

Abstract
Transposon mutagenesis is a method that allows gene disruption via the random genomic insertion of a piece of DNA called a transposon. The protocol below outlines a method for high efficiency transfer between bacterial strains of a plasmid harboring a transposon containing a kanamycin resistance marker. The plasmid-borne transposase is encoded by a variant tnp gene that inserts the transposon into the genome of the recipient strain with very low insertional bias. This method thus allows the creation of large mutant libraries in which transposons have been inserted into unique genomic positions in a recipient strain of either Escherichia coli or Shigella flexneri bacteria. By using bacterial conjugation, as opposed to other methods such as electroporation or chemical transformation, large libraries with hundreds of thousands of unique clones can be created. This yields high-density insertion libraries, with insertions occurring as frequently as every 4-6 base pairs in non-essential genes. This method is superior to other methods as it allows for an inexpensive, easy to use, and high efficiency method for the creation of a dense transposon insertion library. The transposon library can be used in downstream applications such as transposon sequencing (Tn-Seq), to infer genetic interaction networks, or more simply, in mutational (forward genetic) screens.
Keywords: Immunology, Issue 127, Transposon mutagenesis, transposon insertion library, bacterial mutant library, bacterial conjugation, bacterial mating, bacterial sex, Tn10, loss of function mutation, enterobacteria, Escherichia coli, Shigella flexneri
Introduction
The creation of transposon mutagenesis libraries in bacteria is useful for a wide variety of applications, ranging from discoveries of virulence genes in bacterial pathogens1,2, to studies of essential genes3,4,5,6, to the identification of genetic interaction networks7,8,9. Critical to these studies is the ability to create a large library of mutants. The use of transposons (short fragments of DNA that insert randomly into a genome) is a simple means of disrupting gene function, as the insertion of a transposon within the open reading frame or regulatory region of a gene will often disrupt the function or expression of the gene.
Outlined here is a method for the creation of a transposon library in either E. coli or S. flexneri by bacterial conjugation using the plasmid pJA110. There are two main advantages to using this plasmid. The first advantage is that the variant of the Tn10 transposase expressed from the pJA1 plasmid is inducible and has low insertion bias11,12, meaning that the transposon will randomly integrate into the genome when Isopropyl β-D-1-thiogalactopyranoside (IPTG) is added to the media. The transposon contains a kanamycin resistance marker, allowing for the selection of mutants with the transposon inserts into the chromosome of the recipient strain. The second advantage of the pJA1 plasmid is that it contains an R6K mutant origin of replication. The R6K mutant origin of replication requires the lambda pir gene in order for the maintenance of the plasmid13. As the plasmid cannot replicate in pir- strains, it will be lost in the recipient strain (Figure 1). This ensures that Tn10 transposase is removed from the cell and is no longer active, reducing further mutations after the initial transposition event.
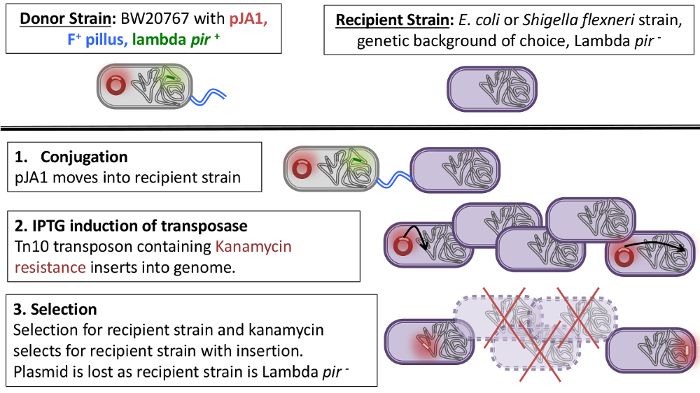
The use of bacterial conjugation to move the pJA1 plasmid from the donor strain to the recipient strain is advantageous for several reasons. Conjugation is simple and inexpensive to perform and does not need special equipment such as an electroporator. Additionally, the high efficiency of bacterial conjugation allows for a very large library (>2 x 105 unique insertions) to be achieved with just a few milliliters (mL) of overnight bacterial culture6. The process takes around two hours of hands-on time along with time for incubation and growth of the bacteria. Langridge et al.14 reported performing 130 electroporations to create a transposon mutagenesis library of a size similar to the one described here8, which is achieved with a single conjugation. The use of 130 electroporations requires labor intensive and time-consuming preparation of electrocompetent cells and the use of many expensive materials (e.g., electroporation cuvettes), at a cost of more than $1,000 USD in consumables alone. Other studies10 have used similar methods, but with different bacterial strains, and achieved far smaller library sizes (5 x 104 colony forming units) than reported here.
