

Abstract
Phospholipase A2 (PLA2) enzymes regulate the formation of eicosanoids and lysophospholipids that contribute to allergic airway inflammation. Secreted PLA2 group X (sPLA2-X) was recently found to be increased in the airways of asthmatics and is highly expressed in airway epithelial cells and macrophages. In the current study, we show that allergen exposure increases sPLA2-X in humans and in mice, and that global deletion of Pla2g10 results in a marked reduction in airway hyperresponsiveness (AHR), eosinophil and T cell trafficking to the airways, airway occlusion, generation of type-2 cytokines by antigen-stimulated leukocytes, and antigen-specific immunoglobulins. Further, we found that Pla2g10–/– mice had reduced IL-33 levels in BALF, fewer type-2 innate lymphoid cells (ILC2s) in the lung, less IL-33–induced IL-13 expression in mast cells, and a marked reduction in both the number of newly recruited macrophages and the M2 polarization of these macrophages in the lung. These results indicate that sPLA2-X serves as a central regulator of both innate and adaptive immune response to proteolytic allergen.
Keywords: Immunology, Inflammation
Keywords: Adaptive immunity, Asthma, Innate immunity
Secreted phospholipase A2 group X (sPLA2-X) plays a key role in controlling both innate and adaptive type-2 immune responses.
Introduction
The secreted phospholipase A2 (sPLA2) family of enzymes has been implicated in inflammatory disorders, including asthma. We focused on sPLA2 group X (sPLA2-X) because we found that the sPLA2-X protein is elevated in the airways of subjects with asthma (1, 2) and is prominently expressed in airway epithelial cells and macrophages (1–3). Of the 10 mammalian sPLA2 enzymes, sPLA2-X has a uniquely high affinity for phosphatidylcholine, the most abundant phospholipid in mammalian cells (4). As sPLA2s hydrolyze membrane-bound phospholipids at the sn2 position, releasing free fatty acids including arachidonic acid (AA) and lysophospholipids (5), this step serves as the rate-limiting step in the formation of cyclooxygenase (COX) and 5-lipoxygenase (5-LOX) derived prostaglandins and leukotrienes (LT). For example, we previously demonstrated that sPLA2-X serves as a key regulator of cysteinyl LT (CysLT) formation by human eosinophils (6, 7). Additionally, sPLA2-X may also act independently of its enzymatic activity as a high-affinity ligand for the phospholipase A2 receptor, PLA2R1, a c-type lectin receptor expressed on human lung macrophages (8) and airway epithelial cells (9). In a T cell–dependent model of OVA sensitization in the presence of the exogenous adjuvant alum, mice lacking the sPLA2-X gene (Pla2g10) had less eosinophilic airway inflammation and reduced airway hyperresponsiveness (AHR), but the effects on T cell activation and allergen-specific IgE were less certain (10). Thus, sPLA2-X may be a central regulator of type-2 immune responses, but the function of this enzyme in a model replicating many of the features of human asthma remains uncertain.
Proteolytic allergens such as house dust mite (HDM) have endogenous components that serve as adjuvants (11, 12), and in such airway sensitization models, there is less dependence on T cells and greater dependence upon epithelial and innate immune mechanisms (13, 14). In particular, the production of epithelial-derived cytokines, including IL-33, has a central role in the response to inhaled allergen by rapidly activating innate immune cells, including type-2 innate lymphoid cells (ILC2s), mast cells, and macrophages. ILC2s and macrophages play a prominent role in animal models of allergic airway disease and are critical to the development of a type-2 immune response (15–20). Thus far, there has been little evidence implicating sPLA2-X as a regulator of these responses; however, a recent paper examining the effect of bee venom PLA2 (bvPLA2) in the periphery demonstrated that bvPLA2 induces the release of IL-33, leading to the generation of ILC2s and subsequent antigen-specific T cell responses (21).
Given the clear evidence implicating sPLA2-X in human asthma and the importance of proteolytic allergens in asthma (22), we examined the function of sPLA2-X in a model of airway sensitization and challenge with HDM utilizing mice globally lacking the Pla2g10 gene. We examined both the innate and adaptive immune response to allergen, including innate immune cell populations, such as ILC2s; recruited macrophages and mast cells; and the polarization of lung and airway macrophages. We also examined alterations in ex vivo antigen-induced changes in type-2 cytokines by lung leukocyte populations and assayed allergen-specific IgG and IgE. To further extend these findings to humans, we assessed the levels of sPLA2-X in airway samples following whole lung allergen challenge and examined the activation of monocyte-derived human macrophages by recombinant sPLA2-X. Our findings indicate that sPLA2-X plays a central role in coordinating innate and adaptive immune responses to an inhaled allergen.
Results
Inhaled proteolytic allergen increases sPLA2-X levels in human and mouse airways.
A potential role of sPLA2-X in human asthma has been identified based on increased levels of the protein in airway fluid and the relationship between the levels of this protein and AHR (1–3). Here, we extend these findings to allergen-induced changes in the levels of this protein in the airways in a group of subjects who underwent inhaled allergen challenge to proteolytic allergens, predominantly HDM (Table 1). Airway exposure to allergen resulted in a time-dependent increase in sPLA2-X protein in induced sputum starting at 7 hours and continuing up to 24 hours (Figure 1A). Similarly, in our mouse model of allergic airway disease using the proteolytic allergen HDM, allergen sensitization and challenge increased Pla2g10 message in lung tissue and sPLA2-X protein in bronchoalveolar lavage fluid (BALF) in WT mice (Figure 1, B and C; see complete unedited blots in the supplemental material). Levels of Pla2g10 message were undetectable in mice lacking Pla2g10 (Figure 1B). Immunostaining for sPLA2-X protein in HDM-exposed animals revealed immunostaining in the airway epithelium and macrophages with characteristics of monocyte-derived macrophages and resident alveolar macrophages (Supplemental Figure 1, A and B; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.94929DS1). Further, quantitative analyses of sPLA2-X protein in lysates of tissues known to express abundant sPLA2-X protein confirmed that our Pla2g10-deficient mice are also deficient in the sPLA2-X protein (Supplemental Figure 1, C and D).
Table 1. Comparison of lung function before and after allergen challenge in subjects with allergic asthma.

Figure 1. Proteolytic allergens increase sPLA2-X in human and mouse airways.

(A) Four individuals with asthma were challenged via the airways with aerosolized allergen, and levels of sPLA2-X protein were measured using time-resolved fluorescence immunoassay (TRFIA) in induced sputum at 7 hours and 24 hours after challenge. Mean ± SEM (n = 4/time point) with 1-way ANOVA. BL, baseline. (B) Whole lung Pla2g10 gene expression from WT and Pla2g10–/– (–/–) mice exposed to saline or HDM (n = 3/group). Mean ± SEM, Mann-Whitney test. (C) BALF sPLA2-X protein levels from WT and Pla2g10–/– mice exposed to HDM were analyzed via Western blot (20 μl/lane) and quantified using densitometry (n = 4 for saline [Sal] and 7 for HDM). Mean ± SEM, Mann-Whitney test. A.U., arbitrary units.
Pla2g10–/– mice are protected from developing allergic airway disease.
To investigate the impact of Pla2g10 gene deletion on the development of airway dysfunction in response to inhaled allergen, we administered HDM in the absence of adjuvant with a single sensitizing dose in the airways, followed by daily allergen challenge for the subsequent 2 weeks to WT and Pla2g10–/– mice (Figure 2A); then, we assessed HDM-induced AHR and inflammatory cell influx into the airways. Inhaled HDM led to a cellular influx in both the lung tissue and in BALF (Figure 2, B and C). There was a significant decrease in the cellular influx in BALF in Pla2g10–/– mice (Figure 2B), while the difference in cellularity in the lung tissue did not reach statistical significance (Figure 2C). Our inhaled HDM model also markedly increased AHR in C57BL6 mice, as evidenced by a marked increase in lung resistance (R) and, to a lesser degree, lung elastance (E) in response to increasing doses of inhaled methacholine (MCh). Mice lacking Pla2g10 exhibited markedly lower levels of AHR measured by R following sensitization and challenge to HDM (Figure 2D). The increase in E seen in WT mice was also significantly attenuated in Pla2g10–/– mice (Figure 2E).
Figure 2. Pla2g10–/– mice are protected from developing airway hyperresponsiveness (AHR) and aberrant lung inflammation.

(A) HDM sensitization and challenge model used to induce allergic airway disease. Animals were instilled with 100 μg HDM (protein) on day 0, and then 25 μg HDM (protein) on days 7–11 and 14–18. On day 21, airway mechanics were assessed and mice were sacrificed. o.p., oropharyngeal administration. (B) Cells in the BALF following HDM sensitization and challenge were isolated and counted using an automated cell counter (n = 3/group for Sal, 7 for WT HDM, and 8 for Pla2g10–/– HDM). Mean ± SEM. (C) Single cell suspensions of lung leukocytes following HDM sensitization and challenge were isolated from digested lung tissue and counted using an automated cell counter (n = 3/group for Sal, 7 for WT HDM, and 8 for Pla2g10–/– HDM). Mean ± SEM. (D and E) Measurement of AHR to increasing doses of methacholine following HDM sensitization and challenge using the forced oscillation technique to measure lung mechanics (n = 3/group for Sal, 10 for WT HDM, and 12 for Pla2g10–/– HDM). Mean ± SEM. Statistical significance was determined by 2-way ANOVA with uncorrected Fisher’s LSD.
