

Abstract
A primary method used to define the presence of neutrophil extracellular traps (NETs) is confocal microscopy. We have modified established confocal microscopy methods to visualize macrophage extracellular traps (METs). These extracellular traps are defined by the presence of extracellular chromatin with co-expression of other components such as granule proteases, citrullinated histones, and peptidyl arginase deiminase (PAD). The expression of METs is generally measured after exposure to a stimulus and compared to un-stimulated samples. Samples are also included for background and isotype control. Cells are analyzed using well-defined image analysis software. Confocal microscopy may be used to define the presence of METs both in vitro and in vivo in lung tissue.
Keywords: Immunology, Issue 128, Macrophage, extracellular traps, confocal microscopy, lung, human, murine
Introduction
Neutrophil extracellular traps (NETs), were first described by Brinkmann et al.1 They are predominantly produced in response to infection (especially to bacteria) and have an important role in host defense1,2. They have also been described to occur in response to non-infectious disease, including vasculitis and systemic lupus erythematosus (SLE); and to the mitogen phorbol 12-myrisate 13-acetate (PMA)2,3. It has recently been recognized that other cell types may also produce extracellular traps, including macrophages. Macrophage extracellular traps (METs) are not yet a well-defined entity in the literature4,5. We have recently established methods to detect the presence of METs both in vitro and in vivo6,7. In this article, the measurement of METs using confocal microscopy will be described.
Key features of NETosis which distinguish it from other cellular pathways (such as apoptosis8) are the extrusion of chromatin in conjunction with: (1) citrullination of histones (H3Cit)9, (2) co-expression of granule proteases10, and (3) involvement of peptidyl arginine deiminase (PAD) 411,12. Macrophages also express H3Cit, granule proteases, and PAD, and these features can be used to define the presence of METs.
METs may have a particularly important role in the lung, as the macrophage is the dominant cell present in the alveoli and airways of the lung and has the initial role in directing the cellular immune response to infection/inflammation. In addition, while much of the lung is empty space (e.g., within the alveoli), the METs are potentially able to expand into the space available, in contrast to solid organs.
The most widely used method to define the presence of NETs is by confocal microscopy. There is not yet a clearly defined way to measure METs. The technique for the measurement of NETs has been adapted to measure the presence of METs in this protocol. The main requirements for this method are access to confocal microscopy and appropriate imaging software for analysis.
Protocol
This protocol follows experiments approved in: (1) humans by the Ethics Committee of Monash Medical Centre, and (2) animals by the Ethics Committee of the University of Melbourne.
1. Bronchoalveolar Lavage (BAL) Macrophages
- Use BAL to obtain lung macrophages in: (1) human subjects by bronchoscopy6, and (2) mice by using intra-tracheal aspiration13. NOTE: Acquisition of the macrophages generally causes some activation of these cells. Macrophages are obtained from patients who require a bronchoscopy to investigate a potential medical problem and not all subjects may be suitable for analysis of METs (this will need to be determined for each individual project).
- Take the BAL solution and spin down (500 x g for 10 min) to form a pellet, wash twice with culture medium (Roswell Park Memorial Institute Medium (RPMI) with 5% fetal calf serum and 1% L-glutamine) at room temperature, and then dilute cells in 4 mL of culture medium.
- Perform a viable macrophage cell count on the BAL solution using Trypan blue exclusion and a hemocytometer. NOTE: BAL solution is obtained from humans and mice as mentioned in step 1.1.
- Add 1 - 4 x 105 macrophages to 24-well plates on coverslips in 500 µL of culture medium per well and leave overnight at 37°C; the cells will adhere to the coverslips. For human samples, add antibiotics (e.g., penicillin and streptomycin, 104 U/mL) to prevent bacterial contamination.
2. Immunofluorescence Labeling/Microscopy of BAL Macrophages
NOTE: Cells are adhered onto coverslips as mentioned above.
Remove the culture medium and wash the cells once with phosphate buffered saline (PBS). Fix cells in 2% paraformaldehyde/periodate/lysine (PLP) fixative for 10 min.