Notes on the donor strain: The donor strain used here is the E. coli strain BW2076715 containing the pJA1 transposon plasmid16. Strain BW20767 can conjugate to other strains, making the transfer of the pJA1 plasmid highly efficient. Also, importantly, BW20767 is lambda pir+. As mentioned previously, the plasmid pJA1 is only able to be maintained in strains containing the lambda pir gene. This strain is kanamycin and ampicillin resistant. The pJA1 plasmid contains the ampicillin resistance marker, and a kanamycin resistance marker is contained within the transposon. This strain is grown with selection on the plasmid using ampicillin at 100 µg/mL. It is also worth noting that other strains harboring constitutively expressed transposase genes are known to be unstable, and while the transposase used here is under inducible control, there remains a low risk of leaky expression. For this reason, it is suggested that the passage of this strain should be minimized and a fresh streak should be taken from a frozen culture for each new library preparation. The donor strain used here is available from our lab upon request.
Notes on the Recipient strain: The recipient strain can be a strain of choice, such as K12-derived lab strains of E. coli or strains of S.flexneri (also, see Discussion). The recipient strain must have an additional antibiotic resistance marker that is not kanamycin resistance, so that the donor strain can be selected against. For the recipient strain, an E. coli MG1655 strain is used here with an introduced spontaneous nalidixic acid mutation. The spontaneous nalidixic acid mutant was selected by plating out 2 mL of overnight culture in 200 µL aliquots onto plates containing nalidixic acid at 30 µg/mL. A single clone of MG1655 was selected that was resistant to nalidixic acid to become the recipient strain. Additionally, the recipient strain must be lambda pir negative, as described above.
Overview: Once bacterial conjugation has taken place and the pJA1 plasmid has moved from the donor strain into the recipient strain, the addition of IPTG to the media will induce the expression of the tnp gene, which is under control of the IPTG-inducible lacIq/Ptac promoter (Figure 1). The tnp gene on pJA1 is a mutant transposase which has a lower frequency of insertion at hot spots6,10,11. Upon addition and induction with IPTG, the transposon is activated and randomly inserted in the genome. The plasmid cannot be maintained in the lambda pir- recipient strain and is lost.
Protocol
CAUTION: If using S. flexneri as a recipient strain, please note that S. flexneri is a human pathogen that can cause gastrointestinal disease. Experiments involving S. flexneri must be performed with appropriate biosafety precautions (designated BSL-2 in Europe and USA or PC2 in New Zealand). All experiments performed in the video associated with this protocol were done using the well-characterized recipient strain of non-pathogenic E. coli MG1655 in a PC1 setting. Work with Shigella flexneri was previously performed in a BSL-2 lab at the Biozentrum in Basel, as described in6.
Note: The following experiment is effectively done twice. The first part (Step 1.1- Step 4.5) is performed to find the best dilution for plating out the library, where the goal is to find a dilution of the conjugation which gives many individual colonies per plate, but not so many that a lawn forms. This is to reduce competition between mutants with significantly reduced fitness and those with more robust fitness. The second part (Steps 5.1- 7.4) is the creation of the final library, where many plates are spread of the appropriate dilution of the library.
1. To Prepare One Day Prior to Experiment
Inoculate, from a frozen stock, the donor strain in 2 mL of LB broth, Millers recipe (NaCl at 10 g/L) media containing ampicillin at 100 µg/mL. The donor strain used here is the E. coli strain BW2076715 harboring the pJA1 plasmid11. The use of ampicillin maintains selection for the pJA1 plasmid.
