

Abstract
Non-canonical nucleic acid secondary structure G-quadruplexes (G4) are involved in diverse cellular processes, such as DNA replication, transcription, RNA processing, and telomere elongation. During these processes, various proteins bind and resolve G4 structures to perform their function. As the function of G4 often depends on the stability of its folded structure, it is important to investigate how G4 binding proteins regulate the stability of G4. This work presents a method to manipulate single G4 molecules using magnetic tweezers, which enables studies of the regulation of G4 binding proteins on a single G4 molecule in real time. In general, this method is suitable for a wide scope of applications in studies for proteins/ligands interactions and regulations on various DNA or RNA secondary structures.
Keywords: Biochemistry, Issue 127, Single-molecule manipulation, magnetic tweezers, G-quadruplexes, folding-unfolding, helicase
Introduction
Four-stranded DNA or RNA G4 structures play critical roles in many important biological processes1. Many proteins are involved in G4 binding and regulation, including telomere binding proteins (telomerase, POT1, RPA, TEBPs, TRF2)1,2, transcription factors (nucleolin, PARP1)3, RNA processing proteins (hnRNP A1, hnRNP A2)4, helicases (BLM, FANCJ, RHAU, WRN, Dna2, Pif1)5, and DNA replication related proteins (Rif1, REV1, PrimPolymerase)6. Protein binding can stabilize or destabilize G4 structures; thus regulating the subsequent biological functions. The stability of G4 was measured by thermal melting using ultraviolet (UV) or circular dichroism (CD) methods7. However, such conditions are not physiological relevant and are difficult to apply to studying the effects of binding proteins7.
The rapid development in single-molecule manipulation technologies has enabled studies of folding and unfolding of a biomolecule, such as a DNA or a protein, at a single-molecule level with nanometer resolution in real time8. Atomic force microscopy (AFM), optical tweezers, and magnetic tweezers are the most commonly used single-molecule manipulation methods. Compared to AFM and optical tweezers9, magnetic tweezers allow stable measurements of folding-unfolding dynamics of a single molecule over days by using an anti-drift technique10,11.
Here, a single-molecule manipulation platform using magnetic tweezers to study the regulation of G4 stability by binding proteins is reported12,13. This work outlines the basic approaches, including sample and flow channel preparation, the setup of magnetic tweezers, and the force calibration. The force control and the anti-drift protocols as described in step 3 allow for long time measurements under various force controls, such as constant force (force clamp) and constant loading rate (force-ramp), and force-jump measurement. The force calibration protocol described in step 4 enables force calibration of < 1 µm short tethers over a wide force range up to 100 pN, with a relative error within 10%. An example of regulation of the stability of the RNA Helicase associated with AU-rich element (RHAU) helicase (alias DHX36, G4R1) that plays essential roles in resolving RNA G4 is used to demonstrate the applications of this platform13.
Protocol
1. Preparation of G4 DNA for Single-molecule Stretching
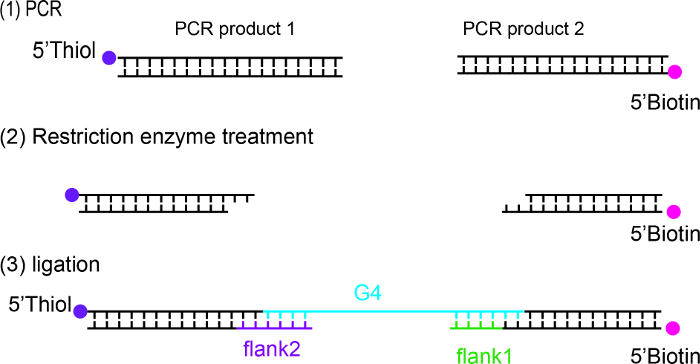
Prepare 5'-thiol labeled and 5'-biotin labeled dsDNA handles by PCR using DDNA polymerase on a lambda phage DNA template using 5'-thiol and 5'-biotin primers14 (Figure 1). Both dsDNA handles have high GC content (> 60%) to prevent DNA melting when DNA is held at high forces or during DNA overstretching transition15.
Purify PCR products using a commercial purification kit and digest with BstXI restriction enzyme according to the manufacturer's protocol.
Ligate G4 forming ssDNA, and flank ssDNA and dsDNA handles using T4 DNA ligase according to the manufacturer's protocol. Purify the ligated product by gel extraction using a commercial purification kit according to the manufacturer's protocol.
2. Preparation of Flow Channel
- Coverslip cleaning and surface functionalization
- Place bottom coverslips (#1.5, 22 mm × 32 mm) and top coverslips (#1.5, 20 mm x 20 mm) into cover glass staining jars (each jar can hold 7 pieces of coverslips, the volume is ~ 20 mL). Rinse the coverslips in the jars with distilled water 2-5 times.
