

Abstract
In vivo microinjection is the most commonly used gene transfer technique for analyzing the gene functions in individual mosquitoes. However, this method requires a more technically demanding operation and involves complicated procedures, especially when used in larvae due to their small size, relatively thin and fragile cuticle, and high mortality, which limit its application. In contrast, viral vectors for gene delivery have been developed to surmount extracellular and intracellular barriers. These systems have the advantages of easy manipulation, high gene transduction efficiency, long-term maintenance of gene expression, and the ability to produce persistent effects in vivo. Mosquito densoviruses (MDVs) are mosquito-specific, small single-stranded DNA viruses that can effectively deliver foreign nucleic acids into mosquito cells; however, the replacement or insertion of foreign genes to create recombinant viruses typically causes a loss of packaging and/or replication abilities, which is a barrier to the development of these viruses as delivery vectors.
Herein, we report using an artificial intronic small-RNA expression strategy to develop a non-defective recombinant Aedes aegypti densovirus (AaeDV) in vivo delivery system. Detailed procedures for the construction, packaging and quantitative analysis of the rAaeDV vectors, and for larval infection are described.
This study demonstrates, for the first time, the feasibility of developing a non-defective recombinant MDV micro RNA (miRNA) expression system, and thus providing a powerful tool for the functional analysis of genes in mosquito and establishing a basis for the application of viral paratransgenesis for controlling mosquito-borne diseases. We demonstrated that Aedes albopictus 1st instar larvae could be easily and effectively infected by introducing the virus into the water body of the larvae breeding site and that the developed rAaeDVs could be used to overexpress or knock down the expression of a specific target gene in larvae, providing a tool for the functional analysis of mosquito genes.
Keywords: Developmental Biology, Issue 128, Mosquito densovirus, gene delivery vector, small RNA, Aedes albopictus, vector control, artificial intron.
Introduction
Mosquito-borne diseases such as malaria, dengue fever, zika fever, and yellow fever, are major international public health problems that continue to account for a significant fraction of the global infectious disease burden1,2. Conventional insecticides, which have been used in response to vectors, are a major component of sustainable integrated mosquito control strategy for the prevention of mosquito-borne diseases. However, such strategies have proven to be relatively ineffective or undesirable due to the associated negative environmental impacts as well as the evolution of resistance in mosquito populations3,4. For these reasons, there is an urgent need for alternative methods of mosquito control, and the use of transgenic methods to produce sterile male mosquitoes and the release of pathogen-resistant mosquitoes have arisen as promising new control strategies. To develop effective new control methods, such as safe and effective approaches for in vivo gene delivery, the comprehensive analysis of mosquito gene function is required.
Direct microinjection of plasmid DNA, double stranded RNA (dsRNA) or small interfering RNA (siRNA) is the most commonly used in vivo gene delivery method in mosquitoes. In fact, the production of transgenic strains of mosquitoes still rely on a process of embryo microinjection5,6,7. However, microinjection has several limitations.First, this technique is technically demanding and involves complicated procedures. Second, injection causes a physical insult to the embryo, larvae, pupae, and adult, which directly affects the viability of the target organism. Third, it is difficult to immobilize mosquito larvae for microinjection because most live in an aquatic habitat and possess a characteristic wriggling movement. Fourth, the size of 1st-2nd instar larvae is 10- to 20-fold smaller than that of 4th instar and older larvae, and the cuticles of the former are more delicate. These features make it difficult to manipulate 1st-2nd instar larvae compared to those in older stages. Combined, these factors contribute to a reduced post-injection survival rate for larvae (approximately 5%) compared to adults8. Viral-based delivery systems have been developed to overcome the associated extracellular and intracellular barriers. These systems have the advantages of easy manipulation, high transduction efficiency, long-term and robust levels of expression, and the ability to produce persistent effects in vivo. Therefore, gene delivery systems utilizing retroviruses, lentiviruses, and adenoviruses have been widely used inmammalian cell lines and model species. The Sindbis viral expression system had been previously used to analyze the gene function in the adult mosquito; besides the biosafety concerns, however, the injection is still a necessary process for viral infection9. Although, the oral delivery by soaking larvae in the dsRNA solution has been reported previously as a feasible delivery method, it is unsuitable for small RNA function analysis10. So, effective viral delivery methods must still be developed for mosquitoes.
Mosquito densoviruses (MDVs) are part of the Densovirinae subfamily of Parvoviridae, and all but one fall within the genus Brevidensovirus11. MDV virions are non-enveloped and consist of a single-stranded DNA (ssDNA) genome and an icosahedral capsid (20 nm in diameter). The viral genome is approximately 4 kb in size and is replicated within the nuclei of host cells. MDVs are relatively stable in the environment and show a narrow host range with high specificity for mosquitoes. These viruses have the potential to spread and persist naturally in mosquito populations and can invade almost all organs and tissues of these insects, including the midgut, Malpighian tubules, fat body, musculature, neurons, and salivary glands12.
