

Abstract
Compared with persons of European descent (ED), persons of African descent (AD) have lower aldosterone (ALDO) levels, with the assumption being that the increased cardiovascular disease (CVD) risk associated with AD is not related to ALDO. However, the appropriateness of the ALDO levels for the volume status in AD is unclear. We hypothesized that, even though ALDO levels are lower in AD, they are inappropriately increased, and therefore, ALDO could mediate the increased CVD in AD. To test this hypothesis, we analyzed data from HyperPATH — 1,788 individuals from the total cohort and 765 restricted to ED-to-AD in a 2:1 match and genotyped for the endothelin-1 gene (EDN1). Linear regression analyses with adjustments were performed. In the total and restricted cohorts, PRA, ALDO, and urinary potassium levels were significantly lower in AD. However, in the AD group, greater ALDO dysregulation was present as evidenced by higher ALDO/plasma renin activity (PRA) ratios (ARR) and sodium-modulated ALDO suppression-to-stimulation indices. Furthermore, EDN1 minor allele carriers had significantly greater ARRs than noncarriers but only in the AD group. ARR levels were modulated by a significant interaction between EDN1 and AD. Thus, EDN1 variants may identify particularly susceptible ADs who will be responsive to treatment targeting ALDO-dependent pathways (e.g., mineralocorticoid-receptor antagonists).
Keywords: Endocrinology, Genetics
Keywords: Cardiovascular disease, Epigenetics, Hypertension
Individuals of African descent that carry the risk allele for endothelin-1 are susceptible to hypertension that may be responsive to mineralocorticoid receptor blockade.
Introduction
Compared with persons of European descent (ED), individuals of African descent (AD) are known to: (i) be more susceptible to cardiovascular disease (CVD); (ii) have a greater frequency of hypertension (HTN) (particularly salt-sensitive HTN); (iii) have more cardiovascular (CV) damage with HTN; (iv) have lower plasma renin activity (PRA); and (v) more resistant HTN (1–6). Of interest, these same characteristics also are associated with increased aldosterone (ALDO) production, e.g., primary aldosteronism. However, several studies have documented that ALDO levels are lower in AD (7–10) compared with ED. Initially, the lower ALDO levels were presumed to be the physiologic consequences of volume expansion with the resultant suppression of renin-angiotensin system (RAS) activity. However, several studies have proposed that the lower ALDO levels may be secondary to a defect in potassium homeostasis or intake. For example, urinary excretion of potassium is significantly lower in AD compared with ED (11), irrespective of sodium (Na+) intake (11–13).
Therefore, the current dogma is that ALDO levels are lower in AD than ED for physiologic reasons, including volume expansion (leading to reduced PRA) and reduced potassium homeostasis excretion, and not associated with the pathogenesis of the HTN, although this conclusion is controversial (6). However, an additional potential mechanism that has been minimally explored relates to endothelin. Endothelin has properties similar to angiotensin II (AngII) with known contractile, proliferative, and ALDO stimulating activities (14–16). Further, some studies have reported an association between endothelin-1 (ET-1) and HTN, although this association has been controversial (17–21). In addition, ET-1 levels have been reported to be 3–4 times higher in AD than ED hypertensives (1). Thus, it is possible that, in AD versus ED, there are increases in ET-1 leading to the following sequence of events: increased ALDO, volume expansion, decreased PRA, and elevated ALDO/PRA ratio (ARR). Finally, these differences might be genetically mediated. There are suggestive data to support this possibility because low renin HTN has been reported to be familially aggregated (22), and, as noted above, low renin HTN is more common in ADs than ED.
These data led us to formulate 2 hypotheses. First, that AD demonstrate dysregulated ALDO secretion compared with ED, assessed by ARR and Sodium-Modulated ALDO Suppression-to-Stimulation Index with AngII infusion (SASSI-II) (23). Second, that genetic variants in ET-1 gene (EDN1) will be associated with inappropriately elevated ALDO. We also determined if ET-1 modulates ALDO levels.
Results
Patient characteristics and analyses of the entire HyperPATH data set.
The demographic characteristics in the entire HyperPATH data set are provided in Supplemental Table 1 (supplemental material available online with this article; https://doi.org/10.1172/jci.insight.95992DS1). Age, blood pressure (BP), BMI, and sex were significantly different between AD and ED, with AD being younger, more commonly female, having a higher BP, and having a greater BMI. AD also had significantly lower liberal (LIB) salt supine serum ALDO, PRA, and 24-hour urine potassium levels as compared with ED (Supplemental Figure 1).
Patient characteristics of the restricted subcohort.
Because of the significant differences in demographics between AD and ED, we then performed a 2:1 study as detailed in Methods. There were no significant differences between AD and ED within this restricted subcohort except BMI (higher in AD) and age (lower in AD) (Table 1).
Table 1. Baseline demographic characteristics and selected clinical values of the subjects in the restricted HyperPATH subcohort to assess the effect of matching in this subcohort.

Comparison by race of ALDO, PRA, and potassium in the restricted subcohort.
As anticipated from the literature, in our cohort on a LIB salt diet, AD had significantly lower serum and urine ALDO and PRA, and had lower urine potassium levels as compared with ED (Figure 1). However, serum potassium levels did not differ by racial group. Similar results were obtained in studies performed on the restricted (RES) salt diet (Figure 2) and after upright posture (Figure 2).
Figure 1. Plasma renin activity, aldosterone and potassium levels in the total HyperPATH Cohort.

Comparison in European descent subjects and African descent subjects of aldosterone (supine serum [A] and 24-hour urine [B]), plasma renin activity (C), and urine potassium levels (D) on a liberal salt diet. Multivariate regression models for plasma renin activity and urine potassium were adjusted for age, sex, BMI, site, and disease state. The models for aldosterone were adjusted for age, sex, BMI, site, disease state, serum cortisol, 24-hour urine potassium, and plasma renin activity. Data are represented in a dot plot with the mean ± SEM. The subjects were from the restricted HyperPATH sub-cohort.
Figure 2. Plasma renin activity, aldosterone and potassium levels in the restricted HyperPATH Cohort.

Comparison in European descent subjects and African descent subjects of aldosterone (supine serum [A] and 24-hour urine [B]), plasma renin activity (C), urine potassium levels (D), aldosterone after 60–90 minutes of upright posture (E), and plasma renin activity after 60–90 minutes of upright posture (F) on a restricted salt diet. Multivariate regression models for plasma renin activity and urine potassium were adjusted for age, sex, BMI, site, and disease state. The models for aldosterone were adjusted for age, sex, BMI, site, disease state, serum cortisol, 24-hour urine potassium, and plasma renin activity. Data are represented in a dot plot with the mean ± SEM. The subjects were from the restricted HyperPATH subcohort.
Repeated-measures ALDO analyses of the restricted subcohort.