- Add ~ 20 mL of 5-40% detergent solution into each jar, and then place in an ultrasonic cleaning bath for 30 min. Rinse with distilled water for > 10 times to remove the detergent.
- Dry the coverslips in the jars in oven (~ 150 °C; CAUTION, hot), or by N2 gas. Store the dried top coverslips in a dry cabinet.
- Use plasma (O2 gas) to clean the coverslips in the jar for 10 min. During the 10 min, prepare 20 mL of 1% (3-Aminopropyl)triethoxysilane (APTES) solution in methanol (CAUTION, toxic/flammable). Use a chemical fume hood to dissolve APTES in methanol. NOTE: Avoid humidity when storing APTES solution. Old APTES often causes problem in surface functionalizing of coverslips.
- Immediately after the plasma cleaning, add all of the 1% APTES solution into the jars and incubate for 1 h. Pour the waste into the waste bottle specific for 1% APTES methanol. Perform the methanol-related steps in the fume hood for flammable chemicals.
- Rinse the jars once with methanol and > 10 times with distilled water, and then dry by oven (~150 °C; CAUTION, hot). Store the APTES-coated bottom coverslips in a dry cabinet if not in use for up to two weeks. NOTE: After each sub-step from 2.1.1 to 2.1.4, the cleaning process can be paused before the next step.
- Assemble the flow channel
- Prepare two "spacers", i.e., parafilm or double-side tape (~ 4 mm × 20 mm) for each channel. Place the two pieces of spacers on a bottom coverslip along the long-edge. Place a top coverslip on the spacer, forming a flow cell in between (~ 10 mm × 20 mm area, Figure 2A, B).
- If parafilm is used as a spacer, place the flow channel on a heater (60-120 °C; CAUTION, hot) for 5-10 s while gently pressing the sides of the top coverslip to stick the two coverslips together by parafilm. The resulting flow channel has a height of ~ 100 µm, and thereby the volume of the channel is ~ 20 µL.
- Seal the long edge of the channel with silicone glue to avoid leakage. Use silicone glue to make a small sink-like structure at each open edge of the flow channel, which serves as an entry and an exit of solution. NOTE: The entry and exit can also be made by other ways, e.g., by adhering small plastic rings using wax.
- Store channels in a dry cabinet for up to 4 weeks.
- Tether DNA onto the bottom surface of the flow channel
- Dilute the amino-coated polystyrene beads (diameter: 3 µm) by 200X in distilled 1X phosphate buffered saline (PBS) buffer. Vortex the bead solution and then flow into channels. Incubate the bead solution in channels for ~ 30 min. Remove any unstuck beads by washing with 200 µL of 1X PBS buffer.
- Adjust (increase/decrease) the incubation time to achieve a surface density of 1-5 beads per 50 mm x 50 mm area. Store the channels deposited with the reference beads for up to 3 days.
- Dilute sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (Sulfo-SMCC) powder into 1X PBS solution (~ 0.5 mg/mL). Vortex the solution and then flow into the channel. NOTE: Prepare fresh Sulfo-SMCC solution before use and add it immediately to the channel to avoid hydrolyzation of the SMCC.
- Incubate the SMCC solution in the channel for 30 min. Remove the SMCC solution by washing with a large amount (1 mL, ~ 50X the channel volume) of 1X PBS solution. NOTE: Wash the excess SMCC carefully. This step is very essential for the binding of DNA on the coverslip.
- DNA tethering
- Dilute the thiol-biotin labeled DNA into 1X PBS, with a resulting DNA concentration of ~ 0.3 nM. Gently pipette to mix the solution, flow the DNA solution into the SMCC-coated channel, and incubate for 30 min at room temperature (~ 23 °C).
- Gently wash away free DNA with 200 µL of blocking solution that contains 1X PBS with 10 mg/mL bovine serum albumin (BSA) and 0.01% 2-Mercaptoethanol.
- Block the channel surface by incubating the channel in blocking solution (10 mg/mL BSA, 0.01% 2-Mercaptoethanol) for > 2 h; after this step, the channel is ready for experiments. The prepared channel can be kept at 4 °C for ~ 1 day. NOTE: The blocking step is important for reducing the non-specific binding of DNA and magnetic beads to the coverslips.
3. Magnetic Tweezers Setup and Identification of Single dsDNA Tether
- Magnetic tweezers setup
- Start the magnetic tweezers control program. Here, the magnetic tweezers were controlled by an in-house-written LabVIEW program.
