

Abstract
The existence of endothelial progenitor cells (EPCs) in peripheral blood and its involvement in vasculogenesis was first reported by Ashara and colleagues1. Later, others documented the existence of similar types of EPCs originating from bone marrow2,3. More recently, Yoder and Ingram showed that EPCs derived from umbilical cord blood had a higher proliferative potential compared to ones isolated from adult peripheral blood4,5,6. Apart from being involved in postnatal vasculogenesis, EPCs have also shown promise as a cell source for creating tissue-engineered vascular and heart valve constructs7,8. Various isolation protocols exist, some of which involve the cell sorting of mononuclear cells (MNCs) derived from the sources mentioned earlier with the help of endothelial and hematopoietic markers, or culturing these MNCs with specialized endothelial growth medium, or a combination of these techniques9. Here, we present a protocol for the isolation and culture of EPCs using specialized endothelial medium supplemented with growth factors, without the use of immunosorting, followed by the characterization of the isolated cells using Western blotting and immunostaining.
Keywords: Developmental Biology, Issue 127, Endothelial progenitor cells, circulating progenitors, late endothelial progenitor cells, blood mononuclear cells, tissue engineering, umbilical cord blood
Introduction
Several investigators have studied the characteristics and potential of human EPCs5,10,11,12,13. EPCs can be described as circulating cells that have the ability to adhere to endothelial tissue in sites of hypoxia, ischemia, injury, or tumor formation and contribute to the formation of new vascular structures4,14. Their observed involvement in neovascularization, in the form of postnatal vasculogenesis, has led to an understanding of the pathophysiology of these cells and their use in therapeutic applications4,15,16. The number of EPCs in an individual has been shown to be correlated with cardiovascular pathology9,15,16,17,18,19,20. Other studies have also differentiated EPCs into a valve fibroblast-like phenotype and proposed that these cells could be used for tissue-engineering heart valves7,21.
The particular cell surface molecules needed to isolate EPCs have not been clearly identified due to discrepancies between investigations4. The adhesion of MNCs to a certain matrix, with exposure to a variety of culture conditions, has been performed by several groups1,17,22,23, suggesting that putative EPCs may display different phenotypic properties. These properties include a lack of phagocytotic ability, tube formation in Matrigel, and the uptake of Dil-acetylated low-density lipoproteins. The high clonogenic and proliferative potential are two properties with which EPCs can be hierarchized5. EPCs can also form in vitro tubules when cocultured with human fetal lung fibroblasts4. These cells are known to express endothelial cell surface markers and to share some of the hematopoietic markers13,24,25. The positively expressed markers that are widely accepted for phenotyping EPCs are CD31, CD34, vascular endothelial growth factor receptor 2 (VEGFR2), von Willebrand Factor (vWF), CD133, c-Kit, and vascular endothelial cadherin (VE-cadherin)4,18. Cells that co-express CD90, CD45, CD14, CD115, or alpha-smooth muscle actin (α-SMA) are not considered to be EPCs because of their limited proliferative potential, ability to phagocytose bacteria, and inability to form de novo human vessels in vivo4,7. This article outlines a modified protocol for the isolation of endothelial progenitor cells from human umbilical cord blood without the need for any cell sorting protocols. For this article, we used CD31, CD34, and VEGFR2 as the positive markers, with α-SMA as the negative indicator.
In this article, we propose a method of isolating and culturing endothelial progenitor cells from umbilical cord blood without cell sorting using specialized endothelial growth medium supplemented with growth factors (EGM). This EGM contains vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF), which enhance the survival, proliferation, and migration of endothelial cells26. It also includes ascorbic acid, which is responsible for maintaining the cobblestone morphology of cells; insulin-like growth factor-1 (IGF-1), which provides angiogenic and migratory function; and heparin, which causes improved long-term stability of growth factors in the medium26. Other growth factors added to the endothelial cell culture medium includes supplementation with epidermal growth factor (EGF), which helps in stimulating cell proliferation and differentiation, and hydrocortisone, which sensitizes the cells to EGF26. We show that the use of this specific growth medium yields higher numbers of EPCs compared to endothelial basal medium (EBM) or Dulbecco's Modified Eagle Medium (DMEM).
Protocol
This research was carried out with the approval of the University of Arkansas Institutional Review Board (Approval number 16-04-722). Umbilical cord blood units were collected in citrate phosphate dextrose (CPD) solution at the Arkansas Cord Blood Bank, and units that did not meet the requirement for storage were donated for research. Cord blood units were couriered to the lab within 24 h of collection at ambient temperatures.
1. Isolation of Endothelial Progenitor Cells from Cord Blood
- Preparation of reagents.
