

Abstract
RNAs are emerging as important biomarkers and therapeutic targets. Thus, there is great potential in developing chemical probes and therapeutic ligands for the recognition of RNA sequence and structure. Chemically modified Peptide Nucleic Acid (PNA) oligomers have been recently developed that can recognize RNA duplexes in a sequence-specific manner. PNAs are chemically stable with a neutral peptide-like backbone. PNAs can be synthesized relatively easily by the manual Boc-chemistry solid-phase peptide synthesis method. PNAs are purified by reverse-phase HPLC, followed by molecular weight characterization by matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF). Non-denaturing polyacrylamide gel electrophoresis (PAGE) technique facilitates the imaging of the triplex formation, because carefully designed free RNA duplex constructs and PNA bound triplexes often show different migration rates. Non-denaturing PAGE with ethidium bromide post staining is often an easy and informative technique for characterizing the binding affinities and specificities of PNA oligomers. Typically, multiple RNA hairpins or duplexes with single base pair mutations can be used to characterize PNA binding properties, such as binding affinities and specificities. 2-Aminopurine is an isomer of adenine (6-aminopurine); the 2-aminopurine fluorescence intensity is sensitive to local structural environment changes, and is suitable for the monitoring of triplex formation with the 2-aminopurine residue incorporated near the PNA binding site. 2-Aminopurine fluorescence titration can also be used to confirm the binding selectivity of modified PNAs towards targeted double-stranded RNAs (dsRNAs) over single-stranded RNAs (ssRNAs). UV-absorbance-detected thermal melting experiments allow the measurement of the thermal stability of PNA-RNA duplexes and PNA·RNA2 triplexes. Here, we describe the synthesis and purification of PNA oligomers incorporating modified residues, and describe biochemical and biophysical methods for characterization of the recognition of RNA duplexes by the modified PNAs.
Keywords: Genetics, Issue 127, PNA, triplex, RNA structure, RNA duplex, RNA-binding ligands, solid-phase peptide synthesis, non-denaturing PAGE, 2-Aminopurine, UV thermal melting, molecular recognition, binding affinity, major groove
Introduction
RNAs are emerging as important biomarkers and therapeutic targets, due to the recent advances in the discoveries of the RNAs' roles in the regulation and catalysis of diverse biological processes1,2,3. Traditionally, antisense strands have been used to bind to ssRNAs through Watson-Crick duplex formation3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27. Recently, triplex-forming peptide nucleic acids (TFPNAs) have been designed to bind to dsRNAs via Hoogsteen hydrogen bonding (Figure 1)3,28,29. dsRNA regions are present in the majority of the traditional antisense-targeted RNAs, including pre-mRNAs and mRNAs, pre- or pri-miRNAs3, and many other non-coding RNAs1,26,27. Targeting dsRNAs through triple helix formation using TFPNAs may be advantageous due to its structure specificity and is of great potential for use in restoring the normal functions of the RNAs, which are dysregulated in diseases, for example.
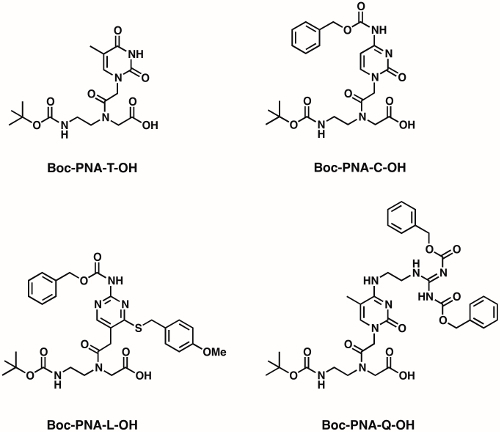
The recently published work by Rozners et al., and us3,28,29,30,31,32,33,34,35,36,37,38,39,40,41, reported the efforts on improving the selective binding of modified TFPNAs towards dsRNAs with enhanced affinity. We have developed synthesis methods for rationally designed PNA monomers (Figure 2) including thio-pseudoisocytosine (L) monomer30 and guanidine-modified 5-methyl cytosine (Q) monomer31. Through various biochemical and biophysical characterization methods, we have demonstrated that relatively short PNAs (6-10 residues) incorporating L and Q residues show improved recognition of Watson-Crick G-C and C-G base pairs, respectively, in dsRNAs. Moreover, compared to unmodified PNAs, PNAs containing L and Q residues show more selective binding towards dsRNA over ssRNA and dsDNA. The guanidine functionality42 in the Q base enables PNAs to enter HeLa cells31.
In our laboratory, we synthesize PNAs by the manual Boc-chemistry (Boc or t-Boc stands for tert- butyloxycarbony (see Figure 2) solid-phase peptide synthesis method4. The synthesis of the PNA monomer with Boc as the amine protecting group is convenient as the Boc group is sterically less bulky in comparison to fluorenylmethyloxycarbonyl (Fmoc) amine protecting group, which may be beneficial during PNA monomer coupling on the solid support. The Boc group is acid-labile and can be easily removed on the solid support by 20-50% trifluoroacetic acid (TFA) in dichloromethane (DCM) during PNA synthesis. An automated peptide synthesizer can be employed to synthesize PNA oligomers; however, 3-5-fold excess of PNA monomer is needed for an automated peptide synthesizer. Manual synthesis requires significantly less PNA monomer (2-3-fold excess), with each coupling easily monitored by the Kaiser test43. Furthermore, many automated synthesizers are not compatible with the Boc strategy synthesis due to the use of corrosive TFA during the Boc removal step.
The PNA oligomers can be purified by reverse-phase high-performance liquid chromatography (RP-HPLC) followed by molecular weight characterization by MALDI-TOF (Figures 3and 4)30,31. We employ non-denaturing PAGE to monitor triplex formation, due to the fact that free RNA duplex constructs and PNA bound triplexes often show different migration rates (Figure 5)30,31. No labeling is needed if efficient post-staining can be achieved for both of the RNA duplex and PNA·RNA2 triplex bands. A relatively small amount of sample is needed for non-denaturing PAGE experiments. However, the loading (incubation) buffers and the running buffers (pH 8.3) may not be the same, resulting in the measurements being limited to the kinetically stable triplexes, because a relatively high pH of 8.3 may significantly destabilize a triplex.
2-Aminopurine is an isomer of adenine (6-aminopurine); the 2-aminopurine fluorescence intensity (with an emission peak at around 370 nm) is sensitive to local structural environment changes, and is suitable for the monitoring of triplex formation with the 2-aminopurine residue incorporated near the PNA binding site (Figure 6)31. Unlike many other dyes that show fluorescence emission in the visible range, 2-aminopurine-labeled RNA can be exposed to room light without photo bleaching. Unlike PAGE experiment in which a running buffer of pH 8.3 is often needed, 2-aminopurine based fluorescence titration allows the measurement of binding in one solution at a specified pH, and thus may allow the measurement and detection of relatively weak and kinetically unstable binding at equilibrium.
UV-absorbance-detected thermal melting experiments allow the measurement of the thermal stability of duplexes (Figure 7)31 and triplexes30,32,44,45. Depending on the length and sequence composition, the melting of triplexes may or may not show a clear transition. Thermodynamic parameters may be obtained if the heating and cooling curves overlap. Accurate thermodynamic parameters can be obtained by isothermal titration calorimetry (ITC)32; however, relatively large amounts of samples are generally required for ITC.
Protocol
1. Manual Solid-phase Peptide Synthesis of PNAs Using Boc Chemistry
NOTE: For the success and ease of the desired PNA oligomer synthesis, all the solvents and reagents should be anhydrous. Add the appropriate molecular sieves (4A, 1-2 mm diameter pellets) and occasionally purge dry nitrogen gas into bottles. For the synthesis of modified PNA monomers, the reported protocols in respective references30,31 can be used. Unmodified PNA monomers can be purchased from commercial sources. In each of the washing steps, the appropriate amount of solvent is added to the resin, forming a slurry, before it is drained off.