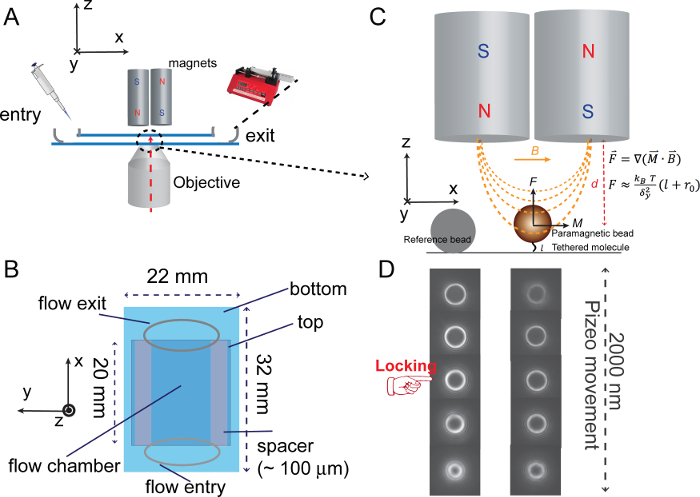
- Align the magnet centers before mounting the channel. Use a 4X objective lens to adjust the x- and y-axis of the magnets in the optical axis of the microscope.
- Use a computer-controlled motorized manipulator to move the magnets along the z-direction (Figure 2A) and set the distance as d = 0 (d: distance between the magnets and coverslip) when the magnets attach to a coverslip on the microscope.
- Program the movement of the magnets through the manipulator to achieve control of the force, including constant force (i.e., constant d) and time varying force F(t) (i.e., time varying d(t)).
- Use bright a light-emitting diode (LED) light source for back-scattered illumination of the bead through the objective. Collect the bead images at a sampling rate of 100 Hz by a charge-coupled device (CCD) camera.
- Sample setup and identify single dsDNA tether
- Tether formation
- Gently flow in 200X diluted M-280, paramagnetic beads in assay solution (100 mM KCl, 2 mM MgCl2, 10 mM Tris-pH 7.4) into a channel. Incubate for 10 min to allow the beads to bind to biotin-labeled DNA molecules immobilized on the SMCC-coated surface through the thiol-labeled end. Gently wash away un-tethered beads using 200 µL of standard reaction solution. NOTE: The M-280 beads show lower non-specific binding to the surface compared to other commercial magnetic beads such as M-270.
- Mount the channel onto the microscope stage. Search for the beads on the bottom surface using the 100X oil immersion objective.
- Select a reference bead on the surface and a moving tethered bead. Build the initial image libraries of both the reference bead and tethered bead at different defocus planes.
- Calibration of beads image
- Before experiments, use an objective piezo actuator to obtain images of both reference and tethered beads at different defocus planes spaced by 20-50 nm, which are stored as two separate bead image libraries and are respectively indexed with the defocus distance (Figure 2D).
- x, y, z position determination
- During experiments, determine the position of the bead in the x-y plane by the bead centroid. Determine the height change of the tethered bead by comparing the current bead image with those stored in the library. Use the auto-correlation function analysis of the power spectrum of the Fourier transform of the bead images16.
- Anti-drift feedback using reference bead
- During experiments, use the piezo to "lock" the distance between the objective and a specific reference bead image stored in the library through a low frequency feedback control, so that the stuck bead image has the best correlation with a specific image stored in the library (Figure 2D).
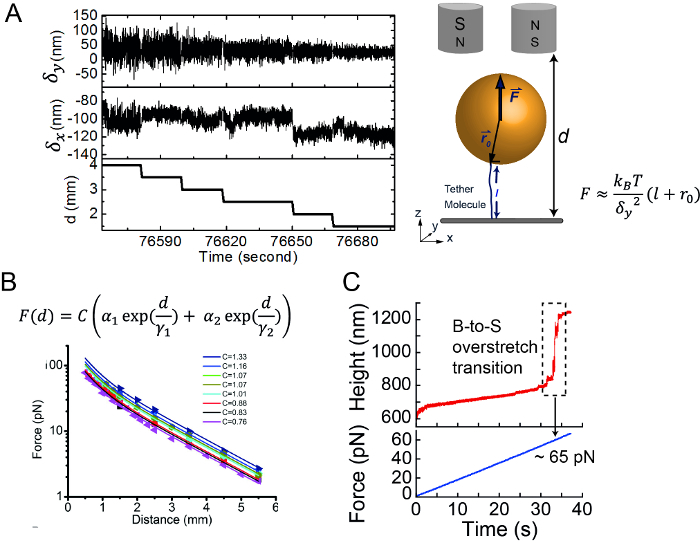
- Determine whether the tether is a single dsDNA molecule by applying ~ 65 pN force Determine a single dsDNA molecule if the tether undergoes the characteristic DNA overstretching transition17,18,19. Repeat the process until a single dsDNA tether is found. (Please refer to the next section for force calibration and DNA overstretching transition).
