

Abstract
Increased sugar consumption is a risk factor for the metabolic syndrome including obesity, hypertriglyceridemia, insulin resistance, diabetes, and nonalcoholic fatty liver disease (NAFLD). Carbohydrate responsive element–binding protein (ChREBP) is a transcription factor that responds to sugar consumption to regulate adaptive metabolic programs. Hepatic ChREBP is particularly responsive to fructose and global ChREBP-KO mice are intolerant to diets containing fructose. It has recently been suggested that ChREBP protects the liver from hepatotoxicity following high-fructose diets (HFrDs). We directly tested this hypothesis using tissue-specific ChREBP deletion. HFrD increased adiposity and impaired glucose homeostasis in control mice, responses that were prevented in liver-specific ChREBP-KO (LiChKO) mice. Moreover, LiChKO mice tolerated chronic HFrD without marked weight loss or hepatotoxicity. In contrast, intestine-specific ChREBP-KO (IChKO) mice rapidly lost weight after transition to HFrD, and this was associated with dilation of the small intestine and cecum, suggestive of malabsorption. These findings were associated with downregulation of the intestinal fructose transporter, Slc2a5, which is essential for fructose tolerance. Altogether, these results establish an essential role for intestinal, but not hepatic, ChREBP in fructose tolerance.
Keywords: Metabolism
Keywords: Intermediary metabolism, Mouse models, Transcription
The transcription factor ChREBP is required in the intestine for fructose tolerance whereas deletion of hepatic ChREBP protects against fructose-induced disease.
Introduction
Diets high in sugar increase risk for the metabolic syndrome including obesity, nonalcoholic fatty liver disease (NAFLD), hypertriglyceridemia, and insulin resistance, which in turn contribute to risk for type 2 diabetes and cardiovascular disease (1–5). Sucrose and high-fructose corn syrup, the 2 major forms of sugar added to Western diets, are composed of approximately equal amounts of glucose and fructose. The fructose component of added sugar may be particularly deleterious when consumed in large amounts. Excessive fructose consumption can quickly cause features of metabolic syndrome in animal models (6). Likewise, fructose, but not glucose overfeeding can exacerbate metabolic syndrome in human subjects (7). The mechanisms by which excessive sugar and fructose overconsumption contribute to distinct components of the metabolic syndrome remain uncertain. Identifying these mechanisms may lead to novel diagnostic and therapeutic strategies.
Carbohydrate responsive element–binding protein (ChREBP, also known as MLXIPL) is a transcription factor expressed in key metabolic tissues that senses intracellular carbohydrate metabolites and regulates metabolic gene expression programs including glycolytic, lipogenic, fructolytic, and gluconeogenic enzymes (8, 9). We and others have shown that hepatic ChREBP is particularly responsive to fructose consumption and that fructose-mediated activation of ChREBP may contribute to hepatic steatosis, hypertriglyceridemia, and both hepatic and peripheral insulin resistance (10–12). Recent data indicate that sugar- or fructose-mediated activation of ChREBP also regulates secretion of hepatokines like FGF21, which contribute to adaptive metabolic responses to sugar consumption, including regulation of macronutrient preference and progression of NAFLD (13–17). Confirming the importance of ChREBP to fructose metabolism, whole-body ChREBP-knockout (ChREBP-KO) mice are intolerant to diets containing fructose and become moribund within 1 to 2 weeks (8). We have previously demonstrated that food intake is curtailed in ChREBP-KO mice within 1 to 2 days after transitioning to a high-fructose diet (HFrD) and the attendant weight loss likely contributes to their morbidity and mortality (11). Recently, Zhang et al., using global ChREBP-KO mice suggested that hepatic ChREBP is required to prevent fructose-induced endoplasmic reticulum (ER) stress and severe hepatotoxicity, and plays a protective role in the progression of steatosis to more advanced forms of NAFLD like nonalcoholic steatohepatitis (NASH) or cirrhosis (18).
As ChREBP is expressed in multiple metabolic tissues including liver, adipose tissue, small intestine, kidney, and skeletal muscle (8), we sought to examine where ChREBP expression is essential for fructose tolerance by conditionally deleting ChREBP in selected tissues. Here, we demonstrate that hepatic ChREBP is not required for fructose tolerance. Liver-specific ChREBP-KO mice (LiChKO) tolerate HFrD without evidence of significant liver inflammation. Rather, intestinal ChREBP is essential for fructose tolerance and intestine-specific ChREBP-KO (IChKO) mice recapitulate the fructose-mediated toxicity observed in global ChREBP-KO mice.
Results
Hepatic ChREBP-KO mice are fructose tolerant.
To determine whether hepatic ChREBP is essential for fructose tolerance, we generated LiChKO mice. We confirmed the loss of ChREBP protein expression in liver, but not adipose tissue of LiChKO mice (Figure 1A), and assessed loss of liver ChREBP function by performing gavage with water or fructose. Hepatic ChREBP is required for basal expression of glycolytic, gluconeogenic, lipogenic, and fructolytic targets and fructose gavage acutely and robustly increased expression of the ChREBP-β isoform as well as ChREBP targets in control, but not LiChKO mice (Figure 1B). In contrast, fructose gavage robustly increased ChREBP-β expression and ChREBP targets in intestine of both control and LiChKO mice (Figure 1B). This demonstrated that fructose can robustly and acutely activate ChREBP and its targets in the small intestine as previously shown in liver (11) and confirmed the specificity of liver ChREBP KO in our model.
Figure 1. Hepatic deletion of ChREBP leads to blunted effects of fructose feeding on body weight and hepatic gene expression.

(A) Western blot for carbohydrate responsive element–binding protein (ChREBP) protein in liver and perigonadal (PG) adipose tissue whole-cell lysates of control (CTL) and liver-specific ChREBP-KO (LiChKO) mice. (B) Eight-week-old female control and LiChKO mice were fasted for 6 hours, and then gavaged with water or fructose and sacrificed 100 minutes later and hepatic and intestinal gene expression was measured (n = 4 per group). (C and D) Control and LiChKO male mice were fed chow or high-fructose diet (HFrD) starting at 7 weeks of age (n = 5 per group) and body weight was measured weekly. (D) Body composition (EchoMRI) was measured after 8 weeks. Body weights are presented as mean ± SEM. All other data are presented as box-and-whisker plots where the line in the box indicates the median, the box extends from the 25th to 75th percentiles, and the whiskers indicate the minimal and maximal values. P values were obtained using 2-way ANOVA. *P < 0.05 compared between water and fructose within the same genotype; #P < 0.05 compared between different genotypes within the same treatment group.