A total of 2,212 ALDO measurements were available from our restricted cohort: 1,504 in ED and 708 in AD. As a formal test of our hypothesis, we examined the effect of race on composite ALDO level in repeated-measures analysis using 4 measurements (RES ALDO supine, RES ALDO stimulated with AngII, LIB ALDO supine, and LIB ALDO stimulated with AngII) adjusted for disease state, age, sex, BMI, site, serum cortisol, PRA, and 24-hour urine potassium levels (Table 2). The composite ALDO measurements were highly significantly lower in AD versus ED (P < 0.0001).
Table 2. The effect of race on aldosterone levels.
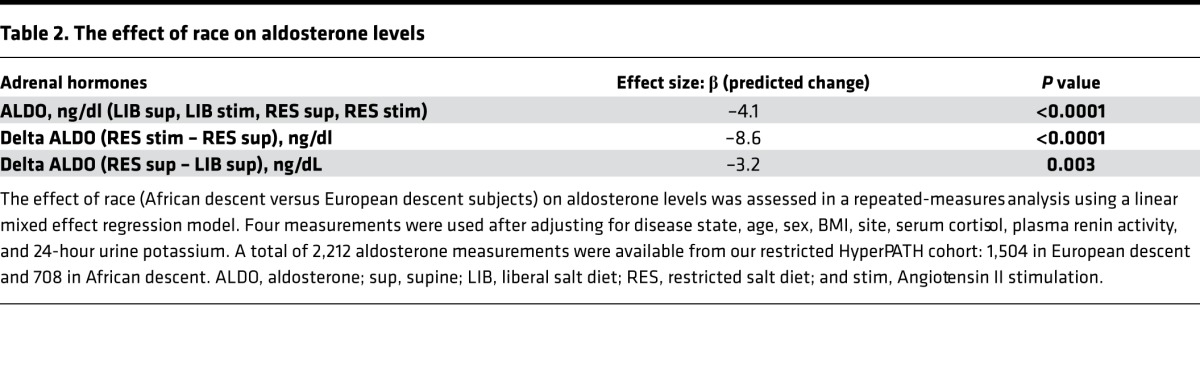
ARR and SASSI-II index of the restricted subcohort.
Although ALDO levels were substantially lower under a variety of conditions in AD versus ED, we next assessed whether its regulation was similar in ADs versus ED using 2 criteria: the ARR and the SASSI-II index. First, we assessed ARR. Whether the subjects were on a LIB salt diet, RES salt diet, or upright posture on a RES salt diet, ARRs were significantly higher in AD than in ED (Figure 3). Next, we determined whether being hypertensive or normotensive influenced the impact of race on ARR; it did not. Both hypertensive and normotensive ADs had significantly higher ARRs than ED; however, within each racial group, ARR did not differ by disease state (Figure 4). Finally, we assessed the levels of SASSI-II as an indication of ALDO dysregulation. The SASSI-II index was significantly increased in AD compared with ED, suggesting relatively increased ALDO dysregulation in AD (Figure 5). The index components (mean ± SEM) were AD, 4.46 ± 0.43 supine LIB salt and 29.98 ± 1.24 RES salt AngII, and ED, 5.15 ± 0.20 supine LIB salt and 38.62 ± 1.04 RES salt AngII (ng/dl).
Figure 3. Aldosterone Renin Ratios in the restricted HyperPATH Cohort.

Comparison in European descent subjects and African descent subjects of aldosterone/renin ratio under 3 conditions: restricted salt diet (A), liberal salt diet (B), and restricted salt diet after 60–90 minutes of upright posture (C). The multivariate regression models were adjusted for age, sex, BMI, site, disease state, serum cortisol, and 24-hour urine potassium. Data are represented in a dot plot with the mean ± SEM. The subjects were from the restricted HyperPATH subcohort (ARR, aldosterone/renin ratio).
Figure 4. Comparison in European descent subjects and African descent subjects of aldosterone/renin ratio was obtained on a restricted salt diet.
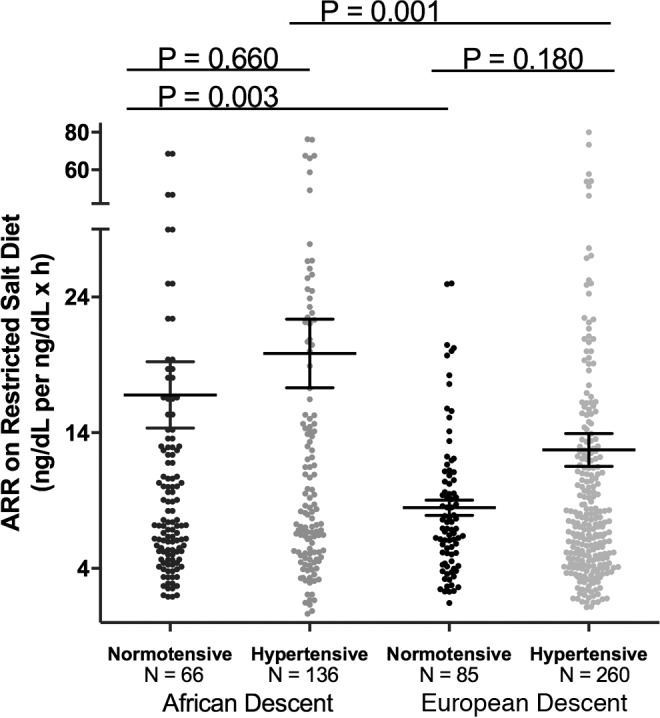
Subjects of African descent had a significantly higher aldosterone/renin ratio than subjects of European descent, regardless of hypertensive status. The multivariate regression models were adjusted for age, sex, BMI, site, serum cortisol, and 24-hour urine potassium. Data are represented in a dot plot with the mean ± SEM. The subjects were from the restricted HyperPATH subcohort. ARR, aldosterone/renin ratio).
Figure 5. Comparison in European descent subjects and African descent subjects of Sodium-Modulated Aldosterone Suppression-to-Stimulation Index with AngII infusion (SASSI-II).