- Loading of the first monomer and capping the excess free primary amines on the resin
- Weigh 30 mg of 4-methylbenzhydrylamine hydrochloride (MBHA·HCl) polystyrene resin (commercially available; loading value 0.7-1.4 mmol/g; 100-200 mesh size), and transfer to a 5 mL solid phase peptide reaction vessel fitted with a stopcock and glass stopper.
- Soak the resin in an appropriate amount of DCM for 1 h, allowing the resin to swell and expose the amines (HCl salt). NOTE: Resin beads should always be fully submerged in solvents throughout the synthesis.
- Drain off the DCM by applying a gentle flow of dry nitrogen gas over the top of the flask. Add 1 mL of 50% (v/v) N,N-diisopropylethylamine (DIPEA) in DCM and leave it for 15 min. This neutralizes the HCl salt attached to the free amines on the resin.
- Repeat step 1.1.3. Meanwhile, weigh 6 µmol of monomer and 6 µmol of (benzotriazol-1-yl-oxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP), and transfer into a 1.5 mL tube. Add 200 µL of dimethylformamide (DMF) and 12 µmol of DIPEA. Vortex the coupling solution for 3-5 min. NOTE: Desired loading value used is 0.2 mmol/g. The first monomer added is the monomer on the C-terminal of the desired PNA sequence. The loading value of the first amino acid can be calculated by the picric acid method46.
- Drain off the DIPEA from the DCM solution. Wash the resin with DCM (x 3), followed by DMF (x 3), and close the stopcock. Add the prepared coupling solution to the resin and shake gently. Push the resin along the inner walls of the vessel into the coupling solution with the use of a clean stainless steel spatula. Attach the glass stopper and secure the vessel in an incubator shaker for 3 h at 40 °C. NOTE: Alternatively, the reaction vessel can be kept steady at room temperature for 6-8 h to allow the completion of the peptide coupling reaction.
- Prepare the capping solution by mixing 240 µmol of acetic anhydride and 360 µmol of DIPEA into 200 µL of DCM. Drain off the coupling solution and wash the resin with DMF (x 3) and DCM (x 3). Add in the capping solution and leave the vessel for 30 min, occasionally shaking the vessel gently. Capping masks the excess free primary amine groups on the resins by acetylation.
- Repeat step 1.1.6. Drain the capping solution and wash the resin with DCM (x 3).
- Remove a small aliquot of resin beads using a thin capillary tube, and place them into a 1.5 mL small glass vial. Perform the Kaiser test43. Add 15 µL of each of the Kaiser test solutions into the glass vial and heat using a heat gun. Observe the color of the beads after heating. The color of the beads should remain unchanged, indicating the lack of free amine groups on the resin. NOTE: The Kaiser test kit is commercially available or can be prepared according to the reported protocol. Solution A: ninhydrin in ethanol; solution B: phenol in ethanol; solution C: potassium cyanide (KCN) in pyridine. CAUTION: KCN is highly toxic; proper protective clothing should be worn and heating should be performed in a well-ventilated fume hood, in the absence of flammable solvent or reagents.
- Repeat step 1.1.6 if the resin beads display blue or faint blue color.
- Removal of N-terminal amine protecting group
- Drain the solvents from the reaction vessel and add a solution of 50% (v/v) of trifluoroacetic acid (TFA) in DCM, ensuring that the resins are fully submerged. Leave the vessel for 15 min, occasionally shaking to facilitate the deprotection of the amine groups. Repeat 2 more cycles. CAUTION: TFA is highly corrosive. Proper protective clothing should be worn when handling.
- Wash the resin with DCM (x 3), DMF (x 3), and DCM (x 3). Add a solution of 5% DIPEA in DCM. Leave the vessel for 15 min. This step activates the free amines by neutralizing the TFA counter anions. Repeat once more.
- Flush out the DIPEA from the DCM solution. Wash the resin with DCM (x 3). Perform the Kaiser test (step 1.1.8). NOTE: Successful deprotection of the amine groups will yield blue discoloration of the beads. Once deprotection is successful, subsequent coupling of monomers by peptide coupling can be carried out.
- Coupling of subsequent monomers
- Weigh 18 µmol of desired monomer (for the Boc-PNA-Q-OH monomer, 13.2 mg of monomer is weighed) and 18 µmol of PyBOP into a 1.5 mL tube. Add 200 µL of DMF and 36 µmol of DIPEA into the 1.5 mL tube. Vortex until all solid compounds are dissolved.
- Wash the resin with DMF (x 3). Add the coupling solution into the reaction vessel and shake gently. Push the resin along the inner walls of the vessel into the coupling solution with the use of a clean stainless steel spatula. Attach the glass stopper and secure the vessel in an incubator shaker for 3 h at 40 °C.
- Drain off the coupling solution and wash the resin with DMF (x 3) and DCM (x 3). Perform step 1.1.8. If the color of the beads remains unchanged, repeat steps 1.2.1-1.3.3 until the desired PNA sequence is completed. If blue discoloration of beads is observed after the coupling of a monomer, repeat steps 1.3.1-1.3.3. If discoloration still persists, perform the capping again (step 1.1.6). NOTE: Extended coupling time (3-12 h) and/or use of excess equivalent of monomer and coupling reagents are recommended if problems with coupling arise.
- Once the desired PNA sequence is completed, wash the resin with DMF (x 3) and DCM (x 3). Completely dry the resin by applying a continuous flow of dry nitrogen gas for 15 min. This dry resin can be split and used to attach lysine or a fluorescent tag such as cyanine 3 (Cy3) and carboxyfluorescein at the N-terminus of the PNA.
- Attachment of lysine or fluorescent tag (Cy3, Cy5, or carboxyfluorescein) to the N-terminus of PNA
- Weigh 10 mg of resin, pre-loaded with the desired PNA sequence. Transfer into a 5 mL reaction vessel. Soak the resin in DCM for 1 h.
- For the attachment of lysine, perform steps 1.2.1-1.3.3, replacing the monomer with either Boc-Lys(Z)-OH or Fmoc-Lys(Boc)-OH.
- For the attachment of carboxyfluorescein, perform steps 1.2.1-1.3.3, with 10-fold excess of 5(6)-carboxyfluorescein as the monomer, and N,N'-diisopropylcarbodiimide (DIPC) and hydroxybenzotriazole (HOBt) as the coupling reagents in DMF. Leave the coupling reaction overnight in the incubator shaker at 40 °C. NOTE: As carboxyfluorescein is light sensitive, the reaction vessel should be covered with aluminum foil.
- For the attachment of Cy3 or Cy5 dye, label by the click chemistry method, perform steps 1.2.1-1.3.3, replacing the monomer with N-Boc-2-propargyl-L-glycine. This functionalizes the N-terminus of the PNA with an alkyne group. Perform the copper-catalyzed click reaction to attach the azide-containing Cy3 or Cy5 fluorescent dye12,47,48,49
- PNA cleavage from solid support, purification, and characterization NOTE: If any amine group is protected with the Fmoc protecting group, first deprotect the amine group by treating with 20% piperdine in DMF solution for 15 min (2 cycles). Wash the resin thoroughly with DMF (x 3) followed by DCM (x 3). Dry the resin completely by applying a continuous flow of dry nitrogen gas for 15 min.
- Transfer 5 mg of dry resin into a small vial. Add 10 µL of thioanisole and 4 µL of 1,2-ethanedithiol, ensuring that the resin is submerged in the reagents. Leave the tube at room temperature for 5 min. NOTE: These reagents act as scavengers, which trap reactive cationic species that are formed during the removal of protecting groups in the PNA. Appropriate scavengers can be chosen based on the side chain protecting groups.