Hereditary fructose intolerance (HFI) is a genetic disease caused by mutations in the key fructolytic enzyme aldolase B (ALDOB) (19). Subjects with this disease develop profound hepatotoxicity when challenged with diets containing fructose and this has been replicated in Aldob-KO mice (20). As hepatic Aldob is downregulated in global ChREBP-KO mice (11), we initially speculated that the fructose intolerance observed in those mice might be analogous to fructose-induced hepatotoxicity observed in HFI. To test this, we subjected LiChKO mice to HFrD. Contrary to our expectations, LiChKO mice tolerated HFrD without difficulty, even though their Aldob expression was lower than that of controls (Figure 1B). LiChKO mice on HFrD gained only slightly less weight than controls over the course of 9 weeks (Figure 1C), and consumed HFrD for 9 months without excess mortality (data not shown). HFrD caused a loss of lean mass in both control and LiChKO mice (Figure 1D). Fat mass doubled in control mice fed HFrD compared with chow, but this increase was not observed in LiChKO mice, thus accounting for their decreased body weight. To investigate the etiology of the diminished accretion of fat in LiChKO mice on HFrD, we performed indirect calorimetry and food intake studies (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.96703DS1). We observed modestly increased energy expenditure in controls fed HFrD compared with controls fed chow and LiChKO fed HFrD. LiChKO fed HFrD ate less than control fed HFrD, which may have contributed to their modest deficit in weight gain and fat mass (Supplemental Figure 2).
We previously demonstrated that hepatic ChREBP plays a central role in mediating hepatocellular hexose-phosphate homeostasis (11). Fructose-induced upregulation of ChREBP induces hepatic glucose-6-phosphatase (G6pc) expression to stimulate glucose production from ingested fructose. However, due to the severe limitations in fructose tolerance in global ChREBP-KO mice, we were previously unable to assess indices of glucose homeostasis and glucose production in mice lacking ChREBP and chronically exposed to HFrD. Since LiChKO mice are tolerant of HFrD, we were able to assess aspects of glucose homeostasis in this model. Neither diet nor genotype affected overnight-fasted blood glucose levels (Figure 2A). However, HFrD increased plasma insulin levels in control mice, consistent with systemic insulin resistance, an effect that was abrogated in LiChKO mice fed HFrD. Hepatic G6pc mRNA levels increased in control mice after fructose administration (Figure 1B), and this was associated with a 2- to 3-fold increase in G6PC protein levels compared with chow (Figure 2B). There was no increase of G6PC in LiChKO mice, and the absence of G6PC upregulation in LiChKO on HFrD likely contributed to marked glycogen accumulation (Figure 2C). To confirm the functional significance of increased G6PC expression, we performed a glycerol tolerance test. The glycemic excursion in HFrD control mice following administration of glycerol exceeded that of chow-fed controls, a response that was prevented in HFrD-fed LiChKO mice (Figure 2D). These results support our prior evidence demonstrating a role for fructose-induced, ChREBP-mediated G6PC upregulation as a determinant of hepatic glucose production that may contribute to hepatic insulin resistance and glucose intolerance.
Figure 2. LiChKO mice demonstrate blunted increase in hepatic glucose production in response to fructose feeding.

Control (CTL) and liver-specific ChREBP-KO (LiChKO) male mice were fed chow or high-fructose diet (HFrD) starting at 7 weeks of age (n = 5 per group). (A) Plasma glucose and insulin were measured in overnight-fasted control and LiChKO mice after 8 weeks of diet. (B) Hepatic expression of G6PC protein was determined in a subset of animals by Western blot with P85 subunit of PI3K used as a loading control (n = 2–3 per group). (C) Liver glycogen content was measured at the end of the study. (D) Glycerol tolerance test after 6 weeks on their respective diets. Glycemia during glycerol tolerance test is presented as mean ± SEM. All other data are presented as box-and-whisker plots where the line in the box indicates the median, the box extends from the 25th to 75th percentiles, and the whiskers indicate the minimal and maximal values. P values were obtained using 2-way ANOVA. *P < 0.05 compared between water and fructose within the same genotype; #P < 0.05 compared between different genotypes within the same treatment group. ChREBP, carbohydrate responsive element–binding protein.
Fructose-induced de novo lipogenesis and liver cholesterol are reduced in LiChKO mice.
The expression of hepatic de novo lipogenesis (DNL) enzymes is coregulated by the transcription factors ChREBP and sterol regulatory element–binding protein 1 (SREBP1, also known as SREBF1). In addition to activation of ChREBP, fructose may enhance lipogenic enzyme expression by promoting cleavage of the immature, inactive form of SREBP1 to the mature active form that localizes to the nucleus (21, 22). We assessed SREBP1 activation by assessing the abundance of the immature, cytosolic SREBP1 protein and the mature, nuclear SREBP1 protein by immunoblotting. The quality of nuclear versus cytosolic fractionation was confirmed by immunoblots for nuclear and cytosolic markers (Supplemental Figure 3). The abundance of immature SREBP1 protein tended to be lower in LiChKO mice and more so in LiChKO mice on HFrD compared with chow (Figure 3, B and C). Normalized to the immature cytosolic SREBP1 protein, fructose feeding increased cleavage of SREBP1 in control mice and tended to increase cleavage in LiChKO mice, but this was insufficient to increase total nuclear SREBP1 protein in LiChKO on HFrD beyond that of chow-fed control mice. Altogether, these results suggest that ChREBP affects SREBP1 abundance, but that fructose-mediated cleavage of SREBP1 is partially independent of ChREBP.
Figure 3. LiChKO mice fail to increase de novo lipogenesis or cholesterol synthesis in response to high-fructose feeding.

