

Abstract
Dopamine (DA) is a modulatory neurotransmitter controlling motor activity, reward processes and cognitive function. Impairment of dopaminergic (DAergic) neurotransmission is strongly associated with several central nervous system-associated diseases such as Parkinson's disease, attention-deficit-hyperactivity disorder and drug addiction1,2,3,4. Delineating disease mechanisms involving DA imbalance is critically dependent on animal models to mimic aspects of the diseases, and thus protocols that assess specific parts of the DA homeostasis are important to provide novel insights and possible therapeutic targets for these diseases.
Here, we present two useful experimental protocols that when combined provide a functional read-out of the DAergic system in mice. Biochemical and functional parameters on DA homeostasis are obtained through assessment of DA levels and dopamine transporter (DAT) functionality5. When investigating the DA system, the ability to reliably measure endogenous levels of DA from adult brain is essential. Therefore, we present how to perform high-performance liquid chromatography (HPLC) on brain tissue from mice to determine levels of DA. We perform the experiment on tissue from dorsal striatum (dStr) and nucleus accumbens (NAc), but the method is also suitable for other DA-innervated brain areas.
DAT is essential for reuptake of DA into the presynaptic terminal, thereby controlling the temporal and spatial activity of released DA. Knowing the levels and functionality of DAT in the striatum is of major importance when assessing DA homeostasis. Here, we provide a protocol that allows to simultaneously deduce information on surface levels and function using a synaptosomal6 DA uptake assay.
Current methods combined with standard immunoblotting protocols provide the researcher with relevant tools to characterize the DAergic system.
Keywords: Neuroscience, Issue 127, Striatum, dopamine, high-performance liquid chromatography (HPLC), synaptosomal dopamine uptake, synaptosomes, dopamine homeostasis, dopamine transporter
Introduction
Dopamine (DA) is a modulatory neurotransmitter critical for motor behaviour, reward and cognitive function1,7,8,9. Imbalances in DA homeostasis are implicated in several neuropsychiatric diseases such as attention-deficit hyperactivity disorder, drug addiction, depression and Parkinson's disease1. DA is released from the presynaptic neuron into the synaptic cleft, where it binds to and activates receptors on the pre- and postsynaptic membrane, thereby further conveying the signal. The level of DA in the synapse after release is spatially and temporally controlled by DAT3,10. The transporter sequesters DA from the extracellular space, and thus sustains physiological DA levels3,11. Genetic removal of DAT in mice causes a hyperdopaminergic phenotype characterized by elevated synaptic DA levels, depletion of intracellular DA pools and profound changes in postsynaptic DAergic signalling10,12.
Here, two separate protocols are presented, one method to measure DA tissue content and another to assess the functionality of DAT. Combined with the surface biotinylation assay described by Gabriel et al.13 these two methods provide information on DA content and functional levels of DAT for a thorough assessment of DA homeostasis. With these methods DA homeostasis of various transgenic mice or disease models can be characterized and described. These tools have been implemented and optimized and are standard use in our laboratories. Current assays have served to investigate the consequences on the DA homeostasis of altering the C-terminal of DAT14 or expressing Cre recombinase under the tyrosine hydroxylase (TH) promoter 5.
Protocol
The guidelines of the Danish Animal Experimentation Inspectorate (permission number: 2017-15-0201-01160) was followed and experiments performed in a fully AAALAC accredited facility under the supervision of a local animal welfare committee.
1. Synaptosomal Dopamine Uptake (Method 1)
NOTE: This protocol is for parallel assessment of two brains, but can be successfully used to perform synaptosomal DA uptake experiments with four brains in parallel.
- Preparations
- Label 48 1.5 mL micro-centrifuge tubes per Table 1 (24 for each brain). Use different colours for the tubes belonging to each brain to prevent confusion between samples
- Label 51 scintillation tubes #1-51.
- Place phosphate buffered saline (PBS) on ice. This will be used to keep brains chilled.
- Turn on the centrifuge to allow to pre-cool to 4 °C.
- Pre-weigh two micro-centrifuge tubes to get the exact tissue weight during the synaptosomal preparation (section 2.6).
- Prepare homogenization buffer by mixing 4 mM HEPES and 0.32 M sucrose. Adjust the pH to 7.4 and keep on ice. NOTE: Homogenization buffer can be kept in aliquots at -20 °C and thawed the day of the experiment.
- Prepare uptake buffer by combining the following: 25 mM HEPES, 120 mM NaCl, 5 mM KCl, 1.2 mM CaCl2, 1.2 mM MgSO4, 1 mM ascorbic acid (0.194 g/L), 5 mM D-glucose (0.991 g/L). Adjust pH to 7.4 with NaOH (leads to a sodium concentration of approximately 130 mM) and keep on ice. NOTE: For 2 brains, prepare 1500 mL uptake buffer. Prepare uptake buffer as a 10x stock solution without ascorbic acid and glucose kept at 4 °C. Make 1x working solution freshly on the day, supplementing with ascorbic acid and glucose. Adjust pH with NaOH to 7.4.
- Prepare uptake buffer + ligand by combining the following: 25 mM HEPES, 120 mM NaCl, 5 mM KCl, 1.2 mM CaCl2, 1.2 mM MgSO4, 1 mM ascorbic acid (0.194 g/L), 5 mM D-glucose (0.991 g/L), 1 μM pargyline, 100 nM desipramine, 10 nM Catechol-O-methyl-transferase (COMT) inhibitor. Adjust to pH 7.4 with NaOH and keep on ice. NOTE: Use 50 mL uptake buffer and add ligand. Pargyline is added to help prevent degradation of DA by oxidation through monoamine oxidase (MAO). COMT inhibitor (RO-41-0960) is added to prevent degradation of DA (catecholamines in general) through COMT. Desipramine is added to inhibit uptake through the norepinephrine and serotonin transporters, and thereby make the uptake results specific for DAT.