Inoculate, from a single colony or frozen stock, the recipient strain in 2 mL of LB media with a resistance marker other than ampicillin or kanamycin. The recipient strain used here is the E. coli "wild type" strain MG165517,18 with a spontaneous resistance mutant to nalidixic acid. It is grown in 30 µg/mL nalidixic acid.
Prepare 20 Luria Broth containing 1.5% agar plates (in 90 mM petri dishes, approximately 12.5 mL). Agar plates lacking antibiotic can be stored at 4 °C for several months until use.
Prepare 20 Luria Broth containing 1.5% agar plates (in 90 mM petri dishes, approximately 12.5 mL) containing both nalidixic acid at 30 µg/mL and kanamycin at 50 µg/mL (LBA Nal30 Kan50). Agar plates containing antibiotics can be stored in the dark at 4 °C for several months until use.
Prepare 200 mL of sterile LB media. LB media can be stored at room temperature for several months.
Prepare 1 mL of 100 mM sterile stock of IPTG, according to manufacturer's instructions.
2. Bacterial Mating or Conjugation
Centrifuge 1 mL of overnight culture of the donor strain for 1 minute at 14,000 x g at room temperature. Discard growth media. This step is done to remove all growth media containing antibiotics.
Resuspend the cell pellet into 110 µL of LB (not containing antibiotics), thus concentrating the culture.
Using sterile forceps, position a sterile nitrocellulose filter (alternatively a sterile piece of blotting paper can be used, see Materials list) onto the middle of an LBA plate (not containing antibiotics). Label the plate "Negative Control: Donor".
Using a pipette, drop 50 µL of the concentrated donor culture onto the filter.
Repeat steps 2.1-4 for the recipient strain, labeling the plate "Negative Control: Recipient."
For the library, drop 50 µL of the donor (from step 2.2) AND 50 µL of the recipient strain (from step 2.5) onto a single sterile filter on an LBA plate (these plates should not contain antibiotics). Label this plate "Library." Mating occurs because the donor and recipient are in close physical contact on the filter.
Place the agar plates containing the filters at 37 °C for 6 h.
3. Activation of pJA1 Transposon by IPTG Induction of the Transposase
Prepare three 15 mL conical vials containing 2 mL of LB with IPTG at a final concentration of 1 mM (i.e., 20 µL of 100 mM stock of IPTG). Label each: Donor Control, Recipient control, and Library.
Remove the plates from the incubator after 6 h of growth at 37 °C (from step 2.4). Some bacterial growth should be visible on the filter.
Using sterile forceps, remove the filter paper from the donor control and place it in the 15 mL conical vial labeled Donor Control (from step 3.1). Do the same for the Recipient Control and Library.
Tap the tubes to get the filter paper to the bottom of the 15 mL conical vial and ensure that it is fully submerged in LB.
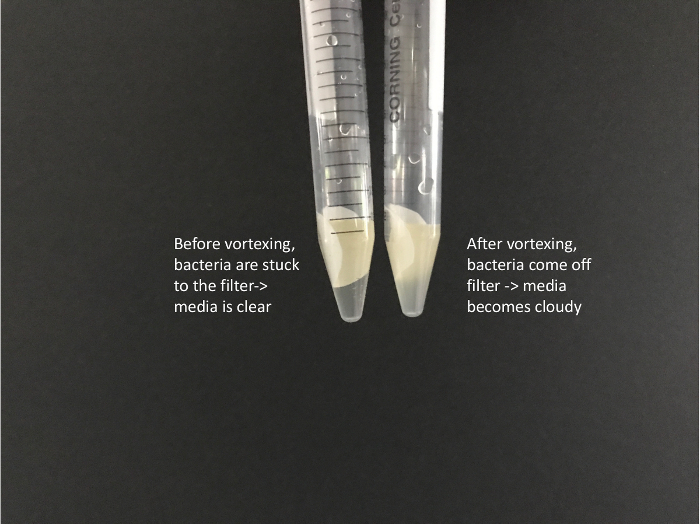
Vortex tubes for at least a full minute on high to disassociate the bacteria from the nitrocellulose filter. The LB should become cloudy with bacterial cells (Figure 2).