(A) Hepatic mRNA expression of lipogenic genes. (B) Western blots of SREBP1 in hepatic cytoplasmic (top) and nuclear (bottom) fractions quantified in (C) were measured in control (CTL) and liver-specific ChREBP-KO (LiChKO) male mice fed standard chow or high-fructose diet (HFrD) starting at 6–8 weeks of age for 10 weeks (n = 4–5 per group). (D) De novo lipogenesis (DNL) rates were measured by incorporation of 3H2O into saponifiable fraction of hepatic lipids in female control and LiChKO mice after 10 weeks of chow or HFrD, (n = 3–4 per group). (E) Hepatic triglycerides, (F) liver mRNA, and (G) hepatic total and free cholesterol were measured in mice as described in A–C. Data are presented as box-and-whisker plots where the line in the box indicates the median, the box extends from the 25th to 75th percentiles, and the whiskers indicate the minimal and maximal values. P values were obtained using 2-way ANOVA. *P < 0.05 compared between water and fructose within the same genotype; #P < 0.05 compared between different genotypes within the same treatment group. ChREBP, carbohydrate responsive element–binding protein.
Consistent with the lipogenic enzyme expression data (Figure 3A), HFrD increased fatty acid synthesis by 70% in control mice (Figure 3D), but not in LiChKO mice. Although DNL may contribute to steatosis, newly synthesized fatty acids constitute a minor fraction of fatty acids in the hepatic triglyceride pool (23). Despite the reduced hepatic DNL in LiChKO mice, hepatic triglyceride content in both control and LiChKO mice increased with HFrD compared with their chow-fed controls (Figure 3E). This indicates that fructose can contribute to steatosis independently of DNL, and ChREBP. It has been suggested that ChREBP suppresses SREBP2, and ChREBP deficiency leads to induction of SREBP2 and its transcriptional targets involved in cholesterol biosynthesis (18). In contrast with this hypothesis, we did not observe an increase in Srebp2 mRNA expression in LiChKO mice (Figure 3F). Moreover, while HFrD induced expression of enzymes involved in cholesterol synthesis, this was dependent on the presence of ChREBP and was not observed in LiChKO mice (Figure 3F). Consistent with this, HFrD increased hepatic total and free cholesterol in control mice but not in LiChKO mice (Figure 3G). Thus, ChREBP appears to enhance rather than repress SREBP2 activity in the setting of HFrD.
LiChKO mice do not develop hepatic inflammation on a HFrD.
To assess whether HFrD caused hepatotoxicity in LiChKO mice, we measured circulating transaminase levels. HFrD did not increase aspartate aminotransferase (AST) in either genotype (Figure 4A), although LiChKO mice fed HFrD had higher serum alanine aminotransferase (ALT) levels than their chow-fed counterparts (Figure 4A). However, the ALT levels remained within the normative range and far below the 100- to 1,000-fold increase in ALT recently reported in global ChREBP-KO mice subjected to several weeks of HFrD (18). We next determined whether hepatic ChREBP was necessary to prevent fructose-induced hepatic ER stress. We found that HFrD had little effect on markers of ER stress measured by quantitative PCR (qPCR) (Figure 4B). Moreover, HFrD tended to decrease expression of activated X-box-binding protein 1 (Xbp1s), a key regulatory factor in the ER stress response, in control livers and this was accentuated in livers from the LiChKO group (Figure 4B). We found no evidence of increased DNA damage–inducible transcript 3 (DDIT3, also known as CHOP) protein, another marker of ER stress, with HFrD in either genotype (Figure 4C). Additionally, we observed no evidence of caspase-3 (CASP3) cleavage, a marker of inflammation-induced apoptosis, in either genotype on HFrD (Figure 4D). Lastly, we observed no microscopic evidence of inflammatory infiltrate in hepatic tissue sections from control or LiChKO mice after 10 weeks on HFrD (Figure 4E). Taken together, these results indicate that hepatic ChREBP is not essential for systemic fructose tolerance, and lack of hepatic ChREBP does not cause fructose-induced hepatic hypercholesterolemia, ER stress, or marked hepatotoxicity, although there is evidence of modest transaminitis of uncertain etiology and unclear significance.
Figure 4. LiChKO mice do not develop ER stress or liver inflammation with high-fructose feeding.

(A) Serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) activity levels, (B) hepatic mRNA expression of genes involved in ER stress, immunoblots for (C) DNA damage–inducible transcript 3 (DDIT3) and (D) caspase-3 (CASP3) performed in the control (CTL) and liver-specific ChREBP-KO (LiChKO) mice described in Figure 3A. (E) Representative images of H&E–stained liver sections taken at ×10 magnification. Data are presented as box-and-whisker plots where the line in the box indicates the median, the box extends from the 25th to 75th percentiles, and the whiskers indicate the minimal and maximal values. P values were obtained using 2-way ANOVA. *P < 0.05 compared between water and fructose within the same genotype; #P < 0.05 compared between different genotypes within the same treatment group. ChREBP, carbohydrate responsive element–binding protein.
Intestinal ChREBP is required for fructose tolerance.
The relatively benign effects of HFrD in LiChKO mice contrasted with the rapid toxicity observed in global ChREBP-KO mice when challenged with HFrD. This suggested that fructose intolerance in global ChREBP-KO mice might be attributed to loss of ChREBP in other tissues. As observed in Figure 1B, ChREBP-β and canonical ChREBP targets including G6pc and Aldob are rapidly induced in the small intestine following fructose gavage. In fact, in control mice, fructose, but not glucose, gavage increased ChREBP targets comparably in the intestine and liver (Supplemental Figure 4 and ref. 11). To determine whether intestinal ChREBP might be important for fructose tolerance, we generated intestine-specific ChREBP-KO mice (IChKO), and confirmed that ChREBP protein is absent in the intestine, but present in the liver of IChKO mice (Figure 5A).
Figure 5. Deletion of ChREBP in the small intestine results in fructose intolerance.

(A) Western blot for carbohydrate responsive element–binding protein (ChREBP) in jejunal and liver whole-cell lysates of control (CTL) and intestine-specific ChREBP-KO (IChKO) mice. (B) Body weight and (C) food intake were measured in 6- to 8-week-old male and female control and IChKO mice fed high-fructose diet (HFrD) for 36 hours (n = 3 per group). (D) Eight-week-old female control and IChKO mice were fasted for 6 hours, and then gavaged with water or fructose and sacrificed 100 minutes later. Jejunal gene expression (n = 3 per group). (E) Representative images of small intestine and cecum in situ and (F) H&E–stained jejunal sections of control and IChKO after 36 hours of HFrD taken at ×10 magnification from mice described in B, as well as (G) serum TNF-α levels, (H) hepatic mRNA expression, and (I) serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) activity. Data are presented as box-and-whisker plots where the line in the box indicates the median, the box extends from the 25th to 75th percentiles, and the whiskers indicate the minimal and maximal values. P values were obtained using 2-way ANOVA. *P < 0.05 compared between water and fructose within the same genotype; #P < 0.05 compared between different genotypes within the same treatment group.