- Prepare uptake buffer + ligand + cocaine by combining the following: 25 mM HEPES, 120 mM NaCl, 5 mM KCl, 1.2 mM CaCl2, 1.2 mM MgSO4, 1 mM ascorbic acid (0.194 g/L), 5 mM D-glucose (0.991 g/L), 1 μM pargyline, 100 nM desipramine, 10 nM COMT inhibitor, 500 μM cocaine. Adjust pH to 7.4 with NaOH and keep on ice. NOTE: Use 7 mL uptake buffer + ligand and add cocaine. Cocaine is added to obtain a background measurement of non-specific DA uptake to subtract from the data, leaving only the DAT-specific uptake (since SERT and NET are inhibited by desipramine as described above). Alternatively, a highly potent and specific DAT inhibitor GBR-12935, can be used instead. Important: Keep everything on ice from now on.
- Synaptosomal preparation
- Sacrifice one mouse at a time by cervical dislocation and decapitation.
- Cut the skin using scissors to reveal the skull and cut off any excess tissue posterior of skull left from decapitation. Cut the skull with small straight scissors along the sutura sagittalis ending up as anteriorly as possible.
- Place the two scissor tips in each eye of the mouse and cut through skull to remove the remainder of the skull and the intact brain. Rapidly remove the brain and place it in ice-cold PBS. This will keep it cold, while the second brain is dissected.
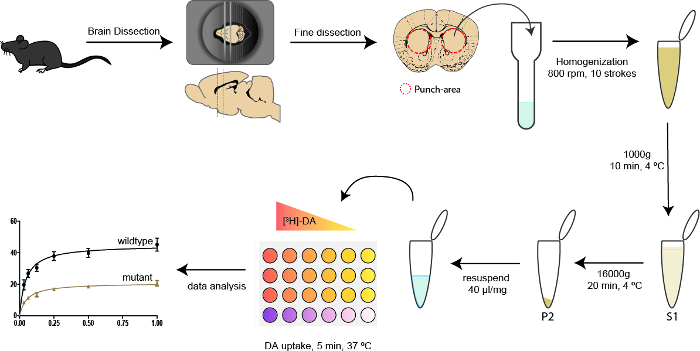
- Using a brain matrix, dissect a 3 mm coronal slice of the striatum (anterior-posterior: +1.5 mm to -1.5 mm) followed by finer dissection with a puncher. See Figure 1 for punch area.
- Keep the tissue on ice at all times moving forward.
- Transfer the tissue to a 1.5 mL micro-centrifuge tube for weighing. NOTE: This weight will be used in step 1.2.14.
- Repeat step 1.2.1-1.2.6 for the second brain.
- Once weight has been obtained for both brains, transfer the tissue to a homogenization glass containing 1 mL of ice-cold homogenization buffer.
- Homogenize the tissue using a motor driven pestle at 800 rpm, 10 even strokes. NOTE: One stroke equals one up and down action, the first stroke should be about 5 s, the following 3-4 s. Homogenization in isotonic medium is important to prevent bursting of the synaptosomes6. Using appropriate force and isotonic medium will allow the presynaptic terminals to reseal enclosing cytoplasm, synaptic vesicles, mitochondria and cytoskeleton15.
- Transfer the homogenate to a 1.5 mL micro-centrifuge tube and collect the remaining homogenate from the homogenization glass by rinsing with 0.5 mL additional homogenization buffer.
- Keep the sample on ice while tissue from the other brain is homogenized. Run the two brains in parallel in steps 1.2.12-1.2.13.
- Pellet the nuclei and cell debris by centrifugation (1,000 x g, 10 min, 4°C).
- Transfer the supernatant (S1) (containing cell membranes and cytoplasm) to new 1.5 mL microcentrifuge tubes and centrifuge at 16,000 x g for 20 min at 4°C.
- Discard the supernatant (containing cytoplasm) and resuspend the pellet (P2) (containing crude synaptosomes) in 40 μL homogenization buffer/ mg tissue (based on the weight obtained in step 1.2.6). Gently resuspend the crude synaptosomal fraction with a p1000 pipette as too much force will break the synaptosomes. NOTE: It is important to end up with at least 280 μL. If not, diluting with more than 40 μL per mg will be necessary. Steps 1.2.1-1.2.13 must be performed as fast as possible and everything has to be kept on ice. As soon as the crude synaptosomes have been resuspended in homogenization buffer they are fairly stable as long as they are kept on ice6,15,16.
2. Uptake Experiment
Add 440 μL uptake buffer + ligand + cocaine to the designated 12 1.5 mL microcentrifuge tubes and 440 μL uptake buffer + ligand to the 36 remaining 1.5 mL microcentrifuge tubes (see Table 1 for the layout). NOTE: Remove them from ice 15 min before the start of the experiment to bring them to room temperature, but keep on ice until then to conserve inhibitors.
- Prepare saturation curve (dopamine (2, 5, 6-[3H]-DA) (3-H dopamine) (91.1 Ci/mmol) at various final concentrations (0.031, 0.0625, 0.125, 0.25, 0.5, and 1.0 μM)).
- Label six 2 mL microcentrifuge tubes as 10, 5, 2.5, 1.25, 0.62, and 0.31.
- Add 750 μL uptake buffer + ligand to the 5 microcentrifuge tubes with lowest number.
- Add 1,455.4 μL uptake buffer + ligand, 14.6 μL dopamine (1 mM), 30 μl 3-H dopamine (11 μM) to the tube labelled 10.
- Transfer 750 μL from the tube labelled 10 to the tube labelled 5. Mix well and transfer 750 μL from this tube to the 2.5 tube. Repeat this dilution with the remaining tubes.
Pre-rinse the glass microfiber filters with 4 mL uptake buffer after placing them on a 12 well filter bucket.
