

Abstract
Elucidation of the binding properties of chromatin-targeting proteins can be very challenging due to the complex nature of chromatin and the heterogeneous nature of most mammalian chromatin-modifying complexes. In order to overcome these hurdles, we have adapted a sequential salt extraction (SSE) assay for evaluating the relative binding affinities of chromatin-bound complexes. This easy and straightforward assay can be used by non-experts to evaluate the relative difference in binding affinity of two related complexes, the changes in affinity of a complex when a subunit is lost or an individual domain is inactivated, and the change in binding affinity after alterations to the chromatin landscape. By sequentially re-suspending bulk chromatin in increasing amounts of salt, we are able to profile the elution of a particular protein from chromatin. Using these profiles, we are able to determine how alterations in a chromatin-modifying complex or alterations to the chromatin environment affect binding interactions. Coupling SSE with other in vitro and in vivo assays, we can determine the roles of individual domains and proteins on the functionality of a complex in a variety of chromatin environments.
Keywords: Genetics, Issue 128, Chromatin, sequential salt extraction, chromatin remodeling complexes, binding profiles, epigenetics, histone marks, histone recognition domains, SWI/SNF, Polybromo-1
Introduction
DNA regulation in eukaryotic cells is an intricate and sophisticated system that is tightly controlled by an assortment of proteins that coordinate responses to intracellular and extracellular stimuli. DNA is wrapped around histone octamers to form nucleosomes, which can be loosely distributed along DNA or compacted into tight coils1. This structural arrangement of DNA and histones is known as chromatin, which is regulated by a network of proteins that read, write, and erase post translational modifications (PTM) on histones2. Some histone PTMs, such as acetylation, change the charge of the amino acid they are deposited on, altering interactions between histones and DNA2. Histone PTMs also serve to recruit transcriptional regulators, chromatin remodelers, DNA damage repair machinery, and DNA replication machinery to specific regions of the genome3.
Most methods for studying chromatin interactions either probe small scale interactions or involve large genome-wide analyses. In vitro binding studies often utilize individual recombinant domains with histone peptides or DNA in assays such as electrophoresis mobility shift assays (EMSA), isothermal titration calorimetry, fluorescence polarization, and peptide pulldowns. Because these assays typically focus on an individual protein domain, they facilitate the understanding of a small piece of the puzzle, but do not allow us to understand the cooperative nature of multi-domain proteins, let alone their role in multi-protein complexes. Another layer of intricacy is added by the heterogeneous composition of most mammalian chromatin-modifying complexes. This protein heterogeneity, in combination with the dynamic nature of the chromatin landscape, makes it challenging to recapitulate the in vivo binding interactions of chromatin proteins to chromatin in vitro.
In vivo methods have made significant advances; however, they are often expensive, time consuming, and technically challenging. Chromatin immunoprecipitation followed by sequencing (ChIP-seq) is very useful for determining the localization of proteins and histone modifications across the genome, however it requires substantial optimization4. Proteins are often crosslinked to chromatin to preserve interactions; however, this can produce artificial interactions and may cause epitope masking5. Furthermore, the immunoprecipitations (IP) require highly specific antibodies, and extensive optimization of DNA shearing and IP conditions by ChIP-qPCR using a known binding site, which is often not available a priori. After optimization of ChIP conditions, processing of the samples is costly and requires several weeks to months to sequence and analyze. Though this method is invaluable for identifying the localization of chromatin bound proteins across the genome, the cost and time commitment make it prohibitive to use this method to generate hypotheses about how small changes may affect global binding properties.
In this paper, we describe how a sequential salt extraction (SSE) assay can be used to examine global binding profiles of chromatin-bound proteins and distinguish how changes in a protein, complex, or global PTM profile can alter interactions. Though salt extractions are a commonly and broadly used technique, we demonstrate how this sequential method is highly reproducible and versatile. SSE allows us to characterize how a single subunit of a complex or even a single domain contributes to the complex's overall affinity for bulk chromatin. SSE can also be used to determine if the binding of a protein is influenced by changes in chromatin landscape, providing interesting hypotheses for histone mark targeting that can be confirmed using ChIP-seq and other genome wide studies.