Using sterile glass beads or a sterile spreader, plate 200 µL of the vortexed bacteria suspension from the donor control onto LBA Nal30 Kan50 plates labeled "Donor Control, 200 µL".
Repeat the step above (3.6) for the recipient control, labeling the plates "Recipient Control, 200 µL".
Repeat the step above (3.7) for the library, labeling the plates "Library, 200 µL".
Make additional dilutions (i.e., 1:5, 1:10 and 1:100 dilutions, using LB not containing antibiotics as a diluent) of the library. Plate 200 µl of undiluted library and additional dilutions onto appropriately labeled LBA Nal30 Kan50 plates.
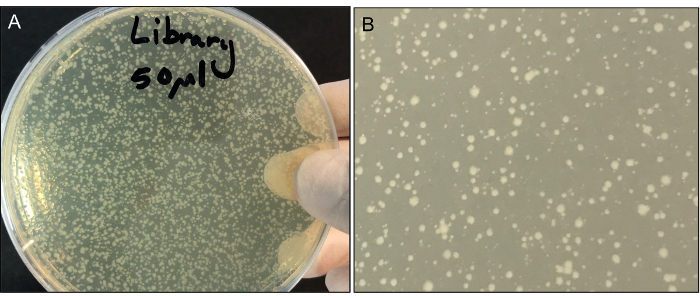
Place plates into the 37 °C incubator for 18 h. Clones with transposons inserted in a gene that causes a large loss of fitness may take longer to grow and appear on the plate. There should be a variety of colony sizes (Figure 3). Donor and recipient strain plates should have no colonies on them.
4. Choose the Appropriate Dilution of the Library to be Used for the Final Mutant Library
Determine a dilution of the library from Step 3.9 that yields many colonies of varying sizes that are adequately spaced. The aim is to get as many colonies as possible on one plate, but not so many that the colonies merge into one another and compete for resources. This number should be approximately 500-2,000 colonies per plate. An example of appropriate colony densities on a plate is shown in Figure 3.
Record the number of colonies on the plates.
Calculate the frequency of conjugation (the number of conjugants per recipient cell). This should be in the range of 1 x 10-4 to 1 x 10-6 19.
Determine the number of mutants desired in the final library based on downstream applications. Many users simply require a library with as many mutants as possible. To generate as many transposon insertions as theoretically possible (one insertion every base pair), aim for approximately the same number of colonies as base pairs in the genome (i.e., ~4.5 x 106 for E. coli) to fully saturate the genome with all possible transposon insertions. It is important to note, many mutants harboring insertions in essential or functionally important genes will not be able to be recovered, as these mutations will be fatal to the recipient.
Calculate how much volume of the initial library is needed to create a library of the desired size. For example, the desired library size is 5 x 104 mutants. The dilution with the best spacing of colonies from Step 3.9 was 200 µL of a 1:10 dilution. The number of colonies on this plate is estimated to be approximately 2,000 colonies. If 5 x 104 mutants are needed and each plate has 2,000 colonies, then 25 plates will be needed at this dilution.
5. Creation of the Final Mutant Library
Repeat steps 1 to 3.8, adjusting the number of plates at step 1.4 as needed (see step 4.4).
At step 3.9, plate the dilution chosen in step 4.1 onto as many plates as needed to create the library of the desired size (see step 4.4).
6. Estimate Library Density
Count how many colonies there are per plate, and thereby get a rough estimate of how dense the library is. For example: A total of 2x105 colonies are plated. The E. coli MG1655 genome is about 4.5 x 106 base pairs. Therefore, a library with about 2 x 105 inserts means that there is an insert on average every 22 base pairs and that each gene should be mutated about 45 times, given that one gene is approximately 1 x 103 base pairs. Insertions in essential genes are likely to be very underrepresented in this library, and thus the density in non-essential genes is likely to be greater than this. This sort of calculation provides one with a broad estimate of transposon density.
7. Pooling Transposon Library and Storage
After counting colonies, add 1 mL of LB (or more, as needed) to the library plate and use a sterile spreader to scrape off bacteria from the plate. Remove the bacterial suspension and place in a 50 mL or 15 mL conical vial. Repeat for all plates.