- Add 100 µL of TFA into the tube containing the resin and scavengers. Gently vortex the mixture and subject to brief centrifugation. Leave the tube at room temperature for 10 min. CAUTION: TFA is highly corrosive. Proper protective clothing should be worn when handling.
- Carefully add 20 µL of trifluoromethanesulfonic acid (TFMSA) to the tube. Gently agitate the reaction mixture before subjecting to brief centrifugation at room temperature. Leave the tube steady at room temperature for 2 h. CAUTION: TFMSA is highly corrosive. Proper protective clothing should be worn when handling.
- Filter off the cleavage cocktail into a 5 mL round-bottom flask (RBF) with the use of a glass Pasteur pipette fitted with cotton. Use a small amount of TFA to wash the resin.
- Purge the dry nitrogen gas to the collected filtrate until all volatile solvents are evaporated. Add 1 mL of cold diethyl ether into the RBF. NOTE: The diethyl ether will cause the PNA to precipitate.
- Rinse the RBF with the diethyl ether several times before transferring the cloudy solution into a 1.5 mL tube. Subject the tube to centrifugation to allow the PNA precipitate to settle. Decant the solvents and add 300-500 µL of autoclaved water to the precipitate. Vortex thoroughly to dissolve the PNA.
- Purify the crude PNA sample via RP-HPLC using water-acetonitrile-0.1% TFA as the mobile phase. Collect the corresponding fractions, evaporate all solvents using a vacuum concentrator before re-dissolving the purified PNA in autoclaved water.
- Characterize the purified PNA via MALDI-TOF analysis with the use of α-cyano-4-hydroxycinnamic acid (CHCA) as the sample crystallization matrix.
- Measure the UV absorbance (260 nm) of the PNA at 65 °C. Calculate the concentration of the PNA with the equation:
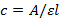 NOTE: Here c is the concentration, A is the absorbance reading obtained, ε is the extinction coefficient of the RNA sequence, and l is the optical path length of the cuvette (1 cm). The extinction coefficient of the PNA sequence is the summation of the extinction coefficient of individual monomers50. The extinction coefficients of adenine, cytosine, guanine and thymine are 15.4, 7.3, 11.7 and 8.8 mL/µmol·cm, respectively. The extinction coefficient of the L and Q monomers used is assumed to be the same as that of the cytosine (C) base.
NOTE: Here c is the concentration, A is the absorbance reading obtained, ε is the extinction coefficient of the RNA sequence, and l is the optical path length of the cuvette (1 cm). The extinction coefficient of the PNA sequence is the summation of the extinction coefficient of individual monomers50. The extinction coefficients of adenine, cytosine, guanine and thymine are 15.4, 7.3, 11.7 and 8.8 mL/µmol·cm, respectively. The extinction coefficient of the L and Q monomers used is assumed to be the same as that of the cytosine (C) base.
2. Non-denaturing PAGE
- Preparation of required buffer solutions
- Prepare Incubation buffer (10 mL) using 116.88 mg NaCl (200 mM), 50 µL of 100 mM ethylenediaminetetraacetic acid (EDTA) (0.5 mM), 200 µL of 1M stock HEPES (20 mM), 9.75 m H2O; adjust buffer to pH 7.5.
- Prepare 1x Tris-Borate-EDTA (TBE) running buffer (1 L) using 100 mL 10x Tris-Borate-EDTA at pH 8.3 and 900 mL H2O.
- Prepare 10% Ammonium persulfate (APS) solution (300 µL) using 30 mg ammonium persulfate, and 300 µL H2O.
- Preparation of 12% polyacrylamide gel
- Clean the well-forming comb, glass casting plates, and spacers with ethanol; the well-forming comb and spacers are 1 mm thick. Set up the gel assembly and seal with gel-sealing tape.
- For a polyacrylamide gel of 22 cm x 16.5 cm x 1 mm dimension, 50 mL of gel solution is adequate. Weigh out 5.7 g of acrylamide and 0.3 g of N,N'-methylenebisacrylamide (19:1) and transfer into a 50 mL centrifuge tube. CAUTION: Acrylamide is carcinogenic. Avoid breathing in dust fumes from acrylamide and ensure that proper protective clothing is worn when handling.
- Dissolve solid compounds in 50 mL 1X TBE running buffer by placing the centrifuge tube into a 50 °C water bath for 15 min or until all solid compounds have dissolved. Carry out centrifugation (3,000 rpm, 5 min, 25 °C) to remove all air bubbles within the solution. Allow the solution to cool to room temperature.
- Add 250 µL of 10% APS solution and 50 µL of tetramethylethylenediamine (TEMED) into the gel solution and mix gently using a spatula. Immediately pour the solution between the glass plates, making sure not to introduce any air bubbles. Insert the well-forming comb and leave the gel setup at room temperature for at least 60 min to allow polymerization to occur, before storing at 4 °C until ready to use.
- Preparation of samples
- Remove required quantity of RNA hairpin (1 µM) from the main stock into a clean 1.5 mL tube. Dry the RNA solution using a vacuum concentrator. NOTE: Each sample contains 1 µM of RNA in 20 µL incubation buffer. Typically, 13 samples of RNA are prepared for one PAGE experiment. The RNA for all samples can be prepared together in a single tube.
- Remove the required volumes of targeted PNA (with the final concentration of up to 50 µM) from the main stock and transfer into separate 1.5 mL tubes. Evaporate the water of the PNA solutions using a vacuum concentrator.
- Add 260 µL of incubation buffer into the 1.5 mL tube containing dried RNA and mix thoroughly to ensure all RNA is dissolved. Subject the RNA to snap cooling: Place the tube into a heat block (preheated to 95 °C) for 5 min. Immediately transfer into an ice bath and leave for 10 min.
- Add 20 µL of RNA into each of the 1.5 mL tubes containing dried PNA and mix thoroughly. Perform annealing: Place the tube containing RNA and PNA mixture into a heat block (preheated to 65 °C) for 10 min. Turn off the power of the heat block and let the samples cool slowly to room temperature. Incubate the samples at 4 °C overnight.
- Running and processing of gel
- Remove the gel-sealing tape on the bottom side of the glass plate. Mount and secure the glass plate onto the vertical gel stand using plastic clamps. NOTE: The gel running is carried out in a cold room at approximately 4 °C. All the equipment, samples, and buffers are cooled at 4 °C before running the gel.
- Fill the lower buffer reservoir with 1x TBE running buffer until the plate is submerged in approximately 1-2 cm of running buffer. Fill the upper buffer reservoir with running buffer until the level of buffer exceeds the top of the gel by 1-2 cm. Slowly and gently remove the well-forming comb, allowing the running buffer to fill the wells.
- Connect the gel stand to a power supply. Pre-run the gel for at least 30 min with a constant voltage of 250 V, which is optimized for a 22 cm x 16.5 cm x 1 mm gel.
- Meanwhile, add 4 µL (20% of sample volume) of 35% glycerol solution to each of the samples and mix gently. Once the pre-run is completed, load 20 µL of each sample (including one RNA alone sample plus samples containing RNA and PNA mixture) carefully into the bottom of the well using a micropipette and gel loading tips, making sure not to introduce any air bubbles. Run the gel at a constant voltage of 250 V, similar to the pre-run, for 5 h.
- Stop the power supply after 5 h and remove the glass plate from the stand. Remove the remaining gel-sealing tape and disassemble the glass plate. Gently remove and immerse the gel into a container filled with 350 mL of deionized water. Carefully add 35 µL of ethidium bromide (10 mg/mL) and place the container on a platform shaker (low speed) for 30 min. CAUTION: Ethidium bromide is a mutagen. Proper protective clothing should be worn when handling.