We originally adapted this method from Wu et al., to examine of the function of Polybromo1 (PBRM1) in the binding of the PBAF chromatin remodeler6,7. Using this technique, we determined the role of PBRM1 for chromatin binding within the PBAF chromatin remodeling complex and then determined the relative contribution of the six individual bromodomains to this function7.
Here we describe how to optimize this method to explore chromatin binding in different cell types, to assess the relative binding affinity of similar chromatin modifying complexes, to examine the displacement of a protein from chromatin by a chemical inhibitor, and to determine the effects of chromatin binding after alterations to the chromatin landscape.
Protocol
1. Preparations
Prepare 100 mL of hypotonic solution Buffer A: 0.3 M sucrose, 60 mM KCl, 60 mM Tris pH 8.0, 2 mM EDTA, and 0.5% NP-40. Store at 4 ºC. NOTE: Some cell lines, such as HEK293T, require less stringent lysing conditions. If nuclei lyse easily, use Modified Buffer A: 25 mM HEPES pH 7.6, 25 mM KCl, 5 mM MgCl2, 0.05 mM EDTA, 0.1% NP-40, and 10% glycerol.
Prepare a 250 mL stock solution of 2x mRIPA solution: 100 mM Tris pH 8.0, 2% NP-40, and 0.5% sodium deoxycholate.
Prepare a 100 mL stock solution of 5 M NaCl.
Using the 2x mRIPA and 5 M NaCl solutions, prepare 50 mL of 1x mRIPA solution for each of the six salt concentrations: 0 mM, 100 mM, 200 mM, 300 mM, 400 mM, and 500 mM NaCl. Prior to starting the experiment, cool all solutions on ice.
Grow cell lines under desired conditions. NOTE: When determining the effects of knocking down a protein, SSE for the knockdown line must be compared to wildtype cells. These SSE must be performed side by side. For examples using chemical inhibitors or stimuli, refer to sections 3 - 5.
2. Basic Sequential Salt Extraction
Harvest 8 million cells of each condition and wash twice with 5 mL of ice cold PBS. NOTE: It is critical to have equal number of cells to have equivalent protein concentrations. The exact number of cells may need to be optimized for individual cell lines or particular proteins. It is important not to have too many, as the increase in protein concentrations will prevent observation of the elution curve, which is dependent on equilibrium binding. However, with too few cells there may not be enough protein to detect the curve, and the chromatin pellet is harder to isolate and can be lost between fractions.
Re-suspend cells in 1 mL of Buffer A + protease inhibitors, transfer into 1.5 mL micro centrifuge tubes, and rotate top over bottom at 4 ºC for 10 min.
Isolate the nuclei by centrifugation at 6000 x g for 5 min at 4 ºC . NOTE: After this step the nuclear envelope should still be intact. If the nuclear envelope is intact, the pellet will re-suspend fully; however, when the envelope is lysed and the chromatin is released, the pellet will not re-suspend. If the nuclear envelope is not intact after Buffer A incubation, use Modified Buffer A.
- Add 200 µL of mRIPA 0 mM NaCl + protease inhibitors to each nuclei pellet before re-suspending pellet. Homogenize each sample by pipetting 15 times with a 1 mL pipette. The pellet should re-suspend easily, however, the nuclei will lyse as it is pipetted and the sample will become thick and sticky and difficult to pipette.
- In order to draw up the pellet into the tip, tap the end of the pipette tip against the bottom of the centrifuge tube. The pellet will not fully dissolve once the DNA is released from the nuclei, but pipetting will break it up.
When all the samples have been homogenized, incubate all the samples on ice for 3 min. NOTE: The incubation time may need to be optimized depending on the protein of interest.
Isolate chromatin pellet by centrifuging the samples for 3 min at 6500 x g at 4 ºC.
Transfer supernatant to a clean 1.5 mL centrifuge tube. This will be the 0 mM fraction. This 0 mM mRIPA solution can act as a wash step to lysis the nuclei and remove any loose proteins not associated with chromatin.
Add 200 µL of mRIPA 100 mM NaCl + protease inhibitors to each chromatin pellet before re-suspending pellet. Break up the chromatin pellet by pipetting the pellet up and down 15 times. NOTE: It is critical to be consistent with the number of times the pellet in pipetted.
Incubate on ice for 3 min. This incubation step allows all samples to reach an equilibrium state, which is particularly important when there are multiple samples.