Add 10 μL of the synaptosomal membrane suspension to the first 24 1.5 mL micro-centrifuge tubes (see Table 1 for the layout) containing 440 μL buffer. Vortex carefully and spin down shortly to ensure that the synaptosomes are submerged in the buffer. Leave at 37 oC for 10 min. NOTE: It is important to be exact on the times during the uptake experiment for fair comparison between brains. Use a stopwatch for precision.
Add 50 μL dopamine of concentration 10 and 5 μM (section 2.2) to the first two columns and leave at 37 oC for 5 min with shaking. After exactly 5 min, stop the reaction by adding 1 mL ice-cold uptake buffer. Do the same with concentration 2.5 and 1.25 μM (section 2.2) and then with concentration 0.62 and 0.31 μM.
Add the samples to the pre-rinsed microfiber filters and wash with 5 x 4 mL ice-cold uptake buffer. When the first 12 samples have been added to the filters, move the filters to scintillation tubes. Repeat this with the next 12 samples.
Repeat step 2.4-2.6 for the next brain.
Prepare 3 max-counting tubes by placing a microfiber filter in the bottom of scintillation tubes 49-51 and adding 25 µL of the maximum dopamine concentration on top (10 μM).
Leave all 51 scintillation tubes in a fume hood for 1 h.
Add 3 mL scintillation fluid to each scintillation tube and shake vigorous on a shaker for 1h.
- Count [3H]-DA in a beta-counter for 1 min.
- Open the program and choose: Label: H-3, Plate/Filter: 4 mL vial, 4 by 6, Assay type: Normal and Counting time: 1 min. NOTE: This step can be performed the following day if more convenient. Leave samples covered with tin foil over night.
- Protein determination
- Use a standard BCA Protein Assay kit to determine protein concentrations of the synaptosomes for adjustments from counts per min (cpm) to fmol/min/μg and for proper comparison of uptake between samples.
3. Data Analysis
Use the data from the uptake experiment to make a saturation curve for each group and calculate the rate of the reaction (Vmax) and the Michaelis Menten constant (KM). NOTE: The KM value reflects the substrate concentration required to reach half of Vmax. Accordingly, Vmax directly reflects the function of DAT (maximum uptake capacity), which depends on the number of DAT on the surface and on how its activity might be modulated by posttranslational modifications and/or interacting proteins as well as on alterations in ion gradients. The KM value is an indirect measure of substrate affinity for the transporter that, importantly also can be modulated by posttranslational modifications and/or interacting proteins. To fully assess functional changes it is therefore important to directly determine surface expression levels by for instance a surface biotinylation assay. A description of dopamine reuptake and an elaboration on meaning of the KM and Vmax values have been reviewed by Schmitz et al.17.
4. High-performance Liquid Chromatography (Method 2)
- Brain dissection
- Label and weigh microcentrifuge tubes; one per region per mouse.
- Sacrifice one mouse at a time by cervical dislocation and decapitation. Rapidly remove the brain as described in section 1.2. Perform further dissection immediately.
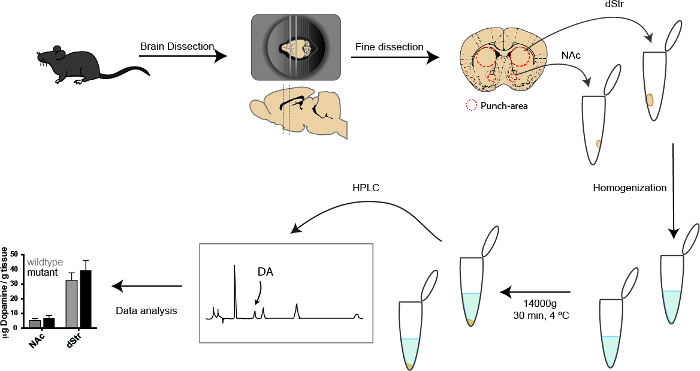
- Using a brain matrix, dissect a 3 mm coronal slice of the striatum followed by finer bilateral dissection of NAc and dStr with a puncher. See Figure 3 for punch area.
- Transfer the tissue to 1.5 mL microcentrifuge tubes and quickly place on dry ice. Weigh the tissue and place it back on the dry ice immediately. NOTE: The tissue weight is important for the concentration calculation later on.
- Repeat steps 4.1.1-4.1.4 with the next mouse. NOTE: Place the tissue at 80oC until further processing or immediately proceed to tissue processing.
5. Tissue Preparation
Turn on centrifuge to allow it to pre-cool to 4 °C
Prepare the homogenization solution (0.1 N perchloric acid (HClO4)) and keep on ice.
Using homogenization solution, prepare a fresh 0.1 pmol/μL dopamine standard from a 1 mM stock solution (a 10,000 x dilution). NOTE: The stock solution is prepared by dissolving dopamine in homogenization buffer to a stock concentration of 1 mM, which can be kept at 20 °C for about one month. If other areas of the brain are of interest, concentration of the standard will have to be modified, since other brain areas including prefrontral cortex, hippocampus, substantia nigra and ventral tegmental area possess up to 100 times less DA compared to striatum.
Add 500 μL homogenization solution to each sample (i.e. NAc and dStr tissue; section 4.1) and homogenize samples using an ultra-homogenizer on full speed for approximately 30 s. Keep the sample in iced water during homogenization. Between each sample, clean the ultra homogenizer with water (full speed for 30 s).
Centrifuge the samples for 30 min at 4 °C, 14,000 x g.
Transfer approximately 200 μL sample to a glass 0.22 μm filter (1 cm diameter) using a 1 mL plastic syringe. NOTE: The use of glass filters is not of important, as long as the filters chosen are 0.22 μm to remove the high molecular weight substances.
Analyze samples by electro-chemical detection (EC)-HPLC methodology18 as described in section 6.