- Dispose the ethidium bromide solution into a designated waste container. Rinse the gel with 1.5 - 2 L of distilled water. Scan the gel using an imager (see Table of Materials) with a green laser of 532 nm and the emission filter set at 610 nm.
- Gel analysis
- Quantify the gel band intensities using a free software, GelQuant.NET (http://www.biochemlabsolutions.com/GelQuantNET.html). Normalize the band intensities according to:
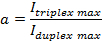 NOTE: Here Iduplex max is the band intensity of the RNA hairpin alone without the addition of PNA, and Itriplex max is the triplex band intensity with the highest concentration of PNA added.
NOTE: Here Iduplex max is the band intensity of the RNA hairpin alone without the addition of PNA, and Itriplex max is the triplex band intensity with the highest concentration of PNA added. - Calculate the fraction of triplex formation according to:
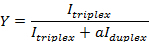
- Plot the fraction of the triplex formation (Y) against the concentration of PNA added (µM). Fit the data to the equation to obtain the dissociation constant (Kd):
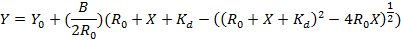 NOTE: Here R0 is the RNA hairpin concentration (1 µM). Here Y0 and B are the initial and the maximum change of the triplex fraction, respectively. Y is the fraction of triplex at varied PNA concentration. X is the total PNA concentration and Kd is the dissociation constant.
NOTE: Here R0 is the RNA hairpin concentration (1 µM). Here Y0 and B are the initial and the maximum change of the triplex fraction, respectively. Y is the fraction of triplex at varied PNA concentration. X is the total PNA concentration and Kd is the dissociation constant.
3. 2-Aminopurine Fluorescence Binding Assay
- Preparation of samples (containing dsRNA)
- Remove the required quantity of 2-aminopurine (2AP) labeled dsRNA (1 µM for each strand) from the main stock into a clean 1.5 mL tube. Evaporate water in the RNA solutions using a vacuum concentrator. NOTE: Each sample contains 1 µM of RNA in 75 µL incubation buffer. Typically, 13 samples of RNA are prepared. RNA for all samples can be prepared together in a single tube.
- Remove the required volumes of the targeted PNA for various concentrations from the main stock and transfer into separate 1.5 mL tubes. Evaporate the water of the PNA solutions using a vacuum concentrator.
- Add 975 µL of incubation buffer into the 1.5 mL tube containing dried RNA and mix thoroughly to ensure all RNA is dissolved. Centrifuge the RNA solution briefly and subject it to annealing: Place the tube into a heat block (preheated to 95 °C) for 10 min. Turn off the power of the heat block and let the samples cool slowly to room temperature.
- Add 75 µL of RNA into each of the 1.5 mL tubes containing dried PNA and mix thoroughly. Leave the samples at room temperature for at least 1 h. Incubate the samples at 4 °C overnight.
- Preparation of samples (containing ssRNA)
- Remove required quantity of 2AP-labeled ssRNA (1 µM) from the main stock into clean 1.5 mL tubes. Extract the required volumes of targeted PNA for various concentrations from the main stock and transfer into the respective 1.5 mL tubes that contain the ssRNA. Dry the RNA and PNA mixture using a vacuum concentrator.
- Add 75 µL of incubation buffer into each of the 1.5 mL tubes and mix thoroughly. Subject the mixture to annealing: Place the tube into a heat block (preheated to 95 °C) for 10 min. Turn off the power and let the samples cool slowly to room temperature. Incubate the samples at 4 °C overnight.
- Measurement and analysis
- Use a fluorescence spectrophotometer to measure the emission over a wavelength range of 330-550 nm. Use an excitation wavelength of 303 nm. NOTE: The samples are measured at room temperature and each sample is measured 3 times, in which the average is taken.
- Transfer 70 µL of incubation buffer into the 1 cm square cuvette. Start the measurement.
- Remove the buffer from the cuvette. Rinse the cuvette with distilled water and purge nitrogen gas to dry. Repeat for all samples. Subtract the buffer measurements from all samples.
- Plot the fluorescence intensity (a.u.) against wavelength (nm). Record the fluorescence intensity at 370 nm for all samples. Plot the fluorescence intensity at 370 nm (a.u.) against the corresponding concentration of the PNA added (µM).
- Fit the data to the equation from step 2.5.2 to obtain the dissociation constant (Kd): Y = Y0 + (B/(2R0))(R0 + X + Kd- ((R0 + X + Kd)2- 4R0X)1/2), where R0 is the 2AP-labeled dsRNA concentration (1 µM). NOTE: Here Y0 and B are the initial and maximum change of fluorescence intensity at 370 nm, respectively. Y is the fluorescence intensity at 370 nm at varied PNA concentration. X is the total PNA concentration and Kd is the dissociation constant.
4. UV-Absorbance-detected Thermal Melting Experiments
- Preparation of samples NOTE: Measure the UV absorbance (260 nm) of the RNA at 95 °C (to make sure the RNA secondary structures are disrupted). Calculate the concentration of the RNA with the equation:
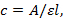 where c is the concentration, A is the absorbance reading obtained, ε is the extinction coefficient of the RNA sequence, and l is the optical path length of the cuvette (1 cm). The extinction coefficient of the RNA is calculated based on a nearest-neighbor model using MeltWin51,52. The program package may be provided upon request.
where c is the concentration, A is the absorbance reading obtained, ε is the extinction coefficient of the RNA sequence, and l is the optical path length of the cuvette (1 cm). The extinction coefficient of the RNA is calculated based on a nearest-neighbor model using MeltWin51,52. The program package may be provided upon request. - Remove the required quantity of ssRNA (5 µM) from the main stock into clean 1.5 mL tubes. Remove the required volumes of targeted PNA (5 µM) from the main stock and transfer into the respective 1.5 mL tubes that contain the ssRNA. Dry the RNA and PNA mixture using a vacuum concentrator.
- Add 130 µL of incubation buffer into each of the 1.5 mL tubes and mix thoroughly. Subject the mixture to annealing: Place the tube into a heat block (preheated to 95 °C) for 10 min. Turn off the power and let the samples cool slowly to room temperature. Incubate the samples at 4 °C overnight.
- Measurement and analysis
- Use a UV-Vis spectrophotometer to measure the absorbance at 260 nm using an 8-microcell cuvette with a path length of 1 cm. Measure the samples' absorbances at increasing temperature from 15 to 95 °C, followed by decreasing temperature from 95 to 15 °C at a ramp rate of 0.5 °C/min.
- Transfer 130 µL of sample into each of the well, making sure that one well contains the incubation buffer. Start the measurement. Repeat as required.
- Normalize the absorbance values with the high temperature reading normalized to unity, and plot the normalized absorbance reading against temperature (°C). Plot the first derivative of the curves. Obtain the melting temperatures by fitting the first derivative curves to a Gaussian function.
