

Abstract
ER stress has emerged as a signaling platform underlying the pathogenesis of various kidney diseases. Thus, there is an urgent need to develop ER stress biomarkers in the incipient stages of ER stress–mediated kidney disease, when a kidney biopsy is not yet clinically indicated, for early therapeutic intervention. Cysteine-rich with EGF-like domains 2 (CRELD2) is a newly identified protein that is induced and secreted under ER stress. For the first time to our knowledge, we demonstrate that CRELD2 can serve as a sensitive urinary biomarker for detecting ER stress in podocytes or renal tubular cells in murine models of podocyte ER stress–induced nephrotic syndrome and tunicamycin- or ischemia-reperfusion–induced acute kidney injury (AKI), respectively. Most importantly, urinary CRELD2 elevation occurs in patients with autosomal dominant tubulointerstitial kidney disease caused by UMOD mutations, a prototypical tubular ER stress disease. In addition, in pediatric patients undergoing cardiac surgery, detectable urine levels of CRELD2 within postoperative 6 hours strongly associate with severe AKI after surgery. In conclusion, our study has identified CRELD2 as a potentially novel urinary ER stress biomarker with potential utility in early diagnosis, risk stratification, treatment response monitoring, and directing of ER-targeted therapies in selected patient subgroups in the emerging era of precision nephrology.
Keywords: Nephrology
Keywords: Cell stress
Cysteine-rich with EGF-like domains 2 (CRELD2) is a mechanistic urinary biomarker for endoplasmic reticulum stress-mediated kidney diseases.
Introduction
Mounting evidence has established the importance of endoplasmic reticulum (ER) stress in the pathogenesis of various glomerular and tubular diseases (1). The ER is the central site for folding, posttranslational modifications and transport of secretory and membrane proteins. A mismatch between the load of unfolded/misfolded proteins and the folding capacity of the ER leads to ER stress (2). The ER responds to stress by activating intracellular signaling pathways, collectively known as the unfolded protein response (UPR), which aligns cellular physiology to the demands imposed by ER stress (2). Dysregulation of the UPR under persistent or intense ER stress eventually results in death of kidney cells.
Nephrotic syndrome (NS), characterized by heavy proteinuria and hypoalbuminemia, is accompanied by an increased risk for infection, venous thromboembolism, and progression to end-stage renal disease (ESRD). Nearly 100% of patients with congenital onset and 44% with infantile onset of NS have podocyte gene mutations, and the overall mutation detection rate is as high as 52% in steroid-resistant pediatric NS patients (3). The leading mutated podocyte genes include laminin β2 (LAMB2) causing congenital NS and Pierson syndrome and collagen IV α chain (COL4A) underlying Alport syndrome (AS) and focal segmental glomerulosclerosis (FSGS) (4). Emerging evidence has demonstrated that podocyte ER stress may be an important functional link from genetic mutations to disease phenotype in primary NS. Recently, we have shown that a C321R missense mutation in LAMB2, which encodes an important constituent of the mature glomerular basement membrane (GBM), induces podocyte ER stress and activates the proapoptotic C/EBP homologous protein (CHOP) pathway in the development of proteinuria (5). In addition, the G1334E mutation in COL4A3 triggers podocyte ER stress and leads to podocytopathy and AS (6). Multiple studies have also linked podocyte ER stress to the pathogenesis of diverse sporadic nephropathies including FSGS, membranous nephropathy, minimal change disease, and diabetic nephropathy (DN) in both experimental models (7–9) and human kidney biopsies (10, 11).
Accumulating evidence has also highlighted a pathogenic role for ER dysfunction and the derangement of ER proteostasis in the onset and progression of renal tubulointerstitial disease, which ultimately advances to ESRD. Autosomal dominant tubulointerstitial kidney disease (ADTKD), due to mutations in the UMOD gene encoding uromodulin, is characterized by gout, alterations in urinary concentration, and progressive loss of kidney function (12, 13). Proteinuria is typically mild or absent. Uromodulin (Tamm-Horsfall protein) is exclusively expressed in the thick ascending limb (TAL) of Henle’s loop. Currently, more than 120 UMOD mutations have been identified (http://www.ukdcure.org/), and most of them are missense mutations (13). In vitro and in vivo studies have shown that UMOD mutations can cause protein misfolding, ER retention, and ER stress activation in TAL cells (14–18). Additionally, tubular ER stress induced by ischemia, albuminuria, and nephrotoxins contributes to a variety of acquired forms of tubular injury (19, 20).
Although multiple lines of evidence from clinical and experimental studies have demonstrated that a maladaptive ER response is mechanistically linked to the pathogenesis of glomerular and tubular disorders, the absence of biomarkers for monitoring podocyte or tubular cell ER stress has hampered early detection and effective therapeutic intervention to restore ER homeostasis. We have previously identified mesencephalic astrocyte-derived neurotrophic factor (MANF) as a urinary ER stress biomarker in mouse models (21). Here, we investigated if another secreted, ER stress–inducible protein, cysteine-rich with EGF-like domains 2 (CRELD2), is also a candidate urinary ER stress biomarker in both mouse models and human patients.
CRELD2 was first identified as a novel ER stress–inducible gene through RNA analysis of Neuro2a mouse neuroblastoma cells treated with thapsigargin (TG) (22). It is a ~50 kDa secretory glycoprotein that predominantly localizes to the ER and Golgi apparatus (22, 23). Its promoter region, which is well conserved among various species, contains a typical ER stress response element (ERSE; CGTGG-N9-ATTGG) that is positively regulated by the ER stress master regulator ATF6 (22). By overexpressing WT and various mutant CRELD2 constructs in HEK293 or COS7 cells, it has been reported that 4 C-terminal amino acids (REDL) play a crucial role in CRELD2 secretion and that Ig binding protein (BiP) and MANF significantly enhance its secretion (23, 24). However, very few studies have been carried out to characterize the intrinsic induction and secretion of CRELD2 in vitro and in vivo. The only published in vivo study has shown that CRELD2 is upregulated and secreted following ER retention of mutant cartilage extracellular matrix proteins in mouse knock-in models of chondrodysplasia resulting from mutations in matrilin-3 (Matn3) or type X collagen (Col10a1) (25). The function of CRELD2 under pathophysiological conditions remains poorly understood. The aim of this study was to determine whether CRELD2 can serve as a sensitive urinary ER stress biomarker for ER stress–mediated kidney diseases by studying a combination of in vitro cell and in vivo mouse models, as well as human patient samples.
Results
CRELD2 is a urinary biomarker for detecting podocyte ER stress in the early stages of podocyte ER stress–induced NS in mice.
In our initial experiments, we determined whether ER stress increases CRELD2 expression and secretion in mouse primary podocytes. Mouse podocytes were treated for 24 hours with tunicamycin (TM), which activates ER stress by blocking N-linked glycosylation in the ER (26), or TG, which inhibits ER calcium uptake and, thus, depletes ER calcium through inhibition of ER calcium ATPase (27). Consistent with prior results obtained from Neuro2a cells (22), both ER stressors induced expression of CRELD2, which was barely detectable in vehicle-treated (DMSO-treated) cells (Figure 1A). As expected, we also noted that TM treatment resulted in expression of an underglycosylated form of CRELD2. Immunofluorescence (IF) of mouse primary podocytes also showed that CRELD2 was upregulated by both ER stressors and that the upregulated CRELD2 in TM- or TG-treated podocytes overlapped exclusively with BiP, a well-known ER stress marker (Figure 1B). We also examined the effects of ER stress on CRELD2 secretion. Western blot (WB) showed that both ER stressors increased CRELD2 secretion into the culture medium by mouse podocytes (Figure 1C), whereas in the absence of ER stress, there was little CRELD2 secretion by mouse podocytes (Figure 1C). These data verify that the upregulation and secretion of CRELD2 induced by ER stress is not a cell-type–specific response.
Figure 1. CRELD2 is a urinary biomarker for detecting podocyte ER stress in the early stages of NS.

Primary podocytes (P1 or P2) were isolated and cultured from WT mice at P27 (A–C). (A) Mouse primary podocytes were treated with DMSO (control), TM (2 μg/ml), or TG (1 μM) for 24 h. Cell lysates were analyzed by WB with the indicated antibodies. (B) Control and TM- or TG-treated mouse podocytes, as described in A, were stained for CRELD2 (red) and BiP (green). Nuclei were counterstained with Hoechst 33342 (blue). Scale bar: 40 μm. (C) Immunoblot analysis for CRELD2 secretion into the media by cultured primary podocytes described in A (20 μl medium each group). (D) Quantitative PCR analysis of CRELD2 mRNA in primary podocytes from Lamb2+/–, Lamb2–/–;Tg-C321R, and Lamb2–/–;Tg-WT mice at P27. CRELD2 expression was normalized as a ratio to mouse podocyte–specific WT-1 mRNA, and the average CRELD2/WT-1 mRNA ratio in Lamb2+/– mice was set as 1 (n = 5 mice per genotype; mean ± SD). *P < 0.05 by 1-way ANOVA. (E) Primary podocyte cell lysates from age-matched Lamb2+/–, Lamb2–/–;Tg-C321R, and Lamb2–/–;Tg-WT mice at P27 were analyzed by WB with the indicated antibodies. (F) Immunoblot analysis for CRELD2 secretion into the media by primary podocytes (20 μl each group) from the indicated genotypes at P27. (G) Urinary CRELD2 excretion was assessed by WB from Lamb2–/–; Tg-C321R mice and their Lamb2+/– littermates and age-matched Lamb2–/–;Tg-WT mice (n = 8, n = 8, and n = 3, respectively). Shown is 1 representative experiment at the indicated ages. (H) Immunoblot analysis of CRELD2 in crude urine specimens from 1 pair of Lamb2+/– and Lamb2–/–;Tg-C321R littermates following the disease course. Shown is 1 representative experiment. Three more independent experiments were performed with similar results. (I) CRELD2 excretion in unprocessed urine specimens was detected by WB from Lamb2–/–;Tg-C321R mice and their Lamb2+/– littermates and Lamb2–/–;Tg-WT mice at 8–16 weeks. The urinary CRELD2 excretion was normalized to urine Cr excretion such that the urine volume applied to the gel reflected the amount of urine containing 1 μg of Cr for G–I. All WB images are representative of at least 3 independent experiments. CRELD2, cysteine-rich with EGF-like domains 2; TM, tunicamycin; TG, thapsigargin; BiP, Ig binding protein; Tg-C321R, Lamb2–/– mice expressing C321R-LAMB2 in podocytes; Tg-WT, Lamb2–/– mice expressing WT LAMB2 in podocytes.