6. High-performance Liquid Chromatography Analysis
Prepare the following solution for mobile phase: Sodium acetate 55 mM, octanesulfonic acid 1 mM, Na2EDTA 0.1 mM, acetonitrile 8 %, acetic acid 0.1 M, pH = 3.2. NOTE: Adjust pH with 0.1 M acetic acid. It is important to be precise with the pH. The solution should be degassed by the on-line degasser (integrated part of the HPLC system). Make 2 L of mobile phase and change it about once a month (change it when the noise gets noticable). Due to fluctuations it takes about 24 h to adjust after changing the mobile phase.
- Set-up of detector:
- Set volt output to 700 mV and temperature for 32 °C on the detector oven; this is very important. Make a range program using the online manual. See the manual for further detail). Add the time program as per Table 2. NOTE: In this study, the program is set to automatically change the current at set retention times, to keep the HPLC at the optimal current and to keep the peaks of the chromatogram as enlarged as possible, but still within view. This has been optimized for striatal brain lysates from mice.
- When the program has been added to the detector, make a 3 point calibration curve, by injecting the standard three times (5, 10 and 15 μL). NOTE: Standard (10 μL (10 μL x 0.1 pmol/μL = 1 pmol DA)) should be injected every tenth sample, and samples calibrated to the nearest standard. Fine tune the system the day prior by adjusting the 3-point standard, and checking if the chromatogram comes out within the expected range. If substantial noise is observed, change the mobile phase. If a peak is higher than 990 mV then re-inject the sample in a lower volume, or make a new range program and a new standard curve. The detection limit is measured as 3 times the noise value in mV to the lowest peak value injected for each standard (NA, DOPAC, DA, 5-HIAA, HVA, 3-MT and 5-HT).
- Set-up system by opening the LC solution and activating analysis 1; this starts in the online mode. Type user ID and password and click ok. Wait for LC solution to connect to instruments. Go to batch table and fill in data information (e.g., sample type: unknown for samples and std for standard, analysis type: IT QT, inj volume: 10, ISTD amt: Level 1 con). Save file (Go to File -> save Batch file as -> Save).
- Make the system inject the first sample by pressing 'Start Batch'. This will result in the injection of 10 μL sample by the autosampler with a solvent delivery module. NOTE: Use a pump flow rate of 0.15 mL/min and a C18 column with reversal phase. Here an amperometric detector for electrochemical detection was used. The glassy carbon electrode should be set to 0.8 V with an Ag/AgCl reference electrode. In order to assay dopamine, use Chromatography 3 µ ODS (3) C18 (DA 2 mm x 100 mm, particle size 3 µm) to achieve chromatographic separation.
- Extract the recorded and calculated peak areas.
- Go to data acquisition to follow the chromatogram when the samples have finished. Go to Lc post run analysis -> chose the file name (the chromatogram will open) -> click on view. On the manual integration bar, add all the peaks to be integrated -> go to data report and print the read out. NOTE: Here the LC solution software was used for data analysis and system control.
- From the peak areas, calculate the concentration of dopamine in a sample, as follows by using the known concentration of the standard:
- Use the peak area of the standard (equals 1 pmol) to acquire factor A.
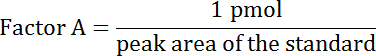
- Use factor A to get the concentration of the samples. Concentration of sample (in pmol per 10 μL) = Factor A × peak area of the sample
- Use this formula to get the sample concentration in ng. Sample concentration pmol / 10 µL x 500 µL x MW of dopamine = sample concentration in ng Sample concentration (in ng) = Sample concentration (in pmol per 10 μL) x 500 μL x MW of dopamine
- Use the sample concentration in ng to get the sample concentration in pg/g tissue (sample concentration in pg / tissue g = pg/g tissue).
Representative Results
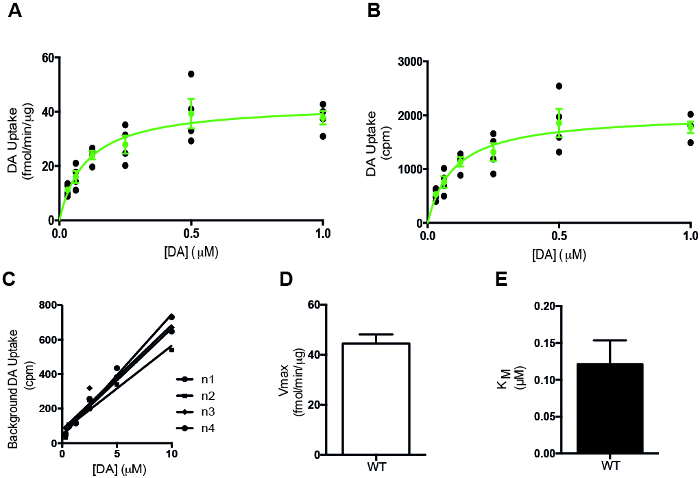
Current DA uptake protocol (Figure 1) includes all steps necessary to assess the functionality of DAT in synaptosomes from mice. Our representative data of the DA uptake method (Figure 2) depicts a saturation curve with unadjusted data (Figure 2B) and adjusted data (Figure 2A). The saturation curve shows uptake from wild type mice. Usually one would make DA uptake for comparison with a mutant mouse, which would lead to a saturation curve for each genotype5. In that case, differences between wild type and mutant mice in 2A and 2B can be explained by multiple factors. The more thorough the experimenter is in following every step of the protocol, the less difference there will be between depicting raw (2B) and protein adjusted (2A) data. The most obvious reasons for differences are i) imprecise weighing of the tissue in step 1.2.6, ii) loss of tissue while transferring to and from homogenization glass in step 1.2.8-1.2.10 and iii) imprecision while transferring supernatant in step 1.2.13 and removing supernatant in step 1.2.14. We recommend performing a preliminary experiment in wild type mice for better precision.
The dopamine EC-HPLC method (Figure 3) includes all steps necessary to asses amount of DA in dStr and NAc of mice. Our representative data (Figure 4 and Table 3) depict the outcome of an experiment performed on wild type mice.