Representative Results
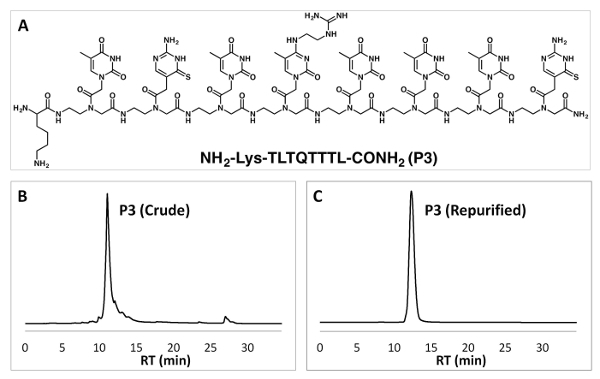
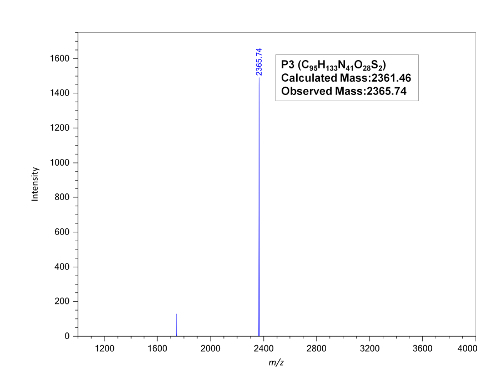
Reverse-phase HPLC allows the purification of PNA oligomers. We can obtain pure PNA oligomers with two rounds of HPLC purification (Figure 3). The identity of the PNAs can be confirmed by MALDI-TOF analysis (Figure 4).
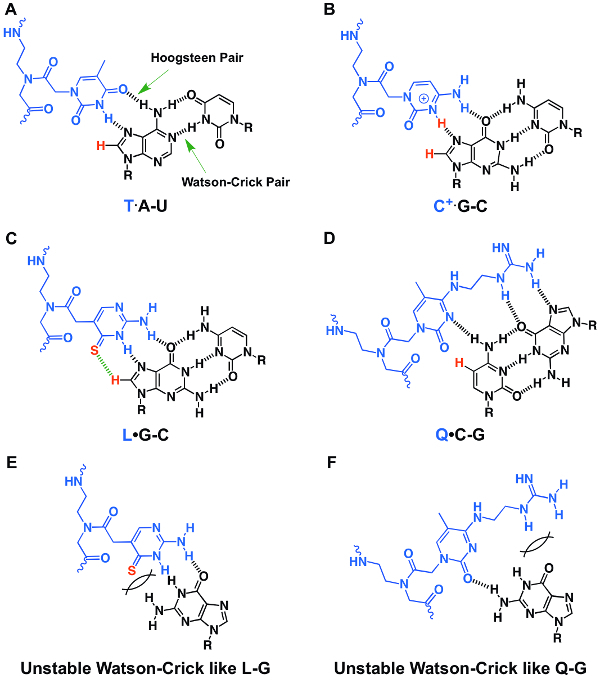
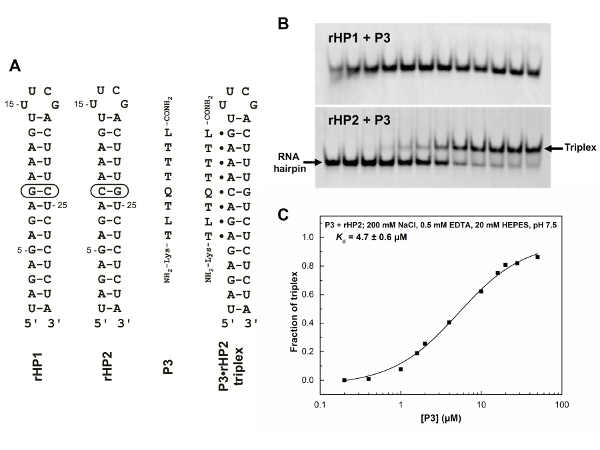
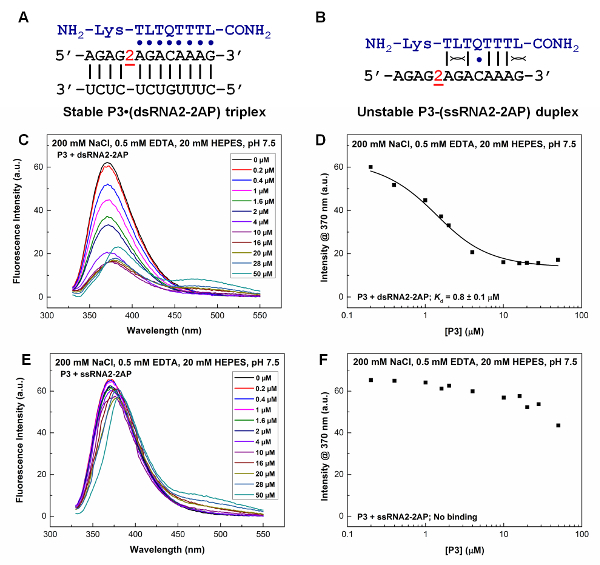
Non-denaturing PAGE is an easy and informative technique for characterizing the binding affinities and specificities of PNA oligomers. We typically use multiple RNA hairpins or duplexes with single base pair mutations to characterize binding properties (Figure 5). The non-denaturing PAGE data shown in Figure 5 clearly suggest that the Q- and L-modified PNA can recognize a dsRNA region with a C-G pair (Figure 5B, bottom panel) but not the one without a C-G pair (Figure 5B, top panel). This specific and enhanced recognition is through the T·A-U, L·G-C, and Q·C-G PNA·RNA2 base triple (Figure 1A, C, D) formation. Various PNAs with single or multiple mutations may also be used to demonstrate the improved binding properties of a modified PNA. We have shown that adding 2 mM Mg2+ in the incubation buffer does not affect the binding significantly31.
We have demonstrated by 2-aminopurine fluorescence titration that a Q- and L-modified PNA binds to a targeted dsRNA region (Figure 6A, 6C, 6D) but not ssRNA (Figure 6B, 6E, 6F). PNA P3 binds to the 2-aminopurine-labeled dsRNA with a Kd value of 0.8 ± 0.1 µM. The fluorescence intensity at 370 nm for the 2-aminopurine-labeled ssRNA remains relatively constant with varied P3 concentration, indicating the lack of binding of PNA P3 to the ssRNA.
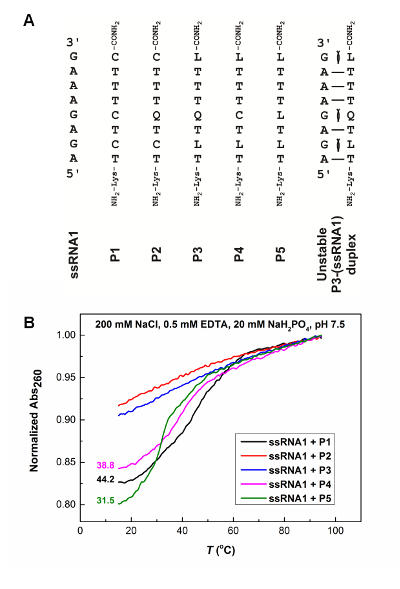
PNAs containing Q residues (P2 and P3) show no thermal melting transitions (Figure 7), suggesting no binding to the ssRNA. This is due to the steric clash present in Watson-Crick like Q-G pair. Compared to unmodified PNA P1, PNAs P4 and P5 containing modified L residues but no Q residues, show decreased melting temperatures for the corresponding RNA-PNA duplexes due to the steric clash present in Watson-Crick like L-G pair. The UV-absorbance-detected thermal melting data are consistent with the 2-aminopurine fluorescence titration data, which also show that a PNA containing Q and L residues does not bind to ssRNA appreciably (Figure 6B, 6E, 6F). Incorporating a Q base is more destabilizing than an L base, as a Q base has a more significant steric clash in the formation of a Watson-Crick-like Q-G pair (Figure 1F) compared to a Watson-Crick-like L-G pair (Figure 1E).
Figure 1: Chemical structures of stable base triple and unstable base pair structures. (A-D) Major-groove PNA·RNA2 base triples of T·A-U (A), C+·G-C (B), L·G-C (C), and Q·C-G (D). (E, F) Unstable Watson-Crick like PNA-RNA base pairs of L-G (E) and Q-G (F). The letter R represents the sugar-phosphate backbone of RNA. Hydrogen bonds are indicated by black dashed lines. The figure is reproduced from reference31. Please click here to view a larger version of this figure.
