

Abstract
Expansion of novel therapeutics to all patients with cystic fibrosis (CF) requires personalized CFTR modulator therapy. We have developed nasospheroids, a primary cell culture–based model derived from individual CF patients and healthy subjects by a minimally invasive nasal biopsy. Confocal microscopy was utilized to measure CFTR activity by analyzing changes in cross-sectional area over time that resulted from CFTR-mediated ion and fluid movement. Both the rate of change over time and AUC were calculated. Non-CF nasospheroids with active CFTR-mediated ion and fluid movement showed a reduction in cross-sectional area, whereas no changes were observed in CF spheroids. Non-CF spheroids treated with CFTR inhibitor lost responsiveness for CFTR activation. However, nasospheroids from F508del CF homozygotes that were treated with lumacaftor and ivacaftor showed a significant reduction in cross-sectional area, indicating pharmacologic rescue of CFTR function. This model employs a simple measurement of size corresponding to changes in CFTR activity and is applicable for detection of small changes in CFTR activity from individual patients in vitro. Advancements of this technique will provide a robust model for individualized prediction of CFTR modulator efficacy.
Keywords: Cell Biology, Pulmonology
Keywords: Epithelial transport of ions and water, Genetic diseases
Ex vivo nasal epithelial nasospheroid model demonstrates CFTR-dependent fluid transport as a surrogate for CFTR activation and has potential for use as a predictive tool for CFTR modulator efficacy.
Introduction
In 1989, the cystic fibrosis transmembrane conductance regulator gene (CFTR) mutation was discovered to be the underlying defect in a life-shortening disease, cystic fibrosis (CF) (1–3). Individuals with CF suffer from abnormal secretions in multiple tissues, including the lungs, pancreas, liver, and intestine, leading to mucus stasis and obstruction. In the lung, the secretions impair proper airway clearance, leading to a cycle of infection, inflammation, progressive bronchiectasis, and decline in lung function that causes substantial mortality (4–8). CF is a highly complex autosomal recessive disease with over 2,000 mutations and genetic variations, of which F508del is the most common (9). Since the discovery of CFTR, this disease has served as a model for the study of lung and epithelial biology and provided insights into mechanisms of a variety of diseases (10). Despite intensive studies, a cure has remained elusive, and until recently, therapies directly targeted to the underlying defect in the CFTR protein were unavailable.
The development of CFTR modulators — correctors that overcome impairments in protein processing and potentiators that enhance CFTR function in situ — has revolutionized the care of patients with CF (11–13). Specifically, ivacaftor (Kalydeco) is targeted to patients with at least 1 copy of a specific type of mutation, called a gating mutation, which results in improper regulation of the ion channel (such as G551D) (14–19). In many cases, this regulation is restored, and patients have had remarkable improvements in lung and pancreatic function. A combination treatment with a corrector (lumacaftor, developed as VX-809) and potentiator (ivacaftor, developed as VX-770) called Orkambi was approved for patients with 2 copies of the most common mutation in CFTR, F508del (20–22). This therapy was designed to target multiple defects of the abnormal F508del CFTR protein and represents the future of CF care: targeting the specific CFTR defect(s) in an individual with a combinatorial regimen. Patients taking this dual therapy exhibited varying responses, although overall, the treatment was less successful than for patients with gating mutations taking ivacaftor alone. Despite the recent expansion of the FDA approval of ivacaftor to CF patients with 1 of 23 additional rare mutations with residual function (23) and numerous modulators in the pipeline (24), there are currently still many patients without successful treatment options for the CFTR basic defect. However, there is no rapid assay available to measure patient-specific responses. Effective therapeutic strategies without understanding the mechanism of defect in hundreds of different rare mutant CFTR alleles appears impossible without a personalized efficacy assay derived from a patient’s own cells. This hypothesis is supported by observations that even the latest regimens do not fully restore CFTR activity, and individuals with similar or the same mutations may respond differently (20, 25–27). Therefore, it is imperative to implement methods to target the therapeutic strategy to the individual, regardless of genotype, genetic context, and environmental background.
Recently, the emergence of organoids derived from rectal biopsies has shown substantial promise as predictive tools (28–36). However, these models are derived from intestinal epithelium and may not predict airway responses to modulators as accurately as models derived from airway epithelium. The use of superficial nasal scrapings, brushings, or excised polyps to obtain epithelial samples for culture has previously been reported in individuals with CF (37–39) and may serve as a surrogate for lower airway epithelial CFTR function. Such samples provide a renewable source of differentiated airway epithelial cells from any individual with CF, regardless of their mutation, that does not require tissue expansion and multiple cell divisions prior to performing the assay. To improve the suitability of these samples for both functional and morphological tests, we have developed an organoid culture that is similar to cultures reported previously, where the apical ciliated membrane orients to the media bath (40–43) but is derived from simple nasal epithelial curettage. This minimally invasive technique does not require polyp excision, which is different from the earlier reports, and utilizes normal nasal epithelial tissue. Very recently, an analogous model was utilized for ex vivo studies of primary ciliary dyskinesia, demonstrating applicability for other diseases (44). This ex vivo assay shows quantifiable response to CFTR activation in non-CF tissues and CF tissues treated with modulators. The simplicity of this assay, which can easily measure fluid transport, may be extended to other diseases resulting from abnormal fluid transport, such as pulmonary edema and acute lung injury, when derived from other epithelial (bronchial and alveolar) cell types.
Results
Nasospheroids form in culture media with the apical membranes facing the growth media (outside) with a definable lumen.