- Prepare EGM by adding endothelial basal medium (EBM) to 10% fetal bovine serum (FBS) supplemented with 20 ng/mL of insulin-like growth factor (IGF), 1 µg/mL ascorbic acid, 5 ng/mL recombinant human epidermal growth factor (rhEGF), 22.5 µg/mL heparin, and 0.5 ng/mL vascular endothelial growth factor (VEGF), along with 10 ng/mL of recombinant human Fibroblast growth factor B (rhFGF-B), 0.2 µg/mL hydrocortisone, and 2% penicillin-streptomycin-glutamine. Sterilize the culture medium using a 0.2 µm polyethersulfone (PES) membrane vacuum filter.
- Prepare rat tail I collagen solution at a concentration of 50 µg/mL using 0.02 N acetic acid. Sterilize this solution using a 0.2 µm syringe filter.
- Pre-coat 6-well plates with 2 mL of prepared rat tail I collagen solution for each well. Place them in an incubator maintained at 5% CO2 and 37 °C for at least 1 h before seeding.
- Dilute approximately 25 mL of cord blood with Hanks Balanced Salt Solution (HBSS) at a 1:1 concentration.
- Prepare radio-immuno precipitation assay (RIPA) Buffer using 150 mM sodium chloride, 1% Triton X-100, 0.5% sodium deoxycholate, 1 mM β-glycerophosphate, 0.1% SDS, and 50 mM Tris (pH 8.0).
- Isolation of endothelial progenitor cells.
- Warm all prepared reagents in a water bath maintained at 37 °C prior to isolation.
- Add 20 mL of density gradient medium reagent to a 50 mL centrifuge tube.
- Carefully layer 20 mL of diluted cord blood in the centrifuge tube containing density gradient medium without breaking this layer by aiming the pipette towards the side walls of the tube.
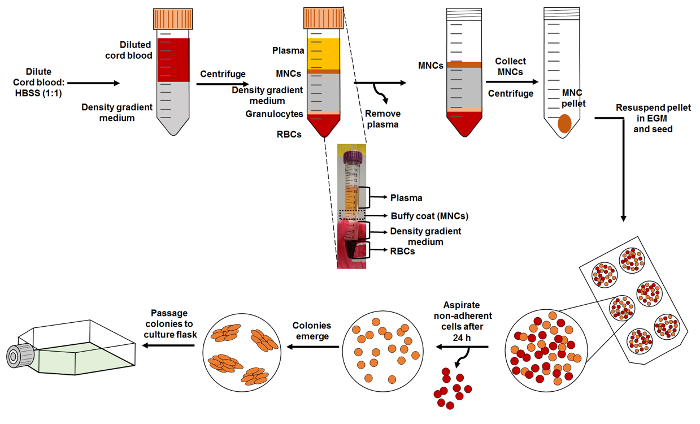
- Separate the components of blood based on their densities (Figure 1) by centrifuging at 800 x g for 30 min at room temperature, with the brakes of the centrifuge rotor off. Do not disturb the different layers.
- Remove the plasma layer by carefully pipetting it out of the tube without disturbing other cell layers until the pipette tip is able to reach the MNC layer (see Figure 1). Discard the removed plasma. NOTE: While using pipette tips to remove the plasma, leave a small layer of plasma above the MNC layer (Figure 1) to reduce disturbances.
- Collect the buffy coat that contains the MNCs using a syringe fitted to an 18-gauge needle and transfer it to a fresh centrifuge tube.
- Add an equal volume of EBM to the collected MNCs. Centrifuge these tubes at 500 x g for 10 min at 4 °C. Discard the supernatant. NOTE: The cell pellet will appear red because of the presence of red blood cells/erythrocytes.
- Add 5 mL of ammonium chloride solution to lyse the red blood cells. Incubate this tube on ice for 5 - 10 min, with occasional shaking. NOTE: Caution should be taken during this step to not exceed the time duration, as it can be harmful to the MNCs.
- Centrifuge the tubes at 500 x g for 10 min at 4 °C and discard the supernatant. If erythrocytes persist, repeat steps 1.2.8 and 1.2.9 until no red color is observed in the pellet.
- Resuspend the pellet in prepared EGM and use the hemocytometer to count the MNCs.
- Aspirate the rat tail I collagen-coated 6-well plate (from step 1.1.3) and wash the wells three times with 1x phosphate-buffered saline (PBS).
- Seed the MNC pellet resuspended in EGM to the collagen plate at a concentration of 1 x 107 MNCs in each well. Place it in the 37 °C, 5% CO2 incubator.