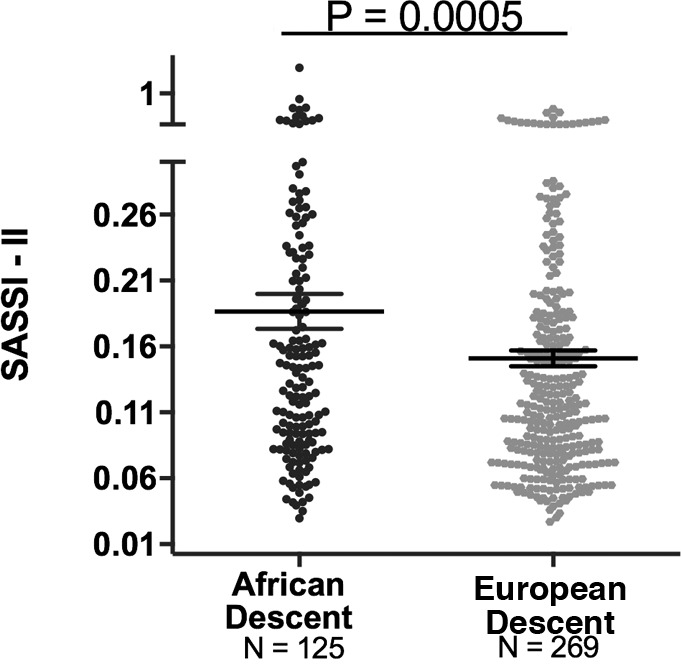
A multivariate linear regression model was used with adjustments for age, sex, BMI, disease state, site, serum cortisol, 24-hour urine potassium, and plasma renin activity (mean ± SEM). The subjects were from the restricted HyperPATH sub-cohort. Data are represented in a dot plot with the mean ± SEM.
Zona glomerulosa cell studies.
In separate experiments, both ET-1 (aa 1–21) and ET-1 precursor (aa 1–38) induced an approximate doubling of ALDO secretion as compared with control conditions. ALDO secretion (mean ± SEM ng/1 × 106 cells) was 32.5 ± 3.8 with ET-1 treatment versus 20.5 ± 1.2 with placebo (P = 0.001), and 410 ± 19 with ET-1 precursor treatment versus 221 ± 17 with placebo (P < 0.0005). The ALDO responses to ET-1 were half or less of the ALDO responses to AngII (161 ± 10 in the ET-1 experiment and 805 ± 22 in the ET-1 precursor experiment).
Relation of EDN1 genotype to ARR in the restricted cohort.
The demographics of the subjects from the restricted cohort who also were genotyped for EDN1 rs5370 are provided in Supplemental Table 2. The relationships of genotype to phenotype and race were assessed in 2 ways. First, the ordered trend relationship between genotype and ARR on a RES salt diet was determined for each race (Figure 6). This trend was significant for AD; the ARR increased as the number of EDN1 risk alleles increased. Second, an interaction term was calculated between race and EDN1 risk alleles and ARR on the LIB diet. For each risk allele, the effect size was +11 ng/dl per ng/dl × hr in AD compared with ED (coefficient 11.75, P = 0.010, 95% confidence intervals [CI], [2.83, 20.7]).
Figure 6. Comparison on aldosterone/renin ratio by endothelin-1 genotype in European descent subjects and African descent subjects.

The trend analyses were adjusted for age, sex, BMI, site, disease state, serum cortisol, 24-hour urine potassium, and plasma renin activity. Data are represented in a dot plot with the mean ± SEM. The subjects were from the restricted HyperPATH subcohort (ARR, aldosterone/renin ratio).
Discussion
In our total HyperPATH cohort study, we confirmed that ALDO, PRA, and potassium levels on a LIB salt diet were lower in AD than ED, as has been shown by others (1–6, 24). Because of limited genotyping data, we also performed analyses in a 2:1 restricted subcohort. In the subcohort, the key demographic characteristics were similar between the 2 races. Again, we confirmed that ALDO, PRA, and potassium levels on a LIB salt diet were lower in AD than ED and extended these conclusions to results of studies on a RES salt intake and after upright posture. Thus, under 3 different conditions, the results were consistent: AD had lower PRA, ALDO, and potassium levels. However, in contrast to some previous studies (1–6) that suggested ALDO and/or an activated mineralocorticoid receptor (MR) are not important in the pathophysiology of CVD in AD, our results — using surrogate markers — led to the opposite conclusion. Two tests of ALDO’s functional status were abnormal: ARR and SASSI-II levels were increased in AD. Thus, using PRA as a surrogate, the level of ALDO is inappropriately increased for the level of salt intake and volume status. This conclusion was the same regardless of whether subjects were on RES or LIB Na+ diets. Further, these racial differences are, in part, secondary to genetic factors driven by the EDN1 gene—in AD, but not in ED. In AD, there is a significant association of ARR with EDN1 genotype. We also documented that ET-1 can stimulate ALDO, confirming previous studies (14). These results suggest that MR or ALDO synthase blockade may be a useful therapeutic modality in AD, specifically in AD that carry the risk allele for EDN1, i.e., precision medicine.
ARR has been used for more than 30 years as a biomarker for abnormal ALDO production relative to the volume state of the individual (25), specifically as a screening test for primary aldosteronism. Indeed, since its introduction, the percent of hypertensives diagnosed with primary aldosteronism has risen dramatically (26). However, in recent years, several studies have suggested that the ARR is also a biomarker of the effect of ALDO on CV homeostasis (27). Thus, the levels of ARR are better indicators of ALDO/MR functional status than the absolute levels of the hormones per se and, therefore, a useful biomarker of CV dysregulation even among normotensives (28). Grim, et al., reported that AD had higher ARR than ED in a study on a LIB diet. They also reported that, regardless of race, hypertensives had higher ratios than normotensives (6). In the present study, we also found that the ARR was significantly increased in AD compared with ED. However, relative to ARR, our findings expand on those by Grim, et al., in 2 respects: i) since the definition of HTN is a moving target, this racial difference was present in both normotensives and hypertensives; and ii) by race, there was no significant difference in ARR between normotensives and hypertensives (Figure 4). Thus, the inclusion of normotensives in this study further strengthens the validity of our hypothesis. Finally, ARR were increased irrespective of dietary salt intake. Even when the renin-angiotensin-ALDO system (RAAS) was activated by Na+ restriction, AD still have increased ARR compared with ED.
More recently, the SASSI-II has been described (23). This index was built on data suggesting that, in CVD — particularly HTN — ALDO responses to physiologic factors that can stimulate or suppress its levels are reduced (29–35). Thus, SASSI-II was developed to assess ALDO’s dynamic responsiveness. The marker was tested in relation to components of the metabolic syndrome, where it not only was highly associated with individual risk factors, but also distinguished between severe and moderate metabolic syndrome and between individuals with and without the metabolic syndrome. SASSI-II predictive power was better than any individual ALDO serum or urine level (23). Thus, SASSI-II is a sensitive marker for the risk of CV dysfunction and disease. In the present study, significantly higher SASSI-II levels were observed in AD versus ED, providing additional support for the hypothesis that AD have an increased ALDO-mediated CV risk than ED, even in the absence of primary aldosteronism or HTN.