Intact MDV genomes can be subcloned into plasmid vectors to produce plasmid-based infectious clones; when these clones are delivered into mosquito cells, the viral genome is extracted from the plasmid vector, and infectious viral particles are produced. Because MDV has a small ssDNA genome, infectious clones are easily constructed and the viral genome can be easily manipulated11,13. These characteristics make MDV a valuable agent for examining mosquito biology. However, because nearly all of the viral genome sequence is essential for viral proliferation, the creation of recombinant virus through the replacement or insertion of foreign genes causes a loss in viral packaging and/or replication abilities, which creates a barrier for the development of MDVs as gene delivery vectors. Herein, we report using an artificial intronic small-RNA expression strategy to develop a non-defective rAaeDV in vivo RNA delivery system that has the advantages of easy plasmid construction and the maintenance of a functional virus that can produce stable and long-term expression in host cells without the need for wild-type virus. Additionally, this method allows for the easy transduction of larvae.
The protocols for the following steps are described in this study: 1) design of rAaeDVs encoding an intronic small-RNA expression cassette, 2) production of recombinant virus particles using the C6/36 packaging cell line, 3) quantitative analysis of cell-free rAaeDV genome copy numbers, and 4) infection of Ae. albopictus larvae by direct introduction of virus into the water body of the larval habitat. Overall, this work demonstrated that specific small RNAs or target genes can be overexpressed or knocked down in mosquito larvae using the developed MDV delivery system.
Protocol
All protocols were approved by the Ethical Committee of Southern Medical University.
1. Designing an Artificial Intron
NOTE: The artificial intron used in this work is 65 bp in length, and the sequence is 5'-GTAAGAGTCGATCGACGCGTTACTAACTGGTACCTCTTCTTTTTTTTTTGATATCCTGCAG-3'.
Place the HpaI restriction site on both ends of artificial intron so that HpaI digestion can release the intron from plasmids (see Figure 1A).
Place the two XbaI restriction sites in the intron so that XbaI digestion can be used to insert small-RNA expression sequences.
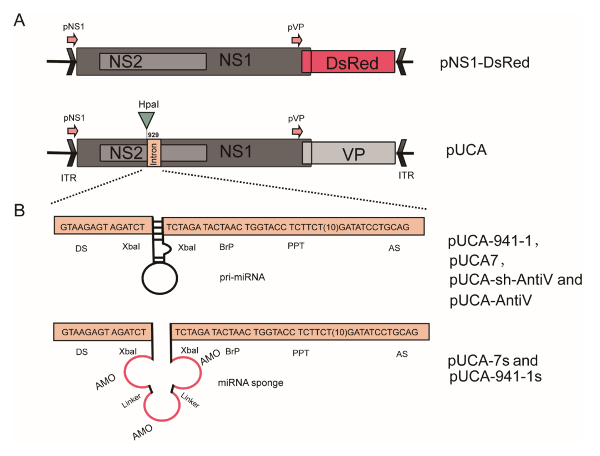
Order the chemical synthesis of full-length DNA sequences of the artificial intron from a DNA manufacturer. The synthetic DNA created is from cloning into a standard pUC19 plasmid. NOTE: The essential components of the artificial intron include several consensus nucleotide elements, including a 5' donor splice site (DS) with the sequence GUAAGAGU, a conserved branch-point sequence (BPS) with the sequence UACUAAC, a poly-pyrimidine tract (PPT) with the sequence UCUUC(U)10, and a 3'-acceptor splice site (AS) with the sequence GAUAUCCUGCAG (Figure 1A). Two XbaI restriction sites were introduced between the DS and BPS that can be used to insert small-RNA expression sequences. The ability to effectively splice this artificial intron from mammalian and mosquito cells has been previously confirmed14.