We next investigated whether CRELD2 could serve as a urinary biomarker to detect podocyte ER stress by utilizing a podocyte ER stress–induced NS model that we have developed (5). We have previously shown that Lamb2–/– mice expressing C321R-LAMB2 in podocytes via the podocyte-specific mouse nephrin promoter (Lamb2–/–;Tg-C321R mice, hereafter referred to as Tg-C321R mice) resemble features of human NS patients carrying the C321R-LAMB2 mutation (5). We have also shown that, in expression-matched Lamb2–/–;Tg-WT mice (hereafter referred to as Tg-WT mice), the expression of WT LAMB2 cDNA in podocytes is sufficient to restore the integrity of the glomerular filtration barrier (GFB) in Lamb2–/– mice (28). Tg-C321R mice begin to develop trace proteinuria by 4 weeks of age before any histological changes, then exhibit mild glomerulosclerosis, diffuse foot process effacement, GBM thickening, and overt proteinuria at approximately 6–8 weeks; they finally die after 10–12 weeks (5). At P24–P28, when Tg-C321R mutants exhibit trace proteinuria without notable renal histological alterations, podocyte ER stress induced by the C321R misfolded protein is evident as demonstrated by the upregulation of BiP and CHOP in Tg-C321R podocytes compared with WT and Tg-WT podocytes (5, 21). In the current study, we expanded upon these findings by determining whether CRELD2 was also induced and secreted by ER-stressed podocytes at the early stage of proteinuria. Primary podocytes were isolated and cultured from Lamb2+/–, Tg-C321R, and Tg-WT mice at P27. Real-time PCR demonstrated that CRELD2 transcript levels were significantly increased in Tg-C321R podocytes compared with the WT and Tg-WT podocytes (Figure 1D). WB also revealed CRELD2 upregulation in Tg-C321R podocytes versus the WT and Tg-WT podocytes (Figure 1E). More importantly, as shown in Figure 1F, secretion of CRELD2 by Tg-C321R podocytes was markedly higher as compared with WT and Tg-WT podocytes. Next, we determined whether CRELD2 secreted from ER-stressed podocytes was detectable in urine. Using urinary creatinine (Cr) concentrations to normalize for sample loading, WB showed that CRELD2 was easily detected in as little as 3 μl of unprocessed urine specimens from C321R mutants in the incipient stage of NS but not from WT or Tg-WT mice (Figure 1G), analogous to the secretion of CRELD2 by cultured podocytes under ER stress (Figure 1F). Furthermore, urinary CRELD2 excretion increased in C321R mutants during disease progression from P27 to 6–12 weeks of age (Figure 1H). In contrast, CRELD2 excretion was not detected in the urine of WT or Tg-WT mice at any time point up to 16 weeks of age examined (Figure 1I). We noted that CRELD2 in mouse urine had faster electrophoretic motility (~37 kDa), which is most likely due to the effect of high concentrations of urinary sodium ions and urea, a well-described phenomenon. Taken together, these data show that CRELD2 cellular secretion and urinary excretion coincide with podocyte ER stress during the development of proteinuria and can be detected at the early stage of the disease.
CRELD2 is an early urinary biomarker for monitoring tubular ER stress in the TM-induced acute kidney injury (AKI) mouse model.
Injection of TM i.p. in mice is a well characterized AKI model caused by renal proximal tubular ER stress (29). We and others have shown that it manifests as extensive tubular dilation, epithelial flattening, tubular basement membrane denudation, and loss of brush borders in proximal tubules, which peaks between 4 and 5 days after injection (21, 29). In comparison, glomeruli and other organs are spared (21, 29). We have also shown that, despite normal renal function and the absence of conspicuous histologic changes on day 1 after TM injection, multiple ER stress response genes including BiP, CHOP, MANF and ER degradation-enhancing α-mannosidase-like protein (EDEM) are upregulated (21). We further investigated whether CRELD2 was induced in ER-stressed renal tubules. Indeed, CRELD2 transcript levels in whole-kidney lysates were significantly increased on day 1 after TM injection versus DMSO vehicle–injected controls or day 5 after injection (Figure 2A). Co-IF staining of CRELD2 and the proximal tubular marker Lotus tetragonolobus lectin (LTL) also showed that CRELD2 immunostaining in proximal tubules was substantially upregulated on day 1 following TM injection compared with controls and subsided by 5 days after injection. Conversely, glomeruli in TM-injected kidneys did not exhibit CRELD2 induction (Figure 2B). WB analysis confirmed that CRELD2 protein expression in kidneys was higher on day 1 compared with controls or day 5 after injection (Figure 2, C and D). Next, we sought to determine whether CRELD2 can be detected in urine on day 1 after treatment with TM. We have previously shown that this time point precedes renal histopathologic changes and upregulation of the early injury marker kidney injury molecule-1 (KIM-1) (21). Indeed, CRELD2 excretion was dramatically increased in urine specimens from TM-injected mice within 24 hours but not from vehicle-injected mice (Figure 2E). Collectively, these data demonstrate that tubular cell ER stress increases urinary CRELD2 excretion prior to any evidence of the subsequent decline in kidney function or histologic changes due to ER stress–induced AKI, thus establishing the excellent early diagnostic capacity of CRELD2 as a biomarker.
Figure 2. Urinary CRELD2 excretion coincides with tubular ER stress in the TM-induced AKI mouse model.

(A) Quantitative PCR analysis of relative transcript levels of CRELD2 in kidneys at the indicated time points following TM injection (1 mg/kg, i.p.) as compared with DMSO vehicle–injected controls. Absolute levels were normalized first to those of GAPDH and then to the levels in the control kidneys (mean ± SEM). ***P < 0.001 by 1-way ANOVA (n = 3 mice per group). (B) Dual IF staining for CRELD2 (green) and LTL (red) on paraformaldehyde-fixed, sucrose-cryoprotected kidney sections from mice treated with vehicle or TM (1 mg/kg) at the indicated time points. Nuclei were counterstained with Hoechst 33342 (blue). G, glomerulus. Scale bars: 40 μm. (C) Whole-kidney lysates from mice treated with or without TM (1 mg/kg) at different time points were analyzed by WB for levels of CRELD2 and β-actin. (D) Quantification of CRELD2 normalized to β-actin in kidney lysates in 5 independent experiments. The average CRELD2/β-actin ratio in vehicle-treated mice was set as 1 (mean ± SEM). *P < 0.05 and **P < 0.01 by 1-way ANOVA. (E) Detection of urinary CRELD2 within 1 day of TM injection. Urine was collected for 24 hours after the mice were injected with vehicle or 1 mg/kg TM (i.p.). Crude urine samples from mice treated with vehicle (n = 8) or TM (n = 8) were analyzed by WB for CRELD2 excretion. The urinary CRELD2 excretion was normalized to 2 μg of urine Cr excretion. TM, tunicamycin; CRELD2, cysteine-rich with EGF-like domains 2; LTL, Lotus tetragonolobus lectin.
CRELD2 is an early mechanistic urinary biomarker for ER-stressed tubular cells in the ischemia-reperfusion–induced AKI mouse model.
We further assessed whether urinary CRELD2 excretion can be utilized to monitor tubular ER stress in another form of tubular cell injury caused by ischemia-reperfusion (I/R). I/R renal injury following hypotension due to various clinical problems and kidney transplantation is a major pathogenic mechanism of AKI. However, the current treatment for AKI is mainly supportive due to a lack of mechanism-based therapies. Emerging evidence has shown that tubular ER stress plays an important role in linking ischemic insult to tubular injury (30, 31). In 9-week-old male C57BL/6J mice subjected to 30 minutes of bilateral kidney ischemia, as illustrated by H&E and periodic acid-Schiff (PAS) staining, early stage of tubular damage manifesting as loss of brush boarders and partial cytoplasmic degeneration was observed in scattered areas of postischemic kidneys as early as 3 hours after reperfusion (arrows, Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.92896DS1). Acute tubular necrosis developed predominantly in the corticomedullary junction area by 9 hours, and more diffuse tubular necrosis occurred by 24 hours after ischemic injury (arrows, Supplemental Figure 1A). On day 3 after reperfusion, tubular regeneration (arrowhead) with much less tubular necrosis (arrows) was detected (Supplemental Figure 1A). In the 15-day reperfusion kidneys, tubular injury had completely resolved (Supplemental Figure 1A). Blood urea nitrogen (BUN) was elevated at 3 hours, peaked at 24 hours, and returned to normal renal function by the 15-day recovery period in the I/R-injured mice compared with sham-operated mice (Supplemental Figure 1B), which was reflective of the histopathologic changes observed (Supplemental Figure 1A). Compared with kidneys undergoing sham operation, postischemic kidneys exhibited significant upregulation of CRELD2 mRNA and protein from 9 hours to 3 days (Figure 3, A–C). Consistent with these results, dual staining of sham-operated and postischemic kidney sections for CRELD2 and LTL demonstrated that CRELD2 induction occurred in sparse proximal tubules mildly by 3 hours, further increased by 9 hours, peaked by 24 hours, declined by 3 days, and resolved by 15 days after the ischemic injury (arrows, Figure 3D). In contrast, the glomeruli were devoid of CRELD2 expression (Supplemental Figure 2). Remarkably, mice with 30 minutes of bilateral ischemia manifested urinary CRELD2 excretion within 3 hours of reperfusion (the first urine collection performed after ischemia) (Figure 3, E and F). The appearance of CRELD2 in the urine persisted for at least 24 hours after ischemic AKI and decreased to normal levels by 3 days of reflow (Figure 3, E and F). In contrast, sham-operated mice did not exhibit CRELD2 urinary excretion (Figure 3, E and F). Meanwhile, we also compared urine CRELD2 with previously established AKI biomarkers, such as KIM-1 and neutrophil gelatinase–associated lipocalin (NGAL). A significant elevation of both KIM-1 and NGAL was noted in kidneys at 9 hours and in the urine at 6 hours after ischemic injury compared with sham-operated mice (Figure 3, B, E and F), indicating that CRELD2 excretion in the urine slightly precedes KIM-1 and NGAL excretion following ischemic AKI. It was also noted that urinary NGAL excretion appeared to persist longer than CRELD2 and KIM-1 excretion (Figure 3, E and F). Together, these results clearly indicate that urinary CRELD2 excretion can serve as a mechanistic biomarker for ER-stressed tubular cells in the early phase of I/R-induced AKI. In addition, CRELD2 excretion in the urine can be utilized to monitor disease progression and recovery in ischemic AKI.