In addition to studies assessing the role of the RAAS in mediating the differences in salt sensitivity of BP between AD and ED, other volume-mediating systems have been evaluated, including ET-1. ET-1 is a peptide with known vasoconstrictor, proliferative, and ALDO stimulatory properties, making it a focus of attention for studies of HTN (14–16). We confirmed ET-1’s ALDO-stimulating properties in isolated glomerulosa cells (14). However, the data relevant to the association of ET-1 levels and HTN are controversial (17–21). In part, the difficulty of finding consistent results may be related to the transient presence of ET-1 in the circulation, as it is as labile as AngII. However, one report is particularly relevant to the present study; Ergul, et al., not only reported a significant association of ET-1 levels and disease state (hypertensive vs. normotensive), but also that this pattern was seen most strikingly among AD and less so in ED (1). Unfortunately, ET-1 serum levels were not available in this study. Thus, we do not know the state of ET-1 levels associated with the risk allele. However, an increase in ALDO in the presence of lower PRA is consistent with another stimulatory factor being present.
A link between genetics, race, and Na+ homeostasis has recently been proposed. Tu and Pratt concluded that the low-renin, salt sensitivity of BP phenotype of AD compared with ED is attributed to alterations in genetic makeup (36). They went on to describe greater renal Na+ absorption among AD compared with ED, thus leading to expanded plasma volume in this population. Findings from a 1979 study by Luft, et al., suggest that the difference in plasma volume, and consequently BP levels, may also be due to the decreased Na+ excretion among AD (37). Several genes have been reported to be associated with BP and race. Lysine-specific demethylase 1 (LSD1) is a histone-modifying enzyme. Polymorphic variants in LSD1 gene were reported to be related to greater salt sensitivity of BP in hypertensives — but only in AD and not ED (38). Additional studies in normotensives suggest that the steepness of the slope of the relationship to BP and age was dependent on polymorphic variants in LSD1 (39). In contrast, polymorphic variants in the β-2 adrenergic receptor are associated with salt-sensitive HTN only in ED (40).
Prior work evaluating effects of EDN1 genotype in humans has repeatedly identified this polymorphism to be of clinical relevance. Its widespread potential effects include modification of risk for HTN in the overweight/obese population (41–43), BP reactivity to stressors (44–46), idiopathic pulmonary arterial HTN (47), pulmonary function in chronic obstructive pulmonary disease (48), severity of obstructive sleep apnea (49), coronary artery disease (50), left ventricular hypertrophy (51), variant angina (52), and hearing impairment (53). Our study expands on this list by documenting that racial differences exists between increased ARR, a likely proximate mediator of the salt-sensitive BP, and EDN1 risk allele carriers in AD but not ED. One potential mechanism would be that the risk allele is associated with increased ET-1 locally in AD, resulting in greater ALDO secretion, volume expansion, suppressed PRA, and salt-sensitive HTN. This provides the first evidence of a potential association between the EDN1 rs5370 SNP and the risk of subclinical hyperaldosteronism in AD. Finally, a previous study reported sex differences in the relationship between systolic BP and a haplotype of EDN1, with males but not females being protected (i.e., having a lower BP; ref. 54). However, there was no main effect of this haplotype on systolic BP, and an interaction with sex was only marginally significant (P = 0.053), particularly given the multiple comparisons in the study. These investigators also assessed the effect of EDN1 rs5370 on systolic BP curves; they did not see an effect. Furthermore, the sample size was substantially smaller than in the current study, the subjects were young, and salt sensitivity of BP was not assessed. Finally, we observed a significant association of salt-sensitive BP and rs5370, even with adjustment for sex.
Conclusions from these data are, however, limited by a small sample size — a concern in many genetic studies. However, the “clean” phenotype in this study compensates, in part, for this issue. Again, because of the sample size, it is not possible with this data set to determine if there are interactions between EDN1 and other genes previously reported to have a racial-specific association, e.g., LSD1. Because of the structure of the HyperPATH cohort, it was not possible to measure ET-1 levels and their potential relationship to genotype, ARR, and/or salt-sensitive BP. Several other factors may, in part, explain the differences between the AD and ED. First, some studies have reported a relationship between the level of stress and the relationship between BP and EDN1 polymorphism and between AD and BP (45, 46, 55, 56). Stress profiles are not present in the HyperPATH data set, and salt sensitivity of BP, ALDO, ARR, or SASSI-II were not performed in these previous studies. Second, 24-hour ambulatory BPs have not been performed in the HyperPATH cohort. Given the likely increase number of “non–BP dippers” in AD versus ED, our striking salt-sensitive BP differences could be even more striking if ambulatory BPs were assessed (57). Third, it has been reported that exercise status can affect rs5370 gene expression in ED but not AD. Exercise status was not measured in the HyperPATH cohort. However, interpretation of the results from this previous study are limited because of the sample size (58). Fourth, other factors (e.g., autoantibodies) could be activating the END1 receptor or the AngII receptor, although there are no data available to support or reject these possibilities. Finally, the present data are limited to salt-sensitive HTN and should be interpreted cautiously when extrapolating the findings to other reported differences in CVD risk between AD and ED.
In conclusion, ALDO levels have been the focus of studies for how ALDO influences CV risk; however, our findings suggest that these levels should be interpreted in the context of PRA levels, as well as the response to salt intake and ALDO responsiveness (via ARR and SASSI-II, respectively) for a more comprehensive picture of ALDO’s potential deleterious properties affecting CV risk. Thus, our findings support the posit that ALDO is an important risk factor for the development of CV disease in AD and likely more so than in ED. In part, at least one mechanism that contributes to ALDO dysregulation in AD is genetic, related to the EDN1. Moving forward, treatment approaches that antagonize ALDO’s actions or target mechanisms that are MR dependent, including the ET-1 system, could have substantial relevance to the reduction of the increased stroke, end-stage renal disease, and heart disease in AD, although additional proof of causality is required
Methods
Participants.
Our initial analyses were performed using all subjects in the HyperPATH cohort (described below). These individuals consumed a LIB salt diet and then, following a supine, overnight fast, had BP, ALDO, and PRA values obtained (1,788 subjects). Since some subjects were from sites outside the US, we are referring to them as ED or AD. Although results from the HyperPATH cohort have been previously reported, the present study investigating ALDO secretion in AD and ED using the present approach and data set has not. This approach was taken to compare results in this cohort with previously studies that had contrasted renin and ALDO levels in ED and AD (1–6). We expanded our analyses to additional factors: first, to assess the impact of Na+ intake on ARR and SASSI-II levels, and second, to determine if there was an association between ARR and EDN1 genotype. Because of limitations in some of the available data, particularly genotype, and due to the fact that the number of ED was 6 times that of the AD, a restricted subcohort that consisted of twice as many ED as AD was used. The ED were matched to the AD using the following demographic characteristics in decreasing order: diastolic BP, sex, age, disease state (hypertensive or normotensive), and BMI. All had tagging SNPs at EDN1.
