldehyde, 0.5% triton X-100 diluted in 1x PBS | 400 mL Neutral Buffered Formalin 10%, 5 ml of Triton X-100 and 595 ml of 1x PBS | Store at RT | This solution is used to fix and permeabilize MDCK cells in viral titration experiments | Prepare the solution in a fume hood to prevent exposure to formaldehyde |
| Blocking solution: 2.5% Bovine Serum Albumin (BSA) in 1x PBS | 2.5 g of BSA in 97.5 mL of 1x PBS | Store at 4 °C | This solution is used as a blocking solution for immunofluorescence assays and ELISAs. | Sterilize by filtration with 0.2 µm filter. |
| Antibody dilution solution (1% BSA in 1x PBS) | 1 g of BSA in 99 mL of 1x PBS | Store at 4 °C | This solution is used for the dilution of primary and secondary antibodies in immunofluorescence assays and ELISAs | Sterilize by filtration with 0.2 µm filter |
| 0.1% crystal violet solution | 1 g of crystal violet in 400 ml of methanol. Add 600 ml of ddH2O | Store at RT | This solution is used to fix and stain MDCK cells in viral neutralization assays | |
| Tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin | Prepare a 1,000x stock solution at 1 mg/ml in ddH2O | Store at -20 °C | The TPCK-trypsin is added in IAV infections | Make 100 µl aliquots |
| RIPA buffer | 10 mM Tris-Cl (pH 8.0), 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl | Store at 4 °C | This solution is used to make cell extracts |
Table 1: Tissue Culture Media and Solutions.
Discussion
The mouse model of IAV is widely used for in vivo studies of IAV pathogenesis, immunogenicity and protection efficacy. The small size of mice makes them easy to manipulate and store as compared to other animal models such as ferrets or guinea pigs. Moreover, the ease in terms of animal cost, housing and reproduction allow their use in pre-clinical vaccination tests in which large numbers of animals are needed. Notably, since mice have been used in multiple research disciplines, several molecular and immunology murine reagents are available to facilitate their use for IAV studies. Furthermore, the minimal host variability and the presence of an immune system that is evolutionarily similar to that present in humans make them a robust small animal model for IAV studies. Finally, the study of host-viral protein interactions and their contribution to viral pathogenesis are currently feasible by the access to KO mice. However, beside the several advantages of using mice for IAV studies, they are not useful for IAV transmission studies. Therefore, ferrets or guinea pigs are more suitable animal models to study IAV transmission. Clinical symptoms in mice infected with IAV usually appear 2 - 3 days post-infection, although this may vary depending on the mouse age, immune status, sex, genetic background and strain; as well as viral strain and dose used. Symptoms include anorexia, lethargy, huddling, ruffled fur, loss of body weight and death16,17.
In this manuscript, we describe how to evaluate viral pathogenesis in mice infected i.n. with IAV by monitoring body weight loss (morbidity), percentage of survival (mortality) and by assessing viral replication in the upper (nasal mucosa) and lower (lungs) respiratory tract (Figure 1and Figure 2). The pathogenesis of IAV is a very important parameter not only in the context of new IAV strains but also in the safe implementation of LAIV vaccine candidates (Figure 3). Seasonal IAVs and IAVs that infect other mammals (like horses, dogs or pigs) do not usually produce signs of disease in mice11,27. In this case, the analysis of viral titers in the lungs or nasal mucosa of infected mice is the main parameter to evaluate viral pathogenesis of IAV strains. Once the lungs and nasal mucosa are collected, they are homogenized and the amount of virus present in these organs can be analyzed by plaque assay, immunofluorescence assay, tissue culture infectious dose 50 (TCID50) or another validated method8,29. We recommend evaluating viral titers using indirect immunofluorescence since the plaque assay requires a higher quantity of MDCK cells (6-well plate format) and, like the TCID50, more time (3 - 4 days) is required to calculate viral titers. However, to perform the indirect immunofluorescence assay, it is necessary to have: 1) primary specific antibodies against an IAV protein (it is recommended to use an antibody against the IAV nucleoprotein, NP, since it is the most abundant viral protein produced during viral infection); 2) secondary antibodies conjugated to a fluorophore; and 3) a fluorescence microscope to count fluorescent positive infected cells.
Since the immune system of mice is evolutionarily similar to the immune system of humans16 and because mice can elicit a strong and protective humoral response against IAV infection, the immunogenicity of IAVs (or vaccine candidates) can be easily evaluated by determining total (ELISA; Figure 4) and neutralizing (HAI, Figure 5) or VNA (Figures 6) antibody responses. To analyze the humoral responses after immunization, mice can be bled by different approved methods such as retro-orbital puncture, tail clip, saphenous vein puncture or submandibular puncture. We chose the submandibular puncture technique26 because the procedure does not require the use of anesthesia and allows us to obtain an acceptable volume of blood for immunological studies; as opposed to the retro-orbital puncture, where mice should be anesthetized, and with the tail clip or saphenous vein puncture, where the blood volume recovered usually is low.