We previously reported that after starting HFrD, whole-body ChREBP-KO mice lost approximately 20% of their body weight within 1 to 2 days. Similarly, IChKO mice approached 20% weight loss within 36 hours after transitioning to HFrD (Figure 5B), accompanied by a greater than 50% reduction in food intake (Figure 5C). Analysis of intestinal gene expression at the end of this period showed marked reductions in the expression of fructolytic, glycolytic, and gluconeogenic enzymes including the intestinal fructose transporter Slc2a5 (also known as Glut5) in IChKO mice (Figure 5D). Barone et al. have previously demonstrated that Slc2a5-KO mice are intolerant to HFrDs and develop a malabsorption syndrome (24). On visual examination, the cecum and small intestine of IChKO mice challenged with HFrD are grossly enlarged, consistent with a malabsorption syndrome (Figure 5E). No overt inflammation or epithelial destruction was noted on histological examination of the small intestine (Figure 5F).
We speculated that intestinal dysfunction might secondarily lead to systemic illness, which might lead to sepsis and cause hepatic inflammation and hepatotoxicity (25, 26). Consistent with this, we observed a significant increase in circulating TNF-α levels in IChKO mice 36 hours after transitioning to HFrD (Figure 5G). We also observed a modest increase in mRNA expression of Bcl2l11, a proapoptotic factor and early marker of sepsis in livers from HFrD-fed IChKO mice (Figure 5H) (25). However, we did not observe evidence of transaminitis at this early time point (Figure 5I).
Twenty percent weight loss is a widely used humane endpoint in mouse toxicity studies and is the limit of weight loss permitted at our institutions. Therefore, we subsequently challenged IChKO mice and controls to HFrD and generated a “survival curve,” euthanizing mice when they lost 20% of their body weight and terminating the remainder of the cohort when more than half the group achieved this threshold (Figure 6A). Over half of the IChKO mice approached 20% weight loss within 5 days. No control mice reached this terminal endpoint. A modest increase in circulating transaminases was observed in IChKO mice compared with controls (Figure 6B). Consistent with their reduced food intake and weight loss, liver total cholesterol and triglycerides were reduced in IChKO mice (Figure 6C). No overt evidence of liver inflammation (Figure 6D), CASP3 cleavage (Figure 6E), nor increased ER stress (Figure 6F) were observed in livers of the IChKO mice. Overall, these results demonstrate that intestinal ChREBP is essential for fructose tolerance and that evidence of a mild transaminitis may begin to develop in IChKO mice secondary to the intestinal dysfunction in the setting of HFrD, but without evidence of overt hepatotoxicity.
Figure 6. Fructose intolerance in IChKO mice occurs without marked liver inflammation.

(A) Ten- to 12-week-old male and female control (CTL) and intestine-specific ChREBP-KO (IChKO) mice were fed high-fructose diet (HFrD) for up to 5 days (N = 7–8 per group). Animals were euthanized when they lost approximately 20% of their body weight and “survival” was plotted. The remaining mice were euthanized at day 5. (B) Serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) activities were measured in blood harvested at euthanasia. (C) Hepatic total cholesterol, free cholesterol, and triglyceride levels, (D) qPCR measurement of hepatic inflammatory markers, and immunoblots in liver samples for (E) caspase-3 (CASP3) and (F) DNA damage–inducible transcript 3 (DDIT3) in mice described in A. Data are presented as box-and-whisker plots where the line in the box indicates the median, the box extends from the 25th to 75th percentiles, and the whiskers indicate the minimal and maximal values. P values were obtained using 2-way ANOVA. *P < 0.05 compared between water and fructose within the same genotype. ChREBP, carbohydrate responsive element–binding protein.
Fructose feeding is well known to enhance intestinal absorption in part through upregulation of SLC2A5 (27), and our results indicate that this is mediated by fructose-induced ChREBP activation in the intestine. Our results also indicate that decreased intestinal SLC2A5 expression in IChKO mice may cause fructose malabsorption and fructose-mediated toxicity. Although Slc2a5 is expressed at low levels in the liver, preliminary ChIP-seq experiments performed to identify ChREBP binding sites in mouse liver suggested the presence of a binding site in an intronic region of the Slc2a5 gene (Chr4:150132688-150132945 aligned to GRCm38/mm10; MAH and LD, unpublished observations). To test whether ChREBP might directly bind the Slc2a5 gene in the intestine, we performed ChIP-PCR for ChREBP at the well-described ChREBP binding site in the promoter of pyruvate kinase liver and red blood cell (Pklr) as well as the putative ChREBP binding site in the Slc2a5 gene in both control and IChKO intestinal samples. ChREBP occupancy was increased 13-fold and 3-fold in Pklr and Slc2a5, respectively, in control mice, but not in IChKO mice, confirming direct binding of ChREBP to regulatory elements in both of these genes in the small intestine (Figure 7, A and B).
Figure 7. ChREBP binds directly to Pklr and Slc2a5 in the jejunum.