Histological evaluation of lung tissue revealed HDM-induced tissue inflammation and leukocyte influx in WT mice that was diminished in Pla2g10–/– mice (Figure 3, A–D). To quantify differences in tissue inflammation, we enumerated leukocyte subsets in BALF and lung tissue lysates by multicolor flow cytometry. In the BALF, the increase in eosinophils and T cells following HDM exposure was markedly reduced in mice lacking Pla2g10 (Figure 3E). Similarly, lung tissue lysates collected after removal of airway cells by BAL had reductions in lung tissue eosinophils and T cells in Pla2g10–/– mice (Figure 3F). There was also a modest increase in neutrophils in HDM-treated mice that was not altered by lack of Pla2g10 in either compartment (Supplemental Figure 2). Collectively, these data indicate that Pla2g10 regulates the development of cellular inflammation and AHR following airway exposure to the proteolytic allergen, HDM.
Figure 3. Leukocyte influx into the airways is decreased in Pla2g10–/– mice.

(A–D) Representative images of lung tissue H&E staining in WT and Pla2g10–/– mice exposed to either saline or HDM as outlined in scheme in Figure 2A. Original magnification, 20×; scale bar: 50 μm. (E and F) Concentration of eosinophils and T cells in (E) BALF and (F) single cell lung suspension following sensitization and challenge with saline or HDM (n = 3/group for Sal, 7 for WT HDM, and 8 for Pla2g10–/– HDM). Cell differentials were determined by multicolor flow cytometry and were defined by the following surface markers: Eosinophils (CD45+CD3–MHCIIloCD11c–SiglecF+), T cells (CD45+CD3+). Statistical significance was determined by 2-way ANOVA with uncorrected Fisher’s LSD.
Pla2g10 deficiency alters eosinophil trafficking to the peribronchial space.
Because eosinophils in close proximity to the airways play a pathogenic role in the development of airway dysfunction in murine models (23–26), we used image analysis on tissue sections immunostained for the eosinophil granule protein major basic protein (MBP). We have previously demonstrated that human sPLA2-X activates CysLT formation in human eosinophils (6) and that endogenous sPLA2-X in eosinophils regulates CysLT formation (7). Immunostaining for MBP was sparse in saline-exposed WT and Pla2g10–/– mice (Figure 4, A and C), but following HDM exposure, eosinophils were observed in the peribronchial space; further, the density of eosinophils surrounding the airways was substantially attenuated in Pla2g10–/– mice (Figure 4, B and D). To quantify these findings, we used segmentation analysis using an automated analysis of the number of MBP-positive cells, as well as the total MBP immunostaining in the peribronchial region of interest, to determine the effect of the Pla2g10 deletion on eosinophil trafficking to this region. The HDM-induced increase in the number of peribronchial eosinophils (Figure 4E) and the percentage of MBP-positive staining surrounding the airways (Figure 4F) was significantly decreased in Pla2g10–/– mice compared with WT mice. To further corroborate the functional significance of these findings, we measured MBP protein in the BALF by Western blot, demonstrating substantially less MBP released into the BALF of Pla2g10–/– mice (Figure 4G; see complete unedited blots in the supplemental material). These data indicate that sPLA2-X plays a key in vivo role in the trafficking of eosinophils to the airways and the activation state of these cells.
Figure 4. Peribronchiolar eosinophilic inflammation is decreased in mice lacking Pla2g10.

(A–D) Representative images of eosinophil major basic protein (MBP) immunostaining in lungs from saline- and HDM-exposed WT and Pla2g10–/– mice. Original magnification, 20×; scale bar: 50 μm. (E) Total number of peribronchial eosinophils and (F) percentage of peribronchial MBP immunostaining per total lung tissue quantified using Visiopharm analysis software (n = 3 mice/group). (G) Western blotting and densitometric analysis of MBP in BALF (20 μl each sample) from saline- and HDM-exposed WT and Pla2g10–/– mice (n = 4/group for Sal and 7/group for HDM). Statistical significance was determined by 2-way ANOVA with uncorrected Fisher’s LSD.
Pla2g10–/– mice have decreased mucus metaplasia and mucus plugging.
To further ascertain the basis for the marked reduction in AHR in mice lacking Pla2g10, we assessed differences in mucus cell hyperplasia and mucus plugging. Recent work has revealed that airway closure and mucus-plugging play a critical role in the pathophysiology of AHR and that the major gel forming mucin, MUC5AC, is essential for AHR (27, 28). Since MCh challenge results in epithelial mucus release (29) and subsequent lavage can potentially remove secreted mucus, we characterized a separate set of mice that did not undergo either MCh challenge or BAL, and we fixed the lung in a manner to examine mucus plugging (27). We found ongoing mucus plugging in the airways of HDM-exposed WT mice (Figure 5A). Quantification of mucus-occluded airways using image analysis revealed a significant decrease in mucus plugging in HDM-exposed Pla2g10–/– mice compared with HDM-exposed WT mice (Figure 5B). Further assessment of epithelial periodic acid Schiff (PAS) staining using image analysis showed significantly elevated mucus content in the epithelium of HDM-exposed WT mice compared with HDM-exposed Pla2g10–/– mice (Figure 5C). These data suggest that HDM-induced mucus production and secretion are regulated in part by sPLA2-X, and this alteration in mucus formation may influence the development of AHR.
Figure 5. Epithelial mucus production in HDM-exposed Pla2g10–/– mice is dampened compared with WT mice.
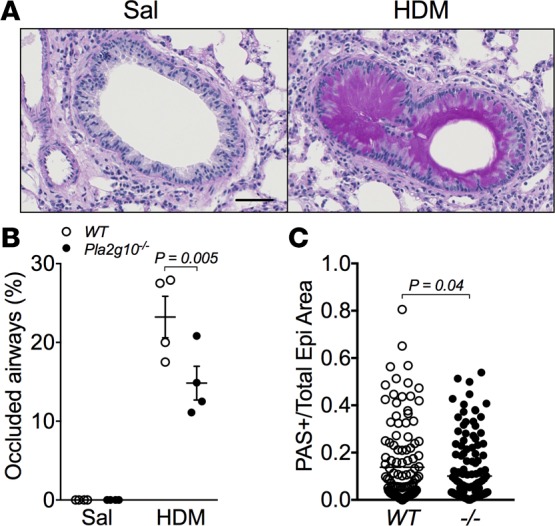
(A) Representative images of PAS staining from a WT mouse exposed to saline or HDM. Original magnification, 20x; scale bar: 50 μm. (B) Percentage of occluded airways in saline- and HDM-exposed mice, quantified based on the extent of PAS-positive staining in the airway lumen (n = 3/group for Sal and 4/group for HDM). Mean ± SEM, 2-way ANOVA with uncorrected Fisher’s LSD. (C) Individual airways were assessed to determine the ratio of epithelial area that stained positive for PAS to the total epithelial area as determined by Visiopharm analysis in HDM-exposed WT and Pla2g10–/– mice (–/–, Pla2g10–/–; n = 122 airways for WT and 149 airways for –/–). Mean ± SEM, unpaired t test.
Lack of Pla2g10 decreases the type-2 immune response to inhaled HDM.
A prior study using Th2 biased model of prolonged exposure to OVA following sensitization in the presence of the exogenous adjuvant alum demonstrated that Pla2g10–/– mice produce significantly less IL-4, IL-5, and IL-13 in the BALF following sensitization and challenge, although the generation of these cytokines by T cells was not altered (10). In our HDM model, the levels of type-2 cytokines were near the level of detection in BALF and were similar in WT and Pla2g10–/– mice (data not shown). To further assess the function of Pla2g10 in the immune response to HDM, we isolated leukocytes from lung tissue and assessed the production of cytokines following 24 hours of cell culture in the presence or absence of allergen. Leukocytes isolated from HDM-sensitized WT lungs displayed a marked increase in type-2 cytokines, including IL-4, IL-5, and IL-13, following HDM stimulation. In this assay, there was a trend toward less IL-4 and IL-5, as well as a significant reduction in IL-13 production by HDM-stimulated leukocytes from HDM-sensitized Pla2g10–/– mice relative to WT mice (Figure 6, A–C). There was also a trend toward less IL-6 production by leukocytes from Pla2g10–/– mice (Figure 6D). We also examined the production of cytokines by splenocytes placed in culture and stimulated with HDM, and we did not observe any alterations in the production of these cytokines in the Pla2g10–/– mice (data not shown), suggesting that the primary effect of Pla2g10 is in the lung in response to an inhaled allergen.
Figure 6. Lack of Pla2g10 alters cytokine and CysLT production.

Concentration of (A) IL-4, (B) IL-5, (C) IL-13, and (D) IL-6 from supernatant of cultured WT or Pla2g10–/– (–/–) lung leukocytes isolated from saline or HDM sensitized mice (Sens.) and stimulated with either saline or HDM. Only IL-13 was significantly lower in Pla2g10–/– mice (n = 6–8 mice/group). Mean ± SEM, 2-way ANOVA with uncorrected Fisher’s LSD. (E) CysLT levels measured in the BALF from HDM sensitized and challenged mice (n = 3 for Sal, 9 for WT HDM, and 11 for Pla2g10–/– HDM). Mean ± SEM, 2-way ANOVA with uncorrected Fisher’s LSD. (F) IL-33 release into the BALF following sensitization and challenge with HDM (n = 5 mice/group). Mean ± SEM, unpaired t test.
Analysis of the BALF from WT and Pla2g10–/– mice was also performed to determine alterations in CysLT formation in the airways. In cellular systems, sPLA2-X regulates the formation of CysLTs, which are important regulators of antigen sensitization (11, 12, 30) and mucus secretion (31–33). We found that exposure to HDM led to abundant CysLT production in the airways of WT mice, a response that was abolished in Pla2g10–/– mice (Figure 6E). We also observed that HDM exposure resulted in elevated Prostaglandin D2 (PGD2) levels in the airways of WT mice, but PGD2 did not increase above baseline in Pla2g10–/– mice (Supplemental Figure 3).