Figure 3. CRELD2 is induced and excreted in the urine in a mouse model of AKI caused by 30 minutes of bilateral kidney ischemia.

(A) Quantitative PCR showed relative transcript levels of CRELD2 in kidneys from sham-operated (n = 4) or I/R-injured mice at 3, 9, and 24 hours, as well as 3 and 15 days, of reflow (n = 3 at each time point). Absolute levels were normalized first to those of GAPDH and then to the levels in the sham-operated kidneys (mean ± SEM). *P < 0.05, **P < 0.01, and ***P < 0.001 by 1-way ANOVA. (B) Representative WB results from sham-operated and postischemic kidneys for levels of CRELD2, KIM-1, NGAL, and β-actin at the indicated time points of reperfusion. (C) Quantification of CRELD2, KIM-1, or NGAL normalized to β-actin in kidney lysates. Absolute levels of CRELD2, KIM-1, or NGAL were normalized first to those of β-actin and then to the levels in the kidneys subjected to sham treatment, at 9 hours after reperfusion, and at 3 hours after reperfusion, respectively (n = 4 mice per time point; mean ± SD). *P < 0.05 and ***P < 0.001 relative to sham-operated kidneys by 1-way ANOVA. (D) Dual IF staining for CRELD2 (green) and LTL (red) on histochoice-fixed paraffin kidney sections from mice with sham operation or following I/R injury at various reperfusion periods as indicated. Nuclei were counterstained with Hoechst 33342 (blue). G, glomerulus. Arrows indicate CRELD2 induction in proximal tubules after ischemic injury. Scale bar: 40 μm. (E) Representative WB analysis of CRELD2, KIM-1, and NGAL in crude urine specimens collected from sham-operated (n = 9) and I/R-injured mice within varying time points after ischemic AKI: 3 hours (n = 5), 6 and 24 hours (n = 10 at each time point), and 3 and 15 days (n = 5 at each time point). The urinary excretion of CRELD2, KIM-1, or NGAL was normalized to 0.5 μg of urine Cr excretion. (F) Quantification of urinary CRELD2, KIM-1 or NGAL excretion. The excretion of individual urinary protein biomarkers in mice within 3 hours of reperfusion was set as 1. *P < 0.05, **P < 0.01, and ***P < 0.001 relative to sham-operated mice by 1-way ANOVA. CRELD2, cysteine-rich with EGF-like domains 2; KIM-1, kidney injury molecule-1; NGAL, neutrophil gelatinase-associated lipocalin; LTL, Lotus tetragonolobus lectin.
CRELD2 is easily detected in the urine after subclinical renal ischemia and urinary CRELD2 levels correlate with the severity of renal ischemia.
To determine the sensitivity of urinary CRELD2 detection in the absence of overt AKI, 9-week-old male C57BL/6J mice underwent 10 minutes of bilateral renal pedicle clamping. Whereas mice subjected to 30 minutes of bilateral ischemia exhibited characteristic histopathologic features of AKI (arrows, Figure 4A and Supplemental Figure 1A), at 24 hours of reflow, tubular injury was absent from mice with mild renal ischemia (Figure 4A). Corresponding to histological changes, mice with bilateral ischemia for 10 minutes displayed BUN levels comparable with control mice, whose levels were significantly lower than the levels in mice with bilateral ischemia for 30 minutes (Figure 4B). Strikingly, CRELD2 was readily detected in unprocessed urine in mice with subclinical renal ischemia within 3 hours of the ischemic insult (in the very first urine collection after ischemia) and persisted for at least 24 hours (Figure 4C). Moreover, mice with 30 minutes of renal ischemia excreted significantly higher levels of CRELD2 as compared with mice with mild ischemia (Figure 4C). In contrast, no correlation between urinary KIM-1 excretion and the duration of renal ischemia at any of the reperfusion periods examined was noted (Figure 4D). NGAL levels in the urine correlated with the severity of renal ischemia at 6 and 24 hours but not at 3 hours of reflow (Figure 4E). These results demonstrate that CRELD2 is a sensitive urinary ER stress biomarker and that its excretion levels correlate with the duration of renal ischemia.
Figure 4. CRELD2 levels in the urine correlate with the duration of renal ischemia.

Mice were exposed to sham treatment or either 10 or 30 minutes of bilateral ischemia. (A) After 24 hours of reperfusion, paraffin sections of control and I/R-injured kidneys were stained with H&E and PAS. Note acute tubular necrosis (arrows) at 24 hours of reflow following 30 minutes of kidney ischemia. Scale bars: 40 μm. (B) BUN levels for the indicated groups: sham-operated mice (n = 10), mice with 10 minutes of bilateral ischemia followed by 24 hours of reperfusion (n = 5), and mice with 30 minutes of bilateral ischemia followed by 24 hours of reperfusion (n = 7). Data represent mean ± SEM, **P < 0.01 by 1-way ANOVA. (C–E) Representative WBs of CRELD2 (C), KIM-1 (D) and NGAL (E) in unprocessed urine samples obtained at reperfusion periods (3, 6 and 24 hours) as shown, after 10 or 30 minutes of bilateral renal vascular pedicles clamping. The urinary excretion of CRELD2 (C), KIM-1 (D), or NGAL (E) was normalized to 1 μg of urine Cr excretion and quantified. I, Ischemia. The average urinary biomarker excretion in mice subjected to bilateral renal ischemia for 10 minutes was set as 1. *P < 0.05, **P < 0.01, and ***P < 0.001 by 2-way ANOVA. PAS, periodic acid-Schiff; BUN, blood urea nitrogen; CRELD2, cysteine-rich with EGF-like domains 2; KIM-1, kidney injury molecule-1; NGAL, neutrophil gelatinase-associated lipocalin.
Urinary CRELD2 elevation within postoperative 6 hours is significantly associated with higher risk of severe AKI and other adverse clinical outcomes after pediatric cardiac surgery.
After we identified CRELD2 as a potentially novel urinary ER stress biomarker in mouse models, our exciting results further prompted us to investigate the utility of CRELD2 in human kidney diseases. I/R-induced AKI, which mechanistically mimics the mouse I/R model, is a common complication of pediatric cardiac surgery and negatively effects short- and long-term outcomes (32). The failure of prior interventional trials to mitigate AKI in cardiac surgery patients has been attributed, in part, to the delays in the diagnosis of AKI. Thus, it presents an urgent need to develop sensitive mechanistic biomarkers for early therapeutic intervention. By utilizing urine samples from NIH-funded Translational Research Investigating Biomarker Endpoints in Acute Kidney Injury (TRIBE-AKI) Consortium (32, 33), we conducted a pilot case-control study of 25 pediatric patients with normal baseline renal function who had received congenital cardiac surgery to evaluate whether early postoperative measure of urine CRELD2 is associated with higher risk of severe AKI postoperatively. After the cardiopulmonary bypass (CPB) surgery, 10 patients developed severe AKI, as defined by either receipt of acute dialysis or postoperative doubling of serum Cr during hospital stay, and 15 patients did not develop AKI, as defined by the percentage change in peak serum Cr < 50% during hospital stay. Two patients who developed severe AKI were excluded due to urine sample unavailable at postoperative 0- to 6-hour time points and missing urine Cr data. Thus, 23 patients were included in the analysis. Demographic and clinical characteristics of enrolled patients were included in Table 1.
Table 1. TRIBE-AKI cohort description.

