

Abstract
We describe a simple, robust and high throughput single molecule flow-stretching assay for studying 1D diffusion of molecules along DNA. In this assay, glass coverslips are functionalized in a one-step reaction with silane-PEG-biotin. Flow cells are constructed by sandwiching an adhesive tape with pre-cut channels between a functionalized coverslip and a PDMS slab containing inlet and outlet holes. Multiple channels are integrated into one flow cell and the flow of reagents into each channel can be fully automated, which significantly increases the assay throughput and reduces hands-on time per assay. Inside each channel, biotin-λ-DNAs are immobilized on the surface and a laminar flow is applied to flow-stretch the DNAs. The DNA molecules are stretched to >80% of their contour length and serve as spatially extended templates for studying the binding and transport activity of fluorescently labeled molecules. The trajectories of single molecules are tracked by time-lapse Total Internal Reflection Fluorescence (TIRF) imaging. Raw images are analyzed using streamlined custom single particle tracking software to automatically identify trajectories of single molecules diffusing along DNA and estimate their 1D diffusion constants.
Keywords: Biochemistry, Issue 128, Single Molecule Imaging, DNA flow stretching, TIRF Imaging, One-step reaction coverslip functionalization, PDMS flow cell, High throughput capability, Single Particle Tracking, One-dimensional diffusion, 1D diffusion constant
Introduction
A long-standing problem in biology is how endogenous proteins that act at specific sites in the genome can locate their DNA targets quickly enough for the organism to survive and respond effectively to its environment. Studies over the past forty years proposed and largely support the hypothesis that the kinetics of DNA target search by a protein can be accelerated by facilitated diffusion in which the protein alternates between 3D diffusion in the bulk and 1D diffusion (including sliding and hopping processes) along the DNA1. It is now known that many proteins involved in gene regulation, nucleic acid metabolism, and other processes are capable of sliding on DNA2,3,4,5,6,7,8,9. Furthermore, recent studies reported that even small peptides can bind to and slide on DNA, with the ability to carry a cargo; for example, a protein molecule or PCR primer, along DNA10,11,12,13,14.
Over the last 15 years, the single molecule flow-stretching assay has been widely used to study binding and diffusion of molecules along DNA2,15,16. In this type of assay, biotinylated double-stranded DNA molecules are immobilized to the surface and a laminar flow is applied to flow stretch the DNA. The > 80% stretched DNAs serve as spatially extended templates for studying binding and transport activity of molecules labeled with fluorophores where the trajectories of single molecules along DNA are tracked by time-lapse fluorescence imaging. In our implementation, optimized for reproducibility and ease of use, this assay consists of five major steps: preparation of biotin-λ-DNA, coverslip functionalization, flow cell construction, fluorescence imaging and data analysis. In previous protocols17, glass coverslips were functionalized by first reacting with (3-Aminopropyl)triethoxysilane (APTES) and then with amine-reactive polyethylene glycol (PEG) reagents (e.g., NHS-PEG-biotin) to form a PEG layer that resists nonspecific adsorption of assay components to the coverslip surface. The quality of functionalized coverslips largely depended on the quality of PEG reagents and reaction conditions at each step. Our protocol describes a simplified functionalization protocol and multiplex flow cell construction which requires no liquid adhesive or curing time on the day flow cells are assembled. We also describe a streamlined and robust data analysis procedure11 that eliminates computationally intensive regression steps by applying a radial symmetry method for centroid localization18 and a covariance-based diffusion constant estimator19.
Here, we report a simple, robust and high throughput single molecule flow stretching assay implementation with significant improvements made in coverslip functionalization, flow cell construction and data analysis. In particular, we developed a one-step coverslip functionalization protocol, in which clean dry coverslips react directly with silane-PEG-biotin. This protocol simplifies the coverslip preparation and improves the reliability of the quality of functionalized surfaces compared to the standard two-step reaction protocol. We describe the use of so-functionalized coverslips with multi-channel PDMS flow cells that enable robust tubing connections to be made without glue. These flow cells further include multiple computer-controlled inlets for each flow chamber which enable automated reagent flow to reduce hands-on time during setup and increased assay throughput.