2. Designing an Artificial Intronic Small RNA Expression Cassette
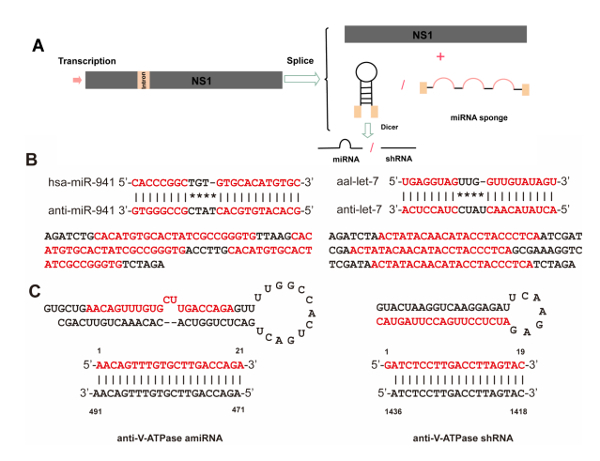
NOTE: For overexpression or knockdown of endogenous miRNA, sequences encoding precursor miRNA or a miRNA sponge were inserted between the DS and BPS. To serve as an example, an endogenous precursor miRNA from Ae. albopictus aal-let-7 was selected to construct recombinant viral vectors for miRNA overexpression and knockdown. An anti-let-7 sponge was designed according to previously described methods15. To knock down target genes in larvae, we designed specific artificial miRNA (amiRNA) or short hairpin RNA (shRNA) sequences using online software16,17; to serve as an example, V-ATPase subunit A mRNA (accession no. AY864912) was selected as a target gene in this study. The amiRNA will be referred to as amiVTP hereafter. All the sequences details of pre-miRNA, sponge, amiRNA, and shRNA are described in Supplemental Table 1.
Order the chemical synthesis of full-length cDNA sequences of the precursor miRNAs, miRNA sponges, and shRNA with XbaI restriction sites on both ends from a DNA manufacturer.
After receiving the ordered DNA, spin the tubes containing DNA blocks at 10,000-14,000 x g for 1 min to ensure that all dried DNA is at the bottom of the tube.
Resuspend the dried DNA in DNase-free water to achieve a final concentration of 10 ng/µL.
Digest the plasmid backbone and DNA sequences with XbaI at 37 °C for 1 h, and then dephosphorylate the 5' ends of the linearized plasmid backbone with calf intestinal alkaline phosphatase (CIAP) according to the manufacturer's protocol.
Inactivate CIAP by heating for 60 min at 65 °C.
Run a 1.0% agarose gel and cut the linearized backbone. Then extract the DNA from the gel slice using a gel extraction kit following the manufacturer's instructions.
Ligate the DNA fragment into the backboneby T4 DNA ligase (5 U/µL) at 22 °C for 1 h.
- Transform the products of the ligation reaction from step 2.7 into competent Escherichia coli (E. coli) cells and plate on a Lysogeny Broth (LB) agar plate containing Ampicillin (at a concentration of 100 µg/mL).
- Perform the transformation through a heat shock method18, and use the appropriate E. coli strain. For example, use E. coli strain DH5α for heat shock transformation.
Pick a single colony from step 2.8 and incubate in a culture of 3 mL enriched medium containing Ampicillin (at a concentration of 100 µg/mL).
Extract the plasmid DNA from the bacterial culture using a mini-prep kit following the manufacturer's instructions. Send the sample to a DNA sequencing company. NOTE: For overexpression or knockdown of endogenous miRNA, the sequences encoding precursor miRNA or a miRNA sponge were inserted between the two XbaI sites. All miRNA expression or sponge constructs must be shorter than 400 bp (10% of the viral genome length) due to the packaging limit of the virus19.
3. Generating a rAeaDV Construct by Cloning a miRNA or shRNA Expression Cassette into a Plasmid Backbone
NOTE: pUCA, an infectious clone consisting of a pUCA plasmid containing the AaeDV genome (3,981 nt)19, was kindly provided by Prof. Jonathan Carlson and has been previously described in detail11. p7NS1-DsRed expresses a non-structural protein 1 (NS1)-red fluorescent protein (DsRed) fusion protein from the p7 promoter and has been described in detail elsewhere14. A human-specific miR-941-1 precursor20 and its sponge, a scrambled shRNA (CGACGACTATCGTGCAATTTCAAGAG AATTGCACGATAGTCGTCG), were also subcloned into AaeDV as a negative control.
Ligate the pre-miRNA, sponge, shRNA or an amiVTP expression cassette into the HpaI site of the AaeDV NS1-coding region in the pUCA plasmid backbone as described in the protocol step 2.4 to 2.7.
Transform the DNA into Stbl3 competent E. coli cells and select a positive clone as described in step 2.8 to 2.10.
Following this, extract the rAaeDV plasmid from the bacterial cells using a maxi prep kit following the manufacturer's instructions. NOTE: Stbl3 or Stbl4 E. coli cells should be utilized for rAaeDV DNA transformation to minimize undesired ITR recombination. A maxi prep should be used to extract the rAaeDV plasmid to obtain a large quantity of DNA (>100 µg). Before generating the virus, the sequence integrity of the rAaeDV plasmid should always be analyzed by DNA sequencing21.