Figure 2: Chemical structures of PNA monomers. Four PNA monomers (T, C, L, and Q) are shown. Please click here to view a larger version of this figure.
Figure 3: Chemical structure of a PNA oligomer and purification by RP-HPLC. (A) Chemical structure of the PNA sequence P3. (B, C) RP-HPLC data of crude PNA P3 (B) and re-purified PNA P3 (C). Please click here to view a larger version of this figure.
Figure 4: MALDI-TOF spectrum of purified PNA P3. Please click here to view a larger version of this figure.
Figure 5: RNA hairpin and PNA sequences and binding characterization by non-denaturing PAGE. (A) RNA hairpins (rHP1 and rHP2), PNA P3, and a PNA·RNA2 triplex formed between PNA P3 and rHP2. (B) Non-denaturing PAGE (12%) results for rHP1 and rHP2 binding to PNA P3. The incubation buffer is 200 mM NaCl, 0.5 mM EDTA, 20 mM HEPES, pH 7.5. The loaded RNA hairpins (rHP1 and rHP2) are at 1 µM in 20 µL. The PNA concentrations in lanes from left to right are 0, 0.2, 0.4, 1, 1.6, 2, 4, 10, 16, 20, 28, and 50 µM. PNA P3 does not bind to rHP1 (top panel) but binds to rHP2 (bottom panel). (C) Kd determination for P3 binding to rHP2. The fraction of triplex formation (Y) was plotted against the PNA concentration. The figure is adapted from reference31. Please click here to view a larger version of this figure.
Figure 6: Fluorescence titration study of PNA P3 binding to 2-aminopurine-labeled RNAs. The 2-aminopurine residue is designated as '2' in the RNA sequence. The incubation buffer is 200 mM NaCl, 0.5 mM EDTA, 20 mM HEPES, pH 7.5. (A) A PNA·RNA2 triplex formed between P3 and a 2-aminopurine-labeled dsRNA (dsRNA2-2AP). (B) A hypothetical PNA-RNA duplex formed between P3 and a 2-aminopurine-labeled ssRNA (ssRNA2-2AP). (C, E) Fluorescence emission spectra for the 2-aminopurine-labeled RNA duplex (1 µM) and ssRNA (1 µM), respectively, with varied P3 concentration at pH 7.5. The peak at around 475 nm is due to the weak fluorescence emission of the L base in the PNA. (D, F) Kd determination based on the plots of 2-aminopurine fluorescence intensity (at 370 nm) of the RNA duplex and ssRNA, respectively, versus PNA P3 concentration. The figure is adapted from reference31. Please click here to view a larger version of this figure.
Figure 7: Thermal melting results for RNA-PNA duplexes. The incubation buffer is 200 mM NaCl, 0.5 mM EDTA, 20 mM NaH2PO4, pH 7.5. All samples contain 5 µM of single-stranded RNA (ssRNA1) and PNA in 130 µL. (A) Single-stranded RNA (ssRNA1), PNAs (P1, P2, P3, P4, and P5) and a hypothetical PNA-RNA duplex formed between PNA P3 and ssRNA1 in a parallel orientation. The steric clash is indicated for the Watson-Crick like Q-G and L-G pairs. (B) Melting curves for different PNAs binding to ssRNA1. The melting temperatures are shown for the curves with melting transitions. The figure is adapted from reference31.
Discussion
RNA duplex-binding PNA oligomers (e.g., 10-mers) are medium-sized molecules and thus can show electrophoretic mobility shift upon binding to RNAs with a comparable or slightly larger size (e.g., 50-mer or smaller). If an RNA is significantly larger than the PNA, titration of PNA into RNA may not work due to a limited gel mobility shift. Thus, the large RNA may be truncated for the non-denaturing PAGE assays. Titration of a large RNA into a fluorophore-labeled PNA allows for the monitoring of the triplex formation by a non-denaturing agarose gel with the sample loaded in the middle of the gel40.
For a titration experiment by non-denaturing PAGE with a constant total concentration of RNA, we typically use an unlabeled RNA concentration of 1 µM for efficient post staining of the free RNA and triplex bands by ethidium bromide. An RNA concentration as low as 0.2 µM may also be sufficient depending on the RNA construct31. The concentration of the unlabeled RNA (0.2 µM) determines that the Kd values that can be accurately measured should be about 0.2 µM or larger. Other staining dyes may be used to enhance the staining efficiency. Alternatively, our unpublished data suggest that Cy3 dye-labeled RNAs can be used in non-denaturing PAGE experiments to measure the tight binding events.
Due to the fact that 2-aminopurine is only moderately fluorescent, 2-aminopurine fluorescence titration is also limited to the measurement of the binding with Kd values close to or above 0.2 µM31. The RNA or the PNA may be labeled with a relatively bright dye for quantifying a relatively tight binding in solution through fluorescence titration, if the binding results in changes in fluorescence signals53,54,55.
The strategy of targeting RNA structures by dsRNA-binding PNAs has been tested for a limited number of RNAs. It is likely that binding properties may vary for dsRNAs with different sequences and base pair compositions. One may always choose the purine-rich strand of a duplex for the design of TFPNAs. It is critical to understand how consecutive Q·C-G triples may affect the stability of a triplex. More extensive sequence-dependent studies are clearly needed to understand the sequence-dependent binding properties of TFPNAs.
The binding affinity of TFPNAs can be further enhanced by increasing the length and/or further modifying the bases and backbones56,57 of TFPNAs. However, a continuous duplex region may not often consist of more than 10 consecutive base pairs without the disruption by non-Watson-Crick structures. One may conjugate TFPNAs with small molecules for the recognition of non-Watson-Crick structures adjacent to dsRNA regions. In principle, a TFPNA-small molecule conjugate is expected to have enhanced binding affinity and specificity compared to a TFPNA or small molecule alone. However, the chemical and physical properties of the linker for the conjugation58,59,60,61,62,63,64 must be optimized.
The fact that TFPNAs can selectively bind to dsRNAs over ssRNAs and dsDNAs suggests that it is possible to develop TFPNAs as very useful chemical probes and potential therapeutic ligands through the regulation of RNA structural dynamics and interactions with proteins and metabolites. Cellular uptake of TFPNAs may be facilitated through the conjugation with cell-penetrating moieties such as small molecules, peptides, and nanoparticles, or complexation with supramolecular structures such as liposomes5,6,12,17,25,31,41,65,66. Further functionalization of TFPNAs with bioimaging tags such as fluorophores and radioisotopes may facilitate the detection, imaging, and targeting of functional RNA structures in living organisms.
Disclosures
A patent application (PAT/179/14/15/PCT) based on the work reported here has been filed.
Acknowledgments
This work was supported by the Singapore Ministry of Education (MOE) Tier 1 (RGT3/13 and RG42/15 to G.C.) and MOE Tier 2 (MOE2013-T2-2-024 and MOE2015-T2-1-028 to G.C.).