4. Magnetic Tweezers Force Calibration
Directly determine the force (up to 100 pN) for long DNA (48,502 bp λ-DNA) molecules using bead fluctuation through:
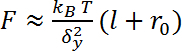 , where kBis the Boltzmann constant, T is the temperature in Kelvin scale, δ2yis the variance of the bead fluctuation in the direction perpendicular to the magnetic field and the force direction. In this direction, the motion of the bead can be described as a pendulum fluctuation with a length of l + r0, where l is the end-to-end distance along the force direction (extension) of the DNA, and r0 is the radius of the bead10 (Figure 3A).
, where kBis the Boltzmann constant, T is the temperature in Kelvin scale, δ2yis the variance of the bead fluctuation in the direction perpendicular to the magnetic field and the force direction. In this direction, the motion of the bead can be described as a pendulum fluctuation with a length of l + r0, where l is the end-to-end distance along the force direction (extension) of the DNA, and r0 is the radius of the bead10 (Figure 3A).Determine the calibration curve for force F as a function of magnets to bead distance d, and F(d) for different beads using the long λ-DNA. Usually, the distance between the magnet and the coverslip is used as the DNA tether length can be ignored. Fit the F-d curve by the double-exponential decay function:
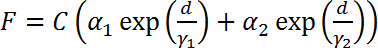 , where the fitting parameters α1, α2, γ1, γ2 are determined by the magnetic tweezers and the parameter C is determined by the heterogeneous property of the magnetic beads. By shifting the C, the F-d curve obtained from different beads can be overlapped10 (Figure 3B).
, where the fitting parameters α1, α2, γ1, γ2 are determined by the magnetic tweezers and the parameter C is determined by the heterogeneous property of the magnetic beads. By shifting the C, the F-d curve obtained from different beads can be overlapped10 (Figure 3B).- Calibration of parameter C for individual magnetic beads for short DNA experiments.
- Determining the force based on the fluctuation requires recording the bead position at a frequency higher than the corner frequency:
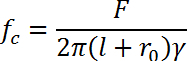 , where γ is the drag coefficient of the bead. For short DNA at high force, it requires recording frequency faster than 100 Hz, therefore, the force cannot be directly measured based on fluctuation with the camera. However, the parameter C can be determined by measuring the force at a low force range (< 15 pN) using fluctuation and fitting the data with a calibrated F-d curve to obtain the parameter C20.
, where γ is the drag coefficient of the bead. For short DNA at high force, it requires recording frequency faster than 100 Hz, therefore, the force cannot be directly measured based on fluctuation with the camera. However, the parameter C can be determined by measuring the force at a low force range (< 15 pN) using fluctuation and fitting the data with a calibrated F-d curve to obtain the parameter C20. - Alternatively, for experiments with dsDNA, determine the parameter C using DNA overstretching transition at a force of ~ 65 pN (Figure 3C)21,22,23 by recording the d position at which dsDNA showed extension jump (0.6X length increase of contour length). After determining the parameter C, calculate the force for the tethered beads.
Control the loading rate by programming the movement of the magnets through the inverse function of F(d). The magnet should approach the sample double exponentially. NOTE: The relative error of force determined by such an extrapolation method is ~ 10%, and is mainly caused by the bead radius heterogeneity20.
5. Single-molecule Manipulation of G4 in the Presence and Absence of Binding Proteins
- Force-ramp experiments
- After identifying the dsDNA tether, perform a force-increase scan at a loading rate of 0.2 pN/s followed by a force-decease scan at -0.2 pN/s. After each stretching cycle, hold the DNA molecule at 1 pN for 30 s to allow the ssDNA to refold to G4.
- Force-jump experiments
- Cycle the force between 54 pN for 30 s under which a folded G4 could be unfolded, and 1 pN for 60s, under which a folded G4-15T could refold.
- Flowing protein solution
- Flow the protein solution when at ~ 20 pN force to avoid attachment of the beads to the coverslip. The DEAH-box RHAU helicase, which showed high specificity to the G4 structure, was used24. The recombinant Drosophila melanogaster RHAU (DmRHAU) helicase was expressed in Escherichia coli and purified as described previously25. The G4 unwinding activity of DmRHAU was assayed on a tetramolecular G4 DNA substrate13.
Analyze the unfolding force using an in-house written Matlab program14.