4. Transfecting C6/36 Cells with rAaeDV Plasmids
Culture C6/36 cells in Roswell Park Memorial Institute (RPMI) 1640 medium containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin in a 28 °C incubator.
On day 0, plate the cells in ten 25 cm2 culture flasks 18-20 h before transfection by splitting 90-95% confluent cells in a 1:2 dilution. NOTE: By day 1, the cells should reach 90-95% confluence.
- On day 1, transfect the cells with the series of rAaeDV plasmids22.
- Dilute 10 µg DNA in 600 µL of RPMI-1640 medium in a microcentrifuge tube and mix gently. Meanwhile, prepare 600 µL of RPMI-1640 medium and 25 µL of transfection reagent per transfection.
- Incubate for 5-10 min at room temperature (RT). Then, add 625 µL of the 1640- transfection reagent mixture to the 1640-plasmid DNA mixture.
- Incubate this mixture for 20-30 min at RT. Before transfection, wash the cells twice with 2 mL of RPMI-1640 medium.
- After the RT incubation (step 4.3.3), add the1640-transfection reagent DNA plasmid mixture to the 25 cm2 culture flask and incubate for 6 h at 28 °C.
- At approximately 6 h post-transfection, remove the transfection media, wash the cells twice with 5 mL of RPMI-1640 medium, and then add RPMI-1640 medium with 10% FBS. NOTE: rAaeDV shows high rates of infection (95%) in Ae. albopictus larvae; however, in our experience, in nearly 50% of infected larvae, the infection was restricted to primary infection sites (e.g., anal papillae and bristle cells) without dissemination23. Therefore, if there is a need to infect larvae in multiple tissues, a defective recombinant virus expressing fluorescent protein should be co-infected to enable the visualization of infection sites. For example, to produce defective recombinant virus expressing DsRed, p7NS1-DsRed should be co-transfected with a helper plasmid into C6/36 cells according to the procedure described above (the cotransfection concentration ratio should be 2:1).
5. Harvesting rAaeDV Virions from Transfected C6/36 Cells
Harvest the cells 5 days after transfection. Dislodge and suspend the cells in the dishes by a 5 mL glass dropper, pipetting up and down with the culture medium. Transfer all cell suspensions to sterile 15-mL tubes.
Freeze at -80 °C or in a dry ice/ethanol bath for 30 min and then thaw at 37 °C. Vortex for 1 min. Repeat the freeze-and-thaw procedure 3 times.
Centrifuge the sample at 4,800 x g for 20 min at 4 °C.Collect the supernatant and discard the pellet. Then, pass the supernatant through a 0.22 µm disposable syringe filter. Aliquot and store the final purified virion stocks at -80 °C. NOTE: Avoid repeated freeze-thaw cycles.
6. Quantifying rAaeDV
7. Mosquito Transduction
Wash 100 1st instar Ae. albopictus larvae three times using deionized water, and then, transfer the larvae into a beaker containing 95 mL deionized water. Add 5 mL of the rAaeDV stocks (final concentration of 1.00 x 1010 copies/mL).
Incubate for 24 h at 28 °C, wash the larvae three times using deionized water, and then transfer the larvae back to the plates and feed regularly.
Monitor the level of target gene expression in the larvae with qPCR or Western blot (representative results are shown in Figures 3 and 4). NOTE: The fluorescence signal detection method can be used to isolate the infected larvae more precisely. For example, to detect fluorescent signals from larvae infected with recombinant virus expressing DsRed, the larvae can be placed on a concavity slide and observed under an inverted fluorescence microscope every 8 h until 2 days post-infection. Once fluorescent larvae are detected, they should be separated into individual plastic test cups to allow for subsequent continuous observation. Larvae with multiple tissues infected can be identified using this method.
Representative Results
Strategies for rAaeDV construction A defective rAaeDV vector was generated to express the DsRed gene in mosquito larvae. The resulting plasmid contained a NS1-DsRed fusion protein cassette with the VP protein deleted (Figure 1A). rAaeDV plasmids containing expression constructs for miRNA, miRNA sponge, shRNA and amiRNA were designed (shown in Figure 1B). For example, a rAaeDV vector was generated to overexpress aal-let-7 in mosquito larvae. The resulting plasmid contained an intronic pre-aal-let-7 cassette in the viral NS1 exon (Figure 1B). A rAaeDV vector containing an intronic aal-let-7 sponge expression cassette in the viral NS1 exon was generated to knock down aal-let-7 expression in mosquito larvae (Figure 1B and Figure 2B). A rAaeDV shRNA vector and a rAaeDV amiRNA vector containing intronic shRNA and pre-amiRNA expression cassettes, respectively, were used to knock down V-ATPase mRNA in mosquito larvae (Figure 2C).