Protocol
1. Preparation of biotin- λ-DNA
NOTE: Biotin-λ-DNA molecules are prepared by ligating a biotin-labeled oligonucleotide, 5'-AGGTCGCCGCCC(A)20-biotin-3' to λ-DNA molecules20.
Prepare 0.1 mM biotin labeled oligo in TE buffer.
Heat 0.5 mg/mL λ-DNA stock at 65 °C for 60 s, and plunge into wet ice right away. NOTE: Fast quench cooling reduces λ-DNA concatemerization.
Pipette 100 µL of the λ-DNA solution into a microcentrifuge tube.
Add 1 µL of the 0.1 mM oligo into 9 µL of TE buffer.
Add 2 µL of the oligo solution to the microcentrifuge tube containing the λ-DNA. Mix thoroughly. NOTE: Oligo is present in approximately a 12-fold molar excess over the complementary λ-DNA end.
Heat the λ-DNA/oligo mixture at 65 °C for 60 s.
Slowly cool the mixture to room temperature. NOTE: This step allows the oligo to anneal to the complementary λ-DNA end.
Place the mixture on ice. Add 11 µL of T4 DNA ligase reaction buffer (final buffer concentration 1x). Mix gently. Then add 2 µL (800 units) of T4 DNA ligase enzyme. Mix gently.
Incubate the mixture for 2 h at 16 °C or overnight at 4 °C.
Purify the product using centrifugal filter tubes with a nominal molecular weight limit of 100 kDa. Specifically, transfer the step 1.9 mixture into a centrifugal filter tube and centrifuge at 14,000 x g for 5 min, wash with 400 µL TE buffer twice, and collect the purified product. NOTE: The as-purified biotin-λ-DNAs in TE buffer are stable at -20 °C for up to one year.
2. Coverslip functionalization
NOTE: Glass coverslips are functionalized by reacting with silane-PEG-biotin. This one-step reaction protocol is simpler and more reliable in our experience than the standard two-step reaction protocol20. Coverslip functionalization with silane-PEG-biotin creates binding sites for streptavidin and minimizes nonspecific DNA and protein adsorption to the coverslip surface.
Sonicate five no. 1 coverslips in 95% ethanol inside a staining jar for 10 min and then rinse with ultrapure water for three times. NOTE: The fifth coverslip provides a spare in case of breakage.
Fill the staining jar with 1 M KOH, sonicate for 10 min and then rinse with ultrapure water three times.
Repeat the 2.1-2.2 cycle twice.
Dry the coverslips under clean dry N2 gas flow.
Further clean and dry the coverslips by applying air plasma treatment at 900 mTorr pressure for 5 min.
Incubate coverslips with 25 mg/mL silane-peg-biotin dissolved in 95% ethanol at room temperature for 2 h. Specifically, sandwich 50 µL silane-peg-biotin solution between two clean dry coverslips, and incubate the coverslip "sandwiches" inside a closed pipette tip box with about 10 mL of water at the bottom (not in contact with the coverslips) to minimize solvent evaporation.
Wash away excess PEG molecules with ultrapure water and dry the PEG coated coverslips under clean dry N2 gas flow. NOTE: The functionalized coverslips can be stored up to three days under ambient conditions.
3. Flow cell construction
NOTE: Flow cells are constructed by sandwiching a double-adhesive tape with pre-cut channels between a PEG functionalized coverslip and a PDMS slab containing inlet and outlet holes. Multiple channels are integrated per flow cell.
Design flow channels in a CAD software and cut the channels (the width, depth and length of each channels are 1, 0.13 and 12 mm, respectively) on a double-adhesive tape by using a tape cutter (typically eight channels are integrated per flow cell). Remove tape residuals to form the flow channels.
To make PDMS slabs, thoroughly mix 45 g of PDMS with 5 g of crosslinking reagent (enough for making twelve 5-mm thick PMDS slabs which can be stored indefinitely under ambient conditions) using a mixer, pour the mixture into two 10 cm Petri dishes, and leave the dishes inside a vacuum chamber until essentially all air bubbles are gone (manually remove air bubbles if any remain when the dishes are removed from the vacuum chamber). Then transfer the dishes into an 80 °C oven until PDMS solidifies (about 2 h).
Cut out a PDMS slab that matches the size of the double-adhesive tape with pre-cut channels. Peel one protection film off the double-adhesive tape and adhere the film to a flat face of the PDMS slab. Punch the outlet and inlet holes on the PDMS slab using a biopsy hole-puncher with a 23 Gauge needle.