Representative Results
The experiment setup for stretching a single G4 molecule is shown in Figure 4. A single-stranded G4 forming sequence spanned between two dsDNA handles was tethered between a coverslip and a paramagnetic bead. To find a single dsDNA tethered bead, an overstretching assay was performed by increasing the force at constant loading rates. Three types of measurements were often used for studying the folding and unfolding of biomolecules: (i) constant force measurement, (ii) force-ramp measurement, and (iii) force-jump measurement. Due to the extremely slow unfolding rates of this G4 structure, the equilibrium folding-unfolding transition could only occur for the time over days, so force-ramp and force-jump measurements were used for characterizing the unfolding kinetics and stability of G4 structures.
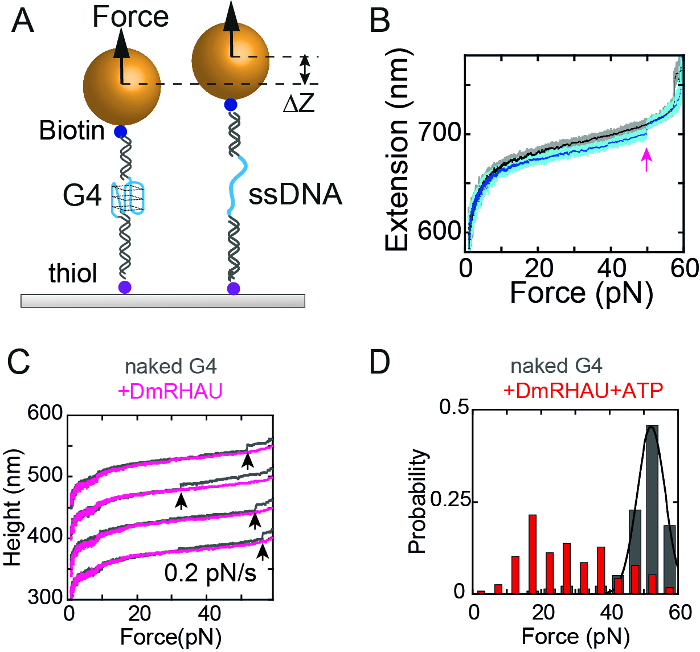
Force-ramp Measurements for G4 Unfolding in the Presence or Absence of RHAU Protein:
Figure 4B shows typical force-extension curves obtained by a force-increase scan at a loading rate of 0.2 pN/s followed by a force-decease scan at -0.2 pN/s. After each stretching cycle, the DNA molecule was held at 1 pN for 30 s to allow ssDNA to refold to G4. The extension jump in the force-increase scan indicated an unfolding transition of G4. When using a non-G4 forming sequence, the sudden extension jump cannot be observed. The unfolding force distribution could be obtained by recording the forces at which unfolding occurred. The unfolding force distribution of G4-15T obtained at 0.2 pN/s showed a single peak at ~ 52 pN. However, in the presence of 10 nM RHAU, G4-15T remained folded during the force-increase scan for up to 60 pN, indicating G4 stabilization by RHAU binding. In the presence of 10 nM RHAU and 1 mM ATP, the unfolding force distribution was shifted to a lower force, indicative of an ATP-dependent destabilization of G4 by RHAU.
Force-jump Measurements of G4 Unfolding in the Presence or Absence of RHAU Protein:
Figure 5 shows a representative trace of the bead height during four force-jump cycles (Figure 5, left panel); the force applied to the molecule was cycled between 54 pN for 30 s under which a folded G4 could be unfolded, and 1 pN for 60 s under which a folded G4-15T could refold. The average lifetime of folded G4-15T at 54 pN was ~ 6.4 s, estimated by fitting the lifetime histogram with an exponential decay function13. After flowing in a solution of 10 nM RHAU helicase without ATP, the extension of tethered DNA remained at a folded level throughout the 30 s holding time at 54 pN, indicating that the RHAU strongly stabilizes the G4-15T structure against mechanical unfolding in the absence of ATP. After flowing in 10 nM RHAU and 1 mM ATP, the extension of the same molecule right after jumping from 1 pN to 54 pN was at the level of unfolded ssDNA, indicating that the RHAU destabilizes the G4 structure in the presence of ATP.