In this manuscript, we described how to evaluate the presence of antibodies against total viral proteins by ELISA using virus infected cell extracts. Since IAV infection induces a protective immunity mediated, mainly, by antibodies against the viral HA (the main antigenic viral target), it is highly recommended to assess the amount of specific antibodies induced against HA by ELISA using recombinant purified HA proteins10. To analyze the presence of neutralizing antibodies, both HAI and VNA methods are accepted but both have advantages and disadvantages. The HAI is rapid (2-3 h) and approved by the Center for Disease and Control (CDC) to evaluate the presence of IAV neutralizing antibodies30. The VNA is more time consuming (3-4 days) but is more specific since it evaluates only antibodies that can neutralize viral infection. Notably, the VNA has been shown to detect stalk-reactive HA neutralizing antibodies31, as well as NA neutralizing antibodies32. Another advantage of the VNA is the assay sensitivity, since less IAV ( 200 FFU) is needed as compared to the HAU assay that requires approximately ~104-105 FFU. Moreover, the HAI assay can be problematic for detecting neutralizing antibodies against avian IAVs due to difficulty with interpreting results33,34,35. Additionally, VNA does not require the use of RBCs for the identification of influenza neutralizing antibodies.
Finally, we describe the experimental procedures to evaluate the three main aspects that have to be considered in developing new IAV vaccines (Figure 3): 1) safety: the vaccine must be safe and not produce disease; 2) immunogenicity: the vaccine must be immunogenic and induce both humoral and cellular protective responses; 3) protection efficacy: the vaccine must induce protection against challenge with WT homologous and/or heterologous viruses. The mouse model is a good choice for the first screening of a new IAV vaccine in vivo because it can evaluate diverse vaccination schemes and conditions, including use of different adjuvants, antigen/vaccine dose, administration and broad protection against challenge with homologous or heterologous IAVs strains16,17. However, before proceeding to human clinical trials, vaccine candidates should at a minimum be tested in a second in vivo model.
Disclosures
The authors have nothing to disclose.
Acknowledgments
Research on influenza virus in LM-S laboratory is partially funded by The New York Influenza Center of Excellence (NYICE), a member of the NIAID Centers of Excellence for Influenza Research and Surveillance (CEIRS). We thank Wendy Bates for her support in the corrections of the manuscript.
References
- Girard MP, Cherian T, Pervikov Y, Kieny MP. A review of vaccine research and development: human acute respiratory infections. Vaccine. 2005;23(50):5708–5724. doi: 10.1016/j.vaccine.2005.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold S, Monto MD. Epidemiology and Virology of Influenza Illness. Am J Manag Care. 2000;6(Suppl 5):255–264. [PubMed] [Google Scholar]
- Palese P, Shaw ML. Orthomyxoviridae: The Viruses and Their Replication. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, editors. Fields Virology. 5th ed. Lippincott Williams and Wilkins; 2007. [Google Scholar]
- Li KS, et al. Genesis of a highly pathogenic and potentially pandemic H5N1 influenza virus in eastern Asia. Nature. 2004;430(6996):209–213. doi: 10.1038/nature02746. [DOI] [PubMed] [Google Scholar]
- Nogales A, Martinez-Sobrido L. Reverse Genetics Approaches for the Development of Influenza Vaccines. Int J Mol Sci. 2017;18(20) doi: 10.3390/ijms18010020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisaki KM, Janoff EN. Influenza in immunosuppressed populations: a review of infection frequency, morbidity, mortality, and vaccine responses. Lancet Infect Dis. 2009;9(8):493–504. doi: 10.1016/S1473-3099(09)70175-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshe RB. Live attenuated versus inactivated influenza vaccine in infants and young children. N Engl J Med. 2007;356(7):685–696. doi: 10.1056/NEJMoa065368. [DOI] [PubMed] [Google Scholar]
- Cox A, Baker SF, Nogales A, Martinez-Sobrido L, Dewhurst S. Development of a mouse-adapted live attenuated influenza virus that permits in vivo analysis of enhancements to the safety of live attenuated influenza virus vaccine. J Virol. 2015;89(6):3421–3426. doi: 10.1128/JVI.02636-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales A, et al. Influenza A Virus Attenuation by Codon Deoptimization of the NS Gene for Vaccine Development. J Virol. 2014;88(18):10525–10540. doi: 10.1128/JVI.01565-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales A, DeDiego ML, Topham DJ, Martinez-Sobrido L. Rearrangement of Influenza Virus Spliced Segments for the Development of Live-Attenuated Vaccines. J Virol. 2016;90(14):6291–6302. doi: 10.1128/JVI.00410-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales A, Huang K, Chauché C, DeDiego ML, Murcia PR, Parrish CR, Martínez-Sobrido L. Canine influenza viruses with modified NS1 proteins for the development of live-attenuated vaccines. Virology. 2016;500(2017):1–10. doi: 10.1016/j.virol.2016.10.008. [DOI] [PubMed] [Google Scholar]