Centrifuge at 6500 x g for 3 min at 4 ºC and transfer supernatant to a clean 1.5 mL tube.
Repeat 2.6 - 2.10 for the remaining salt concentrations. NOTE: After 400 mM NaCl, mRIPA the pellet should be clear and gloopy and will not stay at the bottom of the tube. The pellet can be placed in the lid of the centrifuge tube while the supernatant isolated.
Add 70 µL of 4x protein loading dye to each fraction and load 30 µL of each fraction on to an SDS acrylamide gel for western blot analysis. NOTE: To determine the binding profile of the protein, it is critical to load equivalent volumes of lysate, rather than loading equal protein concentrations.
Perform a standard western blot by transferring onto a membrane and use primary antibodies for proteins of interest.
To quantitate the protein eluted from the chromatin, incubate the blot in infrared fluorescence IRDye secondary antibodies and image blot with an imager. While other methods of development can be used, we recommend fluorescence or infrared imaging, as it is more quantitative in nature.
Use ImageJ or similar software to calculate the intensity for the protein eluted at each salt concentration. By graphing the band intensity against the salt concentration, the elution pattern of your protein of interest can be determined.
3. Sequential Salt Extraction in the Presence of a "Reader" Inhibitor
Harvest two sets of cells (4 million) and isolate the nuclei as in a standard SSE.
Re-suspend both sets in 200 µL of mRIPA 0 mM NaCl and incubate for 3 min. This will allow for the removal of any free protein in the nuclei.
Add 200 µL mRIPA 0 mM NaCl to each pellet. Add the inhibitor (2 µL of 1 mM (+)JQ1) to one sample and DMSO to the control set.
Agitate the pellet by pipetting 15 times and incubate on ice for 5 min.
Centrifuge at 6500 x g for 3 min at 4 ºC and transfer supernatant to a clean 1.5 mL tube.
Repeat 3.3 - 4 for all the salt concentrations and perform a standard western blot.
4. Sequential Salt Extraction in the Presence of a "Writer" Inhibitor
Treat cells with the inhibitor (10 µM SAHA) or DMSO for 3 h.
Harvest 4 million cells for each treatment.
Perform a standard SSE for the samples with the inhibitor added to all the buffers.
5. Sequential Salt Extraction Following DNA Damage
Treat cells with 1 µM doxorubicin for 1 h.
Change the media on the cells and allow them to recover for 3 h.
Harvest 8 million cells and perform the basic SSE.
6. Non-Sequential Salt Extraction
Harvest 12 million cells and wash with PBS.
Divide cells so that there are 2 million cells per micro centrifuge tube.
Re-suspend in 500 µL of Buffer A and incubate for 10 min.
Isolate the nuclei by centrifugation at 6000 x g.
Re-suspend pellets in a 200 µL of each of the mRIPA buffers + NaCl.
Homogenize each sample by pipetting 15 times with a 1 mL pipette tip.
Incubate on ice for 3 min.
Isolate the chromatin by centrifugation at 6500 x g and transfer the supernatant to clean tubes. Add 70 µL of loading dye and run 30 µL on a SDS-page gel for western blot analysis.
Representative Results
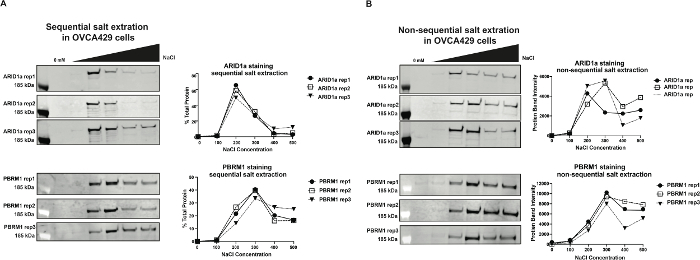
In this paper, we demonstrate the advantages and applications of the commonly used sequential salt extraction (SSE) method that we have adapted from the literature6. In Figure 1, we compare the reproducibility of SSE to extracting proteins non-sequentially by detecting the elution patterns of ARID1a and PBRM1. We consistently observe that ARID1a, a BAF subunit, elutes primarily at 200 mM NaCl and PBRM1, an exclusive PBAF subunit, elutes primarily at 300 mM NaCl. Figure 1a shows three independent replicates of SSE with 8 million OVCA429 cells. Figure 1b depicts a non-sequential salt extraction where nuclei isolated from 2 million cells were re-suspended in a single salt buffer. Though both methods conclude that ARID1a elutes at 200 mM and PBRM1 elutes at 300 mM, the SSE produces a well-defined binding profile and allows for a clear distinction of the binding of these two complexes. Furthermore, in the non-sequential method, the amount of protein eluted in the 400 mM and 500 mM NaCl buffers is lower than the 300 mM and is inconsistent between the three replicates. Though this issue may be resolved with further optimization of incubation time and centrifugation speed, this illustrates the reproducibility advantage of SSE.