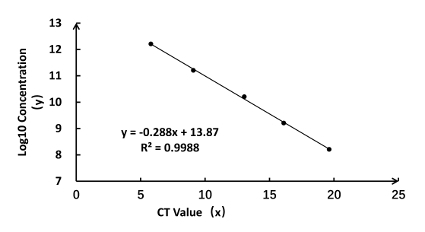
Calculating the rAaeDV titers based on a standard curve that generated with the linear regression analysis The y-axis represents the Log10 value of standard DNA molecular concentration, and the x-axis indicates the CT value. The CT numbers (x-axis) and Log10 (concentration) values (y-axis) displayed a good correlation (R2 = 0.9988) and meet the equation y = - 0.288x + 13.87 (Figure 3). Calculate the rAaeDV titers of samples using the linear equation (Table 1). Using the method described in the protocol (step 6.2 and Table 1), the high titers of rAaeDV (4 mL, >7.65 x 1010 particles/mL) were harvested. Recombinant viruses (VrepUCA-7, VrepUCA-7s, VrepUCA-amiVTP, VrepUCA-shVTP, VrepUCA941-1, VrepUCA-941-1s, and VrepUCA-shct) were generated by transfecting the corresponding infection clones pUCA-7, pUCA-7s, pUCA-amiVTP, pUCA941-1, pUCA941-1s, pUCA-shct into C6/36 cells (sections 3, 4 and 5).
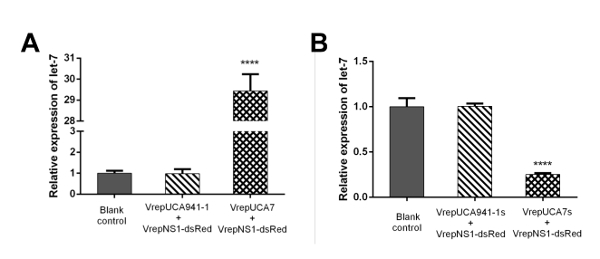
Determining the miRNA overexpression and knockdown efficiency of rAaeDV vectors In the example, 100 1st instar larvae were treated with equal amounts (1.00 x 1010 particles/mL) of rAaeDV containing the intronic pre-let-7 expression vector, the intronic let-7 sponge expression vector, the human-specific pre-miR-941-1 expression vector or the miR-941-1 sponge expression vector by adding the virus into the water body of the larval habitat. Five days after infection, the expression of aal-let-7 was determined by qPCR (Figure 4, n = 3; Supplemental Table 2). The let-7 levels that are displayed as the mean ± SD of three biological replicates in the larvae, were substantially increased or decreased by the pre-let-7-expressing rAaeDV (overexpressed by 2,843.31 ± 80.72%, p <0.0001, t-test) and the let-7-sponge-expressing rAaeDV (downregulated by 74.78 ± 1.60%, p <0.0001, t-test), respectively.
Monitoring the V-ATPase mRNA knockdown efficiency of rAaeDV vectors A total of 100 1st instar larvae were treated with equal amounts (1.00 x 1010 particles/mL) of intronic amiRNA-expressing rAaeDV, intronic shRNA-expressing rAaeDV, human-specific pre-miR-941-1-expressing rAaeDV orscramble shRNA-expressing rAaeDV by adding the virus into the water body of the larval habitat. Five days after infection, the expression of V-ATPase mRNA was determined by qPCR (Figure 5, n = 3; Supplemental Table 2). The V-ATPase level displayed as the mean ± SD of three biological replicates in the larvae was substantially reduced by amiRNA-expressing rAaeDV (downregulated by 43.01 ±6.37%, p = 0.0011, t-test) and intronic shRNA-expressing rAaeDV (downregulated by 72.88 ± 5.74%, p = 0.0001, t-test).
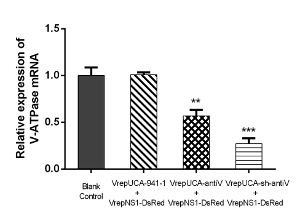
Figure 1: Schematic organization of the recombinant AaeDV plasmids. (A) The pNS and pVP viral promoters drive the expression of the NS1/NS2 genes and VP genes, respectively. In the p7NS1-DsRed plasmid, the DsRed gene was fused to the NS1 gene. (B) pUCA-7, pUCA-7s, pUCA-941-1, pUCA-941-1s, pUCA-shct, pUCA-shVTP and pUCA-amiVTP contain artificial introns, including the aal-let-7, aal-let-7 sponge, hsa-miR-941-1, hsa-miR-941-1 sponge, scrambled shRNA, V-ATPase shRNA and artificial miRNA, respectively, which were cloned into the HpaI site of the NS1 gene. The artificial intron is shown flanked by a splice donor (DS) and an acceptor site (AS) and contains a branch-point domain (BrP), a poly-pyrimidine tract (PPT) and pre-miRNA. The miRNA sponge or artificial miRNA sequence is inserted inside the intron between the 5-splice site and the BrP. Please click here to view a larger version of this figure.