Nasospheroids form spontaneously in 2–5 days from sheets of nonadherent nasal epithelial cells obtained from nasal brushings or curettage, and they can be maintained for at least 12 weeks in serum-free medium (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.95734DS1). These spheroids are explant structures that develop from epithelial sheets that are not disrupted into a single cell suspension (see methods). The formation of spheroids is highly variable, with the diameters ranging from 50–500 μM. In addition, we have observed that, while some subjects (non-CF or CF) only form dozens of nasospheroids, others form thousands, and pediatric CF subjects form frequently hundreds to thousands of nasospheroids. To demonstrate the structures of the organoids, cells were labeled with plasma membrane orange, DRAQ5, and calcein green. Spheroids are composed of a single layer of differentiated pseudostratified epithelial cells containing ciliated and nonciliated cell types (Figure 1, A and B). The apical membranes face the outside, and basolateral membranes line the lumen of the spherical organoids (Figure 1). This orientation has been observed by other groups when nondisrupted epithelial sheets are cultured in suspension (40–44). When CFTR is labeled with a highly specific monoclonal antibody raised against the NBD2 region of CFTR (Ab 528, provided by J.R. Riordan, UNC-Chapel Hill; refs. 45–49) followed by a fluorescently labeled anti-mouse secondary antibody (Alexa Fluor 488) (see Methods), the green fluorescence concentrates to the apical region of cells, facing the outside bath (Figure 1C). Based on the localization of the cilia and CFTR staining, we refer to these structures as apical membrane out (AMO) nasospheroids, in contrast to other reported organoids formed from bronchial (50) or intestinal (28–31) epithelial cells in matrigel that are oriented with the apical, CFTR-containing membrane facing the lumen of the structure. Because of the orientation of AMO nasospheroids, we hypothesized that CFTR activation results in fluid flow from the interior of the structure into the bath and, therefore, reduces the fluid volume inside, leading to shrinking (Figure 1D). To test this hypothesis, we developed a quantification method to assess organoid volume changes following CFTR activation.
Figure 1. Nasospheroids are highly differentiated, hollow spheres with the cilia oriented to the media bath.

(A) Confocal fluorescent microscopy of living nasospheroids treated with calcein green (cytoplasm of living cells), plasma membrane orange (cilia and plasma membranes), and DRAQ5 (1, 5-bis[(2-[di-methylamino]ethyl)amino]-4, 8-dihydroxyanthracene-9, 10-dione, nuclei, in blue). Scale bar: 100 μM. (B) Nasospheroids were fixed in 4% paraformaldehyde and embedded in hydroxyethyl agarose, followed by paraffin-embedding and cryosection to produce thin sections. Processing results in dehydration and reduction in overall size. These sections were stained with H&E as described in Methods and show intact epithelial layer with ciliated and nonciliated cells. Scale bar: 10 μM. (C) Intact nasospheroids were fixed in ice-cold methanol followed by incubation with anti-CFTR antibody and fluorescent-tagged secondary antibody as described in Methods. Nuclei are stained blue with DAPI. Scale bar: 50 μM. (D) Illustration showing expected flow of anions and water with CFTR activation.
Changes in the cross-sectional area of non-CF nasospheroids are CFTR dependent.
First, we tested non-CF spheroids derived from normal human nasal epithelium (Figure 2). Volume changes in similar spheroids have previously been reported in response to fluid transport changes (40–43). To test our hypothesis that CFTR activation leads to fluid movement and consequent volume changes of the nasospheroids, we applied forskolin (10 μM) and 3-isobutyl-1-methylxanthine (IBMX,100 μM), a phosphodiesterase inhibitor that maintains intracellular cAMP levels (51), leading to CFTR activation in a cAMP-dependent manner (Figure 2A). The epithelial sodium channel (ENaC) is also activated by cAMP (52, 53). Because CFTR and the ENaC control ion and fluid movement at the epithelial surface, we inhibited the activity of ENaC on intra-spheroid fluid volumes with amiloride (100 μM) or benzamil (10 μM) (54). We observed that lack of inhibition of ENaC counteracted the fluid transport effects driven by CFTR (Supplemental Figure 2) and, therefore, included inhibition of ENaC as a standard procedure in these assays to isolate CFTR effects. When we measured the cross-sectional area of the spheroids over time, following forskolin + IBMX treatment, we observed a sustained shrinking over time (Figure 2, A and B). The cross-sectional area was measured at 30 and 75 minutes. The area at time 0 was set at 100%, and subsequent images are expressed as a reduction of the area at time 0 (fractional reduction). The average fractional reduction of all non-CF spheroids is shown in Figure 2B. The reduction in size compared with time 0, when stimulated with forskolin is significant (P < 0.001) both at 30 and 75 minutes (Figure 2B).
Figure 2. Non-CF nasospheroids show reduction in cross-sectional area over time when CFTR is stimulated by forskolin.

(A) Representative non–cystic fibrosis (CF) nasospheroids are stained with calcein green. Z-stacks are taken sequentially using confocal microscopy and the maximal projection generated. The same nasospheroids are shown at 30 and 75 minutes. Scale bars: 50 μM. (B) The starting size of each nasospheroid was set at 1, and the fractional reduction of each was calculated at all time points. The average fractional reduction was plotted at each time point. Stimulated nasospheroids were treated with 10 μM forskolin, amiloride, and IBMX. Nonstimulated spheroids were treated with an equal concentration of vehicle dimethyl sulfoxide (DMSO). Stimulated spheroids + inhibitor were treated with CFTRinh-172 at time of testing. Analysis of variance was calculated for the groups. Mean values ± SD are shown.*P < 0.00001 and ‡P < 0.001 for 1-way ANOVA and paired t tests between time points. (C) Each nasospheroid absolute size of maximal projection at each time point was plotted over time for all time points in the “Raw” graph. The “Fitted” graph represents the fitted slopes for each nasospheroid after linear regression (see Methods). (D) Summary data of slopes for all non-CF nasospheroids and non-CF nasospheroids treated with CFTRinh-172. Fitted slopes are represented by a box and dot plot. Each dot is a single nasospheroid. The black box is the mean; the central line is the median; the box borders are the 25th and 75th percentiles. *P < 0.001. (E) Summary data of time-averaged AUC unadjusted for starting size for all non-CF nasospheroids and non-CF nasospheroids treated with CFTRinh-172. AUC is represented by a box plot. *P < 0.0001. n = 9 subjects. A minimum of 3 up to a maximum of 18 nasospheroids were analyzed per subject.
Adjustment for multiple measures and starting size.