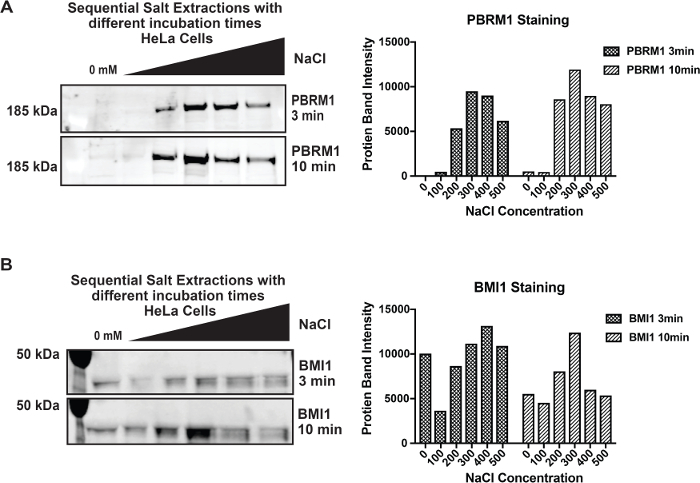
Through our optimization, we found that other chromatin complexes may require more time to be eluted. In Figure 2, we demonstrate that the transcriptional activator, PBAF, elutes from chromatin consistently between SSEs with 3 min and 10 min incubation times. In contrast, elution of the transcriptional repressor, Polycomb Repressive Complex 1 (PRC1), indicated by BMI-1, requires a longer incubation time to be released from chromatin8. This phenomenon could be due to PRC1 being localized in more compact and inaccessible regions of the genome, or could be due to differential binding kinetics for the two complexes.
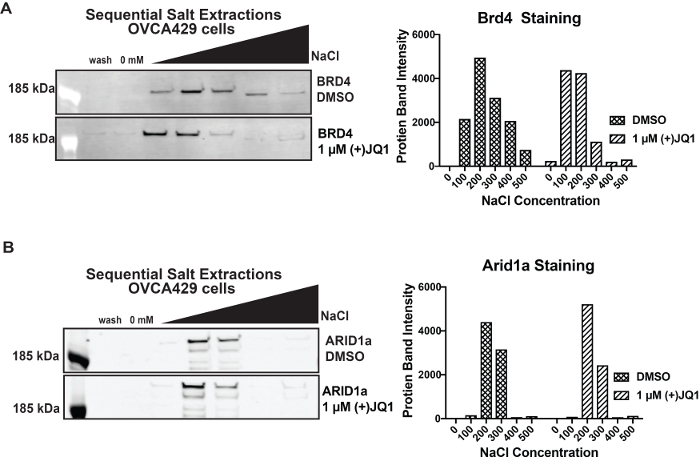
After optimization for a particular protein, SSEs can be used to study how the strength of protein binding changes in different conditions. SSE can be used to examine the effectiveness of an inhibitor of protein-protein interactions. To demonstrate this concept, we utilized (+) JQ1, which is a BRD4 bromodomain inhibitor, to examine how it altered BRD4 binding (Figure 3a)9. We isolated nuclei from 4 million OVCA429 cells. To remove any unbound BRD4, an initial 0 mM wash was performed on both samples. Then a standard SSE was performed with DMSO or 1 µM (+) JQ1 added to each fraction. Samples were incubated for 5 min. We observe that BRD4 elutes earlier in the presence of the inhibitor compared to DMSO. To show that (+) JQ1 is specific for BRD4, we blotted for ARID1a and saw no change in elution (Figure 3b).