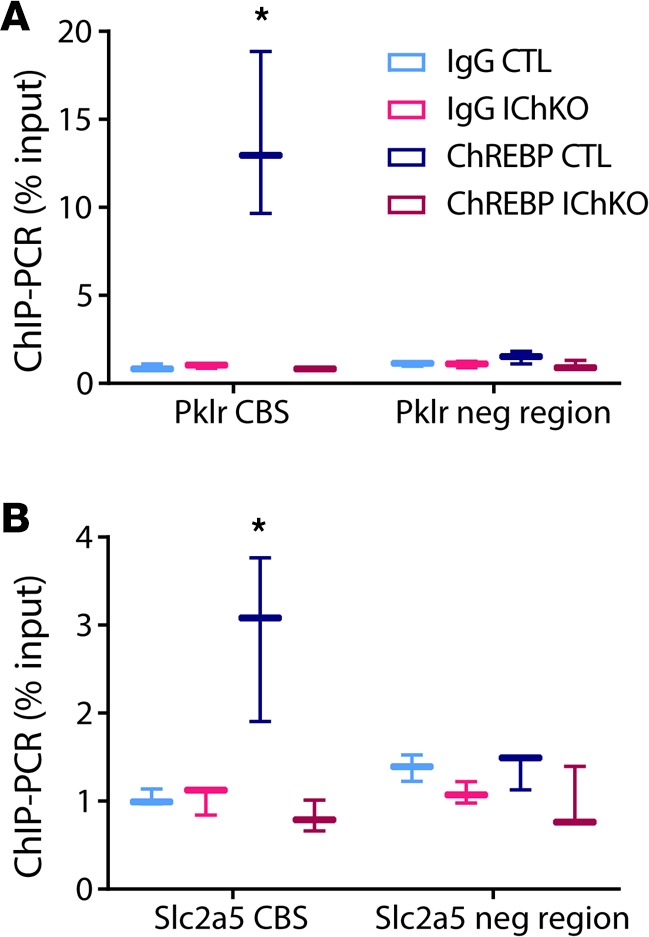
ChIP was performed from jejunal tissue with anti–carbohydrate responsive element–binding protein (anti-ChREBP) antibody or IgG control and qPCR was performed on immunoprecipitated chromatin with primers spanning (A) the E-box in the Pklr promoter and (B) the putative ChREBP binding site (CBS) in the Slc2a5 promoter, and in nonspecific genomic control regions (neg region) in proximity to both regions containing ChREBP response elements (n = 3/group). Data are presented as box-and-whisker plots where the line in the box indicates the median, the box extends from the 25th to 75th percentiles, and the whiskers indicate the minimal and maximal values. P values were obtained using 1-way ANOVA. *P < 0.05 compared with all others.
Discussion
Excessive fructose consumption has been implicated in the growing epidemic of NAFLD (28). Fructose-induced ChREBP activity stimulates expression of DNL enzymes, which are associated with, but may not be necessary for, fructose-induced steatosis as shown here and elsewhere (10). Interestingly, variants in the ChREBP locus that associate with hypertriglyceridemia in genome-wide association studies also associate with increased liver function test abnormalities, suggesting that although ChREBP may not be essential for steatosis, it may participate in the progression from simple steatosis to NASH (29–32). However, it remains unclear whether the variants in ChREBP associated with hypertriglyceridemia and transaminitis might correlate with increased or decreased ChREBP activity and whether increased or decreased ChREBP activity might produce directionally similar effects on circulating triglyceride levels and circulating transaminases in all contexts. For instance, in contrast with the population genetics data, Erion et al. showed that knockdown of ChREBP in rats on HFrD reduced hepatic triglyceride secretion and plasma triglyceride levels without an effect on steatosis, but modestly increased liver function tests (10). Zhang et al. recently showed that global ChREBP-KO mice develop profound hepatic inflammation after 2 weeks on HFrD and concluded that hepatic ChREBP protects against the progression of NAFLD (18). The mechanism proposed to explain this severe inflammation was that hepatic ChREBP suppressed SREBP2 activity, and that absence of ChREBP in the setting of HFrD increased SREBP2 expression and activity, causing hepatic cholesterol accumulation, ER stress, and severe hepatotoxicity (18). Our findings of mild transaminitis without hypercholesterolemia or ER stress in LiChKO mice chronically fed HFrD challenge these conclusions.
The explanation for the marked differences in severity of hepatic inflammation between the study from Zhang et al. compared with our work and that of Erion et al. remains uncertain. It is possible that some of these differences may be related to differences in species and/or genetic background. We studied LiChKO mice on a mixture of C3H/HeJ and C57BL/6J backgrounds because C57BL/6J mice are resistant to fructose-induced obesity and insulin resistance, whereas C3H/HeJ mice are quite sensitive (33). The study by Zhang et al. was performed with mice on a pure C57BL/6J background. However, whether or not the hepatic inflammation phenotype might be unique to the C57BL/6J background, the severe, acute fructose-induced toxicity in global ChREBP KO occurs in both pure C57BL/6J and C3H/HeJ backgrounds (8, 11). Moreover, our results indicate that the fructose-induced toxicity in global ChREBP-KO mice is fully recapitulated in the IChKO mice without overt hepatic inflammation.
The liver inflammation observed in global ChREBP-KO mice on HFrD could potentially be secondary to progressive sepsis resulting from a combination of intestinal dysfunction, reduced food intake, and weight loss beyond that which we were able to achieve in our studies. Sepsis-associated endotoxemia can produce hepatic inflammation and has also been shown to increase hepatic cholesterol synthesis in rodent models, which might reconcile some of Zhang’s observations (34). It also remains possible that the severe inflammation observed by Zhang et al. in global ChREBP-KO mice depends on loss of ChREBP in more than one tissue. Perhaps loss of ChREBP in either liver or intestine alone is insufficient to produce severe liver inflammation in the setting of HFrD, but loss of ChREBP in liver in addition to another tissue might be required for this severe manifestation. Our studies in liver- versus intestine-specific knockout cannot formally exclude these possibilities.
The role of hepatic ChREBP in regulating systemic and hepatic glucose homeostasis and insulin sensitivity remains controversial (see ref. 9 for a recent review of this topic). As an illustration of this controversy, knockdown of hepatic ChREBP on an ob/ob background enhanced systemic insulin sensitivity, whereas overexpression of a constitutively active form of ChREBP in the liver increased steatosis, but also enhanced insulin sensitivity (35, 36). Thus, the effects of hepatic ChREBP on insulin sensitivity and glucose homeostasis may depend on the specific genetic manipulation as well as the nutritional context.
We recently reported that fructose-induced, ChREBP-mediated induction of G6PC is a major determinant of glucose production and this is dominant over insulin’s ability to suppress it (11). Our data here support these conclusions and indicate that loss of hepatic ChREBP protects against fructose-induced insulin resistance. In contrast, Jois et al. recently showed in a distinct genetic model that knocking out hepatic ChREBP caused insulin resistance, in part through effects on brown-fat function (37). These apparently discrepant results may be due to the different dietary interventions used in these 2 studies. Jois et al. studied the loss of hepatic ChREBP in mice fed chow or high-fat diets. They also suggested similar results when their LiChKO mice were studied on a high-carbohydrate diet, but they did not specifically test the effects of fructose. Perhaps more importantly, there are significant differences in the genetic architecture of the 2 liver-specific ChREBP-KO models. In our model, ChREBP’s DNA binding domain is deleted, preventing transactivation by any expressed ChREBP isoform. In contrast, Jois et al. deleted exon 1a which contains the promoter and translational start site for the canonical ChREBP-α isoform. We previously demonstrated that ChREBP can be transcribed from an alternative promoter and alternative exon 1 that skips the canonical exon 1a and can be translated from a start codon in exon 4 (38). Thus, it is likely that hepatic ChREBP in the mouse model reported by Jois et al. is not fully ablated. This may explain why canonical ChREBP transcriptional targets were largely unaffected in their putative liver knockouts, in contrast with the data presented here and the data generated by multiple independent labs using global ChREBP-KO mice, which indicate that ChREBP is required for basal expression of many of its metabolic gene targets in vivo (8, 11, 17, 38). Nevertheless, the results of Jois et al. are interesting and suggest that different ChREBP isoforms may have distinct physiological and pathophysiological functions. The possibility that distinct ChREBP isoforms in different nutritional contexts may have distinct functions may contribute to the controversy as to the role of ChREBP in insulin sensitivity and will require further investigation.