Since we have demonstrated that sPLA2-X is increased in response to HDM, and a similar PLA2 derived from bee venom induces the release of IL-33 from structural cells (21), we measured Il33 mRNA in lung tissue and levels of IL-33 in BALF following sensitization and challenge to HDM. We found that, although Il33 expression was not different between WT and Pla2g10–/– mice exposed to HDM (Supplemental Figure 4), IL-33 protein levels were elevated in the BALF of WT mice exposed to HDM, while the release of IL-33 into the airways in Pla2g10–/– mice was significantly attenuated (Figure 6F). Together, these data show that the absence of sPLA2-X alters the cellular immune response to inhaled allergen, leading to a reduction in leukocyte-derived IL-13, marked reduction in CysLTs and PGD2, and reduced levels of the key epithelial cytokine IL-33.
Pla2g10–/– mice have reduced number of ILC2s in the lung.
Given that airway epithelial cells produce abundant sPLA2-X and Pla2g10–/– mice have diminished type-2 cytokine production in response to OVA (10) and HDM (Figure 6), we hypothesized that lack of sPLA2‑X hinders the ability of these mice to mount an effective innate type-2 immune response to allergen. To address this, we examined the expression the IL-33 receptor (ST2), CD127 (IL-7Rα), and Sca1 on lineage-negative lung leukocytes within the lymphocyte gate (Supplemental Figure 5). The total number of lineage-negative leukocytes (Lin–CD45+) was increased in response to HDM and was reduced in the lungs of Pla2g10–/– mice (data not shown). We found significantly fewer Lin–CD45+ST2+ cells (Figure 7A) and Lin–CD45+ST2+CD127+ cells (Figure 7B) in the lungs of Pla2g10–/– mice compared with WT mice, indicating decreased numbers of ILC2s in the lung. Although we observed a slight decrease in the population of Lin–CD45+ST2+CD127+Sca1+ cells in Pla2g10–/– mice following HDM exposure, these differences did not reach statistical significance (Figure 7C). These data indicate that sPLA2-X serves as an endogenous regulator of ST2+ lung ILC2s in the context of inhaled allergen.
Figure 7. ILC2 polarization is impaired in mice lacking Pla2g10.
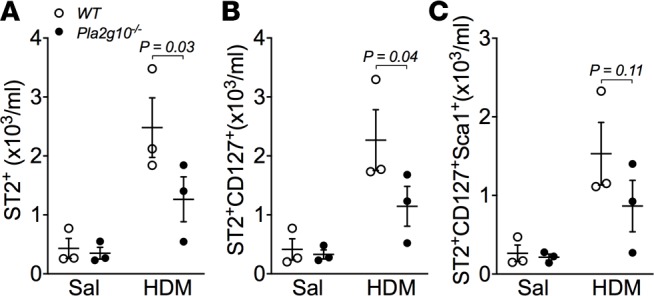
Gating was performed to isolate lineage negative (Lin–), CD45+ leukocytes (Lin–CD45+, see Supplemental Figure 3). Further gating on the Lin–CD45+ leukocyte population was performed to isolate (A) ST2+ cells and (B) ST2+CD127+ cells from WT and Pla2g10–/– mice exposed to saline or HDM. (C) Additional analysis revealed no significant differences in ST2+CD127+Sca1+ cells between WT and Pla2g10–/– mice (n = 3 mice/group). Mean ± SEM, 2-way ANOVA with uncorrected Fisher’s LSD.
Lack of Pla2g10 alters the type-2 polarization of mast cells.
Although ILC2s are critical IL-33–responsive cells in the lung that serve as a major source of IL-13 (15, 34, 35), mast cells also bear the ST2 receptor for IL-33 and serve as a source of IL-13 in the lung (36). Using image analysis, we found that HDM sensitization and challenge led to a marked increase in the number of mast cells in the lung (Figure 8A), but there was no apparent alteration in the number of mast cells in the lung in Pla2g10–/– mice. We examined the activation of BMMCs by IL-33 relative to other relevant stimuli — including IL-25, thymic stromal lymphopoietin (TSLP), IL-13, and HDM — revealing that IL-33 plays a unique role in the induction of IL-13 expression in BMMCs (Figure 8, B and C; see complete unedited blots in the supplemental material). We examined the effects in BMMCs derived from WT and Pla2g10–/– mice and found that the IL-33–induced increase in IL-13 in BMMCs was markedly attenuated in cells derived from Pla2g10–/– mice relative to WT mice (Figure 8C). Since it has been controversial whether mast cells have endogenous sPLA2-X, we conducted Western blots of WT and Pla2g10–/– lungs, revealing a clear band for sPLA2-X in the WT cells, while there was an absence of this band in the Pla2g10–/– mice (Figure 8C, inset). These results reveal that IL-33 plays a unique role in mast cell activation toward a type-2 phenotype and that sPLA2-X plays an essential role in the response to exogenous IL-33 in mast cells.
Figure 8. Lack of Pla2g10 alters mast cell activation.

(A) Lung tissue mast cells were visualized and enumerated using toluidine blue staining in mice exposed to saline or HDM (n = 6 mice for Sal and 8 mice for HDM). Mean ± SEM, Mann-Whitney test. (B) The expression of Il13 in WT BMMCs was evaluated in response to 4-hour stimulation with IL-33 (10 ng/ml), IL-13 (10 ng/ml), or HDM (250 μg/ml) (n = 4/group for Sal, IL-33, and IL-13 and 2 for HDM). (C) WT and Pla2g10–/– BMMC lysates were analyzed for sPLA2-X protein content via Western blot. WT and Pla2g10–/– BMMCs were stimulated with IL-33, IL-25, and TSLP (all at 10 ng/ml), and expression of Il13 was evaluated (n = 6–8/group for Sal and IL-33, 2–4/group for IL-25 and TSLP). Mean ± SEM, 1-way ANOVA with uncorrected Fisher’s LSD.
Lack of Pla2g10 alters macrophage recruitment and phenotype following inhaled allergen.
Because lung macrophages with an alternatively activated or M2 phenotype are implicated in both human asthma and murine models of asthma, we initially assessed the polarization of macrophages in the lung using well-established and potentially novel markers of M2 polarization (37, 38). We found that WT HDM-exposed mice had an increase in the concentration of macrophages in the lung and an increase in the percentage of these cells with positive surface staining for the M2 markers CD71/TfR (transferrin receptor) and CD206/MR (mannose receptor), while the percentage of macrophages bearing these markers was significantly attenuated in HDM-exposed Pla2g10–/– mice (Supplemental Figure 6, A–C).
As recent studies have revealed that recruited macrophages are pathogenic, while resident macrophages have a protective role (39–41), we assessed the impact of Pla2g10 status on these populations and further ascertained the polarization of these subpopulations using mean fluorescent intensity (MFI). We found that HDM exposure did not alter the number of lung-resident macrophages (Figure 9A) but significantly increased the number of recruited macrophages to the lung (Figure 9D); further, we found that Pla2g10–/– mice had fewer recruited macrophages after HDM exposure (Figure 9D). Immunophenotyping of these subpopulations in the lung revealed that the surface expression of the M2 markers CD206/MR and CD71/TfR was higher in both resident and recruited populations after HDM exposure and that Pla2g10–/– mice had less surface expression of these M2 markers in both the resident and recruited macrophage populations (Figure 9, B, C, E, and F). We also assessed the M1 marker ICAM-1 and found a slight decrease in the immunostaining for ICAM-1 in resident macrophages from the Pla2g10–/– mice, while there was no difference in ICAM-1 staining in recruited macrophages from WT and Pla2g10–/– mice (data not shown).
Figure 9. Macrophage polarization and activation are influenced by Pla2g10 status.

(A) Concentrations of resident macrophages in lung tissue from WT and Pla2g10–/– mice were similar after saline and HDM challenge. The surface expression of (B) CD206/MR and (C) CD71/TfR on these resident macrophages was assessed to measure the extent of M2 polarization. (D) HDM-induced recruited macrophages in lung tissue were also evaluated by multicolor flow. In this recruited macrophage population, the surface expression of (E) CD206/MR and (F) CD71/TfR was also evaluated to determine M2 polarization. n = 4–5 mice/group. Mean ± SEM, 2-way ANOVA with uncorrected Fisher’s LSD.
Although recruited and resident macrophages are classically described in the interstitial space of the lung, we assessed these populations in the BALF using a slightly different set of surface markers to identify CD11c+SiglecF+MHCIIlow resident macrophages, and CD11c–SiglecF–MHCIIhigh recruited monocyte-derived macrophages. We found that at baseline, there were fewer resident macrophages in BALF in the Pla2g10–/– mice and that there was a marked increase in recruited macrophages in the BALF after HDM exposure that was markedly attenuated in the Pla2g10–/– mice (Supplemental Figure 6, D and E).
Finally, we isolated lung macrophages after in vivo HDM sensitization and challenge from mouse lung by adherence to plastic, and we stimulated these cells with HDM in vitro for 24 hours. We identified measurable quantities of IFN-γ, IL-10, IL-12p70, IL-1β, IL-5, IL-6, KC, and TNF-α in the supernatant from these cells; the levels of IL-5 and IL-6 in the supernatant tended to be lower in the Pla2g10–/– but did not reach statistical significance (Supplemental Figure 7, A and B). To extend our findings to human disease, we generated monocyte-derived macrophages from the peripheral blood of healthy adults and stimulated these cells with either HDM or recombinant human sPLA2-X (hsPLA2-X) protein. In response to HDM, human macrophages produced a small amount of IL-6, which was augmented with the addition of hsPLA2-X (Supplemental Figure 7C). As macrophages prominently express leukotriene B4 (LTB4) synthase and subsequently produce LTB4, we examined the generation of this eicosanoid in response to HDM and found that, while both HDM and hsPLA2-X led to the generation of LTB4, these effects were not additive (Supplemental Figure 7D). Taken together, these data show for the first time to our knowledge that Pla2g10 plays a key role in the recruitment, polarization, and activation of macrophages in the lung in a manner that is relevant to the development of allergic airways disease.