HyperPATH is an ongoing, international program to study the genetic underpinnings of hormonal mechanisms of HTN and CVD. Five international centers contributed to this data set: Brigham and Women’s Hospital, University of Utah Medical Center, Hospital Broussais (Paris, France), Vanderbilt University (Nashville, Tennessee, USA), and University of La Sapiena in Rome, Italy. The details of this cohort and study design have been previously described in detail (59). In brief, subjects used in this analysis had normal history, physical, except for HTN, although some (<10%) had glucose intolerance and/or mild diabetes mellitus. All subjects were between 18 and 65 years old, and race was self-defined. Normotension was defined as a seated diastolic BP ≤ 80 mm Hg on 3 different dates at least 1 month apart with 3 replicates at each date. HTN was defined as a seated diastolic BP ≥ 100 mm Hg on no medications, ≥ 90 mm Hg on 1 antihypertensive medication at the time of screening, or treatment with ≥ 2 antihypertensive medications. Subjects on ≥ 4 antihypertensive medications at the time of screening were excluded. Secondary forms of HTN were ruled out by comparing normotensives and hypertensives in HyperPATH as follows: normal 24-hour cortisol levels, 24-hour ALDO and supine ALDO levels on LIB salt intake, glucose tolerance test, and physical exam. However, no radiologic studies were performed. All antihypertensive medications were washed out for 1–3 months prior to dietary salt study. Subjects were then studied under 2 dietary conditions, at least 1 week apart: RES and LIB salt intakes. A LIB Na+ diet contained: approximately 200 mmol/d of Na+, 100 mmol/d of potassium, and 20 mmol/d of calcium for 5–7 days. A RES Na+ intake was achieved by providing a diet containing: 10 mmol/d of Na+, 100 mmol/d of potassium, and 20 mmol/d of calcium for 5–7 days. Subjects completed a 24-hour urine collection on the final day of the study diet. To be included in the current analyses, the subjects had to have a RES salt diet Na+ excretion < 25 mmol/day and, on the LIB salt diet, > 160 mmol/day. The subjects then were admitted to the Clinical Research Unit at their respective institutions and kept in a supine posture overnight. After 12 hours of supine, fasting posture, an automated device (Dinamapp, Critikon) was used to obtain BPs. The mean of 3 consecutive readings separated by 5 minutes each were used for analyses. Blood was obtained to measure a variety of hormones (see below), AngII (3 ng/kg/min) was infused for 60 minutes, and repeat blood samples were obtained. On the RES diet, blood samples also were obtained after 60–90 minutes of ambulation.
SASSI-II estimate.
To characterize dynamic ALDO regulation, we determined the SASSI-II by calculating the ratio of supine serum ALDO on the LIB diet to supine serum ALDO after AngII infusion on the RES diet. The AngII infusion while on a RES diet is expected to provide maximal adrenal ALDO stimulation. The SASSI-II index correlates with the Framingham Risk Score and the number of components of the metabolic syndrome (23, 60). Higher values of SASSI-II are indicative of impaired physiologic regulation of ALDO (23, 60).
Laboratory analysis.
Brigham and Women’s Hospital’s Research Assay Core (BRAC), or BRAC’s predecessor, served as the central laboratory for all laboratory processing. All assays used in this study have been extensively reported previously (23, 29, 61). All subjects had PRA and serum ALDO measured in triplicate at each of the intervention points.
Genotyping.
DNA samples were extracted from peripheral leukocytes using DNA extraction procedures from Crimson Biospecimen Core (QIAGEN). Once extracted, DNA samples were stored at –80°C. Genotyping was done at Partners PCPGM Genotyping Facility (Boston, MA, USA) using the Illumina Bead Station Golden Gate platform. Prior reports observed an association between EDN1 rs5370 and HTN (62). Therefore, in evaluating the potential effects of EDN1 genotype, we focused on rs5370. The participants with a risk allele in this study were referred to as subjects carrying the minor allele of rs5370.
Zona glomerulosa cell studies.
Male Wistar rats weighing between 175–250 g were obtained from Charles River Laboratories. All animals were housed in a room lighted 12 hours a day at an ambient temperature of 22°C ± 8°C. Animals were placed on RES rat chow containing 0.03% Na+ (Purina) and had free access to tap water for 5–7 days before sacrifice. Zona glomerulosa (ZG) cells were isolated from adrenal tissues by dissecting the capsular (containing predominantly glomerulosa) portion and the decapsulated portion (fasciculata-reticularis complex) as previously described (40, 63). Cells were incubated for digestion (collagenase 3.7 mg/ml, DNAse 0.05 mg/ml) in Krebs Ringer bicarbonate solution (Sigma-Aldrich) (0.1% BSA, 200 mg glucose/dl, L-glutamine, 3.7 mmol/L of K+) at 37°C for 60 minutes under 95% O2 and 5% CO2. Cells were centrifuged at 700 g at 25°C for 10 minutes, washed, and resuspended for another 30 minutes at 37°C. Final cell suspension was made to yield 2 to 5 × 105 isolated cells/0.5 ml. Cells in Krebs Ringer bicarbonate solution were treated for 1 hour with 1 × 10–8 M ET-1 (aa 1≠21) (MilliporeSigma), 1 × 10–8 M ET-1 precursor (aa 1–38) (American Peptide Company), 1 × 10–8 M AngII (positive control), or placebo. ALDO levels were measured at the end of incubation (ALDO Radioimmunoassay [RIA] kit, IBL International). All experiments were carried out in duplicate cell preps, with each duplicate replicated 2–4 times.
Statistics.
Baseline analyses included the Student t test and χ2. Continuous variables are presented as mean ± SD, 95% CI, or mean ± SEM. Categorical variables are presented as a percentage of the total sample. Normal distribution of dependent variable was tested using Shapiro-Wilk test. Logarithmic transformation was performed for variables that are not normally distributed. The nominal P value was adjusted to 0.01 because of multiple comparisons. All statistical analyses were performed using STATA 14.1. Because of the unique characteristics of our protocol where several measurements are available in the same subject in 4 different intervention settings (RES baseline, RES stimulated with AngII infusion, LIB baseline, and LIB stimulated with AngII), we performed a mixed-model linear regression analysis on serum ALDO, adjusting for confounders: disease state (normotensive or hypertensive), age, sex, site, serum cortisol, PRA, serum potassium, and urinary potassium excretion. These covariates (fixed effects) were chosen for their clinical importance and significance in univariate analyses, whereas identity was the random effect. General linear regression was used to analyze the relationship between other primary endpoints (e.g., LIB PRA Baseline) and race (AD or ED). In addition to known CV risk factors (age, BMI, disease state [hypertensive or normotensive], and sex), ALDO secretagogues (plasma cortisol [surrogate for ACTH], PRA, urine potassium) and site were included in the regression model as appropriate. Also, the interaction of race and EDN1 rs5370 alleles on LIB ARR was determined with the same adjustments as noted above. The model used was: race (0 = ED, 1 = AD) and number of risk alleles (0 = no risk alleles and 1 = at least 1 risk allele).
Study approval.