Methods
Materials.
Glucose, fructose, glycerol, glycogen type III, and amyloglucosidase were purchased from Sigma-Aldrich. Nicotinamide adenine dinucleotide (NAD) was purchased from Roche. Glucose oxidase reagent was purchased from Thermo Fisher Scientific. Insulin ELISAs were purchased from Crystal Chem Inc (Ultra Sensitive Mouse Insulin ELISA). FGF21 ELISAs were purchased from R&D Systems.
Animals and diets.
All studies were carried out using mice obtained from and maintained at 21°C–22°C on a 12-hour light-dark cycle. Floxed ChREBP mice were generated at UT Southwestern Medical Center, using standard gene targeting approaches to introduce a loxP site upstream of exon 9 and another loxP site immediately downstream of exon 15 (details to be described elsewhere by GL). Albumin-Cre mice (stock 003574) and Villin-Cre mice (stock 021504) were obtained from The Jackson Laboratory. LiChKO experiments were performed on a mixed C3H/HeJ and C57BL/6J background and IChKO experiments were performed in a C57BL/6J background. Mice were fed a chow diet (LabDiet 5008 or 5053) or a 60% fructose diet (TD.89247 Harlan Teklad) for indicated durations.
Body weight and metabolic testing.
Body weight was measured on a weekly basis. Analyses were performed using blood samples drawn from overnight-fasted, 5-hour-fasted, or fasted-refed (3 hours) mice as indicated in the legends. Blood was collected from the tail vein or submandibular vein and stored at –80°C prior to analysis. For glycerol tolerance tests, mice were fasted for 5 hours and glycerol (1.5 g/kg body weight) was given via intraperitoneal injection. Blood glucose was measured using a handheld glucometer.
qPCR.
TRI reagent (MRC, catalog TR118) was used for RNA isolation from mouse liver, intestine, and adipose tissue. RNA was reverse transcribed using a SuperScript VILO kit (Invitrogen). Gene expression was analyzed with the ABI Prism sequence detection system (SYBR Green; Applied Biosystems). Gene-specific primers were synthesized by IDT (see Supplemental Table 1). Each sample was run in duplicate, and normalized to Rplp0 RNA.
ChIP-PCR.
Chromatin was isolated using a truChIP Chromatin Shearing Tissue Kit (Covaris) according to the manufacturer’s protocol, with modifications. Briefly, 30 mg of intestine was minced and cross-linked using 2 mM disuccinimidyl glutarate in PBS at room temperature for 45 minutes and washed with PBS. The minced intestine was cross-linked using 1% formaldehyde in Fixing Buffer for 5 minutes, followed by adding Quenching Buffer for 5 minutes to stop cross-linking and washing with PBS. Cross-linked tissue was dounce homogenized in Lysis Buffer with protease inhibitors, incubated for 20 minutes at 4°C with periodic vortexing, and crude nuclei were collected by centrifugation at 1,700 g and washed in Wash Buffer twice. Nuclear pellets were resuspended in SDS Shearing Buffer and sonicated using a Covaris S220 to achieve a DNA fragment size of 200–500 bp. The sonicate was centrifuged to remove debris, and a small aliquot was reverse-crosslinked, and chromatin quantified by Qubit (Thermo Fisher Scientific). Immunoprecipitation reactions were performed using chromatin samples from 3 animals per genotype, each IP set up in duplicate. Approximately 10-μg samples of chromatin were incubated overnight at 4°C with either 1 μg of anti-ChREBP (Novus Biologicals, NB400-135) or anti-rabbit IgG antibodies (Santa Cruz Biotechnology) in 1 ml of ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl pH 8.1, 55 mM NaCl). Immunocomplexes were captured using Protein A Dynabeads (Thermo Fisher Scientific) followed by washes with low-salt, high-salt, and LiCl buffers. Bound chromatin was eluted with 58 μl of digesting buffer (10 mM Tris-Cl pH 8.0, 5 mM EDTA, 300 mM NaCl, 0.1% SDS, 0.7 mg/ml proteinase K, 8.6 μg/ml RNase A) overnight at 65°C. DNA was purified using Agencourt AMPure XPmagnetic beads (Beckman Coulter). Purified DNA was used for qPCR validation with a QuantStudio 6 Flex System (Thermo Fisher Scientific) with SYBR Green master mix. Primer sequences used for Slc2a5 ChREBP binding site: forward — GGGACTGAGAAACATCCGGG, reverse — TGTTGCCCAAGGTGCTGATA; Slc2a5 negative control: forward — TAATCCTTCCCACGGCGTTT, reverse — CCATAGGCCAAGCTTCCAGT; Pklr ChREBP binding site forward — TTTGATCCAGGCTCTGCAGAC, reverse — TCTTGCCAATGGAAGCCTTG; Pklr negative control forward — TGGACATTTGACTCCAGAGC, reverse — AACATGGAGAAGAAGGCAGTG.
Immunoblotting.