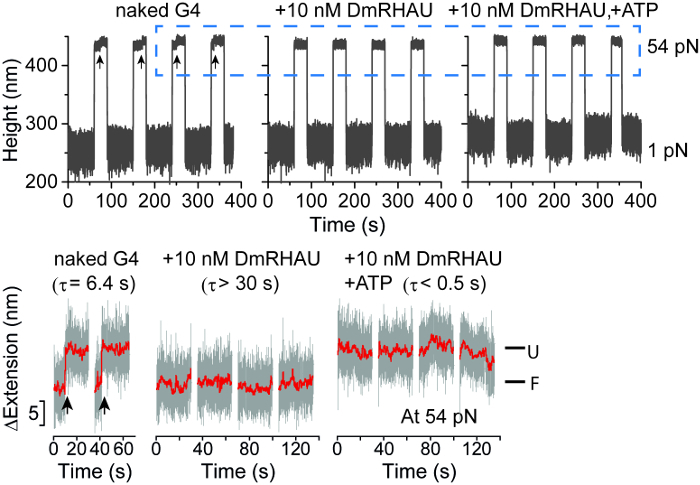
Figure 1: Preparation of G4 DNA for single-molecule stretching experiments. The dsDNA handles were prepared by PCR using 5'-thiol and 5'-biotin primers. PCR products were purified and digested with the BstXI restriction enzyme and were purified by gel extraction. G4 forming ssDNA, two flank ssDNA and dsDNA handle were ligated using T4 DNA ligase. The ligated product was purified by gel extraction. Please click here to view a larger version of this figure.
Figure 2: Sketches of basic magnetic tweezers apparatus. (A) Sketch of a flow channel with a pair of magnets placed above and a microscopy objective placed below. (B) Sketch of an assembled flow channel where the blue colored rectangles represent the top and bottom coverslips, and gray elliptical lines represent the entry and exit of the flow channel. (C) Sketch of the force generation and calibration of the magnetic tweezers setup. (D) Examples of images of both the reference bead and moving bead at different de-focus positions stored in the bead image libraries. The red sign denotes the reference bead image obtained at a particular de-focus plane used for the focal plane locking. Please click here to view a larger version of this figure.
Figure 3: Force calibration of magnetic tweezers. (A) Calibration of force using the fluctuations of bead along a rotation-free y-direction. X-axis is along the same direction as the magnetic field. Force is along the z-axis and is controlled by adjusting the distance, d, between the permanent magnet pair and the coverslip. (B) Calibration of using a long 48,502 bp -DNA. Heterogeneous magnetic bead property can be described by a single-parameter C. This figure has been modified from10. (C) Calibration of parameter C of a magnetic bead attached on a 2 kb-DNA using B to S overstretching transition. Please click here to view a larger version of this figure.
Figure 4: Single-molecule manipulation of a G4. (A) Sketch of a stretching single G4 structure and measuring the unfolding events. (B) Typical example of force-increase (cyan) and force-decrease curve of a tethered G4. This figure has been modified from12. (C) Force-extension curve of G4 in the absence (grey) or presence (pink) of RHAU protein. (D) Unfolding force distribution of G4 in the absence (grey) (n = 140) or presence (red) of RHAU and ATP (n = 117). This figure has been modified from13. Please click here to view a larger version of this figure.
Figure 5: Force-jump measurements of G4 unfolding. (A) Representative time trace of the bead height change during force jump cycles between 1 pN and 54 pN measured without DmRHAU, with 10 nM DmRHAU but without ATP, and with both 10 nM DmRHAU and 1 mM ATP, as indicated in the figure panel. (B) Representative time trace of extension change of a G4 molecule (grey data) after jumping to 54 pN in several force-jump cycles taken from data in (A). The extensions corresponding to folded (F) and unfolded (U) G4 are also indicated; G4 unfolding events are indicated by arrows. This figure has been modified from13. Please click here to view a larger version of this figure.
Discussion
As described above, a platform for studying the mechanical stability of G4 DNA and the interactions of protein to G4 using single-molecule magnetic tweezers is reported. Accompanying the platform, highly efficient protocols of finding G4 DNA tether, and measurement of the folding-unfolding dynamics and stability of the G4 structure with nanometers special resolution are developed. The focal plane locking enables highly stable anti-drift control, which is important for detecting a small structure transition such as G4 (step size ~ 7 nm) and the interactions with proteins in buffer exchange experiments. This platform has recently been employed to studies of the folding and unfolding dynamics of telomeric G414 and c-Myc promoter G412, as well as studies of G4 binding protein RHAU helicase13.
A typical experiment error is the non-specific binding of DNA and beads on the coverslip or multiple DNAs bind to magnetic beads. To reduce the non-specific binding, first, the storage of chemicals such as APTES and SMCC need to be routinely checked. Second, it is important to select a proper type of magnetic bead and block the non-specific binding. Using 10 mg/mL BSA buffer or small DNA oligo, such as poly T, to block the hydrophobic surface of the beads can significantly reduce the non-specific binding. Finally, a single DNA tether can be easily distinguished from multiple DNA tethers by the characteristic DNA overstretching transition17,18,19.