Figure 2: Biogenesis of artificial intronic microRNA (miRNA) and strategy for generating miRNA sponges and artificial miRNAs. (A) Intronic miRNA is co-transcribed within a precursor messenger RNA (pre-mRNA) of NS1, which will be driven by the pNS1 promoter and cleaved out of the pre-mRNA by RNA splicing. Although the exons are ligated to form a mature messenger RNA (mRNA) for NS1 protein synthesis, the spliced intron with the pre-miRNA is further processed into mature miRNA by Dicer. (B) Strategy for generating the anti-let-7 and anti-miR-941-1 constructs. Alignment of anti-let-7 and anti-miR-941-1 sequences with aal-let-7 and hsa-miR-941-1, respectively. Complete matching of the seed regions with the anti-miRNA sequence and tail regions is shown. Both let-7 and miR-941-1 sponges contain three repeat antisense constructs (red letters) that can bind to aal-let-7 and hsa-miR-941-1, respectively. (C) Sequences and predicted precursor structures for the miRNA-based artificial miRNAs and shRNA used in this study. The mature artificial miRNAs and shRNA are shown in red, and their related target mRNA sequences are in blue. Please click here to view a larger version of this figure.
Figure 3: Standard curve for rAaeDV titration and the calculated concentrations of MDVs. (A) Real-time qPCR was performed with a pUCA standard that was diluted by 10 times the magnification ratio, and the CT value was measured on an ABI 7500 as x. Copy number was calculated according to step 6.2 and 6.3 with the following formula: copy number (y) = -0.288x + 10.87; R2 = 0.9988. Please click here to view a larger version of this figure.
Figure 4: Recombinant MDV-mediated aal-let-7 overexpression and knockdown in Ae. albopictus larvae. (A) Analysis of endogenous miRNA expression in larvae after infection with recombinant virus containing the aal-let-7 expression cassette (left panel). (B) The efficiency of the anti-let-7 construct was determined in larvae (right panel). The has-miR-941-1 and has-941-1 sponge (negative control) overexpressed and downregulated the mature let-7 miRNAs, respectively, compared to the basal levels observed in the control group. miRNA abundance was normalized to 5S rRNA, and the data are displayed as the mean ± SD of three biological replicates. T-tests were performed at different levels of significance. The asterisks indicate statistically significant differences between the infected and corresponding mock-treated larvae (four asterisks, p <0.0001). Please click here to view a larger version of this figure.
Figure 5: Analysis of V-ATPase mRNA expression after infection with recombinant virus containing shRNA and amiRNA cassettes. mRNA abundance was normalized to aalrpS7. Error bars represent the standard deviation of the 2−ΔΔCT values for V-ATPase mRNA expression in the Ae. albopictus larvae as evaluated by real-time RT-PCR (**, p <0.01; ***, p <0.001). Please click here to view a larger version of this figure.
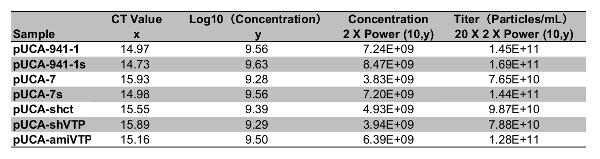 Table 1: The recombinant MDV CT value determined by qPCR corresponds to the x value in the formula, which is used to calculate the y value. The viral genome copy number in each 1 µL sample was obtained by the function power (10, y), which was ultimately multiplied by 40, converting the value to the virus concentration of virus suspension.
Table 1: The recombinant MDV CT value determined by qPCR corresponds to the x value in the formula, which is used to calculate the y value. The viral genome copy number in each 1 µL sample was obtained by the function power (10, y), which was ultimately multiplied by 40, converting the value to the virus concentration of virus suspension.
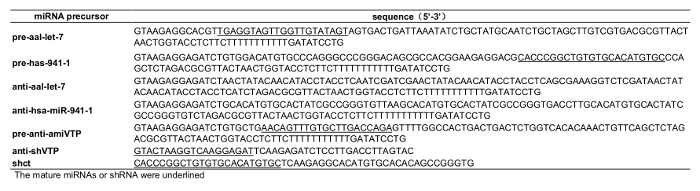 Supplemental Table 1: Sequences of miRNA precursor.