- Garcia-Sastre A, et al. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology. 1998;252(2):324–330. doi: 10.1006/viro.1998.9508. [DOI] [PubMed] [Google Scholar]
- Lowen AC. Blocking interhost transmission of influenza virus by vaccination in the guinea pig model. J Virol. 2009;83(7):2803–2818. doi: 10.1128/JVI.02424-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mubareka S. Transmission of influenza virus via aerosols and fomites in the guinea pig model. J Infect Dis. 2009;199(6):858–865. doi: 10.1086/597073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales A, Baker SF, Martinez-Sobrido L. Replication-competent influenza A viruses expressing a red fluorescent protein. Virology. 2014;476C:206–216. doi: 10.1016/j.virol.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margine I, Krammer F. Animal Models for Influenza Viruses: Implications for Universal Vaccine Development. Pathogens. 2014;3(4):845–874. doi: 10.3390/pathogens3040845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvier N, Lowen AC. Animal Models for Influenza Virus Pathogenesis and Transmission. Viruses. 2010;2(8):1530–1563. doi: 10.3390/v20801530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pica N, Iyer A, Ramos I, Bouvier NM, Fernandez-Sesma A, García-Sastre A, Lowen AC, Palese P, Steel J. The DBA.2 mouse is susceptible to disease following infection with a broad, but limited, range of influenza A and B viruses. J Virol. 2011;85(23):12825–12829. doi: 10.1128/JVI.05930-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Numata K, Ito T, Takagi K, Matsukawa A. Innate immune response in Th1- and Th2-dominant mouse strains. Shock. 2004;22(5):460–466. doi: 10.1097/01.shk.0000142249.08135.e9. [DOI] [PubMed] [Google Scholar]
- Srivastava B, Blazejewska P, Hessmann M, Bruder D, Geffers R, Mauel S, Gruber AD, Schughart K. Host genetic background strongly influences the response to influenza a virus infections. PLoS One. 2009;4(3):e4857. doi: 10.1371/journal.pone.0004857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowen AC, Bouvier NM, Steel J. Transmission in the Guinea Pig Model. Curr Top Microbiol Immunol. 2014;385:157–183. doi: 10.1007/82_2014_390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schickli JH. Plasmid-only rescue of influenza A virus vaccine candidates. Philos Trans R Soc Lond B Biol Sci. 2001;356(1416):1965–1973. doi: 10.1098/rstb.2001.0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Sobrido L, Garcia-Sastre A. Generation of recombinant influenza virus from plasmid DNA. J Vis Exp. 2010. [DOI] [PMC free article] [PubMed]
- National Research Council (U.S.) Committee for the Update of the Guide for the Care and Use of Laboratory Animals., Institute for Laboratory Animal Research (U.S.) Guide for the care and use of laboratory animals. 8th ed. National Academies Press (U.S.); 2011. [Google Scholar]
- Reed LJ, Muench H. A simple method of estimating fifty percent endpoints. The American Journal of Hygiene. 1938;27(3):493–497. [Google Scholar]
- Golde WT, Gollobin P, Rodriguez LL. A rapid, simple, and humane method for submandibular bleeding of mice using a lancet. Lab Animal. 2005;34(9):39–43. doi: 10.1038/laban1005-39. [DOI] [PubMed] [Google Scholar]
- Nogales A. A temperature sensitive live-attenuated canine influenza virus H3N8 vaccine. J Virol. 2016. [DOI] [PMC free article] [PubMed]
- Eisfeld AJ, Neumann G, Kawaoka Y. Influenza A virus isolation, culture and identification. Nat Protoc. 2014;9(11):2663–2681. doi: 10.1038/nprot.2014.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Baker SF, Martinez-Sobrido L, Topham DJ. Induction of CD8 T cell heterologous protection by a single dose of single-cycle infectious influenza virus. J Virol. 2014;88:12006–12016. doi: 10.1128/JVI.01847-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC: Centers for Disease Control and Prevention. Antigenic Characterization. 2017. Available from: http://www.cdc.gov/flu/professionals/laboratory/antigenic.htm.
- He W, Mullarkey CE, Miller MS. Measuring the neutralization potency of influenza A virus hemagglutinin stalk/stem-binding antibodies in polyclonal preparations by microneutralization assay. Methods. 2015. [DOI] [PubMed]
- Gulati U. Antibody epitopes on the neuraminidase of a recent H3N2 influenza virus (A/Memphis/31/98) J Virol. 2002;76(23):12274–12280. doi: 10.1128/JVI.76.23.12274-12280.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beare AS, Webster RG. Replication of avian influenza viruses in humans. Arch Virol. 1991;119:37–42. doi: 10.1007/BF01314321. [DOI] [PubMed] [Google Scholar]
- Rowe T. Detection of antibody to avian influenza A (H5N1) virus in human serum by using a combination of serologic assays. J Clin Microbiol. 1999;37(4):937–943. doi: 10.1128/jcm.37.4.937-943.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson I, Wood JM, Nicholson KG, Zambon MC. Sialic acid receptor specificity on erythrocytes affects detection of antibody to avian influenza haemagglutinin. J Med Virol. 2003;70(3):391–398. doi: 10.1002/jmv.10408. [DOI] [PubMed] [Google Scholar]