Wash cells briefly in PBS, and then permeabilize in 0.2% Tween 20 in PBS for 20 min.
Block cells with 10% chicken sera in 5% bovine serum albumin (BSA) diluted in PBS for 30 min.
Incubate with primary and isotype control antibodies for 1 h at room temperature at 1:100 concentrations (more specific details are listed in Table of Materials) at 37 °C. NOTE: For the BAL samples, a specific marker of macrophages is usually not required.
After the addition of primary antibodies, wash the cells in PBS, and further incubate with the corresponding secondary antibodies for 40 min at room temperature.
Wash the sections in PBS and mount with DAPI-based mounting medium for visualization using a confocal microscope (as mentioned above) at laser excitations 405, 488, 561 and 647 nm and a 40X, 1.0 NA oil objective.
3. Lung Tissue Samples
NOTE: For in vivo studies of lung tissue, study the samples after the relevant exposure (e.g., as described by O'Sullivan et al.7). The exposure that is most relevant for this experiment are infectious microorganisms, particularly bacteria. Mouse strains that could be used for this method are C57BL/6 or BALB/c mice, 10 - 12 weeks old, weight 25 - 30 g, and male.
- Euthanize mice, via an intraperitoneal injection of ketamine (400 mg/kg) and xylazine (40 mg/kg). Open the thoracic cavity and expose the lungs and heart using surgical forceps and scissors.
- Infuse warm PBS using a 21-gauge needle into the right ventricle to wash out the blood from the vessels for 30 s, then infuse 10 mL of 2% PLP fixative for 30 s (into the right ventricle).
- Use warm PBS (42 °C) to lavage the open chest cavity. Intubate the trachea with a 21-gauge needle and a piece of 0.8 mm tubing cut to size, and inject 800 µL of pre-warmed (to 42 °C), low-melting point 3% agarose solution.
- Tie off the trachea with braided silk (4.0 USP). Place the thread around the trachea, just below the insertion point of the cannula, and secure it via a simple overhand knot. To solidify the agarose, administer ice-cold PBS around the lungs. Remove the lungs and heart as a block from the thoracic cavity using both scissors and blunt dissection. Remove each lung individually and then fix them for 16 h in 2% PLP or formalin at 4 °C.
- Store the specimens in PBS at 4 °C until tissue sectioning. These methods for obtaining lung tissue have been previously described13.
- To prepare the lung tissue samples use the following steps.
- Fix lung tissues (resected in step 3.1) tissue in 10% natural-buffered formalin for 24 h at room temperature. Transfer to 75% ethanol and put in a tissue processor on a 12 h cycle, (2.5 h in 75% EtOH, 3 h in absolute EtOH, 3.5 h in solvent 3B, and for 3 h in paraffin).
- Embed in standard paraffin to create formalin-fixed, paraffin-embedded (FFPE) blocks which are stored at room temperature. NOTE: At this stage, it is generally convenient to cut the lung sections for standard slides.
- Cut the FFPE lung sections at 4 - 5 µm thickness using a microtome and mount on charged, coated slides as previously described by O'Sullivan et al.7
- To mount slides, put 3 drops of mounting medium on 60 mm x 24 mm coverslips (size 1.5), invert the slide on the coverslip and push out excess medium. Hermetically seal with nail polish and store at room temperature.
4. Preparation/Microscopy of Lung Tissue Samples
- To de-wax, rehydrate and pretreat the FFPE lung tissue sample with antigen retrieval solution.
- Oven dry the FFPE slides for 60 min at 60 °C. Then put the slides in xylene solution for 30 min and transfer to 70% ethanol solution for 5 min at room temperature. Rinse in tap water.
- Place in heat-proof plastic wrap and subject to HIER-antigen retrieval in a pressure cooker for 10 min in 10 m/mol/L Tris, 1 mmol/EDTA pH 9.0. Cool for 20 min. Wash twice in tap water for 5 min on a rocker, then once with PBS on rocker.
- Block with 10% chicken serum in 5% BSA/PBS for 30 min at room temperature.