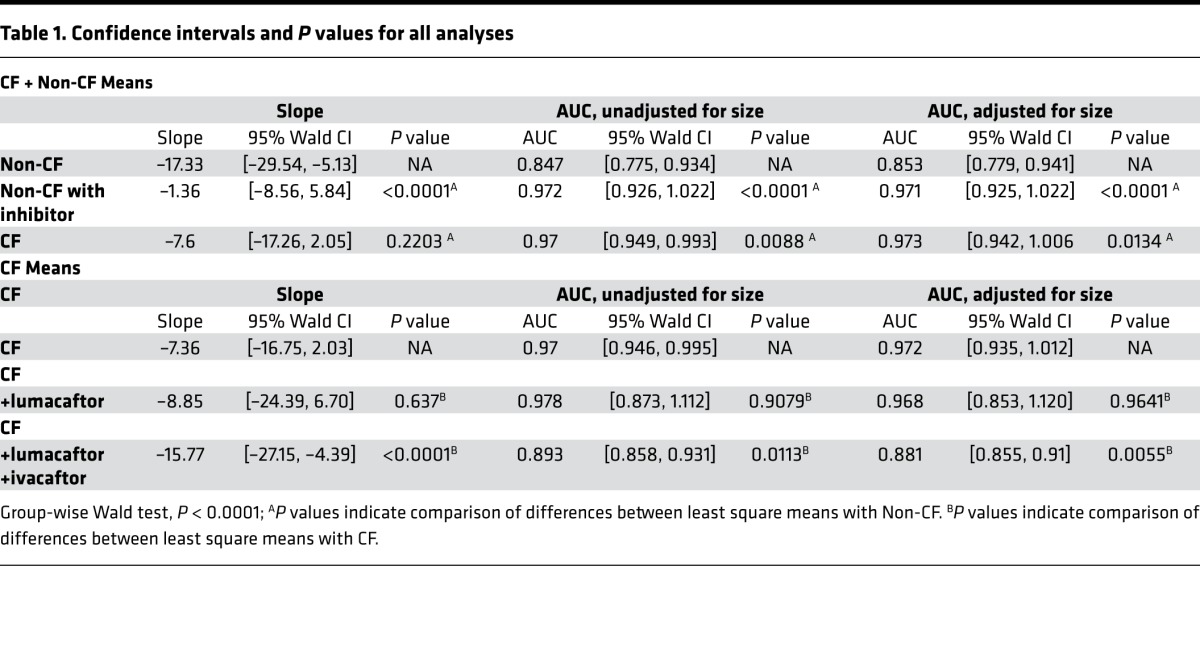
Spheroids have variation in the starting size, rendering the method of analysis shown in Figure 2B simplistic. This method also cannot take into account multiple measurements from a single subject, nor the dependence of observations. Together, these are important attributes of the data that need to be incorporated into analysis, as both influence the statistical inference and conclusions. Therefore, the data were also analyzed using linear modeling of the change in size over time (fitted slopes) and generalized estimating equations (GEE) as described in the methods. The change in area of multiple spheroids are represented by each line over the time course from 0–75 minutes (Figure 2C). The top panel (raw) represents actual area measurements at each time point, whereas the bottom panel (fitted) depicts a calculated reduction in the cross-sectional area of spheroids over time and allows the calculation of the slope. Using this analysis, the estimated mean slope of all spheroids was –17.33 (–29.54, –5.13) (Table 1), showing significant reduction in cross-sectional area over time in normal non-CF spheroids (n = 9 subjects). When we applied CFTR inhibitor 172 (CFTRinh-172) to non-CF spheroids prior to or at the same time as the forskolin + IBMX mixture, we observed significantly diminished reduction of the cross-sectional area over time, with estimated mean slopes of non-CF spheroids treated with the CFTRinh-172 reduced to –1.36 (–8.56, 5.84) (Figure 2E and Table 1). This is consistent with no significant reduction in cross-sectional area (n = 6 subjects, 43 CFTRinh-172–treated observations). Non-CF spheroids that were not stimulated by forskolin demonstrate similar behavior (Figure 2B and Table 1, P < 0.001). Another approach taken was to calculate the AUC analogous to approaches in similar models (29). We first determined the fractional reduction over time relative to the initial size. The AUC was calculated for each curve using trapezoidal rule. Since fractional reduction has value 1 at time 0, AUC is a good measure of change in fractional reduction over time and takes on a value greater than 0: the closer the AUC value is to 0, the more evidence of greater reduction in size. AUC also has the advantage of not assuming a linear relationship, which is a critical assumption when dealing with slopes. On the other hand, AUC is not appropriate when investigating the change over time using the actual size (instead of the fractional change), as it is possible to get the same AUC values for 2 curves that behave differently. In this case, the slope analysis is more appropriate. The estimated mean AUC for non-CF stimulated nasospheroids was 0.847 (0.775, 0.934) (Figure 2E and Table 1), which is significantly different from the estimated mean AUC after CFTRinh-172 treatment (0.972 [0.926, 1.022], P < 0.0001). In addition to the factors incorporated above, the rate of nasospheroid reduction is influenced by each nasospheroid’s baseline area, since physical constraints of large or small nasospheroids may affect the rate of size reduction. To account for this in the analysis of the fractional reduction, we also added the starting size of the nasospheroids, as a covariate in the model, resulting in adjusted model for AUC (Table 1). The result from the adjusted AUC model compared with the unadjusted was similar, and conclusions were the same. These results support the idea that changes in the cross-sectional area and consequently the volume of the spheroids is largely dependent upon CFTR function.
Table 1. Confidence intervals and P values for all analyses.
Nasospheroids derived from CF patients demonstrate diminished volume reduction following CFTR activation.
Stimulation of CF spheroids from F508del homozygotes with forskolin + IBMX showed substantial reduction in cross-sectional area change (Figure 3, B and C, and Table 1). The estimated mean slopes of the fitted line plots were reduced to –7.60 (–17.26, 2.05), similar to what we have seen in non-CF spheroids in the presence of CFTRinh-172, and the unadjusted mean AUC was estimated to be 0.97 (0.946, 0.995) (P = 0.0088 compared with non-CF stimulated). These results further suggest that the absence of substantial volume change after forskolin + IBMX stimulation is due to the loss of functional CFTR. Because these spheroids form sheets of differentiated epithelia with active fluid and ion transport, we hypothesized that the initial sizes of CF spheroids compared with non-CF spheroids might differ, given the differences in fluid and ion transport caused by dysfunctional CFTR in CF epithelia. We analyzed spheroids from CF and non-CF subjects for the distribution of size. We found that CF spheroids and non-CF spheroids had a similar mean size (Figure 3A).
Figure 3. CF nasospheroids show no reduction in cross-sectional area over time when CFTR is stimulated, unless treated with modulators.