Supplemental Table 1: Sequences of miRNA precursor.
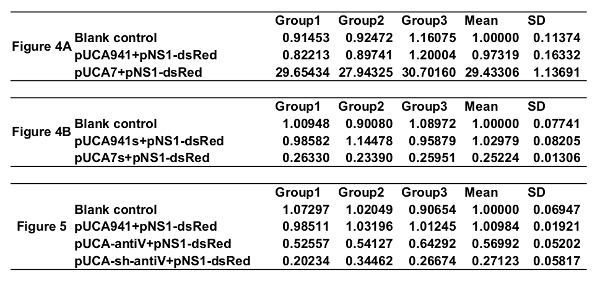 Supplemental Table 2: qPCR data.
Supplemental Table 2: qPCR data.
Discussion
It is important to overcome two of the key barriers that limit rAaeDV construction. The first is the production of defective recombinant virus. It has been reported that MDV can be used as a vector to express appropriately sized foreign genes, such as scorpion insect-specific neurotoxins20 and the GFP protein; however, regardless of the construction strategy, once the ORFs of MDV are inserted or replaced with exogenous genes, virions cannot form independently unless a helper plasmid is cotransfected to supply the missing indispensable viral proteins. The defective virus is unable to engage in secondary transmission, which occurs in vivo in mosquitoes with defective genomes. Our results validate an intronic expression strategy that offers a new paradigm that can overcome the limitations of MDVs with defective genomes to enable the efficient delivery of foreign genes into mosquitoes.
The second is the demanding length limit of the recombinant viral genome imposed by the size of viral capsid. For efficient packaging, the genome size cannot exceed 4,400 nucleotides (110% of the length of the wt MDVs genome). An intronic miRNA expression cassette (including miRNA expression cassette and artificial intron) of no more than 400 nt can be introduced into the AaeDV genome. Although the length of the recombinant virus genome is still suitable for packaging, this delivery system has room for improvement. Because of the limitation in genome length, it is difficult to express genes without defective intronic expression strategies.
It is essential to ensure that C6/36 cells are healthy to enable the successful transfection and packaging of rAaeDV. However, the cationic liposome reagents used here and commercial insect cell specific transfection reagents, do not improve the efficiency of C6/36 cell transfection (40-60%). Therefore, to harvest high-titer recombinant virus, it is necessary to maintain the cells for 5 days after transfection based on previously described growth characteristics14. Additionally, for successful larval infection, it is essential for larvae to be in contact with infectious virus particles for 24-36 h to ensure high infection ratios23.
In conclusion, the intronic expression strategy described here is the first to overcome the barriers imposed by the generation of defective MDV. This strategy enables the production of non-defective recombinant MDVs that can overexpress or downregulate miRNAs and target genes in mosquitoes. This technique provides a tool for the functional analysis of mosquito genes without the use of complicated larvae microinjection procedures and has clear commercial applications. Further studies will expand the application of the described expression strategy to investigate mosquito biology and paratransgenesis for dengue virus control.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
The authors acknowledge financial support from the National Key Research and Development Program of China (2016YFC1200500 to Xiao-Guang Chen), the National Natural Science Foundation of China (81672054 and 81371846), the Research Team Programme of the Natural Science Foundation of Guangdong (2014A030312016), and the Scientific and Technological Programme of Guangzhou (201508020263). We gratefully acknowledge Professor Jonathan Carlson (Colorado State University) for kindly providing the pUCA and p7NS1-GFP plasmids and for critically reading this manuscript.