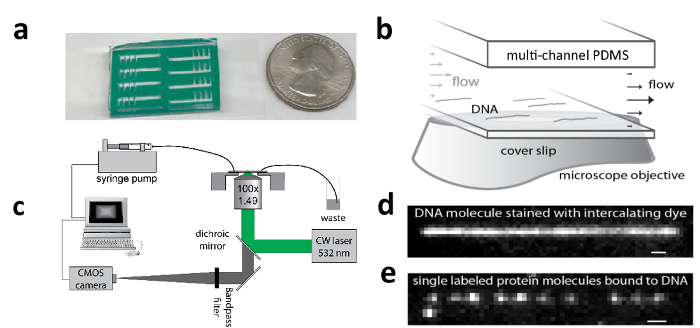
Peel the second protection film off the double-adhesive tape and adhere the PDMS-tape assembly to the functionalized surface of the coverslip (make sure that the coverslip is flat. See a picture of a fully assembled flow cell in Figure 1a).
4. TIRF Imaging
NOTE: Biotin-λ-DNAs are tethered to a flow channel surface and laminar flow is applied to flow stretch the DNA molecules (Figure 1b). The > 80% stretched DNA molecules serve as spatially extended templates for observing the binding and transport activity of fluorescently labeled molecules. The trajectories of single molecules are tracked by time-lapse TIRF imaging (Figure 1c & e).
Fix the flow cell on a microscope stage and load pre-degassed blank buffer (10 mM sodium phosphate, 2 mM NaCl, 50 µM EDTA, 20 mM ethanol, 5% (v/v) glycerol, 0.01% Tween-20, 1% (v/v) β-mercaptoethanol, pH 7.4), 0.2 mg/mL streptavidin solution, 1 mg/mL Bovine Serum Albumin (BSA) solution and 100 pM biotin-λ-DNA solution into four reservoirs that each have a Tygon tubing connected to a solenoid value and another Tygon tubing connected to a PEEK tubing by sleeving the Tygon tubing over the smaller PEEK tubing (prevents backflow of reagents). Degas small volumes of buffer by overnight exposure to vacuum or shorter exposure to vacuum with stirring (until bubbles are no longer visible).
Insert the PEEK tubing of the blank buffer reservoir into one inlet hole of the flow cell. Prime the channel by flowing in blank buffer, and insert three other PEEK tubing into inlet holes The air pressure driven flow of each reagent is controlled by solenoid valves and fully automated via a computer script. Be careful not to induce air bubble formation in the channel.
Connect a Tygon tubing with a short PEEK tubing from the outlet hole to a waste container. The short PEEK tubing at the outlet helps suppress air bubble formation during infusion by maintaining positive pressure inside the channel.
- Tethering biotin- λ-DNA molecules to a flow channel surface.
- Flow in 0.2 mg/mL streptavidin solution, incubate for 5 min and then flow in blank buffer to wash out the unbound streptavidin.
- Flow in 1 mg/mL BSA solution, incubate for 1 min, and then flow in blank buffer to wash out the unbound BSA. NOTE: BSA further suppresses DNA and sample adsorption to the surface.
- Flow in 100 pM biotin-λ-DNA solution, incubate for 10 min and then flow in blank buffer to wash out the unbound DNAs. NOTE: After the DNA is bound to the surface, avoid rapid flows to prevent damage to the tethered DNA molecules.
To check the quality of the functionalized coverslip, flow in DNA staining dye such as Sytox orange under the assay conditions to be used (read 4.6 below), and start fluorescence imaging under 532 nm laser illumination. NOTE: The quality of the functionalized coverslip is typically good and the density of flow stretched DNA molecules will be high. It is essential to have a high enough density of flow stretched DNA molecules so that DNA binding and sliding/1D diffusion events can be observed.
Fine tune the TIRF angle to achieve best signal to noise ratio of images of stretched DNA molecules (Figure 1d).