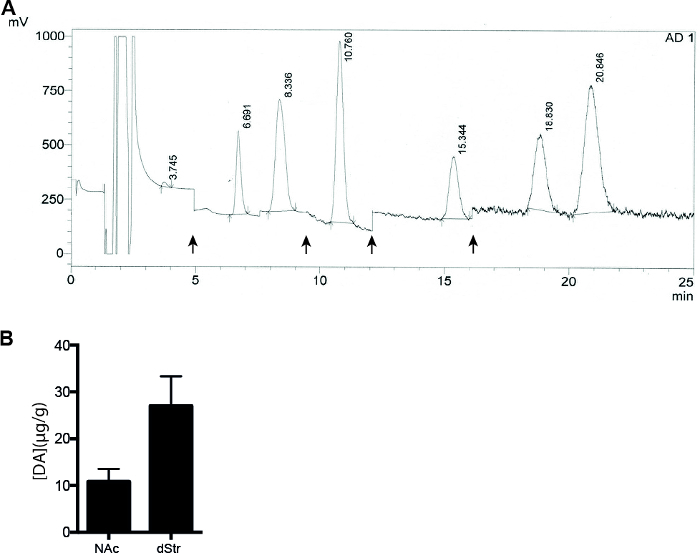
Figure 1: Schematic workflow depicting the different steps in the synaptosomal DA uptake experiment protocol. The mouse is sacrificed by cervical dislocation followed by brain dissection and placement in a brain matrix. A 3 mm thick coronal slice is dissected from the brain, and fine dissection is performed by a bilateral punch of a small area immediately underneath the corpus callosum. Dorsal striatum is homogenized in 1 mL homogenization buffer by mechanical disruption followed by 10 min centrifugation at low speed. Supernatant is transferred to a clean tube and centrifuged at high speed for 20 min. The pellet, containing the synaptosomes, is resuspended and ready for the uptake experiment. Uptake is performed by adding triplicates of the different [3H]-DA concentrations, ranging from a final concentration of 0.031 to 1 μM. In addition, the varying concentrations of tritiated DA are added to a control sample containing 500 μM cocaine. Lastly, the samples are counted in a beta-counter and data analysis is performed revealing the saturation curve of DAT. Please click here to view a larger version of this figure.
Figure 2: Representative results from a synaptosomal DA uptake experiment of four C57Bl/6 wild type mice from a Drd1a-cre mouse strain. (A) Representative data depicting the saturation curve of DA uptake through the DAT of synaptosomal preparations from wild type mice (n = 4). Black dots are the values of the four mice shown separately, the green curve shows how data would normally be depicted by combining data from 4-6 mice per group. This graph combines data from four mice. (B) The saturation graph of the raw data, without adjusting for actual protein concentration of the samples. Essentially, it is the unadjusted version of the data in A. (C) Control data. This graph shows the counts from the cocaine-containing samples, which are used to subtract background from the uptake data. This control is essential to determine the reliability of the uptake data, but is rarely shown in articles. If this data for some reason does not show linearity, it indicates a critical issue with the set-up, which needs to be identified and solved to deduce anything from the data. R squared: n1 = 0.9852, n2 = 0.9584, n3 = 0.9606, n4 = 0.9913. (D) Histogram showing the uptake capacity of DAT (VMAX). VMAX = 43.13 ± 3.2 fmol/min/μg protein). Data are shown as mean ± SEM. (E) Histogram showing the KM for DAT to be 0.1 ± 0.03 μM corresponding well to the rotating disk voltammetry method depicting KM values of DAT to be 0.6 μM19. A KM value of 0.1 μM also corresponds well to the KM values of 0.22 μM that have been obtained by stimulation models of stimulated DA overflow in striatum20,21. Please click here to view a larger version of this figure.
Figure 3: Schematic workflow depicting the different steps in the high-performance liquid chromatography protocol. The mouse is sacrificed by cervical dislocation followed by brain dissection and placement in brain matrix. A 3 mm thick coronal slice is dissected from the brain, and fine dissection is performed by bilaterally punching two smaller areas, dividing the striatum into the dStr and the NAc. Tissue is homogenized, followed by fast centrifugation. A standard with known DA concentrations as well as supernatants from the samples are run in the HPLC, producing chromatograms. The areas of the different peaks are used to calculate the concentration of DA in the samples depicted in a histogram. Please click here to view a larger version of this figure.
Figure 4: Representative results of a high-performance liquid chromatography experiment. (A) HPLC chromatogram for 10 μL sample from dStr injected to a C18 (2 mm x 100 mm) column used for small molecules. Retention times; 3.7 min for Noradrenaline (NA), 6.7 min for dihydroxyphenylacetic acid (DOPAC), 8.3 min for DA, 10.7 min for 5-hydroxyindoleacetic acid (HIAA), 15.3 min for homovanillic acid (HVA), 18.8 min for 3-methoxytyamine (3-MT) and 20.8 min for 5-hydroxtryptamine (5-HT). Only values for the DA peaks are calculated in this study. (B) Histogram showing the concentration of DA in NAc and dStr based on chromatrogram. HPLC analysis of striatal areas including dStr and NAc of C57BL/6 mice (n = 7). Data are shown as mean ± SEM. ![]() Indicates a change in resistance, and has been added to the chromatogram manually. Please click here to view a larger version of this figure.