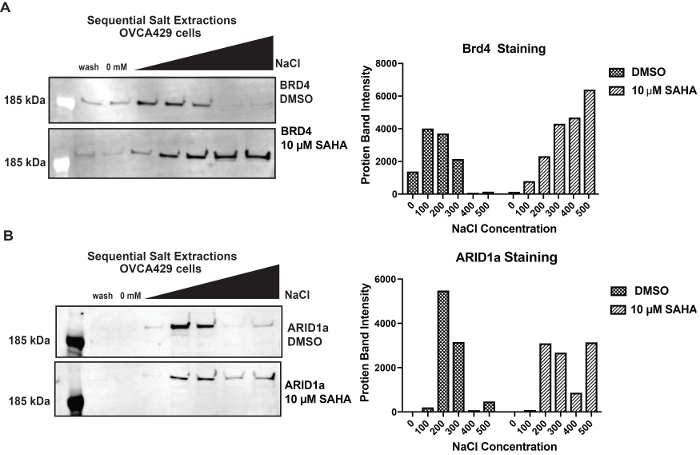
Next we examined how protein binding can be altered when the landscape of the chromatin is modified. BRD4 contains two bromodomains that recognize acetylated lysine residues on histone tails10. To increase the level of histone acetylation, we treated OVCA429 cells with 10 µM of the histone deacetylase inhibitor suberanilohydroxamic acid (SAHA) for 3 h. SAHA (10 µM) was added to all the buffers during the SSE. By increasing the global histone acetylation levels, we observed an increase in BRD4 binding (Figure 4a). When blotting for ARID1a, we observe tighter binding of ARID1a as well, which is not surprising because subunits of the BAF complex, such as BRG1, BRM, and BRD9, all contain bromodomains10,11 (Figure 4b).
Lastly, to exhibit how SSE can be used to look at how protein binding changes when cells experience genomic alterations, we induced double stranded DNA breaks with the topoisomerase II inhibitor, doxorubicin12. We treated HEK293T cells with 1 µM of doxorubicin for an hour and allowed cells to recover for three hours. After performing the SSE, we blotted for PBRM1, which is involved in DNA damage repair13. We observed two peaks for PBRM1 binding: one at 300 mM and one at 500 mM NaCl (Figure 5). This suggests that some of the PBRM1 population is binding normally, but a subset of PBRM1 is more tightly bound to chromatin following DNA damage. This is just one example of how to use SSE to examine how chromatin interactions are altered in response to different external stimuli.
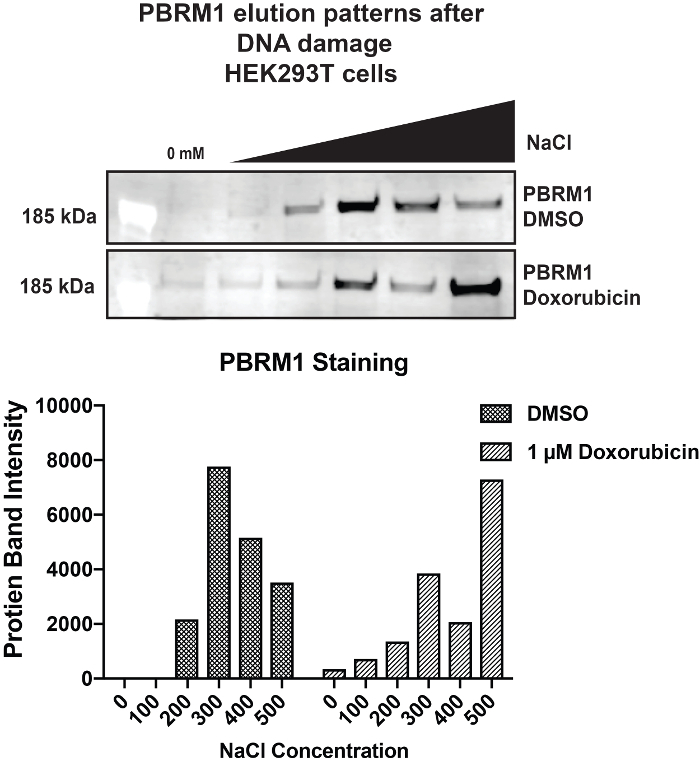
Figure 1: Non-sequential compared to sequential salt extractions in OVCA429 cells A) Immunoblots and quantification of three independent replicates of ARID1a and PBRM1 elution profiles by the sequential salt extraction method. B) Immunoblots and quantification of three independent replicates of ARID1a and PBRM1 elution profiles in the non-sequential salt extraction method. Please click here to view a larger version of this figure.