- To record trajectories of single molecules moving along DNA, repeat steps 4.2-4.4 inside a new channel (without DNA staining dye), then infuse pre-degassed sub-nanomolar fluorophore labeled molecules at a high enough flow rate by using a syringe pump and collect time-lapse fluorescence images with a frame rate of 100 Hz. NOTE: A flow rate equivalent to a Weissenberg number (Wi: the dimensionless product of the longest polymer relaxation time - approximately 0.2 s for λ-DNA - and the shear rate - the change in flow velocity with distance from the coverslip surface) of 500 inside the channel is recommended for single molecule imaging with a frame rate of 100 Hz21.
- Collect 10,000 frames in one Field-Of-View (FOV) to produce a movie, and collect similar movies from multiple (typically ten) FOVs per sample. NOTE: The density of single particles in one FOV should be neither too high - which will complicate data analysis by making object assignment across frames ambiguous, nor too low - which might lead to low occurrence of single molecule events on DNA.
- Optimize the illumination intensity so that molecules bound to the coverslip surface are quickly photo-bleached to suppress background while molecules diffusing on DNA are not bleached too quickly so that longer trajectories can be detected. NOTE: We typically use 200 - 800 W/cm2 power density in the FOV.
At the end of step 4.6, infuse DNA staining dye to confirm that DNA molecules can still be flow-stretched.
Run both positive and negative control samples. NOTE: A good positive control is a tetrapeptide, TetraMethylRhodamine(TMR)-KRRR (TMR is coupled to the N-terminal amine) which has a mean 1D diffusion constant of 10.5 ± 0.7 (standard error) M(bp2/s) at neutral pH11. A good negative control is to measure labeled samples of interest in assay buffer in a channel that has no DNA tethered, where no on-DNA diffusion activity should be detected.
5. Data Analysis:
NOTE: A custom single particle tracking software is used to identify trajectories of single molecules diffusing along DNA and estimate diffusion constants. This software first determines the centroid positions of single particles with high accuracy, identifies particles that are stuck on the coverslip surface, and then links particles in different frames to form time-lapse trajectories. From these trajectories, the 1D diffusion constants are estimated.
To start data analysis, open a scripting software (see table of materials) and go to the directory of the single particle tracking software. Open the script named "largedataprocess3.m". One important parameter to be defined is the threshold value for single particle detection.
To determine the optimal threshold value, run the "Determine_threshold_value.m" script. This script will indicate how the threshold value affects single particle detection.After determining the optimal threshold value, run the "largedataprocess3.m" script.
After completion of single particle tracking analysis, filter the raw trajectories by running the "Trajectory_filtering.m" script. A Graphic User Interface (GUI) is built in to visualize the filtering step.
On the GUI panel, set the minimal displacement along DNA, MinXDisp, to 2 pixels; set the maximal displacement transverse to DNA, MaxYDisp, to 2 pixels; set the minimal number of frames, minFrames, to 10; set the minimal number of states of triplet, minTriplets, to 10; set the minimal diffusion constant along DNA, D_par, to 0; set the maximal estimated diffusion constant transverse to DNA, D_trans, to 10 M(bp2)S-1; set the minimal statistic parameter, Chi2_stat, to -5; set the minimal ratio of displacement along DNA to that transverse to DNA, MinX/Y, to 2. NOTE: All raw trajectories that pass these filtering parameters are listed in Table 1, and those that do not are listed in Table 3.
Click on a trajectory number in Table 1, the trajectory will be displaced in Graphics 1 &2. Click on the "Play" button to play the raw fluorescence images. Click on the "Add_to_Table2" to identify the trajectory as a single molecule sliding trajectory. In the end, all trajectories added to Table2 will be saved when closing the GUI.
To calculate the mean diffusion constants, run the script "Calculate_mean_diffusion_constant.m". The mean diffusion constant is estimated from the set of trajectories that have passed all filtering steps and been added to Table 2.
Representative Results
Figure 2a shows the detected trajectories of pVIc-Cy3B (pVIc: GVQSLKRRRCF; Cy3B is conjugated to the cysteine residue) molecules diffusing on DNA in 2 mM NaCl buffer at pH 6.5. The diffusion of pVIc is bi-directional and driven by ambient thermal energy. From these trajectories, the 1D diffusion constants (D1 values) are estimated for each trajectory (see the histogram in Figure 2b). The mean D1 value of this pVIc conjugate is calculated to be 22.2 ± 0.9 (standard error) M(bp2/s) by averaging each D1 value across trajectories weighted by the number of triplet consecutive position calls contained in each trajectory.