Whole liver tissue lysates were generated by homogenization in RIPA buffer (Cell Signaling Technology, 9806). For nuclear/cytosolic fractionation, 50 mg of frozen liver was minced and subjected to dounce homogenizer in 1 ml of buffer A (1 M HEPES pH 7.5, 5 mM NaF, 10 μM Na2MO4, 0.1 mM EDTA, 1× Protease Inhibitor [Sigma-Aldrich, P8340]). Liver homogenates were incubated on ice for 10 minutes and subsequently NP40 was added at a final concentration of 1%. Lysates were then spun at 1,500 g for 5 minutes and the supernatant representing the cytosolic faction was collected. Nuclear pellets were washed 1 time with buffer A and resuspended in 100 μl of RIPA buffer. Nuclear pellets were sonicated for 10 minutes in a sonicator water bath and 1 M LiCl was added. Lysates were spun at 4,500 g for 10 minutes and the supernatant representing the nuclear extract was collected. Protein concentrations were determined in whole liver lysates, cytosolic, and nuclear fractions using the BCA method. Lysates were then subjected to immunoblotting with the indicated antibodies: ChREBP (Novus Biologicals, NB400-135), ACTB (Santa Cruz Biotechnology, sc-1616), GAPDH (Santa Cruz Biotechnology, sc-47724), LMNB1 (Santa Cruz Biotechnology, sc-6216), G6PC (Abcam, ab83690), p85 (Upstate, 06-496), DDIT3/CHOP (Santa Cruz Biotechnology, sc-7351), CASP3 (Cell Signaling Technology, 9662), and SREBP1 (EMD Millipore, MABS1987). Quantification of indicated blots were performed via imaging on a ChemiDoc XP (Bio-Rad) and use of associated Image Lab software v6.0.
In vivo rate of hepatic lipogenesis.
DNL was measured as previously described (38). Briefly, conscious, ad libitum–fed mice were injected intraperitoneally with 5 mCi of 3H2O and euthanized 1 hour later. Once sacrificed, the liver was frozen in liquid N2 and stored at –80°C for processing. Lipids were extracted by the Folch method. Fatty acids were isolated by saponification and petroleum ether extraction. Incorporation of 3H into fatty acids was measured.
Histological analysis and immunohistochemistry.
A portion of the median hepatic lobe and/or jejunum from each mouse was removed and fixed in 10% formalin at 4°C overnight. Paraffin embedding and sectioning was performed by the Beth Israel Deaconess Medical Center (BIDMC) Histology Core. Sections (5 μm) were stained with hematoxylin and eosin (H&E).
Hepatic triglyceride and cholesterol determination.
Liver neutral lipids were extracted using a modified Folch method. Briefly, approximately 100 mg of liver was homogenized in 4 ml of chloroform/methanol (2:1) and incubated overnight at room temperature. Saline (800 μl of 0.9%) was then added, and each sample was centrifuged at 2,000 g for 10 minutes. The organic phase was removed, dried in a vacuum concentrator, and then dissolved in butanol/(Triton X-100/methanol [2:1]) (30:20). Triglyceride content was assayed with the use of a colorimetric assay (StanBio). Total and free cholesterol were measured using Cholesterol E and Free Cholesterol E Kits (Wako Chemicals).
Glycogen measurement.
Glycogen was extracted from liver samples by incubating liver samples (~10 mg) in 300 μl of 0.5 N KOH at 95°C for 30 minutes with intermittent vortexing. Each sample was then supplemented with 25 μl of 6% sodium sulfate and 750 μl of 100% ethanol and incubated at –80°C for 1 hour. Glycogen was pelleted by centrifugation and washed twice with 70% ethanol. After removing the remaining ethanol by evaporation, 120 μl of amyloglucosidase (2 mg/ml in 0.2 M Na acetate) was added, the samples were vortexed vigorously, and incubated at 55°C for 3 hours. Glucose released from glycogen was measured by the glucose oxidase reaction (Thermo Fisher Scientific) and change in absorption was measured spectrophotometrically at 490 nm. Samples were quantified against defined glycogen standards processed in parallel with the liver samples.
Statistics.
All data are presented as the mean ± SEM. Data sets were analyzed for statistical significance with GraphPad Prism using 2-way ANOVA and individual comparisons with a Tukey’s test. Statistical significance was assumed at P less than 0.05.
Study approval.
All mouse studies were approved by the BIDMC or the Duke University Medical Center Institutional Animal Care and Research Advisory Committee.
Author contributions
MAH, MK, IIA, and SNF designed, performed, and interpreted mouse experiments. SAH, AJF, AS, LD, and HHK assisted with performing and interpreting experiments. GL generated the ChREBP-floxed mice. IIA made figures. MAH conceived of, designed, and supervised the experimental plan, interpreted experiments, and wrote the manuscript. All authors edited the manuscript.
Supplementary Material
Acknowledgments
We thank Christopher Newgard and David D’Alessio for helpful comments. We thank Eleftheria Maratos-Flier for use of the BIDMC indirect calorimeter (Columbus Instruments). This work was supported by NIH grants T32DK007516 (MK), HL20948 (GL), P30DK057521 (MAH), and R01DK100425 (MAH).
Version 1. 12/21/2017
Electronic publication
Footnotes
MK’s present address is School of Biological Sciences, Seoul National University, Seoul, South Korea.
LD’s present address is INSERM U1016, Institut Cochin, Cnrs, UMR8104, Université Paris Descartes Sorbonne Paris Cité, Paris, France.
Conflict of interest: M. A. Herman has received research support from Eli Lilly and Co.
Reference information: JCI Insight. 2017;2(24):e96703. https://doi.org/10.1172/jci.insight.96703.
Contributor Information
Ludivine Doridot, Email: ludivine.doridot@gmail.com.
Ashot Sargsyan, Email: ashot.sargsyan@duke.edu.
Alan J. Fowler, Email: ajf115@georgetown.edu.
Guosheng Liang, Email: guosheng.liang@utsouthwestern.edu.