(A) Baseline area at start of testing is not significantly different in CF compared with non-CF nasospheroids. (B) The starting size of each nasospheroid was set at 1, and the fractional reduction of each was calculated at all time points. The average fractional reduction was calculated and plotted at each time point. Stimulated nasospheroids were treated with 10 μM forskolin, amiloride, and 3-isobutyl-1-methylxanthine (IBMX). Additional nasospheroids were treated with 5 μM lumacaftor (24 hours) and 1 μM ivacaftor, acute treatment. One-way ANOVA was calculated for the groups, P = 0.047. Paired t test between time 0 and time 75 minutes, *P < 0.0001. (C) Summary data of slopes for all CF nasospheroids represented as a box and dot plot. Each dot is a single nasospheroid. The black box is the mean; the central line is the median; the box borders are the 25th and 75th percentiles. *P <0.0001. (D) Summary data of time-averaged AUC unadjusted for starting size for all CF nasospheroids represented as a box plot.*P = 0.0113. n = 3 subjects. Between 3 and 5 nasospheroids were analyzed per subject.
CFTR modulator treatment partially restores cross-sectional area reduction following forskolin + IBMX stimulation of CF nasospheroids.
In the next set of experiments, we treated the CF nasospheroids with a CFTR corrector, lumacaftor (55–57). Following a 24-hour 5 μM lumacaftor treatment, we observed a nonsignificant change in the average fractional size reduction following forskolin + IBMX activation compared with vehicle treated controls (Figure 3A). The average slopes of the fitted lines were estimated to be –8.85 ([–24.39, 6.70], P = 0.6765). Because clinical efficacy was not demonstrated with lumacaftor alone (27), we also evaluated the effect of 24-hour treatment with both lumacaftor and ivacaftor (5 μM), representative of the currently approved clinical regimen for F508del homozygotes using Orkambi (58–62). Combination treatment reveals a synergistic effect with both an increase in average fractional reduction and the resulting estimated mean slope becoming more negative than lumacaftor (–15.77 [–27.15, –4.39]), representing a significant reduction in size (P < 0.0001 compared with untreated CF, Figure 3C and Table 1) and greater restoration of CFTR activity. Utilization of the AUC resulted in similar results when compared with untreated CF nasospheroids (lumacaftor-treated mean AUC of 0.978 [0.8737, 1.112], P = 0.9079; and combination treated mean AUC of 0.893 [0.858, 0.931], P < 0.0113; Figure 3D and Table 1). Adjustment for starting size (Table 1) showed similar findings.
Discussion
As novel CFTR modulators become available, there is an urgency to develop precision tools to determine incremental differences between efficacies of modulators and to optimize combination therapies for individuals. Such tools are essential to determine the efficacy of modulators in patients with rare mutations who might otherwise be ineligible to receive these drugs. Ex vivo models from individual patients, such as the primary spheroids presented in this study, provide a promising approach to personalized medicine for CF. These ex vivo models are derived from primary, terminally differentiated cells from the airway epithelium, and they are not subject to alterations due to culture conditions over several passages. Nasal cells are an easily obtainable and renewable source of respiratory epithelium. Measures of short-circuit current in primary airway epithelia in monolayers are the main test in drug development (57), but these cultures require significant cost, labor, and specialized expertise, making them more suitable for research than as clinical tools. Our model develops quickly in 2–5 days directly from patients’ tissues, has a simple outcome measure, and responds to modulator treatment, providing an approach as a clinical tool to test CFTR modulators. The apical surface is easily accessible, improving the activity of reagents that act directly on the apical surface of epithelia (such as CFTRinh-172). Our data indicate that CFTR activation causes a significant decrease in spheroid volume. The reduction was prevented by CFTRinh-172, suggesting that CFTR is the major player of ion and water transport in this system. Absence of effects of CFTR activation in CF spheroids support this finding.
Spheroids treated with modulators display similar effects to clinical response. Modulator monotherapy treatment (lumacaftor, 25 hours, 5 μM) resulted only in minimal restoration of the volume reduction following CFTR activation, while combination therapy including a potentiator caused a greater response. This incremental finding is consistent with clinical trials that showed no significant improvement in clinical symptoms following lumacaftor administration alone (27) but resulted in significant improvements in percent predicted forced expiratory volume in 1 second (ppFEV1), BMI, and pulmonary exacerbation rate when lumacaftor and ivacaftor (Orkambi) were administered in combination (61–63). Although the number of subjects included in this study is low, our results suggest that this assay may offer a directly observable real-time measure of CFTR activity and may be exploited as a rapid (2- to 5-day) functional assay to test patient-specific CFTR responses to modulators. This assay may be applicable to “n of 1” studies, using a patient’s own cells to test such responses (Supplemental Figure 3). We observed response differences between F508del homozygote individuals when each individual was evaluated separately (Supplemental Figure 3, A–C). In addition, we observe a substantial, albeit nonsignificant, reduction in size with lumacaftor monotherapy in a rare mutation (I618T/F508del, Supplemental Figure 3D). Furthermore, G551D subjects who were on ivacaftor therapy also show a CFTR-specific response (Supplemental Figure 3, E and F). Evaluation of more CF patients with F508del, as well as rare and partial-function mutations and heterozygotes, will allow stratification of the volume reduction response and compare them to clinical markers of drug efficacy. Future studies will also evaluate additional modulator compounds and varied combinatorial regimens.
Our study has several limitations. Variability in size of the nasospheroids complicates analysis of the response. Adjustment of results by initial size and for multiple measures improves analysis, but limitation to a narrower size range for more comparable measurements may further improve the assay. Future studies will focus on determining the most appropriate size range as well as the optimum number of observations to obtain precise and accurate measurements of fluid transport with low variability. This will be necessary to observe incremental responses to CFTR enhancement. Our analysis incorporates both starting size and progression of volume reduction in response to modulators. Our study represents an early stage of a potential in vitro biomarker derived from a patient’s own cells. The use of such a predictive tool for modulator efficacy is essential to bring these drugs to the entire CF population.
Methods
Collection of nasal epithelial cells.
Nine control subjects and 3 CF subjects with known genotypes were recruited to the study. Written informed consent was obtained from all subjects. Nasal epithelial cells were collected under direct visualization using an operating otoscope and 9 mm speculum using a Rhino-Probe curette (Arlington Scientific).
Development of nasal epithelial nasospheroids.