Preoperatively, urinary CRELD2 excretion was absent in 21 participants (Figure 5, A and B). Preoperative mild elevation of urine CRELD2 was noted in 1 non-AKI and 1 severe-AKI patient. Postoperatively, in patients with severe AKI, urine CRELD2 concentration peaked at the first collection (postoperative 0- to 6-hour time points) and declined over the first 3 postoperative days when crude urine specimen normalized to 2 μg of urine Cr excretion were analyzed by WB (arrow, Figure 5A). In marked contrast, in 12 of 15 patients without AKI, urinary CRELD2 levels were undetectable at the first collection (Figure 5, A and B). Although WB is very sensitive, it is semiquantitative and can be cumbersome for a clinical diagnostic lab. An ELISA assay for measuring CRELD2 excretion in human urine samples was performed to achieve absolute quantification and higher throughput. As shown in Figure 5B, the first postoperative urine CRELD2 concentration normalized to urine Cr excretion (urine CRELD2/Cr) was significantly higher in the 8 patients with severe AKI than that in the 15 non-AKI patients (P = 0.002), whereas no difference in serum Cr concentrations on day 1 was observed between the 2 groups (Figure 5C). Serum Cr in severe AKI patients peaked on day 2 after surgery (Figure 5C). Moreover, detectable urine CRELD2 levels at the first urine collection were strongly associated with severe AKI (66% of patients with detectable urine CRELD2 levels versus 14% of patients with undetectable levels within the first postoperative 6 hours developed severe AKI, P = 0.01). Meanwhile, ELISA assays of NGAL, IL-18, and KIM-1 in the urine collected during the postoperative 0–6 hours showed significant elevation of urine NGAL (P = 0.011) and IL-18 (P = 0.013), but not elevation of KIM-1 (P = 0.628), in severe AKI patients versus non-AKI patients (Figure 5D). The average length of hospital stay and ventilation for patients with detectable urinary CRELD2 levels was 15.33 days (±SD, 21.45) and 2.6 days (±SD, 1.6), whereas those for patients with undetectable urinary CRELD2 levels were 4.5 days (±SD, 2.9) and 1.1 days (±SD, 1.1), respectively (P = 0.017 and 0.03, respectively, Figure 5E). Moreover, the postoperative 0- to 6-hour urine CRELD2/Cr values were moderately associated with longer hospital stay and duration of mechanical ventilation (Spearman correlation coefficient 0.55 and 0.54, respectively, P < 0.05) but not with length of intensive care unit (ICU) stay. These data indicate that early postoperative urinary CRELD2 elevation is significantly associated with severe AKI and other adverse outcomes following pediatric cardiac surgery.
Figure 5. Early postoperative measure of urine CRELD2 can stratify pediatric patients for developing severe AKI and other adverse outcomes after cardiopulmonary bypass surgery.

AKI in B–E denotes severe AKI. (A) Representative preoperative (pre-op) and postoperative (post-op) WB analysis of CRELD2 (arrow) in crude urine specimens obtained from non-AKI and severe AKI patients at the indicated time points. Time points 0–6 hours, 6–12 hours, 12–18 hours, day 2, and day 3 are the sample collection times after the CPB surgery. The urinary excretion of CRELD2 was normalized to 2 μg of urine Cr excretion. Three more independent experiments were performed with similar results. (B) ELISA assay of urine CRELD2 concentrations normalized to urine Cr excretion in unconcentrated urine specimen before surgery and at the first postoperative collection (0–6 hours) in severe AKI (n = 8) and non-AKI (n = 15) patients. **P < 0.01 by Wilcoxon rank-sum test. (C) Pre- and post-op serum Cr measurements in severe AKI (n = 8) and non-AKI (n = 15) patients, by Wilcoxon rank-sum test. (D) ELISA assays of urine NGAL, IL-18, and KIM-1 concentrations before surgery and 0–6 hours after surgery in severe AKI (n = 8) and non-AKI (n = 15) patients. *P < 0.05 by Wilcoxon rank-sum test. (E) Length of stay in hospital, ventilation, and stay in ICU in patients with undetectable and detectable urine levels of CRELD2 within postoperative 6 hours, respectively. *P < 0.05 by Wilcoxon rank-sum test. AKI, acute kidney injury; CRELD2, cysteine-rich with EGF-like domains 2; KIM-1, kidney injury molecule-1; NGAL, neutrophil gelatinase-associated lipocalin; ICU, intensive care unit.
Urinary CRELD2 levels are markedly increased in patients with ADTKD caused by mutations in UMOD.
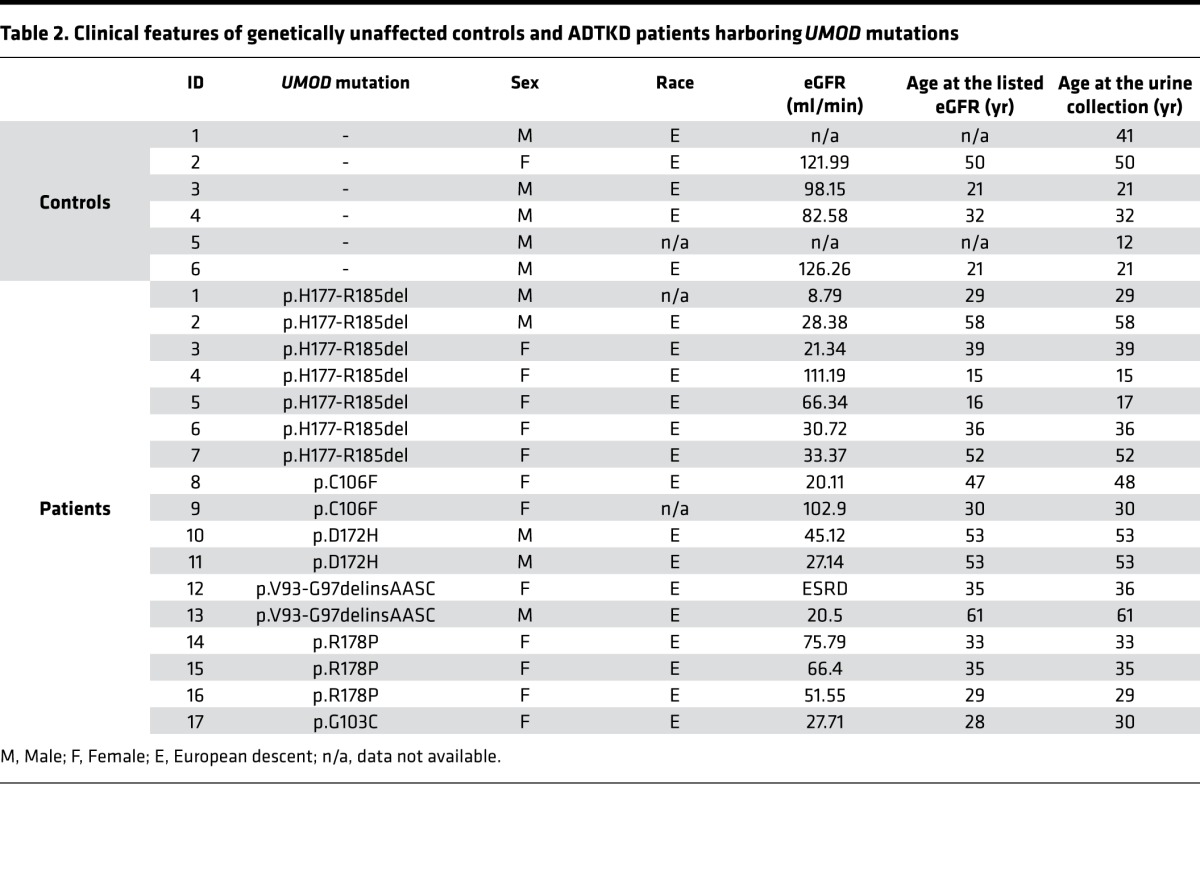
We also validated CRELD2 as a urinary biomarker for detecting tubular ER stress in ADTKD caused by UMOD mutations, a prototypical tubular ER stress disease. Consistent with prior studies of many mutations causing ADTKD-UMOD, trichrome staining of kidney biopsies from patients carrying the UMOD mutation H177-R185 del or W202S revealed significant interstitial fibrosis (Figure 6A). In addition, PAS staining showed tubular dilation, tubular atrophy with tubular basement membrane thickening (arrowheads, Figure 6A), or interstitial inflammatory monocyte infiltration (arrow, Figure 6A). Co-IF staining of CRELD2 and uromodulin in human kidney biopsies were employed to assess protein expression and localization in renal epithelial cells lining the TAL. In agreement with previous studies with other UMOD mutations (14, 15, 17), native uromodulin was enriched at the apical membrane of TAL cells (arrows, Figure 6B). In sharp contrast, mutant H177-R185 del uromodulin exhibited diffuse expression in the cytoplasm of TAL tubules and mutant W202S uromodulin displayed a punctate perinuclear distribution within TAL cells (Figure 6B). Intracellular and intraluminal protein aggregates were also noted in kidney biopsies harboring both mutations, reminiscent of defective intracellular trafficking and ER retention of uromodulin mutants (Figure 6B). Moreover, expression of ER stress response protein CRELD2 was markedly enhanced and completely colocalized with mutant uromodulin in TAL cells (Figure 6B). We subsequently determined whether CRELD2 concentration was elevated in the urine of ADTKD-UMOD patients. CRELD2 was easily detected in 5–25 μl of unconcentrated urine normalized to 10 μg of urine Cr excretion from human ADTKD-UMOD patients (17 affected individuals from 13 families carrying 6 UMOD mutations), whereas urinary CRELD2 excretion was absent from all tested genetically unaffected controls (Figure 6, C and D, and Table 2). Of note, CRELD2 induction was observed in the urine of patients 4, 5, 9, 14, and 15 whose estimated glomerular filtration rates (eGFR) were 111.19, 66.34, 102.9, 75.79, and 66.4 ml/min, respectively (Figure 6, C and D, and Table 2). As expected, all urine samples from these ADTKD-UMOD patients showed trace to mild albuminuria (urinary albumin to Cr ratio [UACR] less than 300 μg/mg — the definition of macroalbuminuria) when urine volume containing 4 μg of Cr was loaded to a gel, and the density of the band was compared with that of 1.2 μg of BSA by Coomassie G-250 stain (Supplemental Figure 3), which excludes the possibility that tubular ER stress was due to heavy albuminuria. Urine CRELD2 concentration in control and ADTKD-UMOD patients was also measured by the ELISA assay (Figure 6E). Since patients with ADTKD-UMOD only affecting TAL segment excreted much less CRELD2 compared with patients with ischemic AKI, proteins in urine volume normalized to 450 g of urine Cr in control, and ADTKD patients were concentrated to ensure that urine CRELD2 concentrations in the individual urine samples were above the detection threshold of the ELISA assay. As illustrated by Figure 6E, urinary CRELD2 levels were significantly elevated in patients with ADTKD-UMOD compared with those in controls. These results demonstrate the superb ability of CRELD2 to discriminate between controls and ADTKD patients with tubular ER stress.