References
- 1.Ma J, et al. Sugar-sweetened beverage, diet soda, and fatty liver disease in the Framingham Heart Study cohorts. J Hepatol. 2015;63(2):462–469. doi: 10.1016/j.jhep.2015.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Green AK, Jacques PF, Rogers G, Fox CS, Meigs JB, McKeown NM. Sugar-sweetened beverages and prevalence of the metabolically abnormal phenotype in the Framingham Heart Study. Obesity (Silver Spring) 2014;22(5):E157–E163. doi: 10.1002/oby.20724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma J, et al. Sugar-sweetened beverage but not diet soda consumption is positively associated with progression of insulin resistance and prediabetes. J Nutr. 2016;146(12):2544–2550. doi: 10.3945/jn.116.234047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malik VS, Popkin BM, Bray GA, Després JP, Willett WC, Hu FB. Sugar-sweetened beverages and risk of metabolic syndrome and type 2 diabetes: a meta-analysis. Diabetes Care. 2010;33(11):2477–2483. doi: 10.2337/dc10-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grundy SM. Metabolic syndrome update. Trends Cardiovasc Med. 2016;26(4):364–373. doi: 10.1016/j.tcm.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 6.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37(12):1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 7.Stanhope KL, et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest. 2009;119(5):1322–1334. doi: 10.1172/JCI37385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci USA. 2004;101(19):7281–7286. doi: 10.1073/pnas.0401516101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abdul-Wahed A, Guilmeau S, Postic C. Sweet sixteenth for ChREBP: established roles and future goals. Cell Metab. 2017;26(2):324–341. doi: 10.1016/j.cmet.2017.07.004. [DOI] [PubMed] [Google Scholar]
- 10.Erion DM, et al. The role of the carbohydrate response element-binding protein in male fructose-fed rats. Endocrinology. 2013;154(1):36–44. doi: 10.1210/en.2012-1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim MS, et al. ChREBP regulates fructose-induced glucose production independently of insulin signaling. J Clin Invest. 2016;126(11):4372–4386. doi: 10.1172/JCI81993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koo HY, Wallig MA, Chung BH, Nara TY, Cho BH, Nakamura MT. Dietary fructose induces a wide range of genes with distinct shift in carbohydrate and lipid metabolism in fed and fasted rat liver. Biochim Biophys Acta. 2008;1782(5):341–348. doi: 10.1016/j.bbadis.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 13.Dushay JR, Toschi E, Mitten EK, Fisher FM, Herman MA, Maratos-Flier E. Fructose ingestion acutely stimulates circulating FGF21 levels in humans. Mol Metab. 2015;4(1):51–57. doi: 10.1016/j.molmet.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fisher FM, et al. A critical role for ChREBP-mediated FGF21 secretion in hepatic fructose metabolism. Mol Metab. 2017;6(1):14–21. doi: 10.1016/j.molmet.2016.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Talukdar S, et al. FGF21 regulates sweet and alcohol preference. Cell Metab. 2016;23(2):344–349. doi: 10.1016/j.cmet.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.von Holstein-Rathlou S, et al. FGF21 mediates endocrine control of simple sugar intake and sweet taste preference by the liver. Cell Metab. 2016;23(2):335–343. doi: 10.1016/j.cmet.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iroz A, et al. A specific ChREBP and PPARα cross-talk is required for the glucose-mediated FGF21 response. Cell Rep. 2017;21(2):403–416. doi: 10.1016/j.celrep.2017.09.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang D, et al. Lipogenic transcription factor ChREBP mediates fructose-induced metabolic adaptations to prevent hepatotoxicity. J Clin Invest. 2017;127(7):2855–2867. doi: 10.1172/JCI89934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ali M, Rellos P, Cox TM. Hereditary fructose intolerance. J Med Genet. 1998;35(5):353–365. doi: 10.1136/jmg.35.5.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santer R, et al. The spectrum of aldolase B (ALDOB) mutations and the prevalence of hereditary fructose intolerance in Central Europe. Hum Mutat. 2005;25(6):594. doi: 10.1002/humu.9343. [DOI] [PubMed] [Google Scholar]
- 21.Haas JT, et al. Hepatic insulin signaling is required for obesity-dependent expression of SREBP-1c mRNA but not for feeding-dependent expression. Cell Metab. 2012;15(6):873–884. doi: 10.1016/j.cmet.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koo HY, Wallig MA, Chung BH, Nara TY, Cho BH, Nakamura MT. Dietary fructose induces a wide range of genes with distinct shift in carbohydrate and lipid metabolism in fed and fasted rat liver. Biochim Biophys Acta. 2008;1782(5):341–348. doi: 10.1016/j.bbadis.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 23.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115(5):1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barone S, et al. Slc2a5 (Glut5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. J Biol Chem. 2009;284(8):5056–5066. doi: 10.1074/jbc.M808128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe E, et al. Sepsis induces extensive autophagic vacuolization in hepatocytes: a clinical and laboratory-based study. Lab Invest. 2009;89(5):549–561. doi: 10.1038/labinvest.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci USA. 1997;94(6):2557–2562. doi: 10.1073/pnas.94.6.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shu R, David ES, Ferraris RP. Dietary fructose enhances intestinal fructose transport and GLUT5 expression in weaning rats. Am J Physiol. 1997;272(3 Pt 1):G446–G453. doi: 10.1152/ajpgi.1997.272.3.G446. [DOI] [PubMed] [Google Scholar]
- 28.Lim JS, Mietus-Snyder M, Valente A, Schwarz JM, Lustig RH. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol. 2010;7(5):251–264. doi: 10.1038/nrgastro.2010.41. [DOI] [PubMed] [Google Scholar]
- 29.Chambers JC, et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet. 2011;43(11):1131–1138. doi: 10.1038/ng.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kathiresan S, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet. 2008;40(2):189–197. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kooner JS, et al. Genome-wide scan identifies variation in MLXIPL associated with plasma triglycerides. Nat Genet. 2008;40(2):149–151. doi: 10.1038/ng.2007.61. [DOI] [PubMed] [Google Scholar]
- 32.Willer CJ, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40(2):161–169. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagata R, et al. Single nucleotide polymorphism (-468 Gly to A) at the promoter region of SREBP-1c associates with genetic defect of fructose-induced hepatic lipogenesis [corrected] J Biol Chem. 2004;279(28):29031–29042. doi: 10.1074/jbc.M309449200. [DOI] [PubMed] [Google Scholar]
- 34.Feingold KR, et al. Effect of endotoxin on cholesterol biosynthesis and distribution in serum lipoproteins in Syrian hamsters. J Lipid Res. 1993;34(12):2147–2158. [PubMed] [Google Scholar]
- 35.Dentin R, et al. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes. 2006;55(8):2159–2170. doi: 10.2337/db06-0200. [DOI] [PubMed] [Google Scholar]
- 36.Benhamed F, et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J Clin Invest. 2012;122(6):2176–2194. doi: 10.1172/JCI41636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jois T, et al. Deletion of hepatic carbohydrate response element binding protein (ChREBP) impairs glucose homeostasis and hepatic insulin sensitivity in mice. Mol Metab. 2017;6(11):1381–1394. doi: 10.1016/j.molmet.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herman MA, et al. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature. 2012;484(7394):333–338. doi: 10.1038/nature10986. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.