Currently, the position determination of the platform is based on the bead imaging analysis, which has a limited spatial resolution of ~ 2 nm. In addition, it has a limited sampling rate of ~ 100 Hz, mainly due to limited acquisition rate of typical CCD cameras and correlation analysis of the images. These limitations can be overcome by using total internal reflection microscopy (TIRF) illumination. It produces a thin layer of evanescent light that exponentially decays with the height from the surface, which can be used to determine the extension change of short molecules at a high signal-to-noise ratio, while avoiding bead image acquisition and analysis26. Another limitation is that it is challenging to directly visualize the tethered molecule or ligand bound to the tethered molecule using fluorescence imaging in the vertical stretching design, which can be overcome by stretching molecules in the focal plane using transverse magnetic tweezers27. Finally, the current design of the reaction channel does not allow rapid solution exchange due to the flow perturbation to the molecule. Furthermore, a large drag force generated at high speed flow may even break the tethers. This limitation can be overcome by manipulating tethers inside the microwells placed on the bottom surface of the channel28.
G4 stabilizing compounds are currently considered as potential therapeutic targets for diseases, including cancer, virus related diseases, and neurodegeneration diseases29,30,31. The protocol can be applied to a wide range of G4 binding proteins2 and small ligands32. In addition to applications in G4 studies, this platform as well as the protocols, in principle, can be used for studies of other nucleic acids secondary structures, including DNA and RNA hairpins, triplex, and i-motif33.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors thank Meng Pan for proofreading the manuscript. This work is supported by Singapore Ministry of Education Academic Research Fund Tier 3 (MOE2012-T3-1-001) to J.Y.; the National Research Foundation through the Mechanobiology Institute Singapore to J.Y.; the National Research Foundation, Prime Minister's Office, Singapore, under its NRF Investigatorship Programme (NRF Investigatorship Award No. NRF-NRFI2016-03 to J.Y.; the Fundamental Research Fund for the Central Universities (2017KFYXJJ153) to H. Y.
References
- Rhodes D, Lipps HJ. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015;43(18):8627–8637. doi: 10.1093/nar/gkv862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazda V, Haronikova L, Liao JC, Fojta M. DNA and RNA quadruplex-binding proteins. Int J Mol Sci. 2014;15(10):17493–17517. doi: 10.3390/ijms151017493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez V, Hurley LH. The C-terminus of nucleolin promotes the formation of the c-MYC G-quadruplex and inhibits c-MYC promoter activity. Biochemistry. 2010;49(45):9706–9714. doi: 10.1021/bi100509s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, et al. telomerase-interacting protein that unfolds telomere G-quadruplex and promotes telomere extension in mammalian cells. Proc Natl Acad Sci U S A. 2012;109(50):20413–20418. doi: 10.1073/pnas.1200232109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza O, Bourdoncle A, Boule JB, Brosh RM, Mergny JL. G-quadruplexes and helicases. Nucleic Acids Res. 2016;44(5):1989–2006. doi: 10.1093/nar/gkw079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavone D, et al. PrimPol Is Required for Replicative Tolerance of G Quadruplexes in Vertebrate Cells. Mol Cell. 2016;61(1):161–169. doi: 10.1016/j.molcel.2015.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane AN, Chaires JB, Gray RD, Trent JO. Stability and kinetics of G-quadruplex structures. Nucleic Acids Res. 2008;36(17):5482–5515. doi: 10.1093/nar/gkn517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodside MT, Block SM. Reconstructing folding energy landscapes by single-molecule force spectroscopy. Annu Rev Biophys. 2014;43:19–39. doi: 10.1146/annurev-biophys-051013-022754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman KC, Nagy A. Single-molecule force spectroscopy: optical tweezers, magnetic tweezers and atomic force microscopy. Nat Methods. 2008;5(6):491–505. doi: 10.1038/nmeth.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, et al. Improved high-force magnetic tweezers for stretching and refolding of proteins and short DNA. Biophys J. 2011;100(2):517–523. doi: 10.1016/j.bpj.2010.12.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, et al. Dynamics of equilibrium folding and unfolding transitions of titin immunoglobulin domain under constant forces. J Am Chem Soc. 2015;137(10):3540–3546. doi: 10.1021/ja5119368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You H, Wu J, Shao F, Yan J. Stability and kinetics of c-MYC promoter G-quadruplexes studied by single-molecule manipulation. J Am Chem Soc. 2015;137(7):2424–2427. doi: 10.1021/ja511680u. [DOI] [PubMed] [Google Scholar]