Figure 6. Detection of CRELD2 in the urine from human ADTKD-UMOD patients caused by UMOD mutations.

(A) Masson’s trichrome and PAS staining of paraffin kidney sections from ADTKD patients carrying UMOD mutation p.H177-R185 del or p.W202S, as well as from normal kidneys after nephrectomy. Scale bar: 40 μm. Arrowheads indicate tbular dilation, tubular atrophy with tubular basement membrane thickening. Arrow indicates interstitial inflammatory monocyte infiltration. (B) Representative IF images of human renal biopsies obtained from patients with p.H177-R185 del or p.W202S UMOD mutation and from normal kidneys, stained for CRELD2 (green) and uromodulin (red) with a nuclear counterstain (Hoechst 33342, blue). Scale bar: 40 μm. Arrows indicate enrichment of native uromodulin at the apical membrane of TAL cells. (C and D) Crude urine samples from human ADTKD-UMOD patients harboring H177-R185 del (C) or other disease-causing mutations including C106F, D172H, V93-G97delinsAASC, R178P, and G103C (D), as well as from genetically unaffected family members, were analyzed by WB for CRELD2 excretion. The urinary CRELD2 excretion was normalized to 10 μg of urine Cr excretion. The control and patient numbers listed in C–D corresponded to the same numbers in Table 2. (E) Dot plot representation of urine CRELD2/Cr values measured by the ELISA in the above 17 ADTKD-UMOD patients harboring various UMOD mutations and 7 genetically unaffected controls. Data represent mean ± SEM, *P < 0.05 by 2-tailed t test. PAS, periodic acid-Schiff; CRELD2, cysteine-rich with EGF-like domains 2; ADTKD, autosomal dominant tubulointerstitial kidney disease.
Table 2. Clinical features of genetically unaffected controls and ADTKD patients harboring UMOD mutations.
Discussion
This proof-of-concept study is the first to our knowledge to show that CRELD2 represents an early, sensitive, noninvasive, and mechanistic biomarker in the urine for ER stress–mediated kidney diseases. Although growing evidence has highlighted that aberrant ER protein proteostasis and ER dysfunction underpin the initiation and/or development of a variety of glomerular and tubular diseases, ER stress biomarkers that can be applied in human kidney disease patients are still lacking. Here, we demonstrate that CRELD2 was rapidly upregulated and secreted when renal cells were subjected to ER stress by utilizing mouse models of podocyte ER stress–induced NS and TM- or I/R-induced AKI. Moreover, CRELD2 was detectable in small aliquots of urine early in the disease course, often preceding clinical or histologic manifestation of the corresponding disease. Finally, we validated the clinical utility of CRELD2 as a urine ER stress biomarker in patients with kidney diseases associated with ER stress. The discriminative ability of urinary CRELD2 excretion to differentiate patients having ER stress from unaffected controls is outstanding.
The use of CRELD2 as an ER stress biomarker in the kidney has many promising applications. Serum Cr — the primary marker of kidney disease and kidney function — only increases after approximately 25% of kidney function has been lost. When elevations of serum Cr occur, irreversible changes such as scarring and fibrosis of the kidney are often present, and treatment is unlikely to restore full kidney function. In contrast, the best markers are those that are associated with the central underlying pathogenesis of the disorder. These biomarkers will be elevated early on in the disease, prior to loss of kidney function and elevations of serum Cr, and treatments that target the same pathogenesis of the disease will have an effect on these markers. Thus, these mechanistic markers will allow the initiation of treatments early on in the disease and will allow one to detect the effect of interventions early on, which will dramatically reduce the amount of follow-up time to determine treatment efficacy and make clinical trials much easier and less expensive to perform. In the current investigation, CRELD2 showed great promise as such a biomarker. Furthermore, the availability of an ELISA assay for measuring urinary excretion of CRELD2 (spot urinary CRELD2/Cr ratio), the key step in the biomarker development pipeline, will increase assay efficiency and enable widespread implementation in large-scale investigations.
An important clinical implication of our findings pertains to determining the pathogenicity and assessing the functional impact of podocyte-specific genetic variations identified in genetic forms of NS and AS. Human genetic studies in the past 2 decades have illuminated podocyte dysfunction as the major contributor to GFB failure in primary NS. Mutations in more than 30 podocyte genes including NPHS1, NPHS2, WT-1, LAMB2, CD2AP, TRPC6, ACTN4, INF2, and COL4A have been implicated as causal factors for primary podocytopathy and GBM nephropathy (34). With the recent advent of next-generation sequencing, a large number of rare (minor allele frequency < 0.5%) missense variants of unknown significance in both known and novel NS-causing podocyte genes are rapidly emerging in thousands of affected patients (35, 36). Due to lack of recurrence and family data for segregation analysis in sporadic patients and patients with de novo mutations, and a sole reliance on bioinformatics-based prediction algorithms, reliably assigning pathogenicity to these genetic variants is challenging (37). In addition, in silico prediction of deleterious or damaging variants may not be completely accurate (38, 39) and, more importantly, cannot provide much mechanistic information. Detection of CRELD2 in urine may serve as a powerful high-throughput tool to functionally screen patients carrying rare genetic variants for causation of ER stress and, thus, may institute the targeted therapy at the early stage.
Another important implication of our study relates to early detection of tubular ER stress, which will be helpful in the development of mechanism-based treatment in ischemic AKI. Approximately 45% of critically ill patients and 20% of hospitalized patients develop AKI (40), which leads to increased hospital stays, infectious complications, and increased mortality, at significant cost (41, 42). In addition, AKI has been associated with future development of chronic kidney disease (43). The leading cause of AKI in native as well as transplanted kidneys is ischemic AKI. A number of biomarkers have been associated with AKI, including NGAL, KIM-1, IL-18, cystatin C, α/π glutathione S-transferase (GST), liver type fatty acid–binding protein 1 (FABP1), and N-acetyl-β-D glucosaminidase (NAG) (44). A unique feature of CRELD2 is its ability to gauge tubular ER stress, an underlying causative mechanism for ischemic AKI. Our preclinical mouse and translational studies in pediatric patients undergoing CPB surgery have demonstrated that CRELD2 can be utilized to stratify the risk of developing AKI after I/R injury. The ultimate utility of CRELD2 may be to identify an early therapeutic window during which we can apply tubular ER stress modulators before a detectable change in renal function occurs. Considering the complexity of pathways involved in ischemic AKI, incorporation of different biomarkers that target distinct pathophysiological mechanisms, together with CRELD2 into the diagnostic or prognostic panel, may be required to profile the multifaceted response of the kidney to insults and, thus, to design combinational mechanism-based treatments in ischemic AKI.
The identification of CRELD2 as an ER stress biomarker for ADTKD-UMOD is extremely important to research in this area, as it will provide a useful tool for clinical trials. ADTKD is a monogenic condition in which ER stress is prominent and leads to tubulointerstitial fibrosis and progression of chronic kidney disease. ADTKD-UMOD is a phenotypically heterogeneous disorder manifested by variable age of disease onset, disease severity, and rate of disease progression among affected individuals within and between families, with patients reaching ESRD between the ages of 25 and 70 years or older (12). Human mature uromodulin, mainly localized at the apical plasma membrane of TAL cells (45), contains a signal peptide, 3 EGF-like domains, a central domain of unknown function, a zona pellucida domain, and a glycosylphosphatidylinositol-anchoring (GPI-anchoring) site (13). Uromodulin has extremely high cysteine content and extensive disulfide bond formation, resulting in extremely slow transit through the ER. After being cleaved by a protease hepsin (46), uromodulin is released to the urine. UMOD mutations are clustered (94%) in exons 3 and 4 encoding for the N-terminal half of the protein, and ~50% of known UMOD mutations affect cysteine (13). All mutations listed in Table 2 involve exon 3. Studies in cellular models and mouse genetic models, as well as findings in patient renal biopsies, have demonstrated that UMOD mutations lead to mutant protein retention and aggregation in the ER, likely due to protein misfolding (14–18). The activation of the UPR leads to progressive damage of TAL, triggering inflammation and interstitial fibrosis (47).
It is important to note that CRELD2 elevation in the urine was detected in ADTKD-UMOD patients with eGFR > 60 ml/min, especially in 2 patients (patients 4 and 9) with normal eGFR. Thus, urinary excretion of CRELD2 may enable identification of disease activity early in the course of disease and allow us to monitor the effects of new therapies, overcoming the challenge posed by the slow rates of rise in serum Cr in these individuals, which is the major reason why currently clinical trials in ADTKD-UMOD cannot be performed. A prospective longitudinal cohort could potentially be assembled in individuals with a UMOD mutation before significant kidney damage has occurred (e.g., patients with an eGFR ≥ 90 ml/min). One could determine whether urine CRELD2 excretion is elevated early in the disease course and correlates with disease onset, severity, and progression, as well as treatment response. Meanwhile, a larger cross-sectional cohort will be carried out to discern whether there is any correlation between urinary CRELD2 levels and clinical phenotypes.
In summary, our study has identified CRELD2 as a sensitive mechanistic urine biomarker in various ER stress–mediated kidney diseases, including glomerular disorders such as hereditary NS, as well as tubulointerstitial disorders such as ischemic AKI and ADTKD-UMOD. We believe that CRELD2 will be very valuable in the study of ER stress not only in these disorders, but in many other kidney disorders, as well. The broad clinical applications of CRELD2 in ER stress–mediated kidney diseases may include early diagnosis, stratification of patients at risk, monitoring treatment response, and guiding the development of ER stress modulators in targeted patient groups. Further studies in a multiinstitutional setting with a larger cohort are required to evaluate the diagnostic accuracy and predictive value of CRELD2.