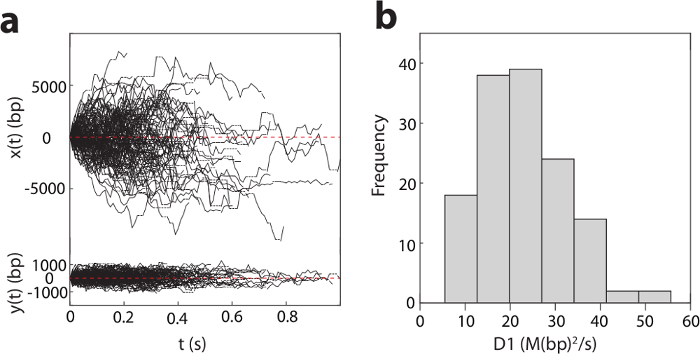
Figure 1: Apparatus for tracking motion of single molecules along DNA. a) An eight-channel PDMS microfluidic chip adhered to a functionalized coverslip to which DNA can be tethered (with USA quarter coin for scale); b) Flow cell zoom schematic showing flow-stretched λ-DNA molecules; c) Schematic of TIRF microscope and flow system for stretching DNA; d) TIRF image of a flow-stretched λ-DNA; e) TIRF image of pVIc-Cy3B (pVIc: GVQSLKRRRCF; Cy3B is conjugated to the cysteine residue) molecules bound to a flow-stretched λ-DNA. This figure has been modified with permission2,12. Please click here to view a larger version of this figure.
Figure 2: One-dimensional diffusion of pVIc (GVQSLKRRRCF) along DNA. a). Diffusion of pVIc along flow stretched λ-DNAs (137 trajectories) in 2 mM NaCl buffer at pH 6.5. x(t) and y(t) are the displacements along and transverse to λ-DNA, respectively. Dotted horizontal lines indicate missing points resulting from dye blinking. b). Histogram of the diffusion constant, D1 for pVIc diffusing along λ-DNAs. Please click here to view a larger version of this figure.
Discussion
When switching from the traditional two-step coverslip functionalization reaction protocol to a one-step reaction protocol, we noticed that the reliability of coverslip functionalization is significantly improved. As the total hands-on time for coverslip preparation is less than thirty min, we recommend preparing fresh coverslips each day single molecule imaging is planned. To minimize deterioration of silane-PEG-biotin, the reagent should be aliquoted and stored under dry N2 gas at -20 °C upon receipt. Here we do not use PEG molecules that produce -COO- or -CH3 terminal groups that have been used in the past20. The negative charges of -COO- groups and the hydrophobic -CH3 groups were utilized to prevent nonspecific adsorption of DNA molecules and specific protein samples to the surface, respectively. We observe very low surface adsorption by small peptide samples on the fully biotin-terminated PEG surface, while silane-PEG-COOH and/or silane-PEG-CH3 might be useful for assays of certain protein samples or when assay conditions (such as pH <7.5) promote DNA-surface interactions.
The shift from glass slide flow cells to PDMS flow cells speeds up the flow cell construction and allows easy integration of multiple channels. We often engineer eight channels per flow cell to enable eight independent experiments per functionalized coverslip. To further increase throughput per coverslip, even larger numbers of micro-fabricated channels could be used.
It is advisable to prevent air bubble formation inside the channel at any point after initial filling. Usually, air bubbles do not cause loss of tethered DNAs (although this is possible), but rather disrupt flow and possibly cause DNAs to stick to the coverslip. Buffer degassing and the addition of small surfactant quantities are typically effective in preventing bubble formation within the tubing and flow channel. In cases where air bubbles are introduced to the tubing, try to flush the bubbles out under a slow flow and make sure that they do not flow through the region where DNAs are tethered.