The corresponding IRBs at Brigham and Women’s Hospital (Boston, Massachusetts, USA), University of Utah School of Medicine (Salt Lake City, Utah, USA), Vanderbilt School of Medicine (Nashville, Tennessee, USA), and the Hôpital Européen Georges Pompidou (Paris, France) approved all studies, and written informed consent was obtained from each participant. All studies were conducted in accordance with the Harvard Medical School institutional guidelines for the humane treatment of animals.
Author contributions
GHW, JSW, and PNH designed the research studies and/or oversaw the HyperPATH protocols in Boston, Massachusetts, and Salt Lake City, Utah, USA. JWT performed the data analyses and wrote the manuscript in collaboration with GHW, TG, GKA, and JRR. WM performed the data analyses. GHW, GKA, and JRR contributed to data interpretation and manuscript preparation. TMY performed the cell-based study.
Supplementary Material
Acknowledgments
We would like to thank the Center for Clinical Investigation and Brigham and Women’s Hospital’s staff, as well as other investigators and staff of the HyperPATH Protocol and participants at each protocol site, including the Clinical Investigation Centre, INSERM CIC 9201, Hôpital Européen Georges Pompidou (Paris, France), University of Utah Medical Center, Vanderbilt University, and University of La Sapiena. We are particularly grateful for the many research subjects who gave their time and effort as participants in the HyperPATH program and the research fellows that cared for them and performed the research protocols that generated the data used in this study. We acknowledge financial assistance, in partial support for these studies, from the following sources: the Brigham and Women’s Hospital, Division of Endocrinology, Diabetes, and Hypertension; Harvard Medical School; NIH grants P50HL055000 (HyperPATH Cohort); UL1RR025758 Harvard Clinical and Translational Science Center, from the National Center for Research Resources; M01-RR02635, Brigham and Women’s Hospital, General Clinical Research Center, from the National Center for Research Resources; NIH grants R01HL086907, R01HL11476, R01HL69208, R01HL4765, R01HL59424, R01HL69208, R01HL096518, R01HL104032, K24 HL103845, and T32HL 007609 from the National Heart, Lung, and Blood Institute; and a grant from the National Center for Advanced Translational Science RM-07-2002.
Version 1. 12/07/2017
Electronic publication
Funding Statement
Brigham and Women’s Hospital, Division of Endocrinology, Diabetes, Hypertension and Harvard Medical School; National Institute of Health Grants P50HL055000 (HyperPATH Cohort); UL1RR025758 Harvard Clinical and Translational Science Center, from the National Center for Research Resources; M01-RR02635, Brigham and Women’s Hospital, General Clinical Research Center, from the National Center for Research Resources; and National Institutes of Health grants R01HL086907, R01HL11476, R01HL69208, R01HL4765, R01HL59424, R01HL69208, R01HL096518, R01HL104032, K24 HL103845 and T32HL 007609 from the National Heart, Lung and Blood Institute and a grant from the National Center for Advanced Translational Science RM-07-2002.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2017;2(23):e95992. https://doi.org/10.1172/jci.insight.95992.
Contributor Information
Tina Gupta, Email: tgupta2@bwh.harvard.edu.
Worapaka Manosroi, Email: worapaka.m@gmail.com.
Gordon H. Williams, Email: gwilliams@partners.org.
References
- 1.Ergul S, Parish DC, Puett D, Ergul A. Racial differences in plasma endothelin-1 concentrations in individuals with essential hypertension. Hypertension. 1996;28(4):652–655. doi: 10.1161/01.HYP.28.4.652. [DOI] [PubMed] [Google Scholar]
- 2.Ferdinand KC, Armani AM. The management of hypertension in African Americans. Crit Pathw Cardiol. 2007;6(2):67–71. doi: 10.1097/HPC.0b013e318053da59. [DOI] [PubMed] [Google Scholar]
- 3.Tu W, Eckert GJ, Pratt JH, Jan Danser AH. Plasma levels of prorenin and renin in blacks and whites: their relative abundance and associations with plasma aldosterone concentration. Am J Hypertens. 2012;25(9):1030–1034. doi: 10.1038/ajh.2012.83. [DOI] [PubMed] [Google Scholar]
- 4.Price DA, Fisher ND. The renin-angiotensin system in blacks: active, passive, or what? Curr Hypertens Rep. 2003;5(3):225–230. doi: 10.1007/s11906-003-0025-x. [DOI] [PubMed] [Google Scholar]
- 5.Sagnella GA. Why is plasma renin activity lower in populations of African origin? J Hum Hypertens. 2001;15(1):17–25. doi: 10.1038/sj.jhh.1001127. [DOI] [PubMed] [Google Scholar]
- 6.Grim CE, et al. Hyperaldosteronism and hypertension: ethnic differences. Hypertension. 2005;45(4):766–772. doi: 10.1161/01.HYP.0000154364.00763.d5. [DOI] [PubMed] [Google Scholar]
- 7.Murro DG, Beavers M, Harshfield GA, Kapuku GK. Aldosterone contributes to elevated left ventricular mass in black boys. Pediatr Nephrol. 2013;28(4):655–660. doi: 10.1007/s00467-012-2367-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fisher ND, Gleason RE, Moore TJ, Williams GH, Hollenberg NK. Regulation of aldosterone secretion in hypertensive blacks. Hypertension. 1994;23(2):179–184. doi: 10.1161/01.HYP.23.2.179. [DOI] [PubMed] [Google Scholar]
- 9.Ripley E, King K, Sica DA. Racial differences in response to acute dosing with hydrochlorothiazide. Am J Hypertens. 2000;13(2):157–164. doi: 10.1016/S0895-7061(99)00168-5. [DOI] [PubMed] [Google Scholar]
- 10.Pratt JH, Jones JJ, Miller JZ, Wagner MA, Fineberg NS. Racial differences in aldosterone excretion and plasma aldosterone concentrations in children. N Engl J Med. 1989;321(17):1152–1157. doi: 10.1056/NEJM198910263211703. [DOI] [PubMed] [Google Scholar]
- 11.Rosa RM, et al. Gastrointestinal and renal excretion of potassium in African-Americans and White Americans. J Hypertens. 2012;30(12):2373–2377. doi: 10.1097/HJH.0b013e32835a27b3. [DOI] [PubMed] [Google Scholar]
- 12.Turban S, Thompson CB, Parekh RS, Appel LJ. Effects of sodium intake and diet on racial differences in urinary potassium excretion: results from the Dietary Approaches to Stop Hypertension (DASH)-Sodium trial. Am J Kidney Dis. 2013;61(1):88–95. doi: 10.1053/j.ajkd.2012.08.036. [DOI] [PubMed] [Google Scholar]