Methods
Antibodies and reagents.
Commercially available antibodies were obtained as follows: goat anti–mouse CRELD2 (AF3686), NGAL (AF1857), and KIM-1 (AF1817) antibodies were from R&D Systems; rabbit anti–mouse CRELD2 antibody crossreacting with human (G-17, sc-86110) was from Santa Cruz Biotechnology Inc.; and rabbit anti–human CRELD2 antibody (LS-C401550) was from LSBio. Rabbit anti–mouse BiP antibody (11587-1-AP) was from Proteintech. Mouse anti–human uromodulin antibody (119-13298) was from RayBiotech. HRP-conjugated anti–mouse β-actin antibody (A3854) was from MilliporeSigma. Alexa 488– and Alexa 594–conjugated secondary antibodies were purchased from Molecular Probes. HRP-conjugated anti-rabbit and anti-goat secondary antibodies were from Santa Cruz Biotechnology Inc. Biotinylated LTL (B-1325) and anti-mouse antibody (BA-9200) were from Vector Laboratories. Alexa 594–conjugated streptavidin (A11227) was purchased from ThermoFisher Scientific. Hoechst 33258 was from ThermoFisher Scientific. Histochoice was purchased from Amresco. Rat collagen I was from Trevigen. TM, TG, and acetic acid were from MilliporeSigma.
Mice.
The Lamb2+/–, WT, and mutant transgenic mice have all been described previously (5, 28). C57BL/6J mice were obtained from The Jackson Laboratory.
TM-induced AKI mouse model.
TM was dissolved in DMSO at 2 mg/ml and injected i.p. at 1 mg/kg into 6- to 10-week-old C57BL/6J mice. Urine was collected from metabolic cages in which the mice were immediately placed after the injection. One day or 5 days after the injection, sera were obtained, and kidneys were harvested. One kidney was snap frozen in dry ice and stored at –70°C until further processing for protein and RNA. The other kidney was fixed in 4% paraformaldehyde (PFA) (pH 7.4) at 4°C and then transferred to 15% and 30% sucrose/PBS for OCT-embedded cryosections.
Renal bilateral I/R mouse model.
C57Bl/6 male mice at age of 9 weeks were subjected to bilateral renal ischemia. Two separate protocols were used: (i) 30 minutes of bilateral ischemia with AKI and (ii) 10 minutes of bilateral mild subclinical ischemia. Briefly, mice were anesthetized with isoflurane, and their body temperature was maintained at 37°C on a heating pad and monitored with a rectal probe throughout surgery. Ischemia was induced by nontraumatic clamping of bilateral renal vascular pedicles for 10 or 30 minutes, during which time the kidneys were kept warm and moist. The clamps were then removed and the kidneys were observed for return of blood flow. Sham-operated C57Bl/6 mice (20 males at age of 9 weeks) underwent an identical procedure without vascular pedicle clamping. Urine was collected from metabolic cages in which the mice were placed immediately after the surgery at different time points. After 3, 9, or 24 hours — as well as 3, 7, or 15 days — of reperfusion, sera were obtained, and kidneys were harvested. One kidney was snap frozen in dry ice and stored at –70°C until further processing for protein and RNA. The other kidney was fixed in histochoice at 4°C and then processed for paraffin blocks. At least 5 mice were examined at each of the reflow periods for both protocols.
Isolation of mouse glomeruli.
Mice were perfused through the heart with magnetic 4.5 μm diameter Dynabeads (Dynabeads M-450 Tosylactivated, Invitrogen). Kidneys were minced into small pieces, digested by collagenase A (Gibco) and DNAse I (MilliporeSigma), filtered, and collected using a magnet. The purity of glomeruli was 95%–100%.
Primary podocyte culture.
Isolated glomeruli from Lamb2+/–, Lamb2–/–;Tg-C321R, and Lamb2–/–;Tg-WT mice at P27 were suspended in DMEM/Ham’s F-12 (2:1) that contained 0.45 μm–filtered 3T3-L1 supernatant, 5% heat-inactivated FBS, insulin-transferrin-sodium selenite (ITS) liquid media supplement, and 100 U/ml penicillin-streptomycin; plated onto collagen I–coated petri dishes; and incubated at 37°C in room air with 5% CO2. After 3 days, cell colonies began to sprout around the glomeruli. These cells (P0) showed an epithelial morphology with a polyhedral shape when confluence was reached and were characterized as podocytes by detection of podocyte-specific markers WT-1, nephrin and podocin by IF staining and WB. Only passages 1–2 podocytes from the indicated genotypes were used in the in vitro studies.
IF staining.
For dual staining of CRELD2 with biotinylated LTL in mouse kidneys, paraffin-embedded sections were used. After dewaxing, PFA-fixed, sucrose-cryoprotected kidney sections from control or TM-treated mice were subjected to antigen retrieval by immersion in 10 mM citric acid buffer (pH 6.0) for 5 minutes at 95°C. Histochoice-fixed kidney sections from sham-operated or I/R-injured mice did not require antigen retrieval. Nonspecific avidin binding was blocked by an avidin/biotin blocking kit (Vector laboratories) before incubating kidney sections with 1% BSA for 30 minutes at room temperature. The slides were stained with a goat anti–mouse CRELD2 antibody (R&D Systems) together with biotinylated LTL overnight at 4°C, followed by an Alexa 488–conjugated anti-goat secondary antibody and Alexa 594–conjugated streptavidin, as well as Hoechst 33342. For CRELD2 staining in sham-operated and postischemic kidneys, the fluorescent signal was further amplified using a Fluorescein tyramide amplification kit (Perkin-Elmer) according to the manufacturer’s instructions. Slides were analyzed under a fluorescence microscope (Nikon Eclipse E800).
Formalin-fixed, paraffin-embedded sections of human kidneys underwent the same process of deparaffinization, antigen retrieval, and blocking. The slides were incubated with a rabbit anti–human CRELD2 antibody (LSBio) in combination with a mouse IgG2b antibody against human uromodulin. A biotinylated anti-mouse antibody was used to amplify fluorescence signals for uromodulin, followed by the corresponding Alexa 488–conjugated anti-rabbit secondary antibody or Alexa 594–labeled streptavidin to detect CRELD2 and uromodulin, respectively. The nuclei were counterstained with Hoechst 33342.
For immunocytochemistry, mouse primary podocytes were seeded on cover slips coated with 5 μg/ml rat collagen I that was dissolved in 0.02 M acetic acid in 24-well plates for 48 hours. After 24-hour treatment with DMSO, TM, or TG, the cells were fixed with 4% PFA for 20 minutes and permeabilized with 1% Triton X-100 for 5 minutes at room temperature, blocked with 1% BSA for 30 minutes at room temperature, and incubated with goat anti–mouse CRELD2 (R&D Systems) and rabbit anti–mouse BiP antibodies for an additional 1 hour at room temperature. The cover slips were washed with PBS and incubated with the corresponding Alexa 594– or Alexa 488–conjugated secondary antibodies and Hoechst 33342 to stain nuclei. The cover slips were then mounted with antiquench solution and visualized using a fluorescence microscope (Nikon Eclipse E800).
Light microscopy.
For light microscopy, mouse kidneys were fixed in 4% PFA or Histochoice, and human kidneys were fixed in 10% formalin, dehydrated through graded ethanols, embedded in paraffin, sectioned at 4 μm, and stained with H&E, PAS, or Masson’s trichrome.
WB analysis.
Isolated mouse glomeruli and cultured mouse primary podocytes were lysed by RIPA buffer (MilliporeSigma) containing protease inhibitor cocktail (Roche Diagnostics). TM untreated and treated kidneys, as well as sham-operated and I/R-injured kidneys, were extracted using the same lysis buffer with the protease inhibitors and homogenized by sonication. The protein concentrations of cell, glomerular, and kidney lysates were determined by Bio-Rad protein assay using BSA as a standard. Urine volume from individual human patients was normalized to urine Cr excretion. Denatured proteins were separated on SDS-PAGE and then transferred to PVDF membranes. Blots were blocked with 5% nonfat milk for 1 hour and then incubated overnight with primary antibodies. The membranes were washed with Tris-buffered saline/Tween buffer and incubated with the appropriate HRP-conjugated secondary antibodies. The proteins were then visualized in an x-ray developer (Alphatek Ax 700LE) using ECL plus detection reagents (GE Healthcare). To ensure equal protein loading, the same blot was stripped with stripping buffer (25 mM glycine + 1% SDS, pH 2.0) and then incubated with a HRP-conjugated anti–mouse β-actin antibody. Relative intensities of protein bands were quantified using ImageJ (NIH) analysis software.
To determine secretion of CRELD2 from mouse primary podocytes, equal numbers of primary podocytes from the indicated genotypes or treatment groups were plated, and their media were harvested after 48 hours of culturing or 24 hours of treatment, respectively. Medium (20 μl) from the respective groups was subjected to the WB analysis. To compare urinary CRELD2 excretion, crude urine samples from the indicated groups that were normalized to urinary Cr were applied to a gel.
mRNA quantification by real-time PCR.
Total RNA from primary podocytes or whole kidneys was extracted using the RNeasy kit (Qiagen) with subsequent DNase I treatment. Cellular or kidney RNA (1 μg) was then reverse-transcribed using an RT-PCR kit (Superscript III; Invitrogen). Gene expression was evaluated by quantitative PCR. cDNA (1 μl) was added to SYBR Green PCR Master Mix (Qiagen) and subjected to PCR amplification (1 cycle at 95°C for 20 seconds, 40 cycles at 95°C for 1 second, and 40 cycles at 60°C for 20 seconds) in an Applied Biosystems 7900HT Fast Real-Time PCR System (Invitrogen). Quantitative PCR (qPCR) was conducted in triplicate for each sample. The sequences of primers were following: mouse CRELD2 forward: 5′ - CAACACGGCCAGGAAGAATTT - 3′, reverse: 5′ - CATGATCTCCAGAAGCCGGAT - 3′; mouse WT-1 forward: 5′ - GAGAGCCAGCCTACCATCC - 3′, reverse: 5′ - GGGTCCTCGTGTTTGAAGGAA - 3′; mouse GAPDH forward: 5′ - TGTAGACCATGTAGTTGAGGTCA - 3′, reverse: 5′ - AGGTCGGTGTGAACGGATTTG - 3′.