References
- Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157(1):77–94. doi: 10.1016/j.cell.2014.03.008. [DOI] [PubMed] [Google Scholar]
- Velagapudi SP, Gallo SM, Disney MD. Sequence-based design of bioactive small molecules that target precursor microRNAs. Nat Chem Biol. 2014;10:291–297. doi: 10.1038/nchembio.1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil KM, Chen G. In: Modified Nucleic Acids in Biology and Medicine. Jurga S, Erdmann VA, Barciszewski J, editors. Springer International Publishing; 2016. pp. 299–317. [Google Scholar]
- Hyrup B, Nielsen PE. Peptide nucleic acids (PNA): synthesis, properties and potential applications. Bioorg Med Chem. 1996;4(1):5–23. doi: 10.1016/0968-0896(95)00171-9. [DOI] [PubMed] [Google Scholar]
- Shiraishi T, Nielsen PE. Improved cellular uptake of antisense peptide nucleic acids by conjugation to a cell-penetrating peptide and a lipid domain. Methods Mol Biol. 2011;751:209–221. doi: 10.1007/978-1-61779-151-2_13. [DOI] [PubMed] [Google Scholar]
- Shiraishi T, Nielsen PE. Nanomolar cellular antisense activity of peptide nucleic acid (PNA) cholic acid ("umbrella") and cholesterol conjugates delivered by cationic lipids. Bioconjugate Chem. 2012;23(2):196–202. doi: 10.1021/bc200460t. [DOI] [PubMed] [Google Scholar]
- Khoo B, Roca X, Chew SL, Krainer AR. Antisense oligonucleotide-induced alternative splicing of the APOB mRNA generates a novel isoform of APOB. BMC Mol Biol. 2007;8:3. doi: 10.1186/1471-2199-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein CA, et al. Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents. Nucleic Acids Res. 2010;38(1):3. doi: 10.1093/nar/gkp841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. 2012;11(2):125–140. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacey E, Rodriguez L, Liu Y, Wolfe MS. Targeting a pre-mRNA structure with bipartite antisense molecules modulates tau alternative splicing. Nucleic Acids Res. 2012;40(19):9836–9849. doi: 10.1093/nar/gks710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenvang J, Petri A, Lindow M, Obad S, Kauppinen S. Inhibition of microRNA function by antimiR oligonucleotides. Silence. 2012;3(1):1. doi: 10.1186/1758-907X-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, et al. Intracellular delivery of antisense peptide nucleic acid by fluorescent mesoporous silica nanoparticles. Bioconjugate Chem. 2014;25(8):1412–1420. doi: 10.1021/bc5002714. [DOI] [PubMed] [Google Scholar]
- Wojtkowiak-Szlachcic A, et al. Short antisense-locked nucleic acids (all-LNAs) correct alternative splicing abnormalities in myotonic dystrophy. Nucleic Acids Res. 2015;43(6):3318–3331. doi: 10.1093/nar/gkv163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenartowicz E, et al. Antisense Oligonucleotides Targeting Influenza A Segment 8 Genomic RNA Inhibit Viral Replication. Nucleic Acid Ther. 2016;26(5):277–285. doi: 10.1089/nat.2016.0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avitabile C, et al. Targeting pre-miRNA by peptide nucleic acids: a new strategy to interfere in the miRNA maturation. Artif DNA PNA XNA. 2012;3(2):88–96. doi: 10.4161/adna.20911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barczak AK, et al. RNA signatures allow rapid identification of pathogens and antibiotic susceptibilities. Proc Natl Acad Sci USA. 2012;109(16):6217–6222. doi: 10.1073/pnas.1119540109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das I, et al. A peptide nucleic acid-aminosugar conjugate targeting transactivation response element of HIV-1 RNA genome shows a high bioavailability in human cells and strongly inhibits tat-mediated transactivation of HIV-1 transcription. J Med Chem. 2012;55(13):6021–6032. doi: 10.1021/jm300253q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabani MM, et al. Efficient inhibition of miR-155 function in vivo by peptide nucleic acids. Nucleic Acids Res. 2010;38(13):4466–4475. doi: 10.1093/nar/gkq160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riguet E, et al. A peptide nucleic acid-neamine conjugate that targets and cleaves HIV-1 TAR RNA inhibits viral replication. J Med Chem. 2004;47(20):4806–4809. doi: 10.1021/jm049642d. [DOI] [PubMed] [Google Scholar]
- Torres AG, et al. Chemical structure requirements and cellular targeting of microRNA-122 by peptide nucleic acids anti-miRs. Nucleic Acids Res. 2012;40(5):2152–2167. doi: 10.1093/nar/gkr885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyay A, Dixit U, Manvar D, Chaturvedi N, Pandey VN. Affinity capture and identification of host cell factors associated with hepatitis C virus (+) strand subgenomic RNA. Mol Cell Proteomics. 2013;12(6):1539–1552. doi: 10.1074/mcp.M112.017020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesolowski D, et al. Basic peptide-morpholino oligomer conjugate that is very effective in killing bacteria by gene-specific and nonspecific modes. Proc Natl Acad Sci USA. 2011;108(40):16582–16587. doi: 10.1073/pnas.1112561108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage BA. The impact of nucleic acid secondary structure on PNA hybridization. Drug Discov Today. 2003;8(5):222–228. doi: 10.1016/s1359-6446(03)02611-4. [DOI] [PubMed] [Google Scholar]
- Thomas SM, et al. Antitumor effects of EGFR antisense guanidine-based peptide nucleic acids in cancer models. ACS Chem Biol. 2013;8(2):345–352. doi: 10.1021/cb3003946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahal R, McNeer NA, Ly DH, Saltzman WM, Glazer PM. Nanoparticle for delivery of antisense gammaPNA oligomers targeting CCR5. Artif DNA PNA XNA. 2013;4(2):49–57. doi: 10.4161/adna.25628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams BD, Parsons C, Walker L, Zhang WC, Slack FJ. Targeting noncoding RNAs in disease. J. Clin. Invest. 2017;127(3):761–771. doi: 10.1172/JCI84424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui M, Corey DR. Non-coding RNAs as drug targets. Nat Rev Drug Discov. 2017;16(3):167–179. doi: 10.1038/nrd.2016.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi G, Zhou Y, Zhong Z, Toh D-FK, Chen G. RNA triplexes: from structural principles to biological and biotech applications. Wiley Interdiscip Rev RNA. 2015;6(1):111–128. doi: 10.1002/wrna.1261. [DOI] [PubMed] [Google Scholar]
- Rozners E. Recent Advances in Chemical Modification of Peptide Nucleic Acids. J Nucleic Acids. 2012;2012:8. doi: 10.1155/2012/518162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi G, Yuan Z, Lu Y, Zhao Y, Chen G. Incorporation of thio-pseudoisocytosine into triplex-forming peptide nucleic acids for enhanced recognition of RNA duplexes. Nucleic Acids Res. 2014;42(6):4008–4018. doi: 10.1093/nar/gkt1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh DK, et al. Incorporating a guanidine-modified cytosine base into triplex-forming PNAs for the recognition of a C-G pyrimidine-purine inversion site of an RNA duplex. Nucleic Acids Res. 2016;44(19):9071–9082. doi: 10.1093/nar/gkw778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Zengeya T, Rozners E. Short peptide nucleic acids bind strongly to homopurine tract of double helical RNA at pH 5.5. J Am Chem Soc. 2010;132(25):8676–8681. doi: 10.1021/ja101384k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta P, Zengeya T, Rozners E. Triple helical recognition of pyrimidine inversions in polypurine tracts of RNA by nucleobase-modified PNA. Chem Commun. 2011;47(39):11125–11127. doi: 10.1039/c1cc14706d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengeya T, Li M, Rozners E. PNA containing isocytidine nucleobase: synthesis and recognition of double helical RNA. Bioorg Med Chem Lett. 2011;21(7):2121–2124. doi: 10.1016/j.bmcl.2011.01.