Indicates a change in resistance, and has been added to the chromatogram manually. Please click here to view a larger version of this figure.
| 10 | 5 | 2.5 | 1.25 | 0.62 | 0.31 |
| 10 | 5 | 2.5 | 1.25 | 0.62 | 0.31 |
| 10 | 5 | 2.5 | 1.25 | 0.62 | 0.31 |
| 10+coc | 5+coc | 2.5+coc | 1.25+coc | 0.62+coc | 0.31+coc |
Table 1: Microcentrifuge tube layout for synaptosomal preparation.
| Time | Range | Filter | Valve | Auto zero | Offset | E cell |
| 0 | 1nA | 0.5 Hz | load | not | 30% | 0.7 V |
| 0.2 | 1nA | 0.5Hz | load | set | 30% | 0.7 V |
| 5 | 500pA | 0.5Hz | load | not | 30% | 0.7 V |
| 5.2 | 500pA | 0.5Hz | load | set | 30% | 0.7 V |
| 9.4 | 200pA | 0.5Hz | load | not | 30% | 0.7 V |
| 9.6 | 200pA | 0.5Hz | load | set | 30% | 0.7 V |
| 12 | 100pA | 0.5Hz | load | not | 30% | 0.7 V |
| 12.2 | 100pA | 0.5Hz | load | set | 30% | 0.7 V |
| 16.2 | 50pA | 0.5Hz | load | not | 30% | 0.7 V |
| 16.4 | 50pA | 0.5Hz | load | set | 30% | 0.7 V |
| End time 25 min |
Table 2: HPLC time program used in this study.
| Peak # | Ret. Time | Area | Height | Area % | Height % |
| 1 | 3,745 | 230451 | 18500 | 0.299 | 0.626 |
| 2 | 6,691 | 5573485 | 382143 | 7,228 | 12,922 |
| 3 | 8,336 | 13209342 | 510378 | 17,131 | 17,258 |
| 4 | 10,760 | 16443198 | 831182 | 21,325 | 28,106 |
| 5 | 15,344 | 7129795 | 282473 | 9,247 | 9,552 |
| 6 | 18,830 | 11279424 | 346248 | 14,628 | 11,708 |
| 7 | 20,846 | 23241754 | 586419 | 30,142 | 19,829 |
| Total | 77107450 | 2957344 | 100,000 | 100,000 |
Table 3: Peak analysis showing area and height of the different peaks used to calculate the concentrations shown in Figure 4B .
Discussion
This manuscript describes useful experimental protocols to delineate DA homeostasis in any mouse model of choice. We provide detailed protocols for measuring levels of DA in brain tissue from mice using HPLC and synaptosomal DA uptake to assess functional DA transport through DAT. The procedures, protocols and limits for the HPLC experiment and synaptosomal DA uptake assay will be elaborated below.
The synaptosomal uptake protocol can provide useful insight to the functionality of DAT. Combined with a surface biotinylation experiment13, knowledge on the total amount, surface level, and functionality of DAT can be obtained. Given the major role of DAT and its influence on DA transmission and its participation in various diseases, it has been a major goal to establish assays that can model DAT function. One of the advantages of the synaptosomal DA uptake experiment is that it can be performed post in vivo manipulations such as upon chemogenetic manipulation as well as different in vivo drug treatments or behavioral training in addition to investigations on genetically modified mice. Drug-treatment can be performed after synaptosomal preparation, instead of in vivo if preferred, making it possible to test the effect of drugs on DAT directly22.
Alternatives to performing synaptosomal DA uptake, is to perform uptake experiments either on DAT transfected cell cultures or in neuronal cultures naturally expressing the transporter. Cell culture assays might be preferred for initial investigations into different modifications of DAT23, whereas neuronal primary cultures with the endogenous transporter may provide a more physiologically trustworthy picture of the transporter function in vivo. Even though neuronal cultures are made directly from animals, there are advantages of using synaptosomes instead. Neuronal cultures are usually made from prenatal or immature neurons, which might influence the function and expression of DAT, whereas synaptosomal preparations represent physiological preparations that can be obtained from adult and even old animals without difficulties6,14.
There are several advantages of using the synaptosomal uptake experiment to investigate function of DAT, but important limitations have to be considered. The synaptosomes have limited viability6. Keeping them on ice is essential to obtain reliable results with low variations. If kept on ice and provided necessary nutrients, purified synaptosomes are viable for hours and take up and release neurotransmitters efficiently15. It is possible to freeze synaptosomes, but the method of freezing is of great importance15. Small variations in the experimental procedure can lead to extensive variations in outcome. Therefore, experimental protocols should be optimized on wild type conditions (e.g. wild type mice) until reproducible results are obtained and then comparisons can be made following various genetic or pharmacological manipulations. Synaptosomal DA uptake assay is an easy, reliable and valid experimental tool to acquire reproducible data with a very low variation in a key parameter in DA homeostasis, DAT functionality (Figure 2A). The limitations are heavily outweighed by the advantages of being able to perform the experiment on preserved nerve endings from adult mice6.
The presently used methods to analyze DA levels in tissue are supported by histochemical methods developed in the 1950's. The significance of developing methods like HPLC to measure DA levels, has been obvious since discovering the substantial decrease of DA in the basal ganglia of Parkinson's patients, thereby founding the principle of treating patients suffering from Parkinson's with L-DOPA24. Since this discovery, more advanced techniques for tissue analysis of DA levels have been developed, but as with any other techniques there are pitfalls. One of the major pitfalls of these techniques is the unstable nature of monoamines (dopamine, noradrenaline and serotonin). How to correctly prepare the tissue preparation to avoid loss of monoamines has been discussed in great detail by Atack et al.25 and will not be discussed further in this article, except to stress the importance of placing the tissue on dry ice directly after dissection and not adding homogenization solution until immediately before the HPLC analysis. From our experience, tissue can be kept at -80 oC for up to one month without any degradation of DA if no solution has been added. Atack et al discuss tissue preparation for methods ranging from the fluoro spectrometric method to advanced HPLC methods allowing a detection limit down to 3 ng/mL tissue25. The method we describe in this paper is based on the same principles. Current advanced technologies enable more refined analyses and detection of DA levels at fmol concentrations. By using a fluorescent HPLC technique, even more robust monoamine analysis can be obtained26. Due to the robustness of the method, HPLC is widely used to obtain information about changes in the levels of monoamines and precursors and metabolites in various brain regions, such as DA in striatum, to validate disease models of Parkinson's in mice, monkeys and minipigs27,28,29. Here, we perform the experiment on tissue from dStr and NAc, but the method is also suitable for other DA-innervated brain areas, such as the prefrontral cortex, hippocampus, substantia nigra and ventral tegmental area 30. In these areas, a more diluted standard sample will be necessary for proper determination of DA levels. Our analysis of DA content show higher levels in striatal subcompartments compared to previous investigations, but this can be explained experimentally. First, we have dissected two parts of the striatum (NAc and dStr) as opposed to investigating the whole striatum, which might account for the difference compared to previous reports12,31. Striatal measurements will have lower DA levels compared to measurements in pure dStr, since levels in NAc are substantially lower compared to dStr. We also have previous studies confirming our DA levels5.