BUN measurement.
BUN was measured by using a QuantiChrom urea assay kit (DIUR-500) (BioAssay Systems).
Urinalysis.
Mouse urine was collected by manual restraint or using a metabolic cage if 24 hours urine collection was required. The mouse urine was centrifuged at 1,800 g for 10 minutes, and human urine was spun at 1,600 g for 10 minutes to remove debris before being frozen at —70°C. Coomassie G-250 stain was used to visualize urinary albumin (VWR). Urinary Cr concentration was quantified by a QuantiChrom Cr assay kit (DICT-500) (BioAssay Systems) or provided by TRIBE-AKI consortium.
Urine protein precipitation.
To measure urine CRELD2 concentrations in ADTKD-UMOD patients, protein from fresh-spun urine normalized to 450 μg of urine Cr excretion was precipitated with ice-cold acetone-methanol with a dilution ratio of 1:15 (v/v), centrifuged at 4,000 g, and washed with fresh acetone-methanol. Precipitated proteins were dissolved in 220 μl assay diluent included in the human CRELD2 ELISA kit for duplicate measurements.
ELISA measurements of human urine biomarkers.
CRELD2 concentrations in human urine samples were determined by an ELISA (RayBiotech). Urinary NGAL, IL-18, KIM-1, and Cr concentrations in the pediatric patients undergoing CPB were measured by TRIBE-AKI consortium. Personnel performing the biomarker measurements including urine CRELD2, NGAL, IL-18, and KIM-1 were blinded to the clinical information of each patient enrolled in TRIBE-AKI.
Statistics.
Statistical analyses were performed using GraphPad Prism software. Data were expressed as mean ± SD or plots. A 2-tailed Student’s t test was used to compare 2 groups. One-way ANOVA with post-hoc Tukey test was used to compare multiple groups. For statistical analysis related to TRIBE-AKI samples, continuous variables were compared with Wilcoxon rank-sum test or 2-sample t test and dichotomous variables with the χ2 test or Fisher’s exact test. P < 0.05 was considered significant.
Study approval.
All animal experiments conformed to the NIH Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011) and were approved by the Washington University Animal Studies Committee. Human urine specimen from severe AKI and non-AKI patients following pediatric CPB surgery were acquired from TRIBE-AKI consortium and approved by an ancillary study. The pediatric patients older than 1 month but younger than 18 years of age undergoing surgery for congenital cardiac lesions at 3 academic medical centers in North America were prospectively enrolled in TRIBE-AKI consortium between July 2007 and December 2009 (32). All human urine samples from ADTKD patients (n = 17) and their genetically unaffected controls (n = 7), as well as their paraffin-embedded slides, were obtained from Wake Forest Cohort under the protocol approved by the IRB of Wake Forest School of Medicine.
Author contributions
YMC conceived, designed, and supervised the study, and wrote the manuscript. YK designed and performed experiments, and contributed to writing the manuscript. SJP, SRM, and CAFM performed experiments and contributed to writing the manuscript. KK and RJP coordinated and supplied patient materials. HTP and CRP assisted with the design of human study, provided patient urine samples from TRIBE-AKI consortium, performed statistical analysis on Figure 5 and Table 1, and contributed to writing the manuscript. HL performed kidney pathology analysis on mouse models and human patients and contributed to writing the manuscript. SK provided patient materials and contributed to writing the manuscript. AJB assisted with the study related to human ADTKD patients and contributed to writing the manuscript. All authors contributed to the review and approval of the manuscript.
Supplementary Material
Acknowledgments
We thank the Mouse Genetics Core, the Washington University Center for Kidney Disease Research (NIH P30DK079333), and Washington University Diabetes Research Center (NIH P30 DK020579) for generating transgenic mice and collecting urine, and the Musculoskeletal Research Center Morphology Core (supported by NIH P30AR057235) for histology. Mice were housed in a facility supported by NIH C06RR015502. SRM was supported by the New Investigator Award from National Kidney Foundation, Midwest Stone Institute grant, and NIH/NIDDK R01DK096177. CAFM was supported by a Brazilian grant from Sao Paaulo Research Foundation – FAPESP (2015/17785-6). SK was supported by the project LQ1604 NPU II from the Ministry of Education, Youth and Sports of the Czech Republic, and NV17-29786A from the Ministry of Health of the Czech Republic. CRP was supported by NIH grants RO1 HL085757 and the O’Brien Kidney Center grant P30DK079310. AJB was supported by NIH grant R21DK106584. YMC was supported by NIH grants R01 DK105056A1, R03DK106451, and K08DK089015; Halpin Foundation-American Society of Nephrology Research grant; Faculty Scholar Award (MD-FR-2013-336) from the Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital; Clinical Scientist Development Award (2015100) from the Doris Duke Charitable Foundation; Career Development Award from the Nephrotic Syndrome Study Network (NEPTUNE); Early Career Development Award from the Central Society for Clinical and Translational Research (CSCTR); and Renal Translational Innovation grant from Washington University Division of Nephrology. YMC is a member of Washington University Institute of Clinical and Translational Sciences (UL1 TR000448). The authors wish to acknowledge the TRIBE-AKI study investigators: Prasad Devarajan (Nephrology and Hypertension, Cincinnati Children’s Hospital Medical Center, Cincinnati, Ohio, USA) and Michael Zappitelli (Department of Pediatrics, Division of Pediatric Nephrology, McGill University Health Centre, Montreal, Quebec, Canada). The authors also thank Jeremiah Morrissey for advice on the urine biomarker development.
Version 1. 12/07/2017
Electronic publication
Funding Statement
We thank the Mouse Genetics Core, the Washington University Center for Kidney Disease Research (NIH P30DK079333), and Washington University Diabetes Research Center (NIH P30 DK020579) for generation of transgenic mice and collecting urine, and the Musculoskeletal Research Center Morphology Core (supported by NIH P30AR057235) for histology. Mice were housed in a facility supported by NIH C06RR015502. S.R.M. was supported by New Investigator Award from National Kidney Foundation, Midwest Stone Institute Grant, and NIH/NIDDK R01DK096177. S.K. was supported by the project LQ1604 NPU II from the Ministry of Education, Youth and Sports of the Czech Republic and The National Center for Medical Genomics (LM2015091). A.J.B. was supported by NIH grant R21DK106584. Y.M.C. was supported by NIH grants K08DK089015 and R03DK106451, Halpin Foundation-American Society of Nephrology Research Grant, Faculty Scholar Award (MD-FR-2013-336) from the Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital, Clinical Scientist Development Award (2015100) from the Doris Duke Charitable Foundation, Career Development Award from the Nephrotic Syndrome Study Network (NEPTUNE), Early Career Development Award from the Central Society for Clinical and Translational Research (CSCTR), and Renal Translational Innovation Grant from Washington University Division of Nephrology. Y.M.C. is a member of Washington University Institute of Clinical and Translational Sciences (UL1 TR000448).
Footnotes
Conflict of interest: Y.M. Chen and Y. Kim are named on a nonprovisional patent application entitled “Methods of detecting biomarkers of endoplasmic reticulum (ER) stress–associated kidney diseases” that has been filed by the Washington University Office of Technology Management (15/664476, filed on July 31, 2017).
Reference information: JCI Insight. 2017;2(23):e92896. https://doi.org/10.1172/jci.insight.92896.
Contributor Information
Yeawon Kim, Email: yeawonkim@wustl.edu.
Sun-Ji Park, Email: sun-ji@wustl.edu.
Kendrah Kidd, Email: kkidd@wakehealth.edu.
Heather Thiessen-Philbrook, Email: heather.philbrook@yale.edu.
Helen Liapis, Email: helen.liapis@arkanalabs.com.
Stanislav Kmoch, Email: skmoch@lf1.cuni.cz.
Anthony J. Bleyer, Email: ableyer@wakehealth.edu.