- After 24 h, aspirate the medium and wash the wells once with EGM. Add 3 mL of EGM medium to each well and place it back in the 37 °C, 5% CO2 incubator.
- Replenish the plate with fresh EGM medium every day for 7 days. After 7 days, change the EGM medium every other day.
- Using a bright-field microscope, take images of the 6-well plate every day to track the progression of endothelial cell colonies. Mark the plate where the colonies arise to keep track of their growth. NOTE: It usually takes between 5 and 9 days for the colonies to appear in culture.
- Expansion of endothelial progenitor cells.
- Coat the T-25 culture flask with rat tail I collagen (50 µg/mL) and place it in the 37 °C, 5% CO2 incubator for at least 60 min. Aspirate and wash the T-25 cell culture flask three times with 1x PBS prior to seeding the cells.
- Trypsinize the endothelial cell colonies when they reach about 3 mm in size using 150 µL of 0.05% Trypsin-EDTA solution in each well. Place the 6-well plate back into the 37 °C, 5% CO2 incubator for 2 - 3 min.
- Gently tap the 6-well plate to dislodge the attached cells. Immediately add 2 mL of EGM and recover the cells in a 15 mL centrifuge tube.
- Count the cells using a hemocytometer and calculate the initial seeding density.
- Spin the 15 mL centrifuge tubes at 400 x g for 5 min. Resuspend the cell pellet in EGM and seed the T-25 flask with ~500,000 cells. Mark this flask as Passage 1 and place the T-25 flask back in the 37 °C, 5% CO2 incubator. NOTE: Cells can be plated in EBM and DMEM to compare the growth with EGM.
- For further passages, when the flask reaches 90% confluence, follow steps 1.3.1 - 1.3.5 and reseed the cells at 104 cells/cm2 on T-75 or T-175 cell culture flasks. Proceed to the characterization of the isolated cells (steps 2 and 3).
2. Characterization of Isolated Cells Using Western Blotting
Add 50 - 100 µL of lysis buffer at every passage when following the expansion steps to characterize the isolated cells. Collect proteins from approximately 500,000 or more cells.
Pipette up and down vigorously to rupture the cells and release the proteins. Collect and transfer the buffer to a microcentrifuge tube.
Spin the microcentrifuge tube at 500 x g and carefully transfer the supernatant to a new microcentrifuge tube. Label and store the tubes at -80 °C for long-term storage.
Quantify the amount of proteins in each tube using either Bradford's or BCA assay27,28,29.
Carry out Western blotting using standard procedures27,28,29. Analyze 5 µg of total protein for the expression of CD31, CD34, VEGFR2, and α-SMA. Use α-tubulin for normalizing the data.
3. Indirect Immunofluorescence
Sonicate 18-mm glass coverslips in 50% ethanol and dry them. Leave the clean coverslips in a 12-well plate overnight in the biosafety hood with a UV light on to sterilize them.
Coat the coverslips with 50 µg/mL of rat tail I collagen and transfer the plate to a 37 °C, 5% CO2 incubator for at least 60 min.
Aspirate the collagen and add 1 mL of 1x PBS to the 12-well plate to wash the coverslips. Aspirate the 1x PBS and repeat twice.
Seed 250,000 cultured EPCs in each well and place them back in the 37 °C, 5% CO2 incubator for at least 3 days before carrying out immunostaining.
Wash the plate 3 times with 1x PBS. Add 1 mL of ice-cold methanol to each well and incubate at room temperature for 15 min to fix the cells.
Carry out immunostaining using a standard protocol27,28,29. Use the antibodies at their appropriate dilutions (e.g., CD31 (1:20), CD34 (1:50), α-SMA (1:100), and VEGFR2 (1:50)).
Take images of the immunostained coverslips using an epifluorescence microscope.