Cells from curettage or brushing of a single donor were collected in RPMI media (Gibco), incubated with DNase I (MilliporeSigma) for 20 minutes, centrifuged at 400 g, and collected as a whole pellet containing sheets of epithelia. This pellet was not disrupted and was seeded onto a collagen-coated dish in bronchial epithelial growth medium (LHC-DMEM, 50:50; BSA, 0.5mg/ml; bovine pituitary extract, 10 μg/ml; insulin, 0.87 μM; transferrin, 0.125 μM; hydrocortisone, 0.21 μM; triiodothyronine, 0.01 μM; epinephrine, 2.7 μM; epidermal growth factor, 25ng/ml; retinoic acid, 5 × 10–8 M; phosphorylethanolamine, 0.5 μM; ethanolamine, 0.5 μM; zinc sulfate, 3 μM; penicillin G sulfate, 100 U/ml; streptomycin sulfate, 100 μg/ml; gentamicin, 50 μg/ml; amphotericin, 0.25 μg/ml; ferrous sulfate, 1.5 × 10–6 M; magnesium chloride, 6 × 10–4 M; calcium chloride, 1.1 × 10–4 M; selenium ,30 nM; manganese, 1 nM; silicone, 500 nM; molybdenum, 1 nM; vanadium, 5 nM; nickel sulfate, 1 nM; tin, 0.5 nM (provided by Scott H. Randell, UNC-Chapel Hill; ref. 64). Propagation of nasal epithelial cells were performed as described, with care not to disrupt the epithelial sheets (65); notably, this method does not disrupt the sheet of epithelia into a single cell suspension, which differs from some other methods. Nasospheroids formed from nonadherent cells in epithelial sheets (Supplemental Figure 1) in suspension in 2–5 days and were removed from collagen-coated dishes into noncoated ultralow attachment dishes. Spheroids were obtained by centrifugation at 25 g and resuspended in culture media.
Histology of nasospheroids.
Nasospheroids were fixed in freshly prepared 4% paraformaldehyde in PBS for 10 minutes at room temperature, centrifuged at 25 g, embedded in Thermo Scientific Richard-Allan Scientific HistoGel (Thermo Fisher Scientific), and embedded in paraffin. Longitudinal serial sections (5 μm) were cut, floated onto charged glass slides (Super-Frost Plus, Thermo Fisher Scientific), and dried overnight at 60°C. Sections were stained with Harris’ acidified hematoxylin with 2 % orange G, and they were counterstained with 0.6 % eosin Y and 1 % phloxine B. Sections were visualized on 40X-900X Phase Contrast Inverted Microscope with 9 MP Camera.
Immunofluorescent labeling of CFTR.
Protocols for labeling CFTR have been previously described (48) and were modified slightly as follows. Intact nasospheroids were fixed with ice-cold methanol. Cells were permeabilized with 0.1% Triton X-100 in PBS for 10 minutes at room temperature. After washing with PBS at room temperature, nasospheroids were incubated with blocking buffer for 1 hour, followed by incubation with blocking buffer containing monoclonal antibody to CFTR that were produced, quality tested, and provided by J.R. Riordan (University of North Carolina-Chapel Hill) through a program of Cystic Fibrosis Foundation Therapeutics (NBD2 antibody 596 and 528; refs. 45–49) and incubated overnight at 4°C. After washing with PBS at room temperature, nasospheroids were incubated in the dark with secondary fluorescent antibodies (Alexa Fluor 488 goat anti–mouse IgG conjugate; Invitrogen/Thermo Fisher Scientific) and visualized.
Confocal microscopy imaging.
Nasospheroids were transferred to glass bottom dishes (#1.5 thickness) and stained with calcein-AM, CellMask Orange (Thermo Fisher Scientific), and DRAQ5 (Biostatus). Imaging completed on Olympus FV1000 confocal microscope system with environmental chamber for temperature (37°C), humidity (55%), and CO2 (5%) control. The chamber was preheated for at least 90 minutes; supplemental humidity was provided by saturated paper tissues or small 0.7% agar blocks near the sample. A 10 × 0.4 NA objective was used for most imaging. Sequential scanned images with excitation at 488, 543, and 638 nm and emission collected at 500–530, 555–625, and 655–755 nm along with DIC using 488 nm were taken. The confocal pinhole was set to 2 Airy units and Z series steps gathered every 13.8 μm from bottom to top of the organoids. Cross-sectional areas were estimated using FIJI (66) by manually outlining perimeters of maximum intensity projections of images. For CFTR stimulation, amiloride (100 μM, MilliporeSigma, A7410) or benzamil (10 μM, MilliporeSigma, B2417) was added to block the ENaC, followed by forskolin (10 μM, MilliporeSigma, F6886), IBMX (100 μM, a phosphodiesterase inhibitor to maintain intracellular cAMP levels, MilliporeSigma, I5879), ivacaftor (5 μM, Selleck Chemicals, S1144) if indicated, and CFTRinh-172 (10 μM, MilliporeSigma, C2992). Preincubation with lumacaftor (5 μM, Selleck Chemicals, S1565) for 24–48 hours was performed in select experiments. Vehicle control used was DMSO (MilliporeSigma, D2650).
Statistics.
To compare spheroid size among the different groups over time using conventional methods of analyses, 1-way ANOVA and 2-tailed Student’s t test were used. The cross-sectional area of each nasospheroid was recorded over time for up to 180 minutes. The trends of cross-sectional area over time were approximated by linear models, and the slope of the fitted line was used as the outcome for each nasospheroid, reducing the outcome over time to a single value. At least 3 time points were required in the calculation of slopes to get a more accurate estimate of the trend. In addition to the slope, we also computed the time averaged AUC using trapezoidal rule of the fractional reduction (FR; which normalizes reductions in size relative to the baseline size) over time as another outcome of interest. Both of these outcomes are useful, since the nasospheroids’ cross-sectional areas were calculated based on actual image sequences, which varied slightly from donor to donor. In addition, these outcomes do not require the data to be collected at the exact same time points nor the same number of time points. In using the slopes of the fitted lines and AUC as outcomes, modeling complications were avoided and data over time were reduced to an interpretable outcome. Note that each donor contributed multiple nasospheroids to the study. As shown by other investigators (67), multiple observations of a bioassay from each subject present a powerful tool to evaluate CFTR activity. In the absence of independent observations due to data from the same donor, the usual methods of analysis (e.g., conventional regression models) are inappropriate because they do not account for the dependence between observations. The risk of incorrectly concluding a significant difference exists with increasing dependence between observations, and this risk can be minimized by properly accounting for within-subject dependency. We address dependence between observations with GEE approach (68). GEEs allow flexible estimation of the correlation structure between observations, are generally robust to the specification of correlation structure, and are unburdened by assumptions of multivariate normality (68). GEEs require the specification of a working correlation matrix, which specifies the nature of the dependency among observations within a subject. For this analysis, the working correlation matrix was specified to be compound symmetric or exchangeable, which assumes equal correlation between any 2 observations from the same subject. Nasospheroids between subjects were treated as independent. Inference was performed with the Wald χ2 statistic, with degrees of freedom equal to the number of parameters; individual parameters may be tested for significance using normal theory. In modeling the slopes, only the group was included in the model. In modeling AUC, we fitted both an unadjusted (group only) and adjusted (group + baseline area) models. For graphical representation, slopes and AUC are presented as box and dot plots. In these plots, each dot is a single nasospheroid. The black box is the mean; the central line is the median; the box borders are the 25th and 75th percentiles. The whiskers extend to 1.5 times the 25th or 75th percentile, respectively. If there are no values beyond 1.5× the interquartile range, then the whiskers extend to the largest (or smallest) value. SAS version 9.4 was employed for all analyses and R version 3.2.0 for various graphics. All P values less than 0.05 were deemed significant. There were no adjustments for multiple comparisons, as this project is still in the exploratory stage.