Lack of Pla2g10 impairs the adaptive immune response to HDM.
In the prior OVA/alum study, Pla2g10 deletion had only a modest effect on OVA-specific IgE, and although there were fewer T helper cells, the activation state of T cells was not altered (10). Recent work has revealed that allergen-specific IgG plays a key role in type-2 biased immune responses (42). To clarify the role of Pla2g10 in the adaptive immune response to proteolytic allergen, we measured total IgE and found that the amount of total IgE was significantly reduced in the Pla2g10–/– mice following HDM exposure (Figure 10A). We found that there was a marked increase in DerP1-specific IgG following HDM exposure in our model that was markedly attenuated in Pla2g10–/– mice (Figure 10B). We were unable to accurately characterize the DerP1-specific IgE in this model because of competition on the assay from the high quantities of DerP1-specific IgG that could not be adequately depleted to ascertain clear results for DerP1-specific IgE. In summary, our data reveal that Pla2g10 plays key roles in both the innate and adaptive immune response to HDM.
Figure 10. Pla2g10–/– mice display an impaired adaptive immune response to HDM.

The serum levels of (A) total IgE and (B) DerP1-specific IgG were assessed by ELISA (n = 3–5 mice/group for Sal and 7–10 mice/group for HDM). A.U., arbitrary units. Mean ± SEM, 2-way ANOVA with uncorrected Fisher’s LSD.
Discussion
The present study is the first to demonstrate a central function of sPLA2-X/Pla2g10 as a regulator of both the innate and adaptive arms of the immune response to an inhaled allergen, HDM, that plays a key role in human asthma. These finding add to an emerging body of knowledge indicating that the innate and adaptive immune responses are tightly linked. Further, our findings are clinically relevant, as we have previously identified sPLA2-X as a target in human airways and further demonstrate here that the enzyme is released in response to inhaled allergen challenge in humans. A previous study with Pla2g10–/– mice in the T cell–dependent model of peritoneal sensitization to OVA in the presence of the adjuvant alum found that mice lacking functional sPLA2-X were protected from developing allergic lung inflammation and displayed a blunted Th2 response — findings that were attributed mainly to the failure of mature T cell trafficking to the lung, as effects on the adaptive immune response and T cell activation were less certain (10). A further study that utilized a human PLA2G10 transgenic mouse showed that OVA-induced lung inflammation could be attenuated with a specific inhibitor of human sPLA2-X (43), suggesting that sPLA2-X could be a viable pharmacological target. However, recent studies using human allergens such as HDM that contain endogenous adjuvants and are not entirely T cell dependent more closely recapitulate the findings in human asthma and involve key functions for innate immune cells including ILC2s, recruited macrophages, and mast cells. The interplay between these innate immune cells that serve as important sources of IL-13 and the adaptive immune response providing the antigen-specific nature of the disease remain incompletely defined. We used an inhaled allergen model with HDM to demonstrate that mice lacking Pla2g10 have less IL-33, CysLTs, and PGD2 in the airways and reduced numbers of ILC2s and recruited macrophages in the lung, and that macrophages from KO mice have less M2 polarization. We found that lung leukocytes from Pla2g10-deficient mice generate less IL-13 in response to antigen restimulation and that Pla2g10-deficient murine mast cells generate less IL-13 following IL-33 stimulation. These alterations in the innate immune response were accompanied by a reduction in the antigen-specific immune response to HDM and reduction in eosinophil and T cell trafficking to the airways, mucus plugging, and AHR, providing evidence of a central role of sPLA2-X in both the innate and adaptive immune response to inhaled allergen and the pathophysiology of airway dysfunction in asthma.
Type-2 inflammation is a hallmark feature of allergic asthma and is generally associated with elevated eosinophils, Th2 polarized CD4+ T cells, and production of type-2 cytokines IL-4, IL-5, and IL-13. Our data revealed robust increases in IL-4, IL-5, and IL-13 in lung leukocytes restimulated with HDM; however, the major effect of the lack of Pla2g10 was on the antigen-stimulated IL-13 levels, while there were lesser effects on IL-4 and IL-5 (Figure 6). These effects are in contrast with the OVA/alum model, where production of all 3 cytokines was dampened in Pla2g10–/– mice (10). In proteolytic allergen models, such as HDM, innate immune cells, particularly ILC2s, serve as a greater source of IL-13 than T cells (15). Other cells, including alternatively activated macrophages (44) and mast cells (36), also serve as key sources of IL-13. We demonstrate that the global lack of Pla2g10 alters the number of ILC2s and recruited macrophages to the lung; we also reveal that mast cells stimulated by IL-33 have a marked increase in IL-13 transcription and that lack of Pla2g10 in these cells leads to a loss of IL-33–induced IL-13 message. These findings suggest that in the lung, endogenous sPLA2-X serves as a central regulator of the innate immune response to inhaled antigen, and that these alterations in innate cell trafficking and proliferation lead to altered airway physiology at least in part through the inhibition of IL-13 production in the lung.
The reduced numbers of ILC2s in the lung that we identified in the present paper is consistent with the findings in the periphery that bvPLA2 induces a subsequent T cell response to antigen in a process that involves the release of IL-33 and the generation of ILC2s (21). We extend these findings to the lung, where we found reduced levels of IL-33 protein in BALF in the absence of changes in Il33 gene expression, as well as reduced numbers of ILC2s. Importantly, we demonstrate here that an endogenous sPLA2 in the lung functions in this manner in response to inhaled allergen. Recent murine models of allergic asthma have revealed the that ILC2s are critical in initiating or augmenting production of type 2–associated cytokines IL-5 and IL-13 independently of antigenic memory (15, 34, 35, 45). In the periphery, it was found that the antigen-specific immune response was dependent upon the IL-33–mediated generation of ILC2s, and we reveal here that the lack of sPLA2-X during the response to inhaled antigen inhibits the generation of DerP1-specific IgG and total IgE. These findings further reinforce the connection between the innate and adaptive arms of the immune response to an inhaled allergen.
In addition to IL-33, which is clearly implicated in the sPLA2-X regulation of ILCs, it is also known that ILC2s express receptors for several lipid mediators that are known to be regulated by this enzyme, including PGD2 (46–48), CysLTs (49), and lipoxin A4 (50), which act to control activation and recruitment of ILC2s. We found a marked reduction in the generation of CysLTs and PGD2 in the airways of Pla2g10-deficient mice, which is particularly important as both IL-33 and the proximal CysLTs LTC4 and LTD4 provide important and nonredundant signals for ILC2 activation (51). These findings indicate that sPLA2-X serves as a central regulator of ILC2s in the lung, a finding that has substantial clinical relevance, given the recent evidence that ILC2s play an active role in the lungs of asthmatics (52–56).
We carefully assessed the morphological distribution of eosinophils in the airways using MBP immunostaining and semiautomated image analysis, and we verified alterations in eosinophil trafficking by multicolor flow cytometry. We clearly demonstrate that the lack of Pla2g10 in this model results in a decrease in trafficking of eosinophils to the lung, particularly a reduction in eosinophils residing in the peribronchial space. Of interest is the relatively modest effect of Pla2g10 deficiency on the allergen-induced production of IL-5 by lung leukocytes, suggesting that alterations in other chemotactic signals for eosinophils are affected by the lack of Pla2g10. The marked decrease in CysLTs may be responsible for these effects as eosinophils express the CysLT1 receptor for LTD4 (57, 58), which mediates eosinophil chemotaxis; further, inhalation of the terminal CysLT LTE4 leads to airway eosinophilia in human subjects (59, 60). Another potential chemotactic signal for eosinophils is PGD2 (61, 62), which was also reduced in the BALF of Pla2g10–/– animals.
Since we have previously demonstrated that the sPLA2-X enzyme is increased in the airways of individuals with asthma (1, 2) and is increased with allergen challenge in humans and in mice, the homeostatic function of the enzyme should be considered, as lipid mediators have both pro- and antiinflammatory effects and the hydrolysis of membranes can directly lead to cell cytotoxicity. For example, other sPLA2s such as sPLA2-IIA, which we have also found to be elevated in the airways of asthmatics (3), have high activity against gram-positive bacteria due in part to its high net-positive charge, allowing for penetration of the bacterial cell wall (63, 64). Although many of the sPLA2s have antibacterial properties, sPLA2-X is relatively unique because it acts strongly on the outer cell membrane of mammalian cells (65) and is generally thought to have less of a role on direct bacterial cytotoxicity (66). Our data indicate that sPLA2-X influences the innate immune responses through activation of macrophages and polarization of both resident and newly recruited macrophages toward an M2 phenotype expressing the transferrin (CD71) and mannose (CD206) receptors. Although M2-polarized macrophages function during the resolution phase of inflammation (37), studies implicate a pathogenic role for M2 macrophages in asthma (67). Prior studies have also demonstrated that sPLA2-V is necessary for the development of allergic inflammation in the HDM (68) and OVA/alum (69) models of allergic airways disease, and that while sPLA2-V is involved in maturation and antigen processing in DCs (68), macrophages expressing sPLA2-V are sufficient to transfer antigen sensitization mice (20). Studies in human macrophages support a role for sPLA2-V in phagocytosis and antigen processing (70). Given our findings of a marked alteration in the adaptive immune response to inhaled allergen, it will be important in future studies to clarify the respective roles of these sPLA2s in macrophage function.