Representative Results
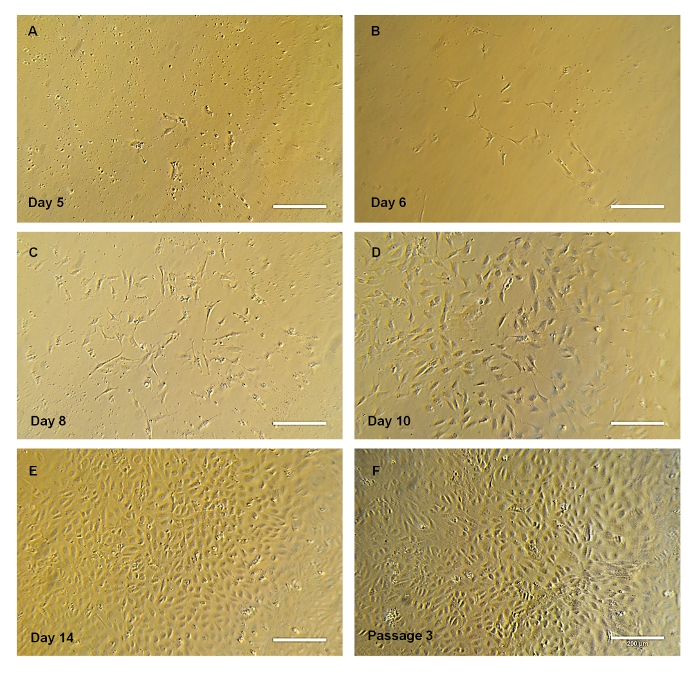
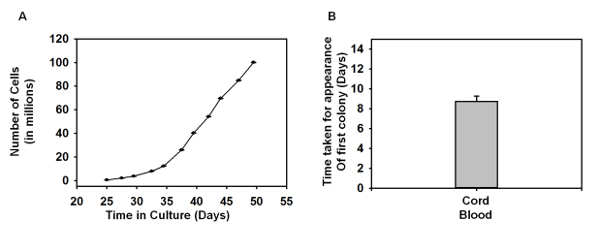
Isolation and Expansion of Endothelial Progenitor Cells: A schematic (Figure 1) is provided depicting the overall protocol. The different blood component layers were observed following density gradient centrifugation of human umbilical cord blood with density gradient medium. Upon seeding MNCs onto the collagen-treated plates, the outgrowth of colonies was first observed between Days 5 and 7 (Figure 2A). These colonies continued to grow and had a spindle-shaped cell morphology (Figure 2A-2D) in the early stages, which later progressed to a cobblestone-like morphology (Figure 2E-2F). Figure 3A shows the total number of cells versus time in culture, and each data point represents the cumulative number of cells harvested at each passage. Figure 3B depicts the average time taken for the first colony to arise in the collagen-treated 6-well plate after seeding the MNCs.
Characterization Using Western Blotting:Figure 4A shows the representative results of the Western blot membrane that was tested against the various antibodies. Our results suggest that the cells were positive for CD31 and CD34. The expression of CD31 (Figure 4B) and CD34 (Figure 4C) appeared to decrease over subsequent passages, whereas VEGFR2 (Figure 4D) was expressed equally in later passages, with the first passage having higher expression. We also observed that the EPCs did not express the mesenchymal cell marker α-SMA (Figure 4E). Human umbilical vein endothelial cells (HUVEC) and valvular interstitial cells (VIC) cell lysates were used as positive and negative controls, respectively. HUVECs are known to express CD31, CD34, and VEGFR2, while VICs express α-SMA.
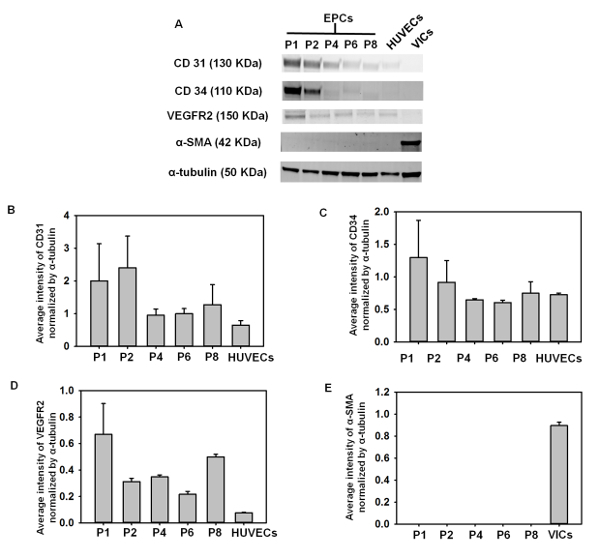
Figure 1: Schematic of EPC Isolation. Start the isolation by diluting cord blood with HBSS and layering it carefully on top of the density gradient medium, as shown. Density gradient centrifugation of the layered blood is carried out to obtain distinct layers of the blood, which is comprised of MNCs (buffy coat), density gradient medium, granulocytes, and RBCs. The subset image provided shows these different layers. MNCs are collected and reseeded onto a collagen-treated cell culture plate. Please click here to view a larger version of this figure.
Figure 2. Progression of the Cell Colony. (A-E) Representative bright-field images of EPC colony progression over time. Note that the colony has spindle-shaped cells at the early stages, adopting the cobblestone morphology. (E) Representative image of EPC cultured in a T-75 flask at passage 3. Scale bar = 200 µm. Please click here to view a larger version of this figure.