Vortex the pooled bacteria suspension for a full minute to homogenize the suspension.
- Add sterile glycerol to a final concentration of 20%.
- Prepare 100 x 20 µL aliquots in 0.25 mL tubes for easy re-growth.
- Prepare at least two or more 1 mL aliquot in cryovials.
Store all aliquots at -80°C
Representative Results
After 18 hours of growth, plates should contain many colonies with a variety of colony sizes(Figure 3). Different colony sizes indicate clones of varying fitness, and are a good sign that the protocol has worked. The donor and recipient controls should have no growth on the plates. Using the protocol above should yield well over 2 x 105 colony forming units (CFU). Scaling up the protocol to five replicate conjugations at once should yield approximately 1 x 106 CFU. In previous work, analysis using next generation sequencing indicated that such a scaled up library should yield >2 x 105 unique insertions6. At very high CFU (i.e., 1 x 106 CFU) the rate of unique insertions is expected to plateau, as all available (non-fatal) insertions become represented.
Figure 1 : Schematic of bacterial conjugation and insertion of transposon into the chromosome. Please click here to view a larger version of this figure.
Figure 2: Image of filters submersed in LB media before and after vortexing. Before vortexing, cells are stuck to the filter and the media is clear. After vortexing, the media becomes cloudy as cells have come off of the filter and are in the media. Please click here to view a larger version of this figure.
Figure 3: (A) Representative plate of transposon library after 18 h of growth. (B) Close up of colony sizes. Please click here to view a larger version of this figure.
Discussion
The protocol described here allows for the construction of a dense insertion library. This method allows for the creation of a transposon library with over 2 x 105 unique transposon mutants using under 5 mL of culture volume6. It is relatively easy to perform, uses reagents available in most basic microbiology labs, is scalable, and requires little in the way of expensive equipment or consumables such as electroporation cuvettes.
A significant benefit of this method is that, in theory, the user has wide latitude in the choice of enterobacterial recipient strains. This paper, as well as others11, use E. coli as a recipient strain, however the pJA1 plasmid has been used successfully with other enterobacterial recipient species such as Shigella flexneri6 and Salmonella enterica serovar Typhimurium strain SL134410. Theoretically, the γ origin of replication (oriR6Kγ) in pJA1 allows this plasmid to be maintained in a broad host range19, allowing that the recipient strain is pir+. Recently, new methods have been described that allow for construction of the pir+ in a range of enterobacterial strains20, giving additional flexibility. Additionally, the300 base pair mob region from the RP4 plasmid in pJA1 allows conjugative transfer of this plasmid to a wide range of gram negative bacterial strains19. Simply put, this method could theoretically be used with a variety of recipient strains, as long as several conditions are met: the strain is pir+, and is marked with an antibiotic resistance other than kanamycin and other than the donor strain.
A critical step in the protocol lies in estimating the proper number of cells to plate out in step 4.1. If colonies are spaced too closely, they compete for nutrients on the plate and less fit mutants are outcompeted. This may lead to a reduction in the total number of mutants. Alternatively, if colonies are spaced too far apart, there will be too few colonies on the plate, and the total number of agar plates needed to achieve a large library becomes burdensome. Therefore, achieving the right balance in terms of colony numbers per plate is important.
It is important to perform the controls listed in the protocol to ensure the steps are working as described. Notably, when using nalidixic acid as a counter-selection against the donor strain, it is important to ensure the negative donor control plates are free of colonies. This is because there may be a low rate (approximately 1 x 10-10)21 of spontaneous resistance to nalidixic acid, yielding false positives. Typically, the rate of conjugation and transposition is approximately 2 x 10-4 19. Therefore, the rate of conjugation and transposition is several orders of magnitude greater than the rate of spontaneous resistance to nalidixic acid. Therefore, the rate of false positives compared to true transposition events is very low and deemed negligible when the protocol is working. However, if rates of conjugation or transposition are significantly reduced, (from low mating efficiency and/or lack of induction of the transposase gene with IPTG) and the protocol is scaled up to compensate for this, then the number of false positives (clones that do not have the transposon inserted) may also increase.