- 13.Palacios C, et al. Racial differences in potassium homeostasis in response to differences in dietary sodium in girls. Am J Clin Nutr. 2010;91(3):597–603. doi: 10.3945/ajcn.2009.28400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cozza EN, Gomez-Sanchez CE, Foecking MF, Chiou S. Endothelin binding to cultured calf adrenal zona glomerulosa cells and stimulation of aldosterone secretion. J Clin Invest. 1989;84(3):1032–1035. doi: 10.1172/JCI114226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rubanyi GM, Polokoff MA. Endothelins: molecular biology, biochemistry, pharmacology, physiology, and pathophysiology. Pharmacol Rev. 1994;46(3):325–415. [PubMed] [Google Scholar]
- 16.Yanagisawa M, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332(6163):411–415. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- 17.Haak T, Jungmann E, Felber A, Hillmann U, Usadel KH. Increased plasma levels of endothelin in diabetic patients with hypertension. Am J Hypertens. 1992;5(3):161–166. doi: 10.1093/ajh/5.3.161. [DOI] [PubMed] [Google Scholar]
- 18.Kohno M, et al. Plasma immunoreactive endothelin in essential hypertension. Am J Med. 1990;88(6):614–618. doi: 10.1016/0002-9343(90)90527-K. [DOI] [PubMed] [Google Scholar]
- 19.Miyauchi T, et al. Age- and sex-related variation of plasma endothelin-1 concentration in normal and hypertensive subjects. Am Heart J. 1992;123(4 Pt 1):1092–1093. doi: 10.1016/0002-8703(92)90734-d. [DOI] [PubMed] [Google Scholar]
- 20.Saito Y, Nakao K, Mukoyama M, Imura H. Increased plasma endothelin level in patients with essential hypertension. N Engl J Med. 1990;322(3):205. doi: 10.1056/nejm199001183220315. [DOI] [PubMed] [Google Scholar]
- 21.Shichiri M, et al. Plasma endothelin levels in hypertension and chronic renal failure. Hypertension. 1990;15(5):493–496. doi: 10.1161/01.HYP.15.5.493. [DOI] [PubMed] [Google Scholar]
- 22.Fisher ND, Hurwitz S, Jeunemaitre X, Hopkins PN, Hollenberg NK, Williams GH. Familial aggregation of low-renin hypertension. Hypertension. 2002;39(4):914–918. doi: 10.1161/01.HYP.0000013784.18175.51. [DOI] [PubMed] [Google Scholar]
- 23.Vaidya A, et al. Abnormal aldosterone physiology and cardiometabolic risk factors. Hypertension. 2013;61(4):886–893. doi: 10.1161/HYPERTENSIONAHA.111.00662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kotchen TA, Kotchen JM, Grim CE, Krishnaswami S, Kidambi S. Aldosterone and alterations of hypertension-related vascular function in African Americans. Am J Hypertens. 2009;22(3):319–324. doi: 10.1038/ajh.2008.327. [DOI] [PubMed] [Google Scholar]
- 25.Hiramatsu K, et al. A screening test to identify aldosterone-producing adenoma by measuring plasma renin activity. Results in hypertensive patients. Arch Intern Med. 1981;141(12):1589–1593. [PubMed] [Google Scholar]
- 26.Mulatero P, et al. Increased diagnosis of primary aldosteronism, including surgically correctable forms, in centers from five continents. J Clin Endocrinol Metab. 2004;89(3):1045–1050. doi: 10.1210/jc.2003-031337. [DOI] [PubMed] [Google Scholar]
- 27.Alvarez-Madrazo S, et al. Familial and phenotypic associations of the aldosterone Renin ratio. J Clin Endocrinol Metab. 2009;94(11):4324–4333. doi: 10.1210/jc.2009-1406. [DOI] [PubMed] [Google Scholar]
- 28.Tomaschitz A, et al. Aldosterone/renin ratio determines peripheral and central blood pressure values over a broad range. J Am Coll Cardiol. 2010;55(19):2171–2180. doi: 10.1016/j.jacc.2010.01.032. [DOI] [PubMed] [Google Scholar]
- 29.Bentley-Lewis R, et al. Body mass index predicts aldosterone production in normotensive adults on a high-salt diet. J Clin Endocrinol Metab. 2007;92(11):4472–4475. doi: 10.1210/jc.2007-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferri C, et al. Relationship between insulin resistance and nonmodulating hypertension: linkage of metabolic abnormalities and cardiovascular risk. Diabetes. 1999;48(8):1623–1630. doi: 10.2337/diabetes.48.8.1623. [DOI] [PubMed] [Google Scholar]
- 31.Garg R, Hurwitz S, Williams GH, Hopkins PN, Adler GK. Aldosterone production and insulin resistance in healthy adults. J Clin Endocrinol Metab. 2010;95(4):1986–1990. doi: 10.1210/jc.2009-2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Price DA, De’Oliveira JM, Fisher ND, Williams GH, Hollenberg NK. The state and responsiveness of the renin-angiotensin-aldosterone system in patients with type II diabetes mellitus. Am J Hypertens. 1999;12(4 Pt 1):348–355. [PubMed] [Google Scholar]
- 33.Shoback DM, Williams GH, Moore TJ, Dluhy RG, Podolsky S, Hollenberg NK. Defect in the sodium-modulated tissue responsiveness to angiotensin II in essential hypertension. J Clin Invest. 1983;72(6):2115–2124. doi: 10.1172/JCI111176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Underwood PC, et al. Nonmodulation as the mechanism for salt sensitivity of blood pressure in individuals with hypertension and type 2 diabetes mellitus. J Clin Endocrinol Metab. 2012;97(10):3775–3782. doi: 10.1210/jc.2012-2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vaidya A, Sun B, Larson C, Forman JP, Williams JS. Vitamin D3 therapy corrects the tissue sensitivity to angiotensin ii akin to the action of a converting enzyme inhibitor in obese hypertensives: an interventional study. J Clin Endocrinol Metab. 2012;97(7):2456–2465. doi: 10.1210/jc.2012-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tu W, Pratt JH. A consideration of genetic mechanisms behind the development of hypertension in blacks. Curr Hypertens Rep. 2013;15(2):108–113. doi: 10.1007/s11906-013-0332-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luft FC, Grim CE, Fineberg N, Weinberger MC. Effects of volume expansion and contraction in normotensive whites, blacks, and subjects of different ages. Circulation. 1979;59(4):643–650. doi: 10.1161/01.CIR.59.4.643. [DOI] [PubMed] [Google Scholar]