Figure 3: Growth Curves of EPCs. (A) The graph depicts the number of cells growing over a period of time, and each of the data point denotes the cell number harvested during each passage (P0 - P10) (n = 4). (B) Time taken for the first EPC colony to appear in the 6-well plate (n = 4). The error bars denote the standard error of the mean (SEM). No statistical significance was found using one-way ANOVA. Please click here to view a larger version of this figure.
Figure 4: Western Blotting. (A) Western blot membrane stained with various antibodies. HUVEC lysate and VIC lysate were used as positive controls. (B) Average intensity of CD31 bands normalized with α-tubulin. (C) Average intensity of CD34 bands normalized with α-tubulin. (D) Average intensity of VEGFR2 bands normalized with α-tubulin. (E) Average intensity of α-SMA bands normalized with α-tubulin (n = 3 - 6). The error bars denote the SEM. No statistical significance was found using one-way ANOVA. Please click here to view a larger version of this figure.
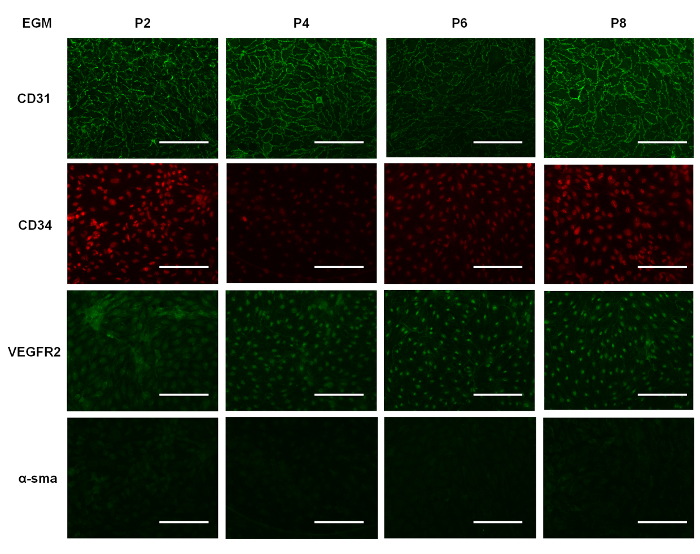
Figure 5: Immunostaining. Representative images of EPCs cultured in EGM for 7 days and immunostained at various passages for CD31, CD34, VEGFR2, and α-SMA. Scale bar - 100 µm. Please click here to view a larger version of this figure.
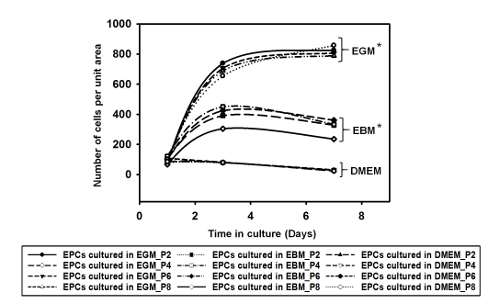 Supplemental Figure 1: The graph depicts the number of cells per unit area over a period of 7 days when cultured in EGM, EBM, and DMEM. EGM showed higher cell growth compared with the media (EBM and DMEM). The data showed statistical significance, with p <0.01, using one-way ANOVA (n = 2). Please click here to view a larger version of this figure.
Supplemental Figure 1: The graph depicts the number of cells per unit area over a period of 7 days when cultured in EGM, EBM, and DMEM. EGM showed higher cell growth compared with the media (EBM and DMEM). The data showed statistical significance, with p <0.01, using one-way ANOVA (n = 2). Please click here to view a larger version of this figure.
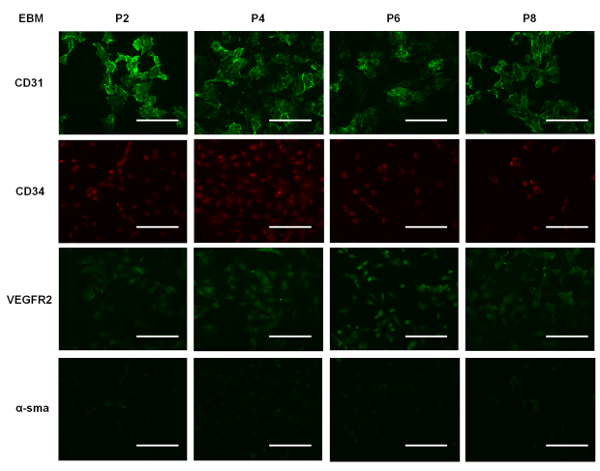 Supplemental Figure 2: Representative images of EPCs cultured in EBM for 7 days and immunostained for CD31, CD34, VEGFR2, and α-SMA. Scale bar = 100 µm. Please click here to view a larger version of this figure.