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta P, Muse O, Rozners E. Recognition of double-stranded RNA by guanidine-modified peptide nucleic acids. Biochemistry. 2012;51(1):63–73. doi: 10.1021/bi201570a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengeya T, Gupta P, Rozners E. Triple-helical recognition of RNA using 2-aminopyridine-modified PNA at physiologically relevant conditions. Angew Chem Int Ed. 2012;51(50):12593–12596. doi: 10.1002/anie.201207925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muse O, et al. Sequence selective recognition of double-stranded RNA at physiologically relevant conditions using PNA-peptide conjugates. ACS Chem Biol. 2013;8(8):1683–1686. doi: 10.1021/cb400144x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnedzko D, Cheruiyot SK, Rozners E. Using Triple Helix Forming Peptide Nucleic Acids for Sequence-selective Recognition of Double-stranded RNA. Curr Protoc Nucleic Acid Chem. 2014;58:61–64. doi: 10.1002/0471142700.nc0460s58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengeya T, Gupta P, Rozners E. Sequence selective recognition of double-stranded RNA using triple helix-forming peptide nucleic acids. Methods Mol Biol. 2014;1050:83–94. doi: 10.1007/978-1-62703-553-8_7. [DOI] [PubMed] [Google Scholar]
- Endoh T, Hnedzko D, Rozners E, Sugimoto N. Nucleobase-Modified PNA Suppresses Translation by Forming a Triple Helix with a Hairpin Structure in mRNA In Vitro and in Cells. Angew Chem Int Ed. 2016;55(3):899–903. doi: 10.1002/anie.201505938. [DOI] [PubMed] [Google Scholar]
- Hnedzko D, McGee DW, Karamitas YA, Rozners E. Sequence-selective recognition of double-stranded RNA and enhanced cellular uptake of cationic nucleobase and backbone-modified peptide nucleic acids. RNA. 2017;23(1):58–69. doi: 10.1261/rna.058362.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexselblatt E, Esko JD, Tor Y. On guanidinium and cellular uptake. J Org Chem. 2014;79(15):6766–6774. doi: 10.1021/jo501101s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser E, Colescott RL, Bossinger CD, Cook PI. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal. Biochem. 1970;34(2):595–598. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- Roberts RW, Crothers DM. Stability and properties of double and triple helices: dramatic effects of RNA or DNA backbone composition. Science. 1992;258(5087):1463–1466. doi: 10.1126/science.1279808. [DOI] [PubMed] [Google Scholar]
- Zhou Y, et al. Recognition of RNA duplexes by chemically modified triplex-forming oligonucleotides. Nucleic Acids Res. 2013;41(13):6664–6673. doi: 10.1093/nar/gkt352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gisin BF. The monitoring of reactions in solid-phase peptide synthesis with picric acid. Anal. Chim. Acta. 1972;58(1):248–249. doi: 10.1016/S0003-2670(00)86882-8. [DOI] [PubMed] [Google Scholar]
- Gogoi K, Mane MV, Kunte SS, Kumar VA. A versatile method for the preparation of conjugates of peptides with DNA/PNA/analog by employing chemo-selective click reaction in water. Nucleic Acids Res. 2007;35(21):139. doi: 10.1093/nar/gkm935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabanpoor F, Gait MJ. Development of a general methodology for labelling peptide-morpholino oligonucleotide conjugates using alkyne-azide click chemistry. Chem Commun. 2013;49(87):10260–10262. doi: 10.1039/c3cc46067c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective "Ligation" of Azides and Terminal Alkynes. Angew Chem Int Ed. 2002;41(14):2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Haaima G, Hansen FH, Christensen L, Dahl O, Nielsen PE. Increased DNA binding and sequence discrimination of PNA oligomers containing 2,6-diaminopurine. Nucleic Acids Res. 1997;25(22):4639–4643. doi: 10.1093/nar/25.22.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder SJ, Turner DH. Methods Enzymol. Vol. 468. Academic Press; 2009. pp. 371–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell JA, Turner DH. Investigation of the Structural Basis for Thermodynamic Stabilities of Tandem GU Mismatches: Solution Structure of (rGAGGUCUC)2 by Two-Dimensional NMR and Simulated Annealing. Biochemistry. 1996;35(45):14077–14089. doi: 10.1021/bi9615710. [DOI] [PubMed] [Google Scholar]
- Sato T, Sato Y, Nishizawa S. Triplex-Forming Peptide Nucleic Acid Probe Having Thiazole Orange as a Base Surrogate for Fluorescence Sensing of Double-stranded RNA. J Am Chem Soc. 2016;138(30):9397–9400. doi: 10.1021/jacs.6b05554. [DOI] [PubMed] [Google Scholar]
- Cheruiyot SK, Rozners E. Fluorescent 2-Aminopyridine Nucleobases for Triplex-Forming Peptide Nucleic Acids. ChemBioChem. 2016;17(16):1558–1562. doi: 10.1002/cbic.201600182. [DOI] [PubMed] [Google Scholar]
- Sato T, Sato Y, Nishizawa S. Optimization of the Alkyl Linker of TO Base Surrogate in Triplex-Forming PNA for Enhanced Binding to Double-Stranded RNA. Chem Eur J. 2017;23(17):4079–4088. doi: 10.1002/chem.201604676. [DOI] [PubMed] [Google Scholar]
- Virta PM, Tahtinen V, Granqvist L, Murtola M, Stromberg R. 19F NMR spectroscopic analysis of the binding modes in triple helical PNA/microRNA-complexes. Chem Eur J. 2017. [DOI] [PubMed]
- Zengeya T, Gindin A, Rozners E. Improvement of sequence selectivity in triple helical recognition of RNA by phenylalanine-derived PNA. Artif DNA PNA XNA. 2013;4(3):69–76. doi: 10.4161/adna.26599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses AC, Huang SW, Schepartz A. Inhibition of Rev.RRE complexation by triplex tethered oligonucleotide probes. Bioorg Med Chem. 1997;5(6):1123–1129. doi: 10.1016/s0968-0896(97)00048-5. [DOI] [PubMed] [Google Scholar]
- Ben Gaied N, Zhao Z, Gerrard SR, Fox KR, Brown T. Potent triple helix stabilization by 5',3'-modified triplex-forming oligonucleotides. ChemBioChem. 2009;10(11):1839–1851. doi: 10.1002/cbic.200900232. [DOI] [PubMed] [Google Scholar]
- Grimm GN, Boutorine AS, Lincoln P, Norden B, Helene C. Formation of DNA triple helices by an oligonucleotide conjugated to a fluorescent ruthenium complex. ChemBioChem. 2002;3(4):324–331. doi: 10.1002/1439-7633(20020402)3:4<324::AID-CBIC324>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Tran T, et al. Targeting the r(CGG) Repeats That Cause FXTAS with Modularly Assembled Small Molecules and Oligonucleotides. ACS Chem Biol. 2014. [DOI] [PMC free article] [PubMed]
- Gianolio DA, Segismundo JM, McLaughlin LW. Tethered naphthalene diimide-based intercalators for DNA triplex stabilization. Nucleic Acids Res. 2000;28(10):2128–2134. doi: 10.1093/nar/28.10.2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumney S, Kool ET. Structural Optimization of Non-Nucleotide Loop Replacements for Duplex and Triplex DNAs. J Am Chem Soc. 1995;117:5635–5646. doi: 10.1021/ja00126a004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford RL, Dervan PB. The reach of linear protein-DNA dimerizers. J Am Chem Soc. 2007;129(45):14026–14033. doi: 10.1021/ja075247b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Bahal R, Gupta M, Glazer PM, Saltzman WM. Nanotechnology for delivery of peptide nucleic acids (PNAs) J Control Release. 2016;240:302–311. doi: 10.1016/j.jconrel.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avitabile C, et al. Incorporation of Naked Peptide Nucleic Acids into Liposomes Leads to Fast and Efficient Delivery. Bioconjugate Chem. 2015;26(8):1533–1541. doi: 10.1021/acs.bioconjchem.5b00156. [DOI] [PubMed] [Google Scholar]