Further evidence that lipid mediators regulate the polarization of macrophages comes from a recent report indicating that, in contrast to the loss of Pla2g10 or Pla2g5, peritoneal macrophages isolated from mice lacking iPLA2β (Pla2g6a) are more readily skewed toward an M2-like phenotype (71). A notable finding in our study was that the lack of Pla2g10 markedly altered the number of recruited macrophages to the lung — a significant finding, given recent studies that have demonstrated that recruited macrophages are central to the promotion of allergic inflammation (39, 72), while resident macrophages, which were not altered by the loss of Pla2g10, tend to suppress allergic inflammation (73, 74). Since our group and others have clearly demonstrated that human and murine macrophages express PLA2G10/Pla2g10 (1, 75, 76), further work will be needed to ascertain whether these functions are due to the loss of sPLA2-X within macrophages or to the lack of macrophage activation by sPLA2-X. In the present study, we found that stimulation of naive human monocyte–derived macrophages with HDM and hsPLA2-X caused an additive effect on IL-6 secretion, while the effects on the eicosanoid LTB4 were not additive. Because HDM is a complex mixture that contains c-type lectins, PAMPs, and DAMPs, as well as endogenous PLA2 activity (77), the results reveal both the importance of sPLA2s in macrophage activation and the complex interaction with heterogeneous allergens such as HDM.
Our results also raise important questions about the cell-specific function of sPLA2-X that cannot be fully addressed using mice with a global deletion of Pla2g10. Cell-specific deletion of Pla2g10 in epithelial cells and myeloid cells will be helpful to ascertain potential beneficial and deleterious effects of the loss of sPLA2-X function to fully appreciate the potential of this pathway as a therapeutic target in humans. Further potential for therapeutic targeting of sPLA2-X can be ascertained using our small-molecule inhibitor of human sPLA2-X, which can be utilized in our transgenic mouse model with the insertion of human PLA2G10 in place of murine Pla2g10 (43). Finally, it will be important to further tease out effects of sPLA2-X and other sPLA2s that, in addition to their enzymatic activity, bind to a C-type lectin receptor that is a paralog of the mannose receptor; we recently demonstrated that this receptor is overexpressed in the epithelium in human asthma and that the loss of this receptor increases AHR in the OVA/alum model (9).
In summary, we have demonstrated that sPLA2-X is increased in the airways, in response to inhaled allergen, in both humans and mice and that the sPLA2-X enzyme in mice plays a critical function in both the innate and adaptive immune response to inhaled allergen and airway dysfunction.
Methods
HDM-induced allergic airway disease.
We used mice with global deficiency of Pla2g10 (Pla2g10–/–) that were described previously (10) and maintained on a C57BL/6 background. WT and Pla2g10–/– mice were bred and maintained under specific pathogen-free conditions at the University of Washington and had ad libitum access to food and water. Female mice between 6 and 8 weeks old were used in all experiments to reduce heterogeneity. To induce allergic airway disease, mice were administered HDM extract (catalog RMB84M, 275533; Greer Laboratories) containing 100 μg on day 1 by oropharyngeal (o.p.) aspiration followed by 25 μg HDM extract by o.p. aspiration on days 8–12 and 15–19. Saline was administered as a control for each experiment. On day 20, mice were intubated via tracheotomy and subsequently ventilated at 200 breaths/minute using a flexiVent ventilator (SCIREQ Inc.). Following increasing doses of inhaled MCh (0, 6.25, 12.5, 25, and 50 mg), lung function was assessed and dynamic R and E were calculated using the single frequency forced oscillation technique (78).
Assessment of airway inflammation.
Postmortem lungs were lavaged via tracheal cannula with ice-cold PBS, and total BALF cell counts were determined using a Cellometer Auto 2000 (Nexcelom Bioscience). BALF was spun down, and cells were aliquoted for cell differential analysis via flow cytometry and the supernatant stored at –80°C with 1× protease inhibitor cocktail.
The left lung was minced in warmed digestion solution (RPMI [Corning Inc.] 1640 with 5% FCS [Gibco Laboratories], 0.5 mg/ml Liberase TL [Roche], and 40 U/ml DNase I [Sigma]). Digestion was stopped with cold HBSS containing 10 mM EDTA, and the digested tissue was pushed through a 40-μm cell strainer and washed several times with wash buffer (RPMI 1640 with 5% FCS). RBC were removed using ACK RBC lysis buffer (eBioscience). Lung digest was resuspended in wash buffer, and an aliquot was removed for cell count and viability using the Cellometer Auto 2000. Cells were centrifuged and resuspended in MACS buffer (HBSS without Ca and Mg, 2 mM EDTA and 0.5% BSA) containing CD45 MACS microbeads (Miltenyi Biotec). CD45+ lung leukocytes were isolated using a Miltenyi LS column and magnetic separator.
Multicolor flow cytometry for cell differentials.
Single cell suspensions from lung leukocytes and BALF cells were stained for cell differentials using a cocktail of the following antibodies: CD11c (N418, eBioscience), Siglec-F (E50-2440, BD Bioscience), Ly-6G (RB6-8C5, eBioscience), CD45 (30-F11, BioLegend), CD3e (145-2C11, eBioscience), and MHCII (M5/114.15.2, BD Biosciences). Cells were blocked using Fc block (BD Biosciences) and incubated with the primary antibody cocktail for 30 minutes at 4°C in the dark. Cells were washed and resuspended in PBS containing eFluor 450 fixable viability dye (eBioscience) and incubated for 15 minutes at 4°C in the dark. Cells were then fixed using 4% paraformaldehyde and stored at 4°C in MACS buffer until analyzed via flow cytometry. Cells were analyzed using a FACS LSR II flow cytometer (Becton Dickinson), and gating was performed using FlowJo software (TreeStar). Single stained controls of lung leukocytes were used to set voltages and for color compensation, and leukocyte populations were determined as previously reported (9). Briefly, our gating strategy consisted of first selecting singlets using forward scatter width and area, followed by selection of viable cells using the fixable viability dye. Leukocytes were then identified using side scatter and CD45. CD45+ cells were gated for CD3+ T cells, and the remaining CD3–CD45+ cells were further subdivided into MHCIIhigh and MHCIIlow populations. In the MHCIIlow population, eosinophils were identified as CD11c–SiglecF+ leukocytes, and neutrophils were identified as CD11c–SiglecF– leukocytes that were also Ly-6G+. In the MHCIIhigh population, CD11c–SiglecF– monocyte-derived macrophages (recruited macrophages) were identified. A population of MHCII-variable cells that expressed CD11c and SiglecF was also identified, consistent with resident macrophages. A similar gating strategy was used to identify leukocyte subsets in the BALF.
Multicolor flow cytometry for macrophage populations.
Single cell suspensions from whole lung digests were stained for the detection of macrophage populations using the following antibodies: CD11b (M1/70, BioLegend), CD11c (N418, BioLegend), Ly6G (1A8, BioLegend), CD45 (30-F11, BioLegend), CD71/TfR (R17217, BioLegend), SiglecF (E50-2440, BD Bioscience), and CD206/MR (C068C2, BioLegend). Cells were blocked using Fc Block (BD Biosciences) and incubated with the primary antibody cocktail for 1 hour at 4°C in the dark. Cells were washed and fixed in 5% formalin and stored at 4°C in MACS buffer until analyzed via flow cytometry. Cells were analyzed using a FACSCanto RUO system (BD Biosciences), and data were analyzed using FlowJo software. For gating, CD45+ leukocytes were selected and Ly6G+ PMNs were excluded. From the CD45+ fraction, alveolar macrophages were identified as CD11c+SiglecF+. After excluding alveolar macrophages and eosinophils (CD11c–SiglecF+), the recruited macrophages were identified as CD11b+CD11cint. Median MFI for CD71/TfR and CD206/MR was determined for each macrophage population, plus isotypes for CD71/TfR and CD206/MR. Data are reported as ΔMFI (calculated as MFI – MFI isotype).
Multicolor flow cytometry for ILC2s.
Single cell suspensions from lung leukocytes and BALF cells were stained for detection of ILC2s using the following antibodies: mouse hematopoietic lineage eFluor 450 cocktail (eBioscience) containing CD3 (17A2), CD45R/B220 (RA3-6B2), CD11b (M1/70), Ter119 (TER-119), and Gr-1/Ly6G (RB6-8C5) with the addition of eFluor 450–conjugated antibodies for CD4 (RM4-5, eBioscience), CD8a (53-6.7, eBioscience), CD11c (N418, eBioscience), CD19 (1D3, eBioscience), and NK1.1 (PK136, eBioscience). Further selection was performed on the lineage negative (Lin–) population using the following antibodies: CD45 (30-F11, eBioscience), T1/ST2 (DJ8, MD Biosciences), Sca-1 (D7, eBioscience), and CD127 (SB/199, BD Pharmingen).
Cells were blocked using Fc block (BD Biosciences) and incubated with the primary antibody cocktail for 30 minutes at 4°C in the dark. Cells were fixed in 4% paraformaldehyde for 5 minutes at room temperature and stored at 4°C in MACS buffer until analyzed via flow cytometry. Cells were analyzed using a FACS LSR II flow cytometer, and gating was performed using FlowJo software. Our gating strategy for ILC2s consisted of selecting singlets using forward scatter width and area, followed by selection of cells that were lineage negative (Lin–) and CD45+. The Lin–CD45+ population was then gated using forward scatter area and side scatter area to isolate lymphocytes. This population was further gated to enumerate ILC2s (Lin–CD45+ST2+, Lin–CD45+ST2+CD127+, and Lin–CD45+ST2+CD127+Sca-1+). Single stained controls of lung leukocytes were used to set voltages and for color compensation and leukocyte populations. Fluorescence minus one (FMO) controls were prepared by omitting CD127, Sca-1, or T1/ST2 antibodies to allow for proper compensation.
Quantitative morphometry of peribronchial eosinophils.