Supplemental Figure 2: Representative images of EPCs cultured in EBM for 7 days and immunostained for CD31, CD34, VEGFR2, and α-SMA. Scale bar = 100 µm. Please click here to view a larger version of this figure.
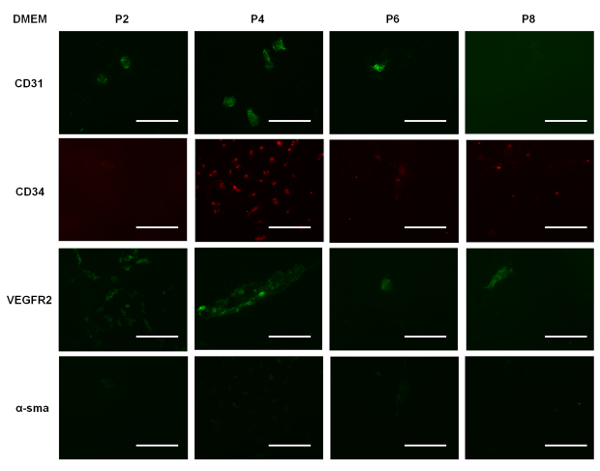 Supplemental Figure 3: Representative images of EPCs cultured in DMEM for 7 days and immunostained for CD31, CD34, VEGFR2, and α-SMA. Scale bar = 100 µm. Please click here to view a larger version of this figure.
Supplemental Figure 3: Representative images of EPCs cultured in DMEM for 7 days and immunostained for CD31, CD34, VEGFR2, and α-SMA. Scale bar = 100 µm. Please click here to view a larger version of this figure.
Discussion
As mentioned earlier, adherent EPCs possess a cobblestone morphology. Our isolated MNCs progressed from a spindle-shaped cell colony (Figure 2A-2D) in the early stages to a cobblestone colony (Figure 2E-2F) over a period of ten days in culture. EPCs have been labeled differently by different research groups, namely as late endothelial progenitor cells10, endothelial colony forming cells5, or endothelial progenitor cells12. It should be noted that these cells are functionally the same and express similar cell surface markers. The number of cells observed during the culture period (Figure 3) increased to almost 108 cells, analogous to previous publications and typical of the high proliferative potential of EPCs3,5. Indeed, it was found that EPCs obtained from human umbilical cord blood showed a high proliferative potential and demonstrated several population doublings before undergoing senescence compared to EPCs isolated from adult human peripheral blood5,10.
Our Western blotting results (Figure 4A) showed the presence of CD31, CD34, and VEGFR2 on the cell lysates obtained from EPCs compared to the positive control, HUVEC. HUVECs express CD31, CD34, and VEGFR2 and activated VIC express α-sSMA. Similar to previous reports10,18, the expression of CD31 decreased with higher passages. The expression pattern was also similar in the case of CD34 and VEGFR2, where both the markers were expressed strongly in the first passage and in relatively lower but equal amounts in later passages. This data shows a trend that is in agreement with previous groups' results, in which immunosorting-based protocols were used for isolating cord blood-derived EPCs12. Generally, EPCs do not express mesenchymal markers and, to verify that the isolated cells were not mesenchymal in nature, we immunolabeled our Western blot membranes with α-SMA. Our results clearly show that there was no α-SMA expression in the cultured EPCs when compared with positive control cell lysates collected from VICs, which exhibit strong expression of α-SMA antibody.
To demonstrate that EGM allows for increased cell proliferation compared to other culture media used for EPCs, we cultured the cells in three different types of media: DMEM, EBM, and EGM (i.e., EBM supplemented with growth factors, as outlined earlier). All were supplemented with 10% FBS. We seeded cells of different passages in each well of a 12-well plate. Supplemental Figure 1 shows the number of cells per unit area in each well over time in culture in the different media formulations. We note that the EPCs cultured in EGM showed a steady increase in cell number, and for cells in EBM, there was a slower rate of increase in cell number during the first three days, which subsequently plateaued. This suggests that the EPCs could still survive in EBM, but the absence of growth factors hindered proliferation. We also note that EPCs cultured in DMEM decline in number, suggesting that this particular medium formulation is not suitable for their survival or proliferation. Our immunostaining data shows that the EPCs cultured in EGM (Figure 5) expressed CD31 more when compared to EPCs cultured in EBM (Supplemental Figure 2) and DMEM (Supplemental Figure 3). Comparing Figure 5 with Supplemental Figure 2 and Supplemental Figure 3, it can be seen that the cells were not contaminated with other cell types. Also, when comparing the immunostained EPCs (Figure 5), we can qualitatively state that the expression of CD31 and CD34 decreased over higher passages, supporting our Western blot data (Figure 4).