A streamlined and efficient data analysis software is critical, due to the huge amount of data generated. We usually collect 700,000 high-resolution images from measurements on one flow cell, which can be processed using our custom single particle tracking software within one day on a desktop computer with a quad-core processor and 32 Gb memory. The false positive rate of detecting 1D diffusion trajectories by the software (step 5.5) is sample dependent and can be quite low given that: 1) The density of single particles in the FOV is low enough so that there is little ambiguity linking particles across frames (protein samples in general are more prone to nonspecific adsorption to the coverslip than peptide samples, and thus, the buffer and the power density for each protein sample needs to be optimized); 2) The signal to noise ratio of single particles is high enough so that the likelihood of false-identification of particles is low. Still, the software can make occasional mistakes, and we recommend manual inspection of the data using the provided GUI to evaluate the software performance. In an application particularly sensitive to false-positive trajectory detection, process parameters including the threshold value, minFrames, minTriplets, and Chi2_stat can be set more stringently at the cost of lower sensitivity for true binding events and potentially greater statistical bias in trajectory identification. We here share and describe our single particle tracking software with the hope of helping standardize data analysis methods and stimulate discussion about best practices.
In our protocol, which requires continuous flow during imaging to maintain the stretched state of the DNA template molecules, the fluid flow could bias the transport of some proteins along DNA templates if they diffuse by a hopping mechanism or if the hydrodynamic radius of the labeled fluorophore is large22,23. While such bias can be measured and corrected in most datasets, the transport of proteins labeled with a small fluorophore and diffusing by a sliding mechanism is not typically biased to a detectable extent by flow2,4,24. Notably, the impact of the flow velocity on the transport of proteins along DNA has been used to study the mechanisms of protein diffusing along DNA23. A closely related alternative approach that enables measurements in the absence of flow utilizes DNA templates with biotin at both termini that are transiently stretched in flow and tethered by both ends to the coverslip such that the DNA molecules remain in an extended configuration when the flow is turned off25,26,27,28. The double-tether approach has the advantage of allowing flow-free transport experiments but requires modifying both ends of the DNA template molecules, careful flow control during tethering, and characterizing the distances between the two tethered sites. In addition, the impact of heterogeneous tensions and DNA conformational dynamics across DNA molecules on the single-molecule assay must be assessed.
Altogether, in addition to studying 1D diffusion of molecules along DNA, the as-reported single molecule assay can benefit many in vitro biochemical and biophysical studies at the single molecule level including DNA target search processes by proteins, enzymatic activities on DNA, and more.
Troubleshooting
Problem 1: The flow channels leak.
Solution: First, make sure that the design of flow channels allows at least 1.5 mm margin for sealing around and between channels; and then, during assembly of the flow cell, make sure that contact surfaces between the coverslip, the double-adhesive tape and the PDMS slab are flat, dry and free of debris, that the pressure applied to form the seal is uniform, and that the sealing operation is performed at room temperature.
Problem 2: The density of tethered DNAs is too low.
Solution: This problem arises when the PEG functionalization efficiency is low or the DNA molecules are not functionalized with biotin. From our experience, the density of tethered DNA should be high for > 90% of the coverslips prepared from fresh or properly stored silane-PEG-biotin reagent. In cases when the density of tethered DNA is low with a proven batch of biotin-λ-DNA, try boosting concentrations of silane-PEG-biotin during PEG functionalization and concentrations of streptavidin and biotin-λ-DNA during DNA tethering. If this fails, replace the PEG and streptavidin reagents.
Problem 3: DNAs stick to the coverslip and cannot be stretched by flow.
Solution: This problem can also arise when the PEG functionalization efficiency is low, and is aggravated by low assay pH. Try flowing in more BSA or raising the pH (eg to 8.0) to help suppress DNA adsorption to the surface (step 4.3.2). If DNA adsorption is a persistent issue under a particular assay condition, try including up to 90% silane-PEG-COOH during PEG functionalization.
DATA ANALYSIS NOTES:
We use custom single particle tracking software to identify trajectories of single molecules diffusing along DNA, from which their 1D diffusion constants, D1 are estimated. To improve data analysis, we implemented the radial symmetry algorithm18 for localizing particles and the covariance-based estimator19 for estimating 1D diffusion constants, which dramatically sped up data analysis without compromising accuracy. We previously validated the custom software by analyzing simulated data11. The major steps are described below.
Particle identification:
This step is for detecting and segmenting particles - diffraction-limited spots - from background. First, spatially band-pass filter the images to enhance the signal of particles in the 3-5 pixel range (which corresponds to the size of the point-spread function on our system) and then threshold based on intensity (see script: spt2_SD_sandbox.m).