For H&E and eosinophil staining, the right lung was inflated to 20 cm H2O pressure, fixed with 4% paraformaldehyde, and embedded in paraffin. Paraffin-embedded lung tissue was cut into 4-μm thick sections and transferred to glass slides for histopathology analysis. For detection of eosinophils, slides were deparaffinized, treated with pepsin (Invitrogen), and blocked with peroxidase. Slides were then blocked using 10% normal goat serum and incubated with rat anti-MBP antibody, provided by James J. Lee (Mayo Clinic, Phoenix, Arizona, USA). MBP staining was visualized using a rabbit anti-rat detection antibody by the DAB method. Stained lung sections were digitalized using a Hamamatsu Nanozoomer whole slide imager, and quantification of MBP immunostaining in the lung was performed using Visiopharm software. To quantify peribronchial eosinophils, we first outlined the submucosal space surrounding the airways to define the peribronchial area. Within this defined area, we then utilized the Visiopharm software to quantify the number of eosinophils using segmentation and total MBP immunostaining using deconvolution.
Image analysis of mucus plugging.
Lung tissue was fixed using an adapted protocol that utilizes methyl Carnoy’s fixative (methacarn) (27). Animals were subjected to the full HDM protocol outlined above; on the final day, animals were euthanized, and the right lung was removed and immediately placed in methacarn. Prior to euthanization, mice were not subjected to MCh challenge, allowing for measurement of baseline epithelial mucus production following HDM exposure and prior to measurements of AHR. Tissues were embedded and prepared as described in the histological analysis section. For analysis of mucus plugging, raw counts of airways were performed and quantified based on the presence or absence of PAS+ staining in the airway lumen. From the raw values, the percentage of occluded airways was determined based on the number of occluded airways versus the total number of airways counted. We quantified the amount of PAS staining in the epithelium relative to the area of the epithelium.
Restimulation of cultured lung leukocytes.
For HDM restimulation of isolated lung leukocytes, 2 × 105 cells were plated in each well of a 96-well tissue culture plate. Cells were simulated with 100 μg/ml HDM or with saline as a control. Following stimulation for 24 hours, the cell culture supernatant was isolated and stored at –80°C with 1× protease inhibitor cocktail (Pierce). Cell culture supernatant was later analyzed for cytokine levels via ELISA.
Generation of mast cells from BM.
BM-derived mast cells (BMMCs) were isolated and expanded as described previously (79). Briefly, the femurs of WT and Pla2g10–/– mice were removed, the epiphyses were cut off, and the marrow was extracted via centrifugation. The collected BM was then pooled for each WT and Pla2g10–/– mouse, and cells were placed into sterile filtered culture medium (IMDM, Penicillin/streptomycin [Corning Inc.] containing 10% FBS, 2 mM L-glutamine [Gibco Laboratories], 1% antibiotic, and 50 μM β-mercapthoethanol [Sigma] and supplemented with 10 ng/ml IL-3 [PeproTech]). Medium was changed every 4 days until mast cells reached maturity at about 6 weeks. Maturity of mast cells was assessed using toluidine blue staining of fixed cytospins.
Cytokine analysis by ELISA.
Murine IL-33 protein content from BALF, lung tissue lysates, and cell culture supernatants was determined by ELISA per manufacturer’s instructions (R&D) and analyzed on a Biotek EL800 platereader. Detection of other murine cytokines (IL-4, IL-5, IL-13, IL-6) was performed using the Mouse V-PLEX Proinflammatory Panel 1 assay and the Mouse IL-13 Ultrasensitive assay per manufacturer’s instructions (Meso Scale Diagnostics), and plates were analyzed using a Sector S 600 plate reader.
Measurement of eicosanoids.
CysLT were measured using a CysLT ELISA kit per manufacturer’s instructions (Cayman Chemical). Briefly, fresh BALF supernatant was combined with 2 volumes of ice-cold methanol containing 0.2% formic acid and incubated on ice for 5 minutes. Samples were centrifuged at 4,000 g for 20 minutes at 4°C, and aliquots were added to 1.5-ml glass vials with Teflon septum–lined screw caps, overlaid with argon and stored at –80°C. CysLT ELISA was then performed following methanol evaporation. PGD2 was assayed in a separate set of samples using the PGD2-MOX ELISA kit per manufacturer’s instructions (Cayman Chemical).
Time-resolved fluorescence immunoassay of sPLA2-X.
Induced sputum samples were obtained from a repository of samples collected at baseline and after whole lung allergen challenges in subjects with allergic asthma (80, 81). Levels of sPLA2-X protein in induced sputum were determined by time-resolved fluorescence immunoassay (3), and recombinant human sPLA2-X was used to generate a linear standard curve (65). Confirmation of Pla2g10–/– mice was performed using the time-resolved fluorescence immunoassay, and a standard curve was generated using recombinant murine sPLA2-X.
Quantitative PCR.
Quantitative PCR (qPCR) was performed using TaqMan primer-probe sets on a Mastercycler ep Gradient S (Eppendorf) and quantified relative to an endogenous housekeeping gene using the ΔCt method. Primer-probe sets were obtained from Applied Biosystems using a FAM probe for Pla2g10 and Il13 and a primer-limited VIC probe for Hprt1 as the housekeeping gene.
Western blotting and densitometry.
BALF was prepared for analysis under reducing and denaturing conditions and probed with antibodies against eosinophil MBP (From James J. Lee, Mayo Clinic, Phoenix, Arizona, USA) or sPLA2-X (82). For detection of MBP and sPLA2-X, an equal volume of BALF was loaded onto a NuPage Bis/Tris 4-12% gel. For detection of sPLA2-X in BMMC lysates, 20 μg protein per sample was loaded onto a NuPage Bis/Tris 4-12% gel. Proteins were transferred onto PVDF membrane, blocked in 5% milk, and probed with an anti-MBP antibody (1:1,000) or anti-sPLA2-X antibody (1:1,000) overnight at 4°C with constant shaking. BMMC lysates were probed with a monoclonal antibody for sPLA2-X (1:1,000; clone EPR11202, Abcam). Membranes were then washed 3 times with TBST. For MBP, membranes were incubated in anti–rat HRP secondary antibody (1:10,000, Santa Cruz Biotechnology Inc.) in 5% milk for 1 hour. For sPLA2-X, membranes were incubated in anti–mouse HRP secondary antibody (1:10,000, Cell Signaling Technology) in 5% milk for 1 hour. Proteins were visualized using LumiGLO chemiluminescent substrate (Cell Signaling Technology), exposed to film, and developed using an automated film developer. Films were subjected to densitometric analysis using ImageJ (v1.47v, NIH, USA).
Statistics.
Differences in levels of IL-33 in the BALF and sPLA2-X protein in murine tissue were analyzed using an unpaired 2-tailed Student’s t test. Differences in Pla2g10 expression in murine lungs and sPLA2-X protein levels in BALF from WT mice assessed via Western blot and densitometric analysis were analyzed using the Mann-Whitney test. Induced sputum sPLA2-X levels were analyzed using the Friedman test with post hoc analysis using Dunn’s multiple comparisons test. Differences in features of inflammation including cell differentials, densitometric analysis of MBP in WT vs. Pla2g10–/– mice, cytokine production from restimulated leukocytes, mast cell gene expression, and immunoglobulin levels were assessed with a 2-way ANOVA with contrasts of allergen treatment and genotype. Differences in AHR to MCh were evaluated with a 2-way ANOVA test with contrasts between MCh dose and genotype. Post hoc analysis was performed at each dose after correction for multiple comparisons with Bonferroni’s test. For all tests, a P value less than 0.05 was considered significant.
Study approval.
All animal studies were approved by the University of Washington IACUC. For human induced sputum samples, written informed consent was obtained from all participants, and the Hamilton Integrated Research Ethics Board approved the study.
Author contributions
Jdn, AMM, GMG, WAA, and TSH conceived and designed studies; JDN, YL, HLO, RCM, DA, FG, GSN, and TSH performed experiments; JDN, YL, HLO, AMM, CWF, MHG, AMP, WAA, and TSH analyzed and interpreted the results of the experiments; JDN and TSH wrote the manuscript; JDN, AMM, CWF, MHG, GMG, AMP, WAA, and TSH edited and revised the manuscript; and all authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
The authors would like to thank the staff at the University of Washington Cell Analysis Facility, especially Michele Black and Thane Mittelstaedt for assistance with flow analysis, and the staff of the University of Washington Histology and Imaging Core, especially Brian Johnson and Megan Larmore for assistance with histological analyses. The authors appreciate the provision of the antibody for eosinophil MBP by James J. Lee from the Mayo Clinic (Phoenix, Arizona, USA).
This work was supported by grants from the NIH (R01HL089215 and K24AI130263 to TSH, R01HL122895 to WAA, and R37HL036235 to MHG).
Version 1. 11/02/2017
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2017;2(21):e94929. https://doi.org/10.1172/jci.insight.94929.
Contributor Information
Ying Lai, Email: ylai@uw.edu.
Herbert Luke Ogden, Email: ogdenhl91@gmail.com.
Dowon An, Email: dowon@uw.edu.
Farideh Ghomashchi, Email: farideh@u.washington.edu.