Some modifications can be made to incubation times in Steps 3.2 and 3.10. Step 3.2 states that conjugation should occur for 6 h, but in our experience, this time step can be varied (i.e., 4-7 h) without changing the results significantly. Additionally, in step 3.10, the length of time the colonies are incubated on the agar plates can also be adjusted. This can be varied depending on the average doubling time or growth rate of the recipient strain. Additionally, in our experience, 18 h yielded a variety of colony sizes, indicating a library of diverse fitness. However, colonies with greatly reduced fitness may take longer to grow and thus may not be visible after 18 h. If this method is being used to find clones of extremely reduced fitness, longer incubation times and fewer colonies on the plate to reduce crowding (i.e., 48 h, 50-300 colonies) may be used.
Additional pitfalls of this method include that it is not possible to use a recipient strain that is already kanamycin resistant. It may be possible to swap out the kanamycin resistance marker in the pJA1 plasmid for an alternative selectable marker, such as chloramphenicol resistance to overcome this hurdle. It may also be worth noting that, in theory, it is possible to use a recipient strain that is ampicillin resistant, as the pJA1 plasmid containing the ampicillin resistance marker is lost shortly after transposition.
The creation of a dense transposon library in the genetic background of choice is potentially advantageous for many downstream applications. For example, a dense transposon library could be used to identify auxotrophic mutants using replica plating22 or to identify mutants that are defective in establishing an infection1,2. More recently, as DNA sequencing costs have dropped and new technologies such as next generation sequencing have become commonplace, transposon libraries have been used with deep DNA sequencing to gain insight into gene essentiality, gene function, and genetic interactions. Some of these methods are reviewed in 23 and include methods such as transposon-directed insertion site sequencing (TraDIS), transposon sequencing (Tn-seq), high-throughput insertion tracking by deep sequencing (HITS), and insertion sequencing (INSeq). All these downstream methods rely on the construction of dense transposon insertion libraries. While other vectors may need to be used for particular downstream methods, the protocol described here gives an overview of the salient procedural points to follow.
Disclosures
The author has nothing to disclose.
Acknowledgments
I thank the George Church Lab for the kind gift of the pJA1 plasmid. I thank Fabienne Hamburger and Alex Boehm from the Urs Jenal Lab at the Biozentrum in Basel for help with bacterial conjugation and for providing the BW20767 strain. I also thank Olin Silander for helpful edits. Funding for this research was provided by funds from Massey University in New Zealand and the Swiss Initiative in Systems Biology (Project "Battle X" awarded to Dirk Bumann) at the University of Basel, Switzerland.
References
- Camacho LR, Ensergueix D, Perez E, Gicquel B, Guilhot C. Identification of a virulence gene cluster of Mycobacterium tuberculosis by signature-tagged transposon mutagenesis. Mol Microbiol. 1999;34(2):257–267. doi: 10.1046/j.1365-2958.1999.01593.x. [DOI] [PubMed] [Google Scholar]
- Hensel M, Shea JE, Gleeson C, Jones MD, Dalton E, Holden DW. Simultaneous identification of bacterial virulence genes by negative selection. Science. 1995;269(5222):400–403. doi: 10.1126/science.7618105. [DOI] [PubMed] [Google Scholar]