Figure 2: Comparison of incubation times of PBAF and PRC1 elution profiles A) Comparison of PBAF (PBRM1) elution profiles with 3 and 10-minute incubation times. B) Comparison of PRC1 (BMI-1) elution profiles with 3 and 10-minute incubation times. Please click here to view a larger version of this figure.
Figure 3: Effectiveness of (+) JQ1 on inhibiting BRD4 binding A) Elution pattern of BRD4 in the presence of 1 µM (+) JQ1 compared to DMSO control in OVCA429 cells. B) Elution profile of ARID1a in the presence of 1 µM (+) JQ1 compared to DMSO control in OVCA429 cells. Band intensity is indicated for 0 mM- 500 mM fraction with DMSO or (+) JQ1. Please click here to view a larger version of this figure.
Figure 4: Alterations in binding when chromatin landscaped is modified A) Comparison of BRD4 elution from OVCA429 cells treated with 10 µM SAHA for 3 hours compared to DMSO treatment. B) Elution profiles of ARID1a from OVCA429 cells treated with10 µM SAHA compared to DMSO. Band intensity is indicated for 0 mM - 500 mM fraction with DMSO or SAHA. Please click here to view a larger version of this figure.
Figure 5: Changes in PBRM1 binding after DNA Damage SSE results for PBRM1 binding from HEK293T cells treated with 1 µM Doxorubicin. Please click here to view a larger version of this figure.
Discussion
Characterization of protein and chromatin interactions through salt extractions is a common method that has been employed for decades14,15; however, it has not been systematically optimized before to reveal its full utility. We demonstrate how this sequential method provides a rapid and inexpensive way to distinguish changes in chromatin binding when the protein or the environment is altered. SSE is highly adaptable and optimizable, and importantly, it is technically simple and requires no specialized equipment. The key aspects that need to be optimized prior to starting are: starting cell number, hypotonic buffer conditions, and incubation time.
The most important aspect of cell number is that it is consistent between all the samples. When looking at BAF complexes, using 8 million cells gives a good profile of how these complexes are binding; however, the number of cells (or the amount of buffer) may need to be adjusted depending on the abundance of the protein of interest. It is important to note that using fewer cells is challenging because at 400 mM and 500 mM NaCl it is hard to visualize the chromatin pellet. It is vital to make sure the pellet is not transferred with the supernatant after 400 mM NaCl.
In order to accurately evaluate nuclear proteins, the nuclei need to stay intact until they are lysed with 0 mM NaCl mRipa solution. Many commonly used hypotonic solutions are too harsh for cell lines such as HEK293T and HeLa cells. For these cell lines, it is recommended to use the modified Buffer A.
Finally, the incubation time may vary depending on the experiment. For BAF subunits the elution pattern does not change between a 3 min and 10 min incubation; however, the elution of the PRC1 complex drastically changes depending on how long the samples are incubated. Additionally, when evaluating the effect of an inhibitor on the binding of its target, the incubation time may need to be optimized depending on inhibitor kinetics.
When performing the experiment, it is important to be as consistent as possible with treatment of the samples. For each fraction, the salt buffer should be added to every sample prior to homogenization so that each sample is equally exposed. The point of pipetting is to break up the chromatin and help release the bound proteins. It is important to make sure the pellet passes through the pipette tip. Especially when using a larger number of cells, the chromatin pellet is challenging to pass through the pipette tip the first few times. We have found that a 1 mL tip works the best for this, as with minor tapping of the tip against the bottom of the micro centrifuge tube, we are able to draw up the pellet into the tip. After passing the chromatin pellet through the tip fifteen times, the pellet should smoothly move through the tip, though it will not dissolve. After incubating the samples on ice and isolating the chromatin by centrifugation, removing the supernatant with a 200 µL pipette tip allows for maximal removal without disrupting the chromatin pellet.