Study approval.
Patients were recruited to the study under protocols approved by the institution’s IRB (F151030001, University of Alabama at Birmingham; 09-0716 and 11-1455, University of North Carolina at Chapel Hill). Written informed consent was obtained from all subjects.
Author contributions
JSG, IGC, NLQ, MC, and MG performed the experiments and analyzed the data. JSG, IJ, and SEB collected and cultured primary cells. JSG, NLQ, IA, and MG discussed the overall design of the experiments. JSG, JML, and IA conducted statistical analysis. JSG, JML, MC, and MG wrote and/or revised the manuscript. All authors read and commented on the manuscript.
Supplementary Material
Acknowledgments
We thank Zsuzsanna Bebok and Steve M. Rowe for editorial assistance; Stephen Mackay, Zhongyu Liu, Matthew Bruehl, and Dezhi Wang for technical assistance; J.R. Riordan for providing anti-CFTR antibodies; Richard C. Boucher for helpful discussion; and M. Kawai for graphic design assistance. This work was supported by the North Carolina Translational and Clinical Sciences Institute 2KR541401 (JSG); the Cystic Fibrosis Foundation GUIMBE14D0 (JSG) and RDP R026-CR11 (to RCB); the Kaul Pediatric Research Institute Pilot Award (JSG); the NIH NIDDK grant P30 DK072482 (to SMR); and the NIH NIDDK grant P30 DK065988 (to RCB).
Version 1. 11/16/2017
Electronic publication
Footnotes
IGC’s present address is: Molecular Biology, Cellular Biology, and Biochemistry program, Boston University, Boston, Massachusetts, USA.
Conflict of interest: M. Gentzsch, J.S. Guimbellot, N.L. Quinney, and S.E. Boyles are an inventors of the technology “airway sphere cultures as diagnostic device to monitor pharmacological responses of ion channels,” which is licensed to Path BioAnalytics. M. Gentzsch, J.S. Guimbellot, N.L. Quinney, and S.E. Boyles could receive royalties related to the license in the future. These relationships have been disclosed to and are under management by the University of North Carolina at Chapel Hill.
Reference information: JCI Insight. 2017;2(22):e95734. https://doi.org/10.1172/jci.insight.95734.
Contributor Information
Michael Chua, Email: michael_chua@med.unc.edu.
Inmaculada Aban, Email: caban@uab.edu.
Ilona Jaspers, Email: ilona_jaspers@med.unc.edu.
Martina Gentzsch, Email: gentzsch@med.unc.edu.
References
- 1.Kerem B, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245(4922):1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- 2.Riordan JR, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245(4922):1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 3.Rommens JM, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245(4922):1059–1065. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- 4.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352(19):1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 5.Boucher RC. New concepts of the pathogenesis of cystic fibrosis lung disease. Eur Respir J. 2004;23(1):146–158. doi: 10.1183/09031936.03.00057003. [DOI] [PubMed] [Google Scholar]
- 6.Davis PB. Cystic fibrosis since 1938. Am J Respir Crit Care Med. 2006;173(5):475–482. doi: 10.1164/rccm.200505-840OE. [DOI] [PubMed] [Google Scholar]
- 7.Peters S. Cystic fibrosis: a review of pathophysiology and current treatment recommendations. S D Med. 2014;67(4):148–151. [PubMed] [Google Scholar]
- 8.Lubamba B, Dhooghe B, Noel S, Leal T. Cystic fibrosis: insight into CFTR pathophysiology and pharmacotherapy. Clin Biochem. 2012;45(15):1132–1144. doi: 10.1016/j.clinbiochem.2012.05.034. [DOI] [PubMed] [Google Scholar]
- 9. Cystic Fibrosis Mutation Database. Cystic Fibrosis Centre at the Hospital for Sick Children in Toronto. http://www.genet.sickkids.on.ca/Home.html Updated April 25, 2011. Accessed October 17, 2017.