References
- Tolle MA. Mosquito-borne diseases. Current problems in pediatric and adolescent health care. 2009;39:97–140. doi: 10.1016/j.cppeds.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Petersen LR, Jamieson DJ, Powers AM, Honein MA. Zika Virus. The New England journal of medicine. 2016;374:1552–1563. doi: 10.1056/NEJMra1602113. [DOI] [PubMed] [Google Scholar]
- Roberts DR, Andre RG. Insecticide resistance issues in vector-borne disease control. The American journal of tropical medicine and hygiene. 1994;50:21–34. doi: 10.4269/ajtmh.1994.50.21. [DOI] [PubMed] [Google Scholar]
- Attaran A, Roberts DR, Curtis CF, Kilama WL. Balancing risks on the backs of the poor. Nature medicine. 2000;6:729–731. doi: 10.1038/77438. [DOI] [PubMed] [Google Scholar]
- Volohonsky G, et al. Transgenic Expression of the Anti-parasitic Factor TEP1 in the Malaria Mosquito Anopheles gambiae. PLoS pathogens. 2017;13:1006113. doi: 10.1371/journal.ppat.1006113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galizi R, et al. A synthetic sex ratio distortion system for the control of the human malaria mosquito. Nature communications. 2014;5:3977. doi: 10.1038/ncomms4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourtzis K, Lees RS, Hendrichs J, Vreysen MJ. More than one rabbit out of the hat: Radiation, transgenic and symbiont-based approaches for sustainable management of mosquito and tsetse fly populations. Acta tropica. 2016;157:115–130. doi: 10.1016/j.actatropica.2016.01.009. [DOI] [PubMed] [Google Scholar]
- Kumar SS, Puttaraju HP. Improvised microinjection technique for mosquito vectors. The Indian journal of medical research. 2012;136:971–978. [PMC free article] [PubMed] [Google Scholar]
- Attardo GM, Higgs S, Klingler KA, Vanlandingham DL, Raikhel AS. RNA interference-mediated knockdown of a GATA factor reveals a link to anautogeny in the mosquito Aedes aegypti. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:13374–13379. doi: 10.1073/pnas.2235649100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AD, Wong S, Ryan CP, Whyard S. Oral delivery of double-stranded RNA in larvae of the yellow fever mosquito, Aedes aegypti: implications for pest mosquito control. Journal of insect science. 2013;13:69. doi: 10.1673/031.013.6901. (Online) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward TW, et al. Aedes aegypti transducing densovirus pathogenesis and expression in Aedes aegypti and Anopheles gambiae larvae. Insect molecular biology. 2001;10:397–405. doi: 10.1046/j.0962-1075.2001.00276.x. [DOI] [PubMed] [Google Scholar]
- Carlson J, Suchman E, Buchatsky L. Densoviruses for control and genetic manipulation of mosquitoes. Advances in virus research. 2006;68:361–392. doi: 10.1016/S0065-3527(06)68010-X. [DOI] [PubMed] [Google Scholar]
- Barreau C, Jousset FX, Bergoin M. Pathogenicity of the Aedes albopictus parvovirus (AaPV), a denso-like virus, for Aedes aegypti mosquitoes. Journal of invertebrate pathology. 1996;68:299–309. doi: 10.1006/jipa.1996.0100. [DOI] [PubMed] [Google Scholar]
- Liu P, et al. Development of non-defective recombinant densovirus vectors for microRNA delivery in the invasive vector mosquito, Aedes albopictus. Scientific reports. 2016;6:20979. doi: 10.1038/srep20979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega MM, Bouamar H. Methods in molecular biology. Vol. 1509. Clifton, N.J: 2017. Guidelines on Designing MicroRNA Sponges: From Construction to Stable Cell Line; pp. 221–233. [DOI] [PubMed] [Google Scholar]
- BLOCKiT RNAi Designer. 2017. Available from: http://rnaidesigner.lifetechnologies.com/rnaiexpress/
- GE Dharmacon siDesign Center siRNA design tool. 2017. Available from: dharmacon.gelifesciences.com/design-center/
- Froger A, Hall JE. Transformation of plasmid DNA into E. coli using the heat shock method. Journal of visualized experiments : JoVE. 2007. p. e253. [DOI] [PMC free article] [PubMed]
- Afanasiev BN, Kozlov YV, Carlson JO, Beaty BJ. Densovirus of Aedes aegypti as an expression vector in mosquito cells. Experimental parasitology. 1994;79:322–339. doi: 10.1006/expr.1994.1095. [DOI] [PubMed] [Google Scholar]
- Hu HY, et al. Evolution of the human-specific microRNA miR-941. Nature communications. 2012;3:1145. doi: 10.1038/ncomms2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences of the United States of America. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalby B, et al. Advanced transfection with Lipofectamine 2000 reagent: primary neurons, siRNA, and high-throughput applications. Methods (San Diego, Calif) 2004;33:95–103. doi: 10.1016/j.ymeth.2003.11.023. [DOI] [PubMed] [Google Scholar]
- Gu JB, Dong YQ, Peng HJ, Chen XG. A recombinant AeDNA containing the insect-specific toxin, BmK IT1, displayed an increasing pathogenicity on Aedes albopictus. The American journal of tropical medicine and hygiene. 2010;83:614–623. doi: 10.4269/ajtmh.2010.10-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Zhang H, Miranda L, Lin S. Serious overestimation in quantitative PCR by circular (supercoiled) plasmid standard: microalgal pcna as the model gene. PloS one. 2010;5:9545. doi: 10.1371/journal.pone.0009545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, et al. Preparation of rAAV9 to Overexpress or Knockdown Genes in Mouse Hearts. Journal of visualized experiments : JoVE. 2016. [DOI] [PMC free article] [PubMed]