References
- 1.Hallstrand TS, Chi EY, Singer AG, Gelb MH, Henderson WR. Secreted phospholipase A2 group X overexpression in asthma and bronchial hyperresponsiveness. Am J Respir Crit Care Med. 2007;176(11):1072–1078. doi: 10.1164/rccm.200707-1088OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hallstrand TS, et al. Regulation and function of epithelial secreted phospholipase A2 group X in asthma. Am J Respir Crit Care Med. 2013;188(1):42–50. doi: 10.1164/rccm.201301-0084OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hallstrand TS, et al. Relationship between levels of secreted phospholipase A2 groups IIA and X in the airways and asthma severity. Clin Exp Allergy. 2011;41(6):801–810. doi: 10.1111/j.1365-2222.2010.03676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bezzine S, et al. Exogenously added human group X secreted phospholipase A(2) but not the group IB, IIA, and V enzymes efficiently release arachidonic acid from adherent mammalian cells. J Biol Chem. 2000;275(5):3179–3191. doi: 10.1074/jbc.275.5.3179. [DOI] [PubMed] [Google Scholar]
- 5.Murakami M, Taketomi Y, Miki Y, Sato H, Hirabayashi T, Yamamoto K. Recent progress in phospholipase A2 research: from cells to animals to humans. Prog Lipid Res. 2011;50(2):152–192. doi: 10.1016/j.plipres.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Lai Y, et al. Eosinophil cysteinyl leukotriene synthesis mediated by exogenous secreted phospholipase A2 group X. J Biol Chem. 2010;285(53):41491–41500. doi: 10.1074/jbc.M110.153338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hallstrand TS, et al. Endogenous secreted phospholipase A2 group X regulates cysteinyl leukotrienes synthesis by human eosinophils. J Allergy Clin Immunol. 2016;137(1):268–277.e8. doi: 10.1016/j.jaci.2015.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Granata F, et al. Activation of cytokine production by secreted phospholipase A2 in human lung macrophages expressing the M-type receptor. J Immunol. 2005;174(1):464–474. doi: 10.4049/jimmunol.174.1.464. [DOI] [PubMed] [Google Scholar]
- 9.Nolin JD, et al. Identification of Epithelial Phospholipase A2 Receptor 1 as a Potential Target in Asthma. Am J Respir Cell Mol Biol. 2016;55(6):825–836. doi: 10.1165/rcmb.2015-0150OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henderson WR, et al. Importance of group X-secreted phospholipase A2 in allergen-induced airway inflammation and remodeling in a mouse asthma model. J Exp Med. 2007;204(4):865–877. doi: 10.1084/jem.20070029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barrett NA, Maekawa A, Rahman OM, Austen KF, Kanaoka Y. Dectin-2 recognition of house dust mite triggers cysteinyl leukotriene generation by dendritic cells. J Immunol. 2009;182(2):1119–1128. doi: 10.4049/jimmunol.182.2.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barrett NA, et al. Dectin-2 mediates Th2 immunity through the generation of cysteinyl leukotrienes. J Exp Med. 2011;208(3):593–604. doi: 10.1084/jem.20100793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat Med. 2009;15(4):410–416. doi: 10.1038/nm.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hammad H, et al. Inflammatory dendritic cells--not basophils--are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. J Exp Med. 2010;207(10):2097–2111. doi: 10.1084/jem.20101563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klein Wolterink RG, et al. Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur J Immunol. 2012;42(5):1106–1116. doi: 10.1002/eji.201142018. [DOI] [PubMed] [Google Scholar]
- 16.Gold MJ, et al. Group 2 innate lymphoid cells facilitate sensitization to local, but not systemic, TH2-inducing allergen exposures. J Allergy Clin Immunol. 2014;133(4):1142–1148. doi: 10.1016/j.jaci.2014.02.033. [DOI] [PubMed] [Google Scholar]
- 17.Li BW, et al. T cells are necessary for ILC2 activation in house dust mite-induced allergic airway inflammation in mice. Eur J Immunol. 2016;46(6):1392–1403. doi: 10.1002/eji.201546119. [DOI] [PubMed] [Google Scholar]
- 18.Vroman H, et al. Development of eosinophilic inflammation is independent of B-T cell interaction in a chronic house dust mite-driven asthma model. Clin Exp Allergy. 2017;47(4):551–564. doi: 10.1111/cea.12834. [DOI] [PubMed] [Google Scholar]
- 19.Liu CF, Drocourt D, Puzo G, Wang JY, Riviere M. Innate immune response of alveolar macrophage to house dust mite allergen is mediated through TLR2/-4 co-activation. PLoS One. 2013;8(10):e75983. doi: 10.1371/journal.pone.0075983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohta S, Imamura M, Xing W, Boyce JA, Balestrieri B. Group V secretory phospholipase A2 is involved in macrophage activation and is sufficient for macrophage effector functions in allergic pulmonary inflammation. J Immunol. 2013;190(12):5927–5938. doi: 10.4049/jimmunol.1203202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palm NW, Rosenstein RK, Yu S, Schenten DD, Florsheim E, Medzhitov R. Bee venom phospholipase A2 induces a primary type 2 response that is dependent on the receptor ST2 and confers protective immunity. Immunity. 2013;39(5):976–985. doi: 10.1016/j.immuni.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Platts-Mills TA, Erwin EA, Heymann PW, Woodfolk JA. Pro: The evidence for a causal role of dust mites in asthma. Am J Respir Crit Care Med. 2009;180(2):109–113. doi: 10.1164/rccm.200811-1756PR. [DOI] [PubMed] [Google Scholar]
- 23.Webb DC, McKenzie AN, Matthaei KI, Rothenberg ME, Foster PS. Distinct spatial requirement for eosinophil-induced airways hyperreactivity. Immunol Cell Biol. 2001;79(2):165–169. doi: 10.1046/j.1440-1711.2001.00989.x. [DOI] [PubMed] [Google Scholar]
- 24.Shen HH, et al. A causative relationship exists between eosinophils and the development of allergic pulmonary pathologies in the mouse. J Immunol. 2003;170(6):3296–3305. doi: 10.4049/jimmunol.170.6.3296. [DOI] [PubMed] [Google Scholar]
- 25.Song DJ, et al. Anti-Siglec-F antibody reduces allergen-induced eosinophilic inflammation and airway remodeling. J Immunol. 2009;183(8):5333–5341. doi: 10.4049/jimmunol.0801421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacobsen EA, Lee NA, Lee JJ. Re-defining the unique roles for eosinophils in allergic respiratory inflammation. Clin Exp Allergy. 2014;44(9):1119–1136. doi: 10.1111/cea.12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evans CM, et al. The polymeric mucin Muc5ac is required for allergic airway hyperreactivity. Nat Commun. 2015;6:6281. doi: 10.1038/ncomms7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bossé Y, Riesenfeld EP, Paré PD, Irvin CG. It’s not all smooth muscle: non-smooth-muscle elements in control of resistance to airflow. Annu Rev Physiol. 2010;72:437–462. doi: 10.1146/annurev-physiol-021909-135851. [DOI] [PubMed] [Google Scholar]
- 29.Singer M, et al. A MARCKS-related peptide blocks mucus hypersecretion in a mouse model of asthma. Nat Med. 2004;10(2):193–196. doi: 10.1038/nm983. [DOI] [PubMed] [Google Scholar]
- 30.Kim DC, et al. Cysteinyl leukotrienes regulate Th2 cell-dependent pulmonary inflammation. J Immunol. 2006;176(7):4440–4448. doi: 10.4049/jimmunol.176.7.4440. [DOI] [PubMed] [Google Scholar]
- 31.Marom Z, Shelhamer JH, Bach MK, Morton DR, Kaliner M. Slow-reacting substances, leukotrienes C4 and D4, increase the release of mucus from human airways in vitro. Am Rev Respir Dis. 1982;126(3):449–451. doi: 10.1164/arrd.1982.126.3.449. [DOI] [PubMed] [Google Scholar]
- 32.Coles SJ, et al. Effects of leukotrienes C4 and D4 on glycoprotein and lysozyme secretion by human bronchial mucosa. Prostaglandins. 1983;25(2):155–170. doi: 10.1016/0090-6980(83)90101-6. [DOI] [PubMed] [Google Scholar]
- 33.Mehrotra AK, Henderson WR. The role of leukotrienes in airway remodeling. Curr Mol Med. 2009;9(3):383–391. doi: 10.2174/156652409787847209. [DOI] [PubMed] [Google Scholar]
- 34.Barlow JL, et al. Innate IL-13-producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. J Allergy Clin Immunol. 2012;129(1):191–8.e1. doi: 10.1016/j.jaci.2011.09.041. [DOI] [PubMed] [Google Scholar]
- 35.Halim TY, Krauss RH, Sun AC, Takei F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity. 2012;36(3):451–463. doi: 10.1016/j.immuni.2011.12.020. [DOI] [PubMed] [Google Scholar]
- 36.Price AE, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci USA. 2010;107(25):11489–11494. doi: 10.1073/pnas.1003988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnston LK, Rims CR, Gill SE, McGuire JK, Manicone AM. Pulmonary macrophage subpopulations in the induction and resolution of acute lung injury. Am J Respir Cell Mol Biol. 2012;47(4):417–426. doi: 10.1165/rcmb.2012-0090OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Becker L, et al. Unique proteomic signatures distinguish macrophages and dendritic cells. PLoS One. 2012;7(3):e33297. doi: 10.1371/journal.pone.0033297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zasłona Z, et al. Resident alveolar macrophages suppress, whereas recruited monocytes promote, allergic lung inflammation in murine models of asthma. J Immunol. 2014;193(8):4245–4253. doi: 10.4049/jimmunol.1400580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bedoret D, et al. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergy in mice. J Clin Invest. 2009;119(12):3723–3738. doi: 10.1172/JCI39717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams JW, Tjota MY, Sperling AI. The contribution of allergen-specific IgG to the development of th2-mediated airway inflammation. J Allergy (Cairo) 2012;2012:236075. doi: 10.1155/2012/236075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Henderson WR, et al. Blockade of human group X secreted phospholipase A2 (GX-sPLA2)-induced airway inflammation and hyperresponsiveness in a mouse asthma model by a selective GX-sPLA2 inhibitor. J Biol Chem. 2011;286(32):28049–28055. doi: 10.1074/jbc.M111.235812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Byers DE, Holtzman MJ. Alternatively activated macrophages and airway disease. Chest. 