- Glass JI, Assad-Garcia N, et al. Essential genes of a minimal bacterium. PNAS. 2006;103(2):425–430. doi: 10.1073/pnas.0510013103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama NR, Shepherd B, Falkow S. Global transposon mutagenesis and essential gene analysis of Helicobacter pylori. J Bacteriol. 2004;186(23):7926–7935. doi: 10.1128/JB.186.23.7926-7935.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerley BJ, Rubin EJ, Camilli A, Lampe DJ, Robertson HM, Mekalanos JJ. Systematic identification of essential genes by in vitro mariner mutagenesis. PNAS. 1998;95(15):8927–8932. doi: 10.1073/pnas.95.15.8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed NE, Bumann D, Silander OK. Combining Shigella Tn-seq data with gold-standard E. coli gene deletion data suggests rare transitions between essential and non-essential gene functionality. BMC Microbiology. 2016;16(1):1–14. doi: 10.1186/s12866-016-0818-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Opijnen T, Bodi KL, Camilli A. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nature Methods. 2009;6(10):767–772. doi: 10.1038/nmeth.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langridge GC, Phan MD, et al. Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Research. 2009;19(12):2308–2316. doi: 10.1101/gr.097097.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawronski JD, Wong SMS, Giannoukos G, Ward DV, Akerley BJ. Tracking insertion mutants within libraries by deep sequencing and a genome-wide screen for Haemophilus genes required in the lung. PNAS. 2009;106(38):16422–16427. doi: 10.1073/pnas.0906627106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K, Kim CC, Falkow S. Microarray-based detection of Salmonella enterica serovar Typhimurium transposon mutants that cannot survive in macrophages and mice. Infect and Immun. 2005;73(9):5438–5449. doi: 10.1128/IAI.73.9.5438-5449.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badarinarayana V, Estep PW, et al. Selection analyses of insertional mutants using subgenic-resolution arrays. Nature biotechnol. 2001;19(11):1060–1065. doi: 10.1038/nbt1101-1060. [DOI] [PubMed] [Google Scholar]
- Bender J, Kleckner N. IS10 transposase mutations that specifically alter target site recognition. The EMBO Journal. 1992;11(2):741–750. doi: 10.1002/j.1460-2075.1992.tb05107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafferman A, Helinski DR. Structural properties of the beta origin of replication of plasmid R6K. J Biol Chem. 1983;258(7):4083–4090. [PubMed] [Google Scholar]
- Langridge GC, Phan MD, et al. Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Research. 2009;19(12):2308–2316. doi: 10.1101/gr.097097.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf WW, Jiang W, Daniels LL, Kim SK, Haldimann A, Wanner BL. Conditionally replicative and conjugative plasmids carrying lacZ alpha for cloning, mutagenesis, and allele replacement in bacteria. Plasmid. 1996;35(1):1–13. doi: 10.1006/plas.1996.0001. [DOI] [PubMed] [Google Scholar]
- Badarinarayana V, Estep PW, et al. Selection analyses of insertional mutants using subgenic-resolution arrays. Nature biotechnol. 2001;19(11):1060–1065. doi: 10.1038/nbt1101-1060. [DOI] [PubMed] [Google Scholar]
- Guyer MS, Reed RR, Steitz JA, Low KB. Identification of a Sex-factor-affinity Site in E. coli as γδ. Cold Spring Harbor Symposia on Quantitative Biology. 1981;45(0):135–140. doi: 10.1101/sqb.1981.045.01.022. [DOI] [PubMed] [Google Scholar]
- Blattner FR, Plunkett G, et al. The Complete Genome Sequence of Escherichia coli K-12. Science. 1997;277(5331):1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- Alexeyev MF, Shokolenko IN. Mini-Tn10 transposon derivatives for insertion mutagenesis and gene delivery into the chromosome of gram-negative bacteria. Gene. 1995;160(1):59–62. doi: 10.1016/0378-1119(95)00141-r. [DOI] [PubMed] [Google Scholar]
- Kvitko BH, Bruckbauer S, et al. A simple method for construction of pir+ Enterobacterial hosts for maintenance of R6K replicon plasmids. BMC Res Notes. 2012;5(1):157. doi: 10.1186/1756-0500-5-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick JE, Yu DS, et al. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature. 2009;461(7268):1243–1247. doi: 10.1038/nature08480. [DOI] [PubMed] [Google Scholar]
- Jacobs MA, Alwood A, et al. Comprehensive transposon mutant library of Pseudomonas aeruginosa. PNAS. 2003;100(24):14339–14344. doi: 10.1073/pnas.2036282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Opijnen T, Camilli A. Transposon insertion sequencing: a new tool for systems-level analysis of microorganisms. Nature Rev Microbiol. 2013;11(7):435–442. doi: 10.1038/nrmicro3033. [DOI] [PMC free article] [PubMed] [Google Scholar]