Centroid assignment
Segmenting the image only localizes molecules to pixel-level spatial resolution. To localize molecules to much higher precision, employ the Radial Symmetry super resolution algorithm18 to images filtered as above. (This method is based on an analytical calculation and thus vastly reduces computational burden compared with other popular high-accuracy methods such as 2D Gaussian fitting (see script: spt2_SD_sandbox.m).)
Particle tracking:
To track the trajectory of a particle through time, we must re-identify the same particles as they move between frames. We consider the initiation, blinking, and termination stages to be components of a trajectory. We constrain linkage of particles in current frames to those appearing in previous frames by the maximum displacement allowed per frame. In cases where multiple linkages are possible, the Jonker-Volgenant algorithm is used to yield globally optimal linkage sets (see script: traceTraj4_kx.3.m).
D1 estimation:
To estimate D1 values from trajectories, we use the Covariance-Based Estimator (CVE)19. Based on an analytical calculation, this method is more computationally efficient and statistically rigorous than other commonly used methods such as regression of mean squared displacements (see script: runspt5_SD.m).
Trajectory Filtering:
Some trajectories contain artifacts such as linkage to surface-bound molecules and must be removed. We implemented several features such as minimum displacement parallel to DNA, maximum displacement transverse to DNA, minimal length of trajectory, minimal number of triplet consecutive detected positions, and minimum probability score (see Xiong et al11 for definition) that can be filtered to isolate a minimally biased set of trajectories likely to represent molecules diffusing on DNA (see script: sptViewFig2_update3x.m).
Disclosures
The Broad Institute may elect to file patent applications that include aspects of the work presented here.
Acknowledgments
We thank Tony Kulesa, Robin Kirkpatrick and Evangelos Gatzogiannis for assistance with initial instrumentation setup and testing. We further thank Tony Kulesa for significant help writing and evaluating the customized single particle tracking software. This work was supported by startup funding and a Career Award at the Scientific Interface (to PCB) from the Burroughs Wellcome Foundation.
References
- Berg OG, Winter RB, von Hippel PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry. 1981;20:6929–6948. doi: 10.1021/bi00527a028. [DOI] [PubMed] [Google Scholar]
- Blainey PC, van Oijen AM, Banerjee A, Verdine GL, Xie XS. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. P Natl Acad Sci USA. 2006;103:5752–5757. doi: 10.1073/pnas.0509723103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leith JS, et al. Sequence-dependent sliding kinetics of p53. P Natl Acad Sci USA. 2012;109:16552–16557. doi: 10.1073/pnas.1120452109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blainey PC, et al. Regulation of a Viral Proteinase by a Peptide and DNA in One-dimensional Space IV. Viral proteinase slides along DNA to locate and process its substrates. J Biol Chem. 2013;288:2092–2102. doi: 10.1074/jbc.M112.407460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziano V, et al. Regulation of a Viral Proteinase by a Peptide and DNA in One-dimensional Space II. Adenovirus proteinase is activated in an unusual one-dimensional biochemical reaction. J Biol Chem. 2013;288:2068–2080. doi: 10.1074/jbc.M112.407312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziano V, et al. Regulation of a viral proteinase by a peptide and DNA in one-dimensional space: I. binding to DNA and to hexon of the precursor to protein VI, pVI, of human adenovirus. J Biol Chem. 2013;288:2059–2067. doi: 10.1074/jbc.M112.377150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baniecki ML, McGrath WJ, Mangel WF. Regulation of a viral proteinase by a peptide and DNA in one-dimensional space: III. atomic resolution structure of the nascent form of the adenovirus proteinase. J Biol Chem. 2013;288:2081–2091. doi: 10.1074/jbc.M112.407429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabata H, et al. Visualization of single molecules of RNA polymerase sliding along DNA. Science. 1993;262:1561–1563. doi: 10.1126/science.8248804. [DOI] [PubMed] [Google Scholar]