References
- 1.Inagi R, Ishimoto Y, Nangaku M. Proteostasis in endoplasmic reticulum--new mechanisms in kidney disease. Nat Rev Nephrol. 2014;10(7):369–378. doi: 10.1038/nrneph.2014.67. [DOI] [PubMed] [Google Scholar]
- 2.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 3.Hinkes BG, et al. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2) Pediatrics. 2007;119(4):e907–e919. doi: 10.1542/peds.2006-2164. [DOI] [PubMed] [Google Scholar]
- 4.Gast C, et al. Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2016;31(6):961–970. doi: 10.1093/ndt/gfv325. [DOI] [PubMed] [Google Scholar]
- 5.Chen YM, Zhou Y, Go G, Marmerstein JT, Kikkawa Y, Miner JH. Laminin β2 gene missense mutation produces endoplasmic reticulum stress in podocytes. J Am Soc Nephrol. 2013;24(8):1223–1233. doi: 10.1681/ASN.2012121149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pieri M, et al. Evidence for activation of the unfolded protein response in collagen IV nephropathies. J Am Soc Nephrol. 2014;25(2):260–275. doi: 10.1681/ASN.2012121217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cybulsky AV, Takano T, Papillon J, Bijian K. Role of the endoplasmic reticulum unfolded protein response in glomerular epithelial cell injury. J Biol Chem. 2005;280(26):24396–24403. doi: 10.1074/jbc.M500729200. [DOI] [PubMed] [Google Scholar]
- 8.Cybulsky AV, Takano T, Papillon J, Khadir A, Liu J, Peng H. Complement C5b-9 membrane attack complex increases expression of endoplasmic reticulum stress proteins in glomerular epithelial cells. J Biol Chem. 2002;277(44):41342–41351. doi: 10.1074/jbc.M204694200. [DOI] [PubMed] [Google Scholar]
- 9.Inoki K, et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest. 2011;121(6):2181–2196. doi: 10.1172/JCI44771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bek MF, et al. Expression and function of C/EBP homology protein (GADD153) in podocytes. Am J Pathol. 2006;168(1):20–32. doi: 10.2353/ajpath.2006.040774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Markan S, et al. Up regulation of the GRP-78 and GADD-153 and down regulation of Bcl-2 proteins in primary glomerular diseases: a possible involvement of the ER stress pathway in glomerulonephritis. Mol Cell Biochem. 2009;324(1-2):131–138. doi: 10.1007/s11010-008-9991-2. [DOI] [PubMed] [Google Scholar]
- 12.Eckardt KU, et al. Autosomal dominant tubulointerstitial kidney disease: diagnosis, classification, and management--A KDIGO consensus report. Kidney Int. 2015;88(4):676–683. doi: 10.1038/ki.2015.28. [DOI] [PubMed] [Google Scholar]
- 13.Rampoldi L, Scolari F, Amoroso A, Ghiggeri G, Devuyst O. The rediscovery of uromodulin (Tamm-Horsfall protein): from tubulointerstitial nephropathy to chronic kidney disease. Kidney Int. 2011;80(4):338–347. doi: 10.1038/ki.2011.134. [DOI] [PubMed] [Google Scholar]
- 14.Bernascone I, et al. A transgenic mouse model for uromodulin-associated kidney diseases shows specific tubulo-interstitial damage, urinary concentrating defect and renal failure. Hum Mol Genet. 2010;19(15):2998–3010. doi: 10.1093/hmg/ddq205. [DOI] [PubMed] [Google Scholar]
- 15.Bernascone I, et al. Defective intracellular trafficking of uromodulin mutant isoforms. Traffic. 2006;7(11):1567–1579. doi: 10.1111/j.1600-0854.2006.00481.x. [DOI] [PubMed] [Google Scholar]
- 16.Kemter E, et al. Type of uromodulin mutation and allelic status influence onset and severity of uromodulin-associated kidney disease in mice. Hum Mol Genet. 2013;22(20):4148–4163. doi: 10.1093/hmg/ddt263. [DOI] [PubMed] [Google Scholar]
- 17.Rampoldi L, et al. Allelism of MCKD, FJHN and GCKD caused by impairment of uromodulin export dynamics. Hum Mol Genet. 2003;12(24):3369–3384. doi: 10.1093/hmg/ddg353. [DOI] [PubMed] [Google Scholar]
- 18.Vylet’al P, et al. Alterations of uromodulin biology: a common denominator of the genetically heterogeneous FJHN/MCKD syndrome. Kidney Int. 2006;70(6):1155–1169. doi: 10.1038/sj.ki.5001728. [DOI] [PubMed] [Google Scholar]
- 19.Bando Y, et al. ORP150/HSP12A protects renal tubular epithelium from ischemia-induced cell death. FASEB J. 2004;18(12):1401–1403. doi: 10.1096/fj.03-1161fje. [DOI] [PubMed] [Google Scholar]
- 20.El Karoui K, et al. Endoplasmic reticulum stress drives proteinuria-induced kidney lesions via Lipocalin 2. Nat Commun. 2016;7:10330. doi: 10.1038/ncomms10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim Y, et al. Mesencephalic Astrocyte-Derived Neurotrophic Factor as a Urine Biomarker for Endoplasmic Reticulum Stress-Related Kidney Diseases. J Am Soc Nephrol. 2016;27(10):2974–2982. doi: 10.1681/ASN.2014100986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oh-hashi K, Koga H, Ikeda S, Shimada K, Hirata Y, Kiuchi K. CRELD2 is a novel endoplasmic reticulum stress-inducible gene. Biochem Biophys Res Commun. 2009;387(3):504–510. doi: 10.1016/j.bbrc.2009.07.047. [DOI] [PubMed] [Google Scholar]
- 23.Oh-hashi K, Kunieda R, Hirata Y, Kiuchi K. Biosynthesis and secretion of mouse cysteine-rich with EGF-like domains 2. FEBS Lett. 2011;585(15):2481–2487. doi: 10.1016/j.febslet.2011.06.029. [DOI] [PubMed] [Google Scholar]
- 24.Oh-hashi K, Norisada J, Hirata Y, Kiuchi K. Characterization of the Role of MANF in Regulating the Secretion of CRELD2. Biol Pharm Bull. 2015;38(5):722–731. doi: 10.1248/bpb.b14-00825. [DOI] [PubMed] [Google Scholar]
- 25.Hartley CL, et al. Armet/Manf and Creld2 are components of a specialized ER stress response provoked by inappropriate formation of disulphide bonds: implications for genetic skeletal diseases. Hum Mol Genet. 2013;22(25):5262–5275. doi: 10.1093/hmg/ddt383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olden K, Pratt RM, Jaworski C, Yamada KM. Evidence for role of glycoprotein carbohydrates in membrane transport: specific inhibition by tunicamycin. Proc Natl Acad Sci USA. 1979;76(2):791–795. doi: 10.1073/pnas.76.2.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thastrup O, Cullen PJ, Drøbak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci USA. 1990;87(7):2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miner JH, Go G, Cunningham J, Patton BL, Jarad G. Transgenic isolation of skeletal muscle and kidney defects in laminin beta2 mutant mice: implications for Pierson syndrome. Development. 2006;133(5):967–975. doi: 10.1242/dev.02270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zinszner H, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12(7):982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dong B, et al. Ischemia/reperfusion-induced CHOP expression promotes apoptosis and impairs renal function recovery: the role of acidosis and GPR4. PLoS ONE. 2014;9(10):e110944. doi: 10.1371/journal.pone.0110944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang JR, et al. Ischemia-reperfusion induces renal tubule pyroptosis via the CHOP-caspase-11 pathway. Am J Physiol Renal Physiol. 2014;306(1):F75–F84. doi: 10.1152/ajprenal.00117.2013. [DOI] [PubMed] [Google Scholar]
- 32.Parikh CR, et al. Postoperative biomarkers predict acute kidney injury and poor outcomes after pediatric cardiac surgery. J Am Soc Nephrol. 2011;22(9):1737–1747. doi: 10.1681/ASN.2010111163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parikh CR, et al. Performance of kidney injury molecule-1 and liver fatty acid-binding protein and combined biomarkers of AKI after cardiac surgery. Clin J Am Soc Nephrol. 2013;8(7):1079–1088. doi: 10.2215/CJN.10971012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lovric S, Ashraf S, Tan W, Hildebrandt F. Genetic testing in steroid-resistant nephrotic syndrome: when and how? Nephrol Dial Transplant. 2016;31(11):1802–1813. doi: 10.1093/ndt/gfv355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sampson MG, et al. Using Population Genetics to Interrogate the Monogenic Nephrotic Syndrome Diagnosis in a Case Cohort. J Am Soc Nephrol. 2016;27(7):1970–1983. doi: 10.1681/ASN.2015050504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trautmann A, et al. Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort. Clin J Am Soc Nephrol. 2015;10(4):592–600. doi: 10.2215/CJN.06260614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sampson MG, Pollak MR. Opportunities and Challenges of Genotyping Patients With Nephrotic Syndrome in the Genomic Era. Semin Nephrol. 2015;35(3):212–221. doi: 10.1016/j.semnephrol.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gillies CE, Robertson CC, Sampson MG, Kang HM. GeneVetter: a web tool for quantitative monogenic assessment of rare diseases. Bioinformatics. 2015;31(22):3682–3684. doi: 10.1093/bioinformatics/btv432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grimm DG, et al. The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Hum Mutat. 2015;36(5):513–523. doi: 10.1002/humu.22768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li PK, Burdmann EA, Mehta RL, World Kidney Day Steering Committee 2013. Acute kidney injury: global health alert. Kidney Int. 2013;83(3):372–376. doi: 10.1038/ki.2012.427. [DOI] [PubMed] [Google Scholar]
- 41.Himmelfarb J, Ikizler TA. Acute kidney injury: changing lexicography, definitions, and epidemiology. Kidney Int. 2007;71(10):971–976. doi: 10.1038/sj.ki.5002224. [DOI] [PubMed] [Google Scholar]
- 42.Singbartl K, Kellum JA. AKI in the ICU: definition, epidemiology, risk stratification, and outcomes. Kidney Int. 2012;81(9):819–825. doi: 10.1038/ki.2011.339. [DOI] [PubMed] [Google Scholar]
- 43.Bydash JR, Ishani A. Acute kidney injury and chronic kidney disease: a work in progress. Clin J Am Soc Nephrol. 2011;6(11):2555–2557. doi: 10.2215/CJN.09560911. [DOI] [PubMed] [Google Scholar]
- 44.Siew ED, Ware LB, Ikizler TA. Biological markers of acute kidney injury. J Am Soc Nephrol. 2011;22(5):810–820. doi: 10.1681/ASN.2010080796. [DOI] [PubMed] [Google Scholar]
- 45.Pennica D, et al. Identification of human uromodulin as the Tamm-Horsfall urinary glycoprotein. Science. 1987;236(4797):83–88. doi: 10.1126/science.3453112. [DOI] [PubMed] [Google Scholar]
- 46.Brunati M, et al. The serine protease hepsin mediates urinary secretion and polymerisation of Zona Pellucida domain protein uromodulin. Elife. 2015;4:e08887. doi: 10.7554/eLife.08887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schaeffer C, Creatore A, Rampoldi L. Protein trafficking defects in inherited kidney diseases. Nephrol Dial Transplant. 2014;29 Suppl 4:iv33–iv44. doi: 10.1093/ndt/gfu231. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.