Every assay has its limitations. Assays are developed in an attempt to model and provide information on specific aspects of a cellular process, and they might leave out possible important details or provide a too generalized picture of the real world process. An important limitation to consider when choosing HPLC, is that it only provides a snapshot of the neurotransmitter levels. However, neurotransmitter levels are prone to fluctuate over a day, week or month32,33, which emphasizes the need to obtain samples at a narrow time window, instead of comparing samples taken hours, days or months apart as though they were taken within the same hour. However, HPLC data can provide useful information on DA content and reveal aberrant altered levels such as those demonstrated by the DAT-KO and DAT-KD transgenic mouse lines, where genetic deletion or knock-down of DAT significantly influences DA homeostasis by perturbing DA reuptake. These data furthermore demonstrate that striatal DA pools primarily consist of sequestered DA rather than de-novo synthesized DA and that replenishment of intracellular striatal DA pools is critically dependent on the reuptake process12,34. One important pitfall to consider is that tissue dissection may limit a more specific and accurate description of the DA content in a brain area at any given time. The variation in DA concentration in different brain areas varies greatly. Therefore, the accuracy of dissection is of great importance, which can be improved by dissecting smaller areas to ensure only tissue from the area of interest is included.
A more functional measure of the endogenous DA pools can be analyzed using microdialysis. This has been developed and pioneered by Ungerstedt et al. using the vertical microdialysis probe35. The microdialysis technique makes it possible to measure monoamine concentrations in the brain of freely moving animals and in different brain structures. Another advantage of the microdialysis technique over the tissue sampling through HPLC is the option to measure and follow monoamine changes over a large window of time. This is a considerable advantage compared to sampling of brain tissue where only one time point is possible per animal as in the HPLC protocol. While, microdialysis can provide insight into the release of DA, tissue sampling followed by HPLC will instead reveal changes in the endogenous pools and vesicular DA. To obtain real-time information on DA release kinetics in various brain areas, methods like fast-scan cyclic voltammetry36,37 or high-speed chronoamperometry38 can be implemented.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by the UCPH 2016 Program of Excellence (U.G., A.R., K.J.), the Lundbeck Foundation (M.R.) the Lundbeck Foundation Center for Biomembranes in Nanomedicine (U.G.), the National Institute of Health Grants P01 DA 12408 (U.G.), the Danish Council for independent Research - Medical Sciences (U.G.).
References
- Tritsch NX, Sabatini BL. Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron. 2012;76:33–50. doi: 10.1016/j.neuron.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier EA, et al. A biochemical and functional protein complex involving dopamine synthesis and transport into synaptic vesicles. J Biol Chem. 2010;285:1957–1966. doi: 10.1074/jbc.M109.054510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen AS, et al. SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol Rev. 2011;63:585–640. doi: 10.1124/pr.108.000869. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Caron MG. Monoamine transporters: from genes to behavior. Annu Rev Pharmacol Toxicol. 2003;43:261–284. doi: 10.1146/annurev.pharmtox.43.050802.112309. [DOI] [PubMed] [Google Scholar]
- Runegaard AH, et al. Preserved dopaminergic homeostasis and dopamine-related behaviour in hemizygous TH-Cre mice. Eur J Neurosci. 2017;45:121–128. doi: 10.1111/ejn.13347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker VP, Michaelson IA, Kirkland RJ. The separation of synaptic vesicles from nerve-ending particles ('synaptosomes') Biochem J. 1964;90:293–303. doi: 10.1042/bj0900293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornykiewicz O. Dopamine (3-hydroxytyramine) and brain function. Pharmacol Rev. 1966;18:925–964. [PubMed] [Google Scholar]
- Schultz W. Behavioral dopamine signals. Trends Neurosci. 2007;30:203–210. doi: 10.1016/j.tins.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Torres GE, Amara SG. Glutamate and monoamine transporters: new visions of form and function. Curr Opin Neurobiol. 2007;17:304–312. doi: 10.1016/j.conb.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Jones SR, et al. Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc Natl Acad Sci U S A. 1998;95:4029–4034. doi: 10.1073/pnas.95.7.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel LR, Wu S, Melikian HE. Brain slice biotinylation: an ex vivo approach to measure region-specific plasma membrane protein trafficking in adult neurons. J Vis Exp. 2014. [DOI] [PMC free article] [PubMed]
- Rickhag M, et al. A C-terminal PDZ domain-binding sequence is required for striatal distribution of the dopamine transporter. Nat Commun. 2013;4:1580. doi: 10.1038/ncomms2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkley PR, Jarvie PE, Robinson PJ. A rapid Percoll gradient procedure for preparation of synaptosomes. Nat Protoc. 2008;3:1718–1728. doi: 10.1038/nprot.2008.171. [DOI] [PubMed] [Google Scholar]