2011;140(3):768–774. doi: 10.1378/chest.10-2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klein Wolterink RG, et al. Essential, dose-dependent role for the transcription factor Gata3 in the development of IL-5+ and IL-13+ type 2 innate lymphoid cells. Proc Natl Acad Sci USA. 2013;110(25):10240–10245. doi: 10.1073/pnas.1217158110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xue L, et al. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J Allergy Clin Immunol. 2014;133(4):1184–1194. doi: 10.1016/j.jaci.2013.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wojno ED, et al. The prostaglandin D2 receptor CRTH2 regulates accumulation of group 2 innate lymphoid cells in the inflamed lung. Mucosal Immunol. 2015;8(6):1313–1323. doi: 10.1038/mi.2015.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang JE, Doherty TA, Baum R, Broide D. Prostaglandin D2 regulates human type 2 innate lymphoid cell chemotaxis. J Allergy Clin Immunol. 2014;133(3):899–901.e3. doi: 10.1016/j.jaci.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doherty TA, Khorram N, Lund S, Mehta AK, Croft M, Broide DH. Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J Allergy Clin Immunol. 2013;132(1):205–213. doi: 10.1016/j.jaci.2013.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barnig C, et al. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci Transl Med. 2013;5(174):174ra26. doi: 10.1126/scitranslmed.3004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.von Moltke J, O’Leary CE, Barrett NA, Kanaoka Y, Austen KF, Locksley RM. Leukotrienes provide an NFAT-dependent signal that synergizes with IL-33 to activate ILC2s. J Exp Med. 2017;214(1):27–37. doi: 10.1084/jem.20161274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mjösberg JM, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. 2011;12(11):1055–1062. doi: 10.1038/ni.2104. [DOI] [PubMed] [Google Scholar]
- 53.Bartemes KR, Kephart GM, Fox SJ, Kita H. Enhanced innate type 2 immune response in peripheral blood from patients with asthma. J Allergy Clin Immunol. 2014;134(3):671–678.e4. doi: 10.1016/j.jaci.2014.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Christianson CA, et al. Persistence of asthma requires multiple feedback circuits involving type 2 innate lymphoid cells and IL-33. J Allergy Clin Immunol. 2015;136(1):59–68.e14. doi: 10.1016/j.jaci.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kabata H, Moro K, Koyasu S, Asano K. Group 2 innate lymphoid cells and asthma. Allergol Int. 2015;64(3):227–234. doi: 10.1016/j.alit.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 56.Smith SG, et al. Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J Allergy Clin Immunol. 2016;137(1):75–86.e8. doi: 10.1016/j.jaci.2015.05.037. [DOI] [PubMed] [Google Scholar]
- 57.Figueroa DJ, et al. Expression of the cysteinyl leukotriene 1 receptor in normal human lung and peripheral blood leukocytes. Am J Respir Crit Care Med. 2001;163(1):226–233. doi: 10.1164/ajrccm.163.1.2003101. [DOI] [PubMed] [Google Scholar]
- 58.Zhu J, et al. Localization and upregulation of cysteinyl leukotriene-1 receptor in asthmatic bronchial mucosa. Am J Respir Cell Mol Biol. 2005;33(6):531–540. doi: 10.1165/rcmb.2005-0124OC. [DOI] [PubMed] [Google Scholar]
- 59.Mulder A, Gauvreau GM, Watson RM, O’byrne PM. Effect of inhaled leukotriene D4 on airway eosinophilia and airway hyperresponsiveness in asthmatic subjects. Am J Respir Crit Care Med. 1999;159(5 Pt 1):1562–1567. doi: 10.1164/ajrccm.159.5.9810095. [DOI] [PubMed] [Google Scholar]
- 60.Gauvreau GM, Parameswaran KN, Watson RM, O’Byrne PM. Inhaled leukotriene E(4), but not leukotriene D(4), increased airway inflammatory cells in subjects with atopic asthma. Am J Respir Crit Care Med. 2001;164(8 Pt 1):1495–1500. doi: 10.1164/ajrccm.164.8.2102033. [DOI] [PubMed] [Google Scholar]
- 61.Hirai H, et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med. 2001;193(2):255–261. doi: 10.1084/jem.193.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Monneret G, Gravel S, Diamond M, Rokach J, Powell WS. Prostaglandin D2 is a potent chemoattractant for human eosinophils that acts via a novel DP receptor. Blood. 2001;98(6):1942–1948. doi: 10.1182/blood.V98.6.1942. [DOI] [PubMed] [Google Scholar]
- 63.Weiss JP. Molecular determinants of bacterial sensitivity and resistance to mammalian Group IIA phospholipase A2. Biochim Biophys Acta. 2015;1848(11 Pt B):3072–3077. doi: 10.1016/j.bbamem.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beers SA, Buckland AG, Koduri RS, Cho W, Gelb MH, Wilton DC. The antibacterial properties of secreted phospholipases A2: a major physiological role for the group IIA enzyme that depends on the very high pI of the enzyme to allow penetration of the bacterial cell wall. J Biol Chem. 2002;277(3):1788–1793. doi: 10.1074/jbc.M109777200. [DOI] [PubMed] [Google Scholar]
- 65.Singer AG, et al. Interfacial kinetic and binding properties of the complete set of human and mouse groups I, II, V, X, and XII secreted phospholipases A2. J Biol Chem. 2002;277(50):48535–48549. doi: 10.1074/jbc.M205855200. [DOI] [PubMed] [Google Scholar]
- 66.Koduri RS, et al. Bactericidal properties of human and murine groups I, II, V, X, and XII secreted phospholipases A(2) J Biol Chem. 2002;277(8):5849–5857. doi: 10.1074/jbc.M109699200. [DOI] [PubMed] [Google Scholar]
- 67.Girodet PO, et al. Alternative Macrophage Activation Is Increased in Asthma. Am J Respir Cell Mol Biol. 2016;55(4):467–475. doi: 10.1165/rcmb.2015-0295OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Giannattasio G, Fujioka D, Xing W, Katz HR, Boyce JA, Balestrieri B. Group V secretory phospholipase A2 reveals its role in house dust mite-induced allergic pulmonary inflammation by regulation of dendritic cell function. J Immunol. 2010;185(7):4430–4438. doi: 10.4049/jimmunol.1001384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Henderson WR, et al. Key role of group v secreted phospholipase A2 in Th2 cytokine and dendritic cell-driven airway hyperresponsiveness and remodeling. PLoS One. 2013;8(2):e56172. doi: 10.1371/journal.pone.0056172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rubio JM, Rodríguez JP, Gil-de-Gómez L, Guijas C, Balboa MA, Balsinde J. Group V secreted phospholipase A2 is upregulated by IL-4 in human macrophages and mediates phagocytosis via hydrolysis of ethanolamine phospholipids. J Immunol. 2015;194(7):3327–3339. doi: 10.4049/jimmunol.1401026. [DOI] [PubMed] [Google Scholar]
- 71.Ashley JW, et al. Polarization of Macrophages toward M2 Phenotype Is Favored by Reduction in iPLA2β (Group VIA Phospholipase A2) J Biol Chem. 2016;291(44):23268–23281. doi: 10.1074/jbc.M116.754945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gundra UM, et al. Alternatively activated macrophages derived from monocytes and tissue macrophages are phenotypically and functionally distinct. Blood. 2014;123(20):e110–e122. doi: 10.1182/blood-2013-08-520619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Upham JW, Strickland DH, Bilyk N, Robinson BW, Holt PG. Alveolar macrophages from humans and rodents selectively inhibit T-cell proliferation but permit T-cell activation and cytokine secretion. Immunology. 1995;84(1):142–147. [PMC free article] [PubMed] [Google Scholar]
- 74.Holt PG, et al. Downregulation of the antigen presenting cell function(s) of pulmonary dendritic cells in vivo by resident alveolar macrophages. J Exp Med. 1993;177(2):397–407. doi: 10.1084/jem.177.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Giannattasio G, et al. Expression of phospholipases A2 in primary human lung macrophages: role of cytosolic phospholipase A2-alpha in arachidonic acid release and platelet activating factor synthesis. Biochim Biophys Acta. 2009;1791(2):92–102. doi: 10.1016/j.bbalip.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shridas P, Bailey WM, Talbott KR, Oslund RC, Gelb MH, Webb NR. Group X secretory phospholipase A2 enhances TLR4 signaling in macrophages. J Immunol. 2011;187(1):482–489. doi: 10.4049/jimmunol.1003552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jarrett R, et al. Filaggrin inhibits generation of CD1a neolipid antigens by house dust mite-derived phospholipase. Sci Transl Med. 2016;8(325):325ra18. doi: 10.1126/scitranslmed.aad6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bates JH, Wagers SS, Norton RJ, Rinaldi LM, Irvin CG. Exaggerated airway narrowing in mice treated with intratracheal cationic protein. J Appl Physiol. 2006;100(2):500–506. doi: 10.1152/japplphysiol.01013.2005. [DOI] [PubMed] [Google Scholar]
- 79.Kalesnikoff J, Galli SJ. Antiinflammatory and immunosuppressive functions of mast cells. Methods Mol Biol. 2011;677:207–220. doi: 10.1007/978-1-60761-869-0_15. [DOI] [PubMed] [Google Scholar]
- 80.Pizzichini E, et al. Indices of airway inflammation in induced sputum: reproducibility and validity of cell and fluid-phase measurements. Am J Respir Crit Care Med. 1996;154(2 Pt 1):308–317. doi: 10.1164/ajrccm.154.2.8756799. [DOI] [PubMed] [Google Scholar]
- 81.Gauvreau GM, Watson RM, O’Byrne PM. Kinetics of allergen-induced airway eosinophilic cytokine production and airway inflammation. Am J Respir Crit Care Med. 1999;160(2):640–647. doi: 10.1164/ajrccm.160.2.9809130. [DOI] [PubMed] [Google Scholar]
- 82.Degousee N, et al. Groups IV, V, and X phospholipases A2s in human neutrophils: role in eicosanoid production and gram-negative bacterial phospholipid hydrolysis. J Biol Chem. 2002;277(7):5061–5073. doi: 10.1074/jbc.M109083200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.