To reiterate, the primary goal of our proposed protocol is to demonstrate that one can isolate endothelial progenitor cells by culturing blood MNCs in specialized selective media. This is to show that EPCs can be isolated without sophisticated equipment and procedures, unlike other methods that involve the use of flow cytometry - either immediately after MNC isolation1,3,20,30 or after the colonies have grown into a monolayer - followed by replating4,10,12,13,23. The EPC colonies usually appear between days 5 and 9. Our method is thus a faster and more cost-efficient method for isolating EPCs. The primary limitation of this technique is that the expression of CD31 and CD34 decreases with subsequent passages, which could suggest that the cells are losing their progenitor phenotype. Thus, it is recommended that one should use cells obtained from passage 5 or lower to conduct experiments if using this protocol.
Precautions should be taken after the density centrifugation to not disturb the different layers when taking the centrifuged tubes back into the hood and also when removing the plasma. Another critical step pertains to erythrocyte lysis, where a longer incubation time could potentially damage the MNC. It is thus recommended not to repeat this step more than twice. Indeed, since the medium aspiration is carried out 24 h after plating, trace amounts of erythrocytes are removed, as they are non-adherent. Alternative substrates, like fibronectin or gelatin, while not tested by us, can also be used according to the specific protocols for creating cell culture coatings. By employing minor modification in the initial steps, researchers may isolate non-adherent EPCs30.
In conclusion, we have reported a method of isolating EPCs from umbilical cord blood. It is important to note that the EPCs do not express markers like α-SMA, suggesting that they are not mesenchymal-like cells. Indeed, α-SMA serves as an important marker when trying to study the endothelial-mesenchymal transition (endo-MT) of EPCs. Endo-MT is a cellular transdifferentiation process during which endothelial cells are known to lose their specific markers and to transform to a mesenchymal-like cell phenotype. It has been shown that EPCs can undergo this transition in the presence of transforming growth factor-β7 and can release extracellular membrane proteins on their tissue-engineered scaffolds8. All these observations indicate the promise of EPCs for use as an autologous cell source to create a heart valve or other cardiovascular tissue-engineered constructs.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This material is based upon work supported by the National Science Foundation under Grant No. CMMI-1452943 and by the University of Arkansas Honors College. We would also like to acknowledge the Arkansas Cord Blood Bank for providing us with cord blood units.
References
- Asahara T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275(5302):964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- Lin Y, Weisdorf DJ, Solovey A, Hebbel RP. Origins of circulating endothelial cells and endothelial outgrowth from blood. J Clin Invest. 2000;105(1):71–77. doi: 10.1172/JCI8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, et al. Evidence for circulating bone marrow-derived endothelial cells. Blood. 1998;92(2):362–367. [PubMed] [Google Scholar]
- Hirschi KK, Ingram DA, Yoder MC. Assessing identity, phenotype, and fate of endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2008;28(9):1584–1595. doi: 10.1161/ATVBAHA.107.155960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram DA, et al. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood. 2004;104(9):2752–2760. doi: 10.1182/blood-2004-04-1396. [DOI] [PubMed] [Google Scholar]
- Yoder MC, et al. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007;109(5):1801–1809. doi: 10.1182/blood-2006-08-043471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sales VL, et al. Transforming growth factor-beta1 modulates extracellular matrix production, proliferation, and apoptosis of endothelial progenitor cells in tissue-engineering scaffolds. Circulation. 2006;114:193–199. doi: 10.1161/CIRCULATIONAHA.105.001628. 1 Suppl. [DOI] [PubMed] [Google Scholar]
- Sales VL, et al. Endothelial Progenitor Cells as a Sole Source for Ex Vivo Seeding of Tissue-Engineered Heart Valves. Tissue Eng Pt A. 2010;16(1):257–267. doi: 10.1089/ten.tea.2009.0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew A, Barry F, O'Brien T. Endothelial progenitor cells: diagnostic and therapeutic considerations. Bioessays. 2006;28(3):261–270. doi: 10.1002/bies.20372. [DOI] [PubMed] [Google Scholar]