- Harada Y, et al. Single-molecule imaging of RNA polymerase-DNA interactions in real time. Biophysical journal. 1999;76:709–715. doi: 10.1016/S0006-3495(99)77237-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangel WF, McGrath WJ, Xiong K, Graziano V, Blainey PC. "Molecular sled"- vehicle of 11-amino acids that facilitates biochemical interactions via sliding components along DNA. Nature Communications. 2016. [DOI] [PMC free article] [PubMed]
- Xiong K, Blainey PC. Molecular sled sequences are common in mammalian proteins. Nucleic Acids Res. 2016;44:2266–2273. doi: 10.1093/nar/gkw035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong K, Erwin GS, Ansari AZ, Blainey PC. Sliding on DNA: from peptides to small molecules. Angew. Chem. Int. Ed. 2016;55:15110–15114. doi: 10.1002/anie.201606768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkin A, et al. Speeding up biomolecular interactions by molecular sledding. Chem Sci. 2016;7:916–920. doi: 10.1039/c5sc03063c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zheng L, Meng Z, Balinin K, Loznik M, Herrmann ,A. Accelerating chemical reactions by molecular sledding. Chem Commun (Camb) 2017;47:6331–6334. doi: 10.1039/c7cc02500a. [DOI] [PubMed] [Google Scholar]
- van Oijen AM, et al. Single-molecule kinetics of lambda exonuclease reveal base dependence and dynamic disorder. Science. 2003;301:1235–1238. doi: 10.1126/science.1084387. [DOI] [PubMed] [Google Scholar]
- Fazio T, Visnapuu ML, Wind S, Greene EC. DNA curtains and nanoscale curtain rods: high-throughput tools for single molecule imaging. Langmuir. 2008;24:10524–10531. doi: 10.1021/la801762h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulczyk AW, Tanner NA, Loparo JJ, Richardson CC, Oijen AMV. Direct Observation of Enzymes Replicating DNA Using a Single-molecule DNA Stretching Assay. J Vis. Expt. 2010. p. e1689. [DOI] [PMC free article] [PubMed]
- Parthasarathy R. Rapid, accurate particle tracking by calculation of radial symmetry centers. Nat Methods. 2012;9:724. doi: 10.1038/nmeth.2071. [DOI] [PubMed] [Google Scholar]
- Vestergaard CL, Blainey PC, Flyvbjerg H. Optimal estimation of diffusion coefficients from single-particle trajectories. Physical review. E, Statistical, nonlinear, and soft matter physics. 2014;89:022726. doi: 10.1103/PhysRevE.89.022726. [DOI] [PubMed] [Google Scholar]
- Schroeder CM, Blainey PC, Kim S, Xie XS. Hydrodynamic Flow-stretching Assay for Single-Molecule Studies of Nucleic Acid-Protein Interactions. Cold Spring Harbor Laboratory Press; 2008. [Google Scholar]
- Blainey PC. Single-Molecule Studies of Protein-DNA Interaction: Diffusive Search and Sequence-Dependent Motors. Harvard University; 2007. [Google Scholar]
- Gorman J, Greene EC. Visualizing one-dimensional diffusion of proteins along DNA. Nat Struct Mol Biol. 2008;15:768–774. doi: 10.1038/nsmb.1441. [DOI] [PubMed] [Google Scholar]
- Lin YH, et al. Using the Bias from Flow to Elucidate Single DNA Repair Protein Sliding and Interactions with DNA. Biophysical journal. 2009;96:1911–1917. doi: 10.1016/j.bpj.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blainey PC, et al. Nonspecifically bound proteins spin while diffusing along DNA. Nat Struct Mol Biol. 2009;16:1224–1234. doi: 10.1038/nsmb.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuculis L, Abil Z, Zhao H, Schroeder CM. Direct observation of TALE protein dynamics reveals a two-state search mechanism. Nat Commun. 2015;6:7277. doi: 10.1038/ncomms8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuculis L, Abil Z, Zhao HM, Schroeder CM. TALE proteins search DNA using a rotationally decoupled mechanism. Nat Chem Biol. 2016;12:831–837. doi: 10.1038/nchembio.2152. [DOI] [PubMed] [Google Scholar]
- Yardimci H, Loveland AB, van Oijen AM, Walter JC. Single-molecule analysis of DNA replication in Xenopus egg extracts. Methods. 2012;57:179–186. doi: 10.1016/j.ymeth.2012.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene EC, Wind S, Fazio T, Gorman J, Visnapuu ML. DNA Curtains for High-Throughput Single-Molecule Optical Imaging. Method Enzymol. 2010;472:293–315. doi: 10.1016/S0076-6879(10)72006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]