- 38.Brown JM, et al. Human interventions to characterize novel relationships between the renin-angiotensin-aldosterone system and parathyroid hormone. Hypertension. 2014;63(2):273–280. doi: 10.1161/HYPERTENSIONAHA.113.01910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krug AW, et al. Lysine-specific demethylase-1 modifies the age effect on blood pressure sensitivity to dietary salt intake. Age (Dordr) 2013;35(5):1809–1820. doi: 10.1007/s11357-012-9480-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pojoga L, et al. Beta-2 adrenergic receptor diplotype defines a subset of salt-sensitive hypertension. Hypertension. 2006;48(5):892–900. doi: 10.1161/01.HYP.0000244688.45472.95. [DOI] [PubMed] [Google Scholar]
- 41.Tiret L, et al. The Lys198Asn polymorphism in the endothelin-1 gene is associated with blood pressure in overweight people. Hypertension. 1999;33(5):1169–1174. doi: 10.1161/01.HYP.33.5.1169. [DOI] [PubMed] [Google Scholar]
- 42.Jin JJ, et al. Association of endothelin-1 gene variant with hypertension. Hypertension. 2003;41(1):163–167. doi: 10.1161/01.HYP.0000043680.75107.CF. [DOI] [PubMed] [Google Scholar]
- 43.Asai T, et al. Endothelin-1 gene variant associates with blood pressure in obese Japanese subjects: the Ohasama Study. Hypertension. 2001;38(6):1321–1324. doi: 10.1161/hy1101.095333. [DOI] [PubMed] [Google Scholar]
- 44.Iglarz M, et al. Preproendothelin-1 gene polymorphism is related to a change in vascular reactivity in the human mammary artery in vitro. Hypertension. 2002;39(2):209–213. doi: 10.1161/hy0202.103442. [DOI] [PubMed] [Google Scholar]
- 45.Treiber FA, et al. Endothelin-1 gene Lys198Asn polymorphism and blood pressure reactivity. Hypertension. 2003;42(4):494–499. doi: 10.1161/01.HYP.0000091266.41333.15. [DOI] [PubMed] [Google Scholar]
- 46.Gregoski MJ, Barnes VA, Tingen MS, Dong Y, Zhu H, Treiber FA. Differential Impact of Stress Reduction Programs upon Ambulatory Blood Pressure among African American Adolescents: Influences of Endothelin-1 Gene and Chronic Stress Exposure. Int J Hypertens. 2012;2012:510291. doi: 10.1155/2012/510291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vadapalli S, Rani HS, Sastry B, Nallari P. Endothelin-1 and endothelial nitric oxide polymorphisms in idiopathic pulmonary arterial hypertension. Int J Mol Epidemiol Genet. 2010;1(3):208–213. [PMC free article] [PubMed] [Google Scholar]
- 48.Kaparianos A, Argyropoulou E, Efremidis G, Flordellis C, Spiropoulos K. Decline in FEV1 related to genetic polymorphisms (+138insA/delA and Lys198Asn) of the endothelin-1 gene in COPD. A pilot study. Eur Rev Med Pharmacol Sci. 2010;14(8):705–719. [PubMed] [Google Scholar]
- 49.Diefenbach K, et al. Endothelin-1 gene variant Lys198Asn and plasma endothelin level in obstructive sleep apnea. Cardiology. 2009;112(1):62–68. doi: 10.1159/000137702. [DOI] [PubMed] [Google Scholar]
- 50.Popov AF, et al. Impact of endothelin-1 Lys198Asn polymorphism on coronary artery disease and endorgan damage in hypertensives. Coron Artery Dis. 2008;19(7):429–434. doi: 10.1097/MCA.0b013e32830936e5. [DOI] [PubMed] [Google Scholar]
- 51.Castro MG, et al. Screening of the endothelin1 gene (EDN1) in a cohort of patients with essential left ventricular hypertrophy. Ann Hum Genet. 2007;71(Pt 5):601–610. doi: 10.1111/j.1469-1809.2007.00351.x. [DOI] [PubMed] [Google Scholar]
- 52.Lee J, Cheong SS, Kim J. Association of endothelin-1 gene polymorphisms with variant angina in Korean patients. Clin Chem Lab Med. 2008;46(11):1575–1580. doi: 10.1515/CCLM.2008.313. [DOI] [PubMed] [Google Scholar]
- 53.Uchida Y, Sugiura S, Nakashima T, Ando F, Shimokata H. Endothelin-1 gene polymorphism and hearing impairment in elderly Japanese. Laryngoscope. 2009;119(5):938–943. doi: 10.1002/lary.20181. [DOI] [PubMed] [Google Scholar]
- 54.Dong Y, Wang X, Zhu H, Treiber FA, Snieder H. Endothelin-1 gene and progression of blood pressure and left ventricular mass: longitudinal findings in youth. Hypertension. 2004;44(6):884–890. doi: 10.1161/01.HYP.0000147824.08621.a6. [DOI] [PubMed] [Google Scholar]
- 55.Tobe SW, et al. The impact of endothelin-1 genetic analysis and job strain on ambulatory blood pressure. J Psychosom Res. 2011;71(2):97–101. doi: 10.1016/j.jpsychores.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 56.Rabineau KM, Treiber FA, Poole J, Ludwig D. Interactive effects of anger expression and ET-1 Lys198Asn polymorphism on vasoconstriction reactivity to behavioral stress. Ann Behav Med. 2005;30(1):85–89. doi: 10.1207/s15324796abm3001_10. [DOI] [PubMed] [Google Scholar]
- 57.Gregoski MJ, et al. Interactive influences of ethnicity, endothelin-1 gene, and everyday discrimination upon nocturnal ambulatory blood pressure. Ann Behav Med. 2013;45(3):377–386. doi: 10.1007/s12160-013-9472-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rankinen T, et al. Effect of endothelin 1 genotype on blood pressure is dependent on physical activity or fitness levels. Hypertension. 2007;50(6):1120–1125. doi: 10.1161/HYPERTENSIONAHA.107.093609. [DOI] [PubMed] [Google Scholar]
- 59.Underwood PC, et al. Nonmodulation as the mechanism for salt sensitivity of blood pressure in individuals with hypertension and type 2 diabetes mellitus. J Clin Endocrinol Metab. 2012;97(10):3775–3782. doi: 10.1210/jc.2012-2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brown JM, et al. Aldosterone dysregulation with aging predicts renal vascular function and cardiovascular risk. Hypertension. 2014;63(6):1205–1211. doi: 10.1161/HYPERTENSIONAHA.114.03231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chamarthi B, Williams JS, Williams GH. A mechanism for salt-sensitive hypertension: abnormal dietary sodium-mediated vascular response to angiotensin-II. J Hypertens. 2010;28(5):1020–1026. doi: 10.1097/HJH.0b013e3283375974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Panoulas VF, et al. Polymorphisms of the endothelin-1 gene associate with hypertension in patients with rheumatoid arthritis. Endothelium. 2008;15(4):203–212. doi: 10.1080/10623320802228708. [DOI] [PubMed] [Google Scholar]
- 63.Braley LM, Menachery AI, Brown EM, Williams GH. Comparative effect of angiotensin II, potassium, adrenocorticotropin, and cyclic adenosine 3’,5’-monophosphate on cytosolic calcium in rat adrenal cells. Endocrinology. 1986;119(3):1010–1019. doi: 10.1210/endo-119-3-1010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.