- 10.Pearson H. Human genetics: One gene, twenty years. Nature. 2009;460(7252):164–169. doi: 10.1038/460164a. [DOI] [PubMed] [Google Scholar]
- 11.Bell SC, De Boeck K, Amaral MD. New pharmacological approaches for cystic fibrosis: promises, progress, pitfalls. Pharmacol Ther. 2015;145:19–34. doi: 10.1016/j.pharmthera.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 12.Bertoncini E, Colomb-Lippa D. Pulmonology: CFTR modulators for cystic fibrosis. JAAPA. 2013;26(2):59–60. doi: 10.1097/01720610-201302000-00013. [DOI] [PubMed] [Google Scholar]
- 13.Derichs N. Targeting a genetic defect: cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis. Eur Respir Rev. 2013;22(127):58–65. doi: 10.1183/09059180.00008412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu H, et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J Cyst Fibros. 2012;11(3):237–245. doi: 10.1016/j.jcf.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 15.Kotha K, Clancy JP. Ivacaftor treatment of cystic fibrosis patients with the G551D mutation: a review of the evidence. Ther Adv Respir Dis. 2013;7(5):288–296. doi: 10.1177/1753465813502115. [DOI] [PubMed] [Google Scholar]
- 16.Kapoor H, Koolwal A, Singh A. Ivacaftor: a novel mutation modulating drug. J Clin Diagn Res. 2014;8(11):SE01–SE05. doi: 10.7860/JCDR/2014/6486.5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Molloy K, McElvaney NG. Ivacaftor: from bench to bedside... and back again. Am J Respir Crit Care Med. 2014;190(2):128–129. doi: 10.1164/rccm.201406-1122ED. [DOI] [PubMed] [Google Scholar]
- 18.Wainwright CE. Ivacaftor for patients with cystic fibrosis. Expert Rev Respir Med. 2014;8(5):533–538. doi: 10.1586/17476348.2014.951333. [DOI] [PubMed] [Google Scholar]
- 19.Whiting P, et al. Ivacaftor for the treatment of patients with cystic fibrosis and the G551D mutation: a systematic review and cost-effectiveness analysis. Health Technol Assess. 2014;18(18):1–106. doi: 10.3310/hta18180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boyle MP, et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med. 2014;2(7):527–538. doi: 10.1016/S2213-2600(14)70132-8. [DOI] [PubMed] [Google Scholar]
- 21.Jones AM, Barry PJ. Lumacaftor/ivacaftor for patients homozygous for Phe508del-CFTR: should we curb our enthusiasm? Thorax. 2015;70(7):615–616. doi: 10.1136/thoraxjnl-2015-207369. [DOI] [PubMed] [Google Scholar]
- 22.Wainwright CE, et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220–231. doi: 10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. FDA Approves KALYDECO® (ivacaftor) for More Than 900 People Ages 2 and Older with Cystic Fibrosis Who Have Certain Residual Function Mutations. Vertex Pharmaceuticals Incorporated. https://www.vrtx.com/story/kalydeco-approved-900-people-cf-ages-2-who-have-certain-residual-function-mutations Published May 17, 2017. Accessed October 17, 2017.
- 24.Pettit RS, Fellner C. CFTR Modulators for the Treatment of Cystic Fibrosis. P T. 2014;39(7):500–511. [PMC free article] [PubMed] [Google Scholar]
- 25.Ramsey BW, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663–1672. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Awatade NT, et al. Measurements of Functional Responses in Human Primary Lung Cells as a Basis for Personalized Therapy for Cystic Fibrosis. EBioMedicine. 2015;2(2):147–153. doi: 10.1016/j.ebiom.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clancy JP, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax. 2012;67(1):12–18. doi: 10.1136/thoraxjnl-2011-200393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dekkers JF, van der Ent CK, Beekman JM. Novel opportunities for CFTR-targeting drug development using organoids. Rare Dis. 2013;1:e27112. doi: 10.4161/rdis.27112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dekkers JF, et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med. 2013;19(7):939–945. doi: 10.1038/nm.3201. [DOI] [PubMed] [Google Scholar]
- 30.Okiyoneda T, et al. Mechanism-based corrector combination restores ΔF508-CFTR folding and function. Nat Chem Biol. 2013;9(7):444–454. doi: 10.1038/nchembio.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwank G, et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013;13(6):653–658. doi: 10.1016/j.stem.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 32.Beekman JM. Individualized medicine using intestinal responses to CFTR potentiators and correctors. Pediatr Pulmonol. 2016;51(S44):S23–S34. doi: 10.1002/ppul.23553. [DOI] [PubMed] [Google Scholar]
- 33.Dekkers JF, et al. Optimal correction of distinct CFTR folding mutants in rectal cystic fibrosis organoids. Eur Respir J. 2016;48(2):451–458. doi: 10.1183/13993003.01192-2015. [DOI] [PubMed] [Google Scholar]
- 34.Dekkers R, et al. A bioassay using intestinal organoids to measure CFTR modulators in human plasma. J Cyst Fibros. 2015;14(2):178–181. doi: 10.1016/j.jcf.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 35.Noordhoek J, Gulmans V, van der Ent K, Beekman JM. Intestinal organoids and personalized medicine in cystic fibrosis: a successful patient-oriented research collaboration. Curr Opin Pulm Med. 2016;22(6):610–616. doi: 10.1097/MCP.0000000000000315. [DOI] [PubMed] [Google Scholar]
- 36.Zomer-van Ommen DD, et al. Limited premature termination codon suppression by read-through agents in cystic fibrosis intestinal organoids. J Cyst Fibros. 2016;15(2):158–162. doi: 10.1016/j.jcf.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 37.Wiszniewski L, et al. Long-term cultures of polarized airway epithelial cells from patients with cystic fibrosis. Am J Respir Cell Mol Biol. 2006;34(1):39–48. doi: 10.1165/rcmb.2005-0161OC. [DOI] [PubMed] [Google Scholar]
- 38.de Courcey F, et al. Development of primary human nasal epithelial cell cultures for the study of cystic fibrosis pathophysiology. Am J Physiol, Cell Physiol. 2012;303(11):C1173–C1179. doi: 10.1152/ajpcell.00384.2011. [DOI] [PubMed] [Google Scholar]
- 39.Mosler K, et al. Feasibility of nasal epithelial brushing for the study of airway epithelial functions in CF infants. J Cyst Fibros. 2008;7(1):44–53. doi: 10.1016/j.jcf.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 40.Pedersen PS, Frederiksen O, Holstein-Rathlou NH, Larsen PL, Qvortrup K. Ion transport in epithelial spheroids derived from human airway cells. Am J Physiol. 1999;276(1 Pt 1):L75–L80. doi: 10.1152/ajplung.1999.276.1.L75. [DOI] [PubMed] [Google Scholar]