Though SSE requires technical consistency, it is simple, straightforward, and can be performed with common laboratory resources. It is important to note that the true power of this method is when it is coupled with other phenotypical examinations. For example, using this technique, we compared the binding profiles of PBRM1 when each of its six bromodomains were mutated, we determined that all but one of the bromodomains were involved in the binding of PBRM1 to varying degrees7. Intriguingly, we found that the bromodomains that were the most important for binding were also essential for PBRM1 regulation of gene expression and control cell proliferation7. We also validated the changes in binding of these mutants to a discrete genomic locus by quantitative ChIP-qPCR7.
We have shown only a few examples of how this technique can be used to study protein-chromatin interactions; however, in our lab, we have found that SSE is a versatile tool for investigating a wide array of questions regarding chromatin reader proteins. In combination with other analyses, this technique facilitates our ability elucidate the functionality of the components comprising these elaborate protein complexes. By understanding the importance of individual domains, we can identify whether they are potential therapeutic targets for the development of an inhibitor that blocks chromatin association.
We have demonstrated how SSE can be expanded to evaluate the effectiveness of a small molecule inhibitor on protein binding to chromatin. Additionally, by inhibiting epigenetic writers and erasers, SSE can determine the relationship between PTM levels and reader proteins. We have also shown the SSE can be used to determine changes in binding in response to external stimuli such as DNA damage. Though this is a simple technique, when applied in the proper conditions, it can greatly advance our knowledge of chromatin and its regulatory proteins.
Disclosures
The authors have no competing financial interests.
Acknowledgments
This work was supported by a V scholar award (V2014-004) and a V scholar plus award (D2016-030) from the V Foundation for Cancer Research, and an American Cancer Society Institutional Research Grant (ACS IRG Grant 58-006-53) to the Purdue University Center for Cancer Research. E. G. P. was supported by the Borch Graduate Endowment Award to the Purdue University Medicinal Chemistry and Molecular Pharmacology Department.
References
- Ramakrishnan V. Histone structure and the organization of the nucleosome. Annu. Rev. Biophys. Biomol. Struct. 1997;26:83–112. doi: 10.1146/annurev.biophys.26.1.83. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Rese. 2011;21(3):381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman CA, Lalonde M-E, Côté J, Kutateladze TG. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol. 2012;19(12):1218–1227. doi: 10.1038/nsmb.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey TS. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat Rev Genet. 2012;13(12):840–852. doi: 10.1038/nrg3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Henikoff S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife. 2017;6 doi: 10.7554/eLife.21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SY, Lee AY, Lai HT, Zhang H, Chiang CM. Phospho switch triggers brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol Cell. 2013;49(5):843–857. doi: 10.1016/j.molcel.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter EG, Dykhuizen EC. Individual bromodomains of Polybromo-1 contribute to chromatin association and tumor suppression in clear cell renal carcinoma. J Biol Chem. 2017;292(7):2601–2610. doi: 10.1074/jbc.M116.746875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerppola TK. Polycomb group complexes--many combinations, many functions. Trends cell biol. 2009;19(12):692–704. doi: 10.1016/j.tcb.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149(1):214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez R, Zhou M-M. The role of human bromodomains in chromatin biology and gene transcription. Curr opin drug disc dev. 2009;12(5):659–665. [PMC free article] [PubMed] [Google Scholar]
- Yang F, Teves SS, Kemp CJ, Henikoff S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim. Biophys. Acta - Rev Cancer. 2014;1845(1):84–89. doi: 10.1016/j.bbcan.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakarougkas A, et al. Requirement for PBAF in Transcriptional Repression and Repair at DNA Breaks in Actively Transcribed Regions of Chromatin. Mol Cell. 2014;55(5):723–732. doi: 10.1016/j.molcel.2014.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Park J-H, Kim S, Park E-J, Yun Y, Kwon J. A proteomics approach for the identification of nucleophosmin and heterogeneous nuclear ribonucleoprotein C1/C2 as chromatin-binding proteins in response to DNA double-strand breaks. Biochem J. 2005;388(Pt 1) doi: 10.1042/BJ20042033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson MM, Boner W, Morgan IM. TopBP1 Regulates Human Papillomavirus Type 16 E2 Interaction with Chromatin. J Virol. 2007;81(8):4338–4342. doi: 10.1128/JVI.02353-06. [DOI] [PMC free article] [PubMed] [Google Scholar]