- You H, Lattmann S, Rhodes D, Yan J. RHAU helicase stabilizes G4 in its nucleotide-free state and destabilizes G4 upon ATP hydrolysis. Nucleic Acids Res. 2017;45(1):206–214. doi: 10.1093/nar/gkw881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You H, et al. Dynamics and stability of polymorphic human telomeric G-quadruplex under tension. Nucleic Acids Res. 2014;42(13):8789–8795. doi: 10.1093/nar/gku581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Chen H, Marko JF, Yan J. Two distinct overstretched DNA states. Nucleic Acids Res. 2010;38(16):5594–5600. doi: 10.1093/nar/gkq309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosse C, Croquette V. Magnetic tweezers: micromanipulation and force measurement at the molecular level. Biophys. J. 2002;82(6):3314–3329. doi: 10.1016/S0006-3495(02)75672-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, et al. Transition dynamics and selection of the distinct S-DNA and strand unpeeling modes of double helix overstretching. Nucleic Acids Res. 2011;39(8):3473–3481. doi: 10.1093/nar/gkq1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen H, Fu H, Doyle PS, Yan J. Two distinct overstretched DNA structures revealed by single-molecule thermodynamics measurements. Proc Natl Acad Sci U S A. 2012;109(21):8103–8108. doi: 10.1073/pnas.1109824109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, et al. Revealing the competition between peeled ssDNA, melting bubbles, and S-DNA during DNA overstretching by single-molecule calorimetry. Proc Natl Acad Sci U S A. 2013;110(10):3865–3870. doi: 10.1073/pnas.1213740110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, et al. Improved High-Force Magnetic Tweezers for Stretching and Refolding of Proteins and Short DNA. Biophys. J. 2011;100(2):517–523. doi: 10.1016/j.bpj.2010.12.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu HX, et al. Transition dynamics and selection of the distinct S-DNA and strand unpeeling modes of double helix overstretching. Nucleic Acids Res. 2011;39(8):3473–3481. doi: 10.1093/nar/gkq1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen H, Fu H, Doyle PS, Yan J. Two distinct overstretched DNA structures revealed by single-molecule thermodynamics measurements. Proc. Natl. Acad. Sci. U.S.A. 2012;109(21):8103–8108. doi: 10.1073/pnas.1109824109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, et al. Revealing the competition between peeled ssDNA, melting bubbles, and S-DNA during DNA overstretching by single-molecule calorimetry. Proc. Natl. Acad. Sci. U.S.A. 2013;110(10):3865–3870. doi: 10.1073/pnas.1213740110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughn JP, et al. The DEXH protein product of the DHX36 gene is the major source of tetramolecular quadruplex G4-DNA resolving activity in HeLa cell lysates. J Biol Chem. 2005;280(46):38117–38120. doi: 10.1074/jbc.C500348200. [DOI] [PubMed] [Google Scholar]
- Giri B, et al. G4 resolvase 1 tightly binds and unwinds unimolecular G4-DNA. Nucleic Acids Res. 2011;39(16):7161–7178. doi: 10.1093/nar/gkr234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vlaminck I, Dekker C. Recent advances in magnetic tweezers. Annu Rev Biophys. 2012;41:453–472. doi: 10.1146/annurev-biophys-122311-100544. [DOI] [PubMed] [Google Scholar]
- Yan J, Skoko D, Marko JF. Near-field-magnetic-tweezer manipulation of single DNA molecules. Phys Rev E Stat Nonlin Soft Matter Phys. 2004;70(1 Pt 1):011905. doi: 10.1103/PhysRevE.70.011905. [DOI] [PubMed] [Google Scholar]
- Le S, et al. Disturbance-free rapid solution exchange for magnetic tweezers single-molecule studies. Nucleic Acids Res. 2015;43(17):e113. doi: 10.1093/nar/gkv554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neidle S. Quadruplex Nucleic Acids as Novel Therapeutic Targets. J Med Chem. 2016;59(13):5987–6011. doi: 10.1021/acs.jmedchem.5b01835. [DOI] [PubMed] [Google Scholar]
- Simone R, Fratta P, Neidle S, Parkinson GN, Isaacs AM. G-quadruplexes: Emerging roles in neurodegenerative diseases and the non-coding transcriptome. FEBS Lett. 2015;589(14):1653–1668. doi: 10.1016/j.febslet.2015.05.003. [DOI] [PubMed] [Google Scholar]
- Balasubramanian S, Hurley LH, Neidle S. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat Rev Drug Discov. 2011;10(4):261–275. doi: 10.1038/nrd3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato J, et al. Toward the Development of Specific G-Quadruplex Binders: Synthesis, Biophysical, and Biological Studies of New Hydrazone Derivatives. J Med Chem. 2016;59(12):5706–5720. doi: 10.1021/acs.jmedchem.6b00129. [DOI] [PubMed] [Google Scholar]
- Wells RD. Non-B DNA conformations, mutagenesis and disease. Trends Biochem Sci. 2007;32(6):271–278. doi: 10.1016/j.tibs.2007.04.003. [DOI] [PubMed] [Google Scholar]