- 41.Pedersen PS, Holstein-Rathlou NH, Larsen PL, Qvortrup K, Frederiksen O. Fluid absorption related to ion transport in human airway epithelial spheroids. Am J Physiol. 1999;277(6 Pt 1):L1096–L1103. doi: 10.1152/ajplung.1999.277.6.L1096. [DOI] [PubMed] [Google Scholar]
- 42.Pedersen PS, Braunstein TH, Jørgensen A, Larsen PL, Holstein-Rathlou NH, Frederiksen O. Stimulation of aquaporin-5 and transepithelial water permeability in human airway epithelium by hyperosmotic stress. Pflugers Arch. 2007;453(6):777–785. doi: 10.1007/s00424-006-0157-3. [DOI] [PubMed] [Google Scholar]
- 43.Pedersen PS, Procida K, Larsen PL, Holstein-Rathlou NH, Frederiksen O. Water permeability in human airway epithelium. Pflugers Arch. 2005;451(3):464–473. doi: 10.1007/s00424-005-1484-5. [DOI] [PubMed] [Google Scholar]
- 44.Marthin JK, Stevens EM, Larsen LA, Christensen ST, Nielsen KG. Patient-specific three-dimensional explant spheroids derived from human nasal airway epithelium: a simple methodological approach for ex vivo studies of primary ciliary dyskinesia. Cilia. 2017;6:3. doi: 10.1186/s13630-017-0049-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cui L, et al. Domain interdependence in the biosynthetic assembly of CFTR. J Mol Biol. 2007;365(4):981–994. doi: 10.1016/j.jmb.2006.10.086. [DOI] [PubMed] [Google Scholar]
- 46.Gentzsch M, Cui L, Mengos A, Chang XB, Chen JH, Riordan JR. The PDZ-binding chloride channel ClC-3B localizes to the Golgi and associates with cystic fibrosis transmembrane conductance regulator-interacting PDZ proteins. J Biol Chem. 2003;278(8):6440–6449. doi: 10.1074/jbc.M211050200. [DOI] [PubMed] [Google Scholar]
- 47.Kreda SM, et al. Characterization of wild-type and deltaF508 cystic fibrosis transmembrane regulator in human respiratory epithelia. Mol Biol Cell. 2005;16(5):2154–2167. doi: 10.1091/mbc.E04-11-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kreda SM, Gentzsch M. Imaging CFTR protein localization in cultured cells and tissues. Methods Mol Biol. 2011;742:15–33. doi: 10.1007/978-1-61779-120-8_2. [DOI] [PubMed] [Google Scholar]
- 49.Gentzsch M, et al. Restoration of R117H CFTR folding and function in human airway cells through combination treatment with VX-809 and VX-770. Am J Physiol Lung Cell Mol Physiol. 2016;311(3):L550–L559. doi: 10.1152/ajplung.00186.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu X, Peters-Hall JR, Bose S, Peña MT, Rose MC. Human bronchial epithelial cells differentiate to 3D glandular acini on basement membrane matrix. Am J Respir Cell Mol Biol. 2011;44(6):914–921. doi: 10.1165/rcmb.2009-0329OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rückes C, et al. Amiloride-sensitive Na+ channels in human nasal epithelium are different from classical epithelial Na+ channels. Biochem Biophys Res Commun. 1997;237(3):488–491. doi: 10.1006/bbrc.1997.7106. [DOI] [PubMed] [Google Scholar]
- 52.Snyder PM, Olson DR, Kabra R, Zhou R, Steines JC. cAMP and serum and glucocorticoid-inducible kinase (SGK) regulate the epithelial Na(+) channel through convergent phosphorylation of Nedd4-2. J Biol Chem. 2004;279(44):45753–45758. doi: 10.1074/jbc.M407858200. [DOI] [PubMed] [Google Scholar]
- 53.Yang LM, Rinke R, Korbmacher C. Stimulation of the epithelial sodium channel (ENaC) by cAMP involves putative ERK phosphorylation sites in the C termini of the channel’s beta- and gamma-subunit. J Biol Chem. 2006;281(15):9859–9868. doi: 10.1074/jbc.M512046200. [DOI] [PubMed] [Google Scholar]
- 54.Hirsh AJ, et al. Evaluation of second generation amiloride analogs as therapy for cystic fibrosis lung disease. J Pharmacol Exp Ther. 2004;311(3):929–938. doi: 10.1124/jpet.104.071886. [DOI] [PubMed] [Google Scholar]
- 55.Loo TW, Bartlett MC, Clarke DM. Corrector VX-809 stabilizes the first transmembrane domain of CFTR. Biochem Pharmacol. 2013;86(5):612–619. doi: 10.1016/j.bcp.2013.06.028. [DOI] [PubMed] [Google Scholar]
- 56.Ren HY, et al. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol Biol Cell. 2013;24(19):3016–3024. doi: 10.1091/mbc.E13-05-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Van Goor F, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci USA. 2011;108(46):18843–18848. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rehman A, Baloch NU, Janahi IA. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373(18):1783. doi: 10.1056/NEJMc1510466. [DOI] [PubMed] [Google Scholar]
- 59.Cholon DM, Esther CR, Gentzsch M. Efficacy of lumacaftor-ivacaftor for the treatment of cystic fibrosis patients homozygous for the F508del-CFTR mutation. Expert Rev Precis Med Drug Dev. 2016;1(3):235–243. doi: 10.1080/23808993.2016.1175299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Elborn JS, et al. Efficacy and safety of lumacaftor/ivacaftor combination therapy in patients with cystic fibrosis homozygous for Phe508del CFTR by pulmonary function subgroup: a pooled analysis. Lancet Respir Med. 2016;4(8):617–626. doi: 10.1016/S2213-2600(16)30121-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Milla CE, et al. Lumacaftor/Ivacaftor in Patients Aged 6-11 Years with Cystic Fibrosis and Homozygous for F508del-CFTR. Am J Respir Crit Care Med. 2017;195(7):912–920. doi: 10.1164/rccm.201608-1754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Konstan MW, et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): a phase 3, extension study. Lancet Respir Med. 2017;5(2):107–118. doi: 10.1016/S2213-2600(16)30427-1. [DOI] [PubMed] [Google Scholar]
- 63.Wainwright CE, et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220–231. doi: 10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Randell SH, Fulcher ML. Epithelial cell culture protocols. New York, NY: Humana Press; 2012. [Google Scholar]
- 65.Muller L, Brighton LE, Carson JL, Fischer WA, 2nd, Jaspers I. Culturing of human nasal epithelial cells at the air liquid interface. J Vis Exp. 2013;(80):50646. doi: 10.3791/50646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wine JJ, et al. In vivo readout of CFTR function: ratiometric measurement of CFTR-dependent secretion by individual, identifiable human sweat glands. PLoS ONE. 2013;8(10):e77114. doi: 10.1371/journal.pone.0077114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zeger SL, Liang KY. Longitudinal data analysis for discrete and continuous outcomes. Biometrics. 1986;42(1):121–130. doi: 10.2307/2531248. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.