- Hur J, et al. Characterization of two types of endothelial progenitor cells and their different contributions to neovasculogenesis. Arterioscler Thromb Vasc Biol. 2004;24(2):288–293. doi: 10.1161/01.ATV.0000114236.77009.06. [DOI] [PubMed] [Google Scholar]
- Ingram DA, Caplice NM, Yoder MC. Unresolved questions, changing definitions, and novel paradigms for defining endothelial progenitor cells. Blood. 2005;106(5):1525–1531. doi: 10.1182/blood-2005-04-1509. [DOI] [PubMed] [Google Scholar]
- Melero-Martin JM, et al. In vivo vasculogenic potential of human blood-derived endothelial progenitor cells. Blood. 2007;109(11):4761–4768. doi: 10.1182/blood-2006-12-062471. [DOI] [PubMed] [Google Scholar]
- Melero-Martin JM, Bischoff J. Chapter 13. An in vivo experimental model for postnatal vasculogenesis. Methods Enzymol. 2008;445:303–329. doi: 10.1016/S0076-6879(08)03013-9. [DOI] [PubMed] [Google Scholar]
- Yoder MC. Human endothelial progenitor cells. Cold Spring Harb Perspect Med. 2012;2(7):006692. doi: 10.1101/cshperspect.a006692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddique A, Shantsila E, Lip GYH, Varma C. Endothelial progenitor cells: what use for the cardiologist. J Angiogenes Res. 2010;2(6) doi: 10.1186/2040-2384-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camci-Unal G, et al. Surface-modified hyaluronic acid hydrogels to capture endothelial progenitor cells. Soft Matter. 2010;6(20):5120–5126. doi: 10.1039/c0sm00508h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JM, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348(7):593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- Young PP, Vaughan DE, Hatzopoulos AK. Biologic properties of endothelial progenitor cells and their potential for cell therapy. Prog Cardiovasc Dis. 2007;49(6):421–429. doi: 10.1016/j.pcad.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta JL, Szwedo J. Circulating endothelial progenitor cells, microparticles and vascular disease. J Hypertens. 2010;28(8):1611–1613. doi: 10.1097/HJH.0b013e32833bcfe9. [DOI] [PubMed] [Google Scholar]
- Nevskaya T, et al. Circulating endothelial progenitor cells in systemic sclerosis are related to impaired angiogenesis and vascular disease manifestations. Ann Rheum Dis. 2007;66:67–67. [Google Scholar]
- Cebotari S, et al. Clinical application of tissue engineered human heart valves using autologous progenitor cells. Circulation. 2006;114:132–137. doi: 10.1161/CIRCULATIONAHA.105.001065. 1 Suppl. [DOI] [PubMed] [Google Scholar]
- Ito H, et al. Endothelial progenitor cells as putative targets for angiostatin. Cancer Res. 1999;59(23):5875–5877. [PubMed] [Google Scholar]
- Vasa M, et al. Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation. 2001;103(24):2885–2890. doi: 10.1161/hc2401.092816. [DOI] [PubMed] [Google Scholar]
- Wu X, et al. Tissue-engineered microvessels on three-dimensional biodegradable scaffolds using human endothelial progenitor cells. Am J Physiol Heart Circ Physiol. 2004;287(2):480–487. doi: 10.1152/ajpheart.01232.2003. [DOI] [PubMed] [Google Scholar]
- Boyer M, et al. Isolation of endothelial cells and their progenitor cells from human peripheral blood. J Vasc Surg. 2000;31(1):181–189. doi: 10.1016/s0741-5214(00)70080-2. Pt 1. [DOI] [PubMed] [Google Scholar]
- Huber B, Czaja AM, Kluger PJ. Influence of epidermal growth factor (EGF) and hydrocortisone on the co-culture of mature adipocytes and endothelial cells for vascularized adipose tissue engineering. Cell Biol Int. 2016;40(5):569–578. doi: 10.1002/cbin.10595. [DOI] [PubMed] [Google Scholar]
- Sturdivant NM, Smith SG, Ali SF, Wolchok JC, Balachandran K. Acetazolamide Mitigates Astrocyte Cellular Edema Following Mild Traumatic Brain Injury. Sci Rep. 2016;6:33330. doi: 10.1038/srep33330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam NT, Muldoon TJ, Quinn KP, Rajaram N, Balachandran K. Valve interstitial cell contractile strength and metabolic state are dependent on its shape. Integr Biol (Camb) 2016;8(10):1079–1089. doi: 10.1039/c6ib00120c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon I, et al. Valve interstitial cell shape modulates cell contractility independent of cell phenotype. J Biomech. 2016;49(14):3289–3297. doi: 10.1016/j.jbiomech.2016.08.013. [DOI] [PubMed] [Google Scholar]
- Cockshell MP, Bonder CS. Isolation and Culture of Human CD133+ Non-adherent Endothelial Forming Cells. Bio-Protocol. 2016;6(7) [Google Scholar]