- Whittaker VP. Thirty years of synaptosome research. J Neurocytol. 1993;22:735–742. doi: 10.1007/BF01181319. [DOI] [PubMed] [Google Scholar]
- Schmitz Y, Benoit-Marand M, Gonon F, Sulzer D. Presynaptic regulation of dopaminergic neurotransmission. J Neurochem. 2003;87:273–289. doi: 10.1046/j.1471-4159.2003.02050.x. [DOI] [PubMed] [Google Scholar]
- Yang L, Beal MF. Determination of neurotransmitter levels in models of Parkinson's disease by HPLC-ECD. Methods Mol Biol. 2011;793:401–415. doi: 10.1007/978-1-61779-328-8_27. [DOI] [PubMed] [Google Scholar]
- Earles C, Schenk JO. Rotating disk electrode voltammetric measurements of dopamine transporter activity: an analytical evaluation. Anal Biochem. 1998;264:191–198. doi: 10.1006/abio.1998.2850. [DOI] [PubMed] [Google Scholar]
- Wu Q, Reith ME, Kuhar MJ, Carroll FI, Garris PA. Preferential increases in nucleus accumbens dopamine after systemic cocaine administration are caused by unique characteristics of dopamine neurotransmission. J Neurosci. 2001;21:6338–6347. doi: 10.1523/JNEUROSCI.21-16-06338.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonfuss D, Reum T, Olshausen P, Fischer T, Morgenstern R. Modelling constant potential amperometry for investigations of dopaminergic neurotransmission kinetics in vivo. J Neurosci Methods. 2001;112:163–172. doi: 10.1016/s0165-0270(01)00465-4. [DOI] [PubMed] [Google Scholar]
- Hoover BR, Everett CV, Sorkin A, Zahniser NR. Rapid regulation of dopamine transporters by tyrosine kinases in rat neuronal preparations. J Neurochem. 2007;101:1258–1271. doi: 10.1111/j.1471-4159.2007.04522.x. [DOI] [PubMed] [Google Scholar]
- Hansen FH, et al. Missense dopamine transporter mutations associate with adult parkinsonism and ADHD. J Clin Invest. 2014;124:3107–3120. doi: 10.1172/JCI73778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damier P, Hirsch EC, Agid Y, Graybiel AM. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson's disease. Brain. 1999;122(Pt 8):1437–1448. doi: 10.1093/brain/122.8.1437. [DOI] [PubMed] [Google Scholar]
- Atack CV. The determination of dopamine by a modification of the dihydroxyindole fluorimetric assay. Br J Pharmacol. 1973;48:699–714. doi: 10.1111/j.1476-5381.1973.tb08259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshitake T, et al. High-sensitive liquid chromatographic method for determination of neuronal release of serotonin, noradrenaline and dopamine monitored by microdialysis in the rat prefrontal cortex. J Neurosci Methods. 2004;140:163–168. doi: 10.1016/j.jneumeth.2004.04.041. [DOI] [PubMed] [Google Scholar]
- Decressac M, Mattsson B, Lundblad M, Weikop P, Bjorklund A. Progressive neurodegenerative and behavioural changes induced by AAV-mediated overexpression of alpha-synuclein in midbrain dopamine neurons. Neurobiol Dis. 2012;45:939–953. doi: 10.1016/j.nbd.2011.12.013. [DOI] [PubMed] [Google Scholar]
- Huot P, Johnston TH, Koprich JB, Fox SH, Brotchie JM. L-DOPA pharmacokinetics in the MPTP-lesioned macaque model of Parkinson's disease. Neuropharmacology. 2012;63:829–836. doi: 10.1016/j.neuropharm.2012.06.012. [DOI] [PubMed] [Google Scholar]
- Mikkelsen M, et al. MPTP-induced Parkinsonism in minipigs: A behavioral, biochemical, and histological study. Neurotoxicol Teratol. 1999;21:169–175. doi: 10.1016/s0892-0362(98)00037-3. [DOI] [PubMed] [Google Scholar]
- Salvatore MF, Pruett BS, Dempsey C, Fields V. Comprehensive profiling of dopamine regulation in substantia nigra and ventral tegmental area. J Vis Exp. 2012. [DOI] [PMC free article] [PubMed]
- Van Dam D, et al. Regional distribution of biogenic amines, amino acids and cholinergic markers in the CNS of the C57BL/6 strain. Amino Acids. 2005;28:377–387. doi: 10.1007/s00726-005-0208-7. [DOI] [PubMed] [Google Scholar]
- Barth C, Villringer A, Sacher J. Sex hormones affect neurotransmitters and shape the adult female brain during hormonal transition periods. Front Neurosci. 2015;9(37) doi: 10.3389/fnins.2015.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corthell JT, Stathopoulos AM, Watson CC, Bertram R, Trombley PQ. Olfactory bulb monoamine concentrations vary with time of day. Neuroscience. 2013;247:234–241. doi: 10.1016/j.neuroscience.2013.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang X, et al. Hyperactivity and impaired response habituation in hyperdopaminergic mice. Proc Natl Acad Sci U S A. 2001;98:1982–1987. doi: 10.1073/pnas.98.4.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerstedt U, Pycock C. Functional correlates of dopamine neurotransmission. Bull Schweiz Akad Med Wiss. 1974;30:44–55. [PubMed] [Google Scholar]
- Wickham RJ, Park J, Nunes EJ, Addy NA. Examination of Rapid Dopamine Dynamics with Fast Scan Cyclic Voltammetry During Intra-oral Tastant Administration in Awake Rats. J Vis Exp. 2015. p. e52468. [DOI] [PMC free article] [PubMed]
- Phillips PE, Robinson DL, Stuber GD, Carelli RM, Wightman RM. Real-time measurements of phasic changes in extracellular dopamine concentration in freely moving rats by fast-scan cyclic voltammetry. Methods Mol Med. 2003;79:443–464. doi: 10.1385/1-59259-358-5:443. [DOI] [PubMed] [Google Scholar]
- Callaghan PD, Irvine RJ, Daws LC. Differences in the in vivo dynamics of neurotransmitter release and serotonin uptake after acute para-methoxyamphetamine and 3,4-methylenedioxymethamphetamine revealed by chronoamperometry. Neurochem Int. 2005;47:350–361. doi: 10.1016/j.neuint.2005.04.026. [DOI] [PubMed] [Google Scholar]