- Stain the tissue.
- To define METs in macrophages, add primary antibodies (MMP-9, H3Cit, and F4/80) in 1% BSA/PBS for 16 h at 4 °C at 1/100 dilution (specific details about antibody amounts are listed in Table of Materials). Wash two times with PBS for 5 min on a rocker.
- Achieve fluorescent detection by incubation with corresponding secondary antibodies in 1% BSA/PBS for 40 min at room temperature. Antibodies are: (1) chicken anti-mouse 488 (green), (2) chicken anti-goat 594 (red), and (3) donkey anti-mouse 647 (far red).
- Wash the sections in PBS and mount with DAPI-containing mounting medium for chromatin staining.
- Obtain fluorescent images using a confocal laser scanning head attached to an inverted microscope.
- Excite the preparation with 405, 488, 561, and 647 nm lasers. Capture single plane 512 x 512-pixel images by clicking on the line-sequential, leveling button (2 line averaging) using 20X 0.1 NA air and 40X 1.0 NA oil objectives.
- Obtain at least ten fields of view per section for analysis and data for each result (i.e., 10 high-power fields of view (HFOV) for the control, 10 for the stimulated sample, etc., for each mouse). NOTE: To obtain a representative sample of the whole field, a matrix system can be used in which the field is divided into different sections and for each different sample the same matrix is used to select fields of view (e.g., the field can be divided into 9 sections, and from the center and the 4 corners, two HFOV are taken).
5. Three-dimensional (3-D) Imaging of METs
NOTE: As METs are 3-D structures, by taking multiple z-stack images, which are reconstituted, may give valuable information.
Section the lung specimens from paraffin blocks, at 200 µm using a vibratome at an amplitude set at 1.8 mm and speed of 0.1-0.5 mm/s.
Block lung tissue with 20% of the respective serum (10% fetal calf serum and 10% chicken serum) and then permeabilize in PBS supplemented and 3.75% Triton-X 100 for 7 h at room temperature under gentle agitation.
Stain the sections for 16 h at 4 °C with the relevant primary antibodies (MMP-9, H3Cit, and F4/80) in 200 µL volumes in microcentrifuge tubes (antibody concentrations are listed in Table of Materials).
Wash the sections in PBS, 3 times for 10 min before incubating with relevant secondary antibodies at room temperature for 1 h.
Stain the lung sections for DAPI (1:5,000) in solution for 20 min, before adding the cover slips to the microscope slides in PBS.
Obtain fluorescence images using an inverted confocal microscope (40x 1.3 NA) equipped with 405 nm, 440 nm, 473 nm, 543 nm, and 635 nm lasers.
Record 3-D z-stacks of 0.54 µm thickness with optimal z-sectioning for the 40x 1.3 NA oil objective. NOTE: The software used will vary depending on the individual microscope. An example of how the software can be used for one microscope is provided in Supplementary File 1.
6. Image Analysis
NOTE: The analysis of samples requires the use of specific imaging analysis software and examples are listed in Table of Materials. While the analysis of results will depend on the specific program used, the listed points below are important.
- Analysis of BAL samples
- Define the number of macrophages by selecting the blue channel for DAPI and assigning a minimum size for the cells (e.g., > 18 µm in diameter). Using the relevant software, measure the total number of macrophages per field.
- Count the number of cells that have the features of a MET by the presence of extracellular chromatin detected by DAPI with co-expression of other markers (e.g., MMP12, PAD2, and H3Cit). NOTE: The number of markers that can be analyzed depends on the number of lasers available, but ideally in addition to DAPI, at least two other markers should be included. Other less specific parameters for MET expression (e.g., number of cells with extracellular chromatin and an enlarged nucleus) can be used.
- To compare the MET expression in BAL samples (between the control, smoke, and smoke/DNase treated groups), analyze a minimum of 100 BAL macrophages per sample.
- To measure secondary antibody and isotype control levels of fluorescence; measure a minimum of 100 cells in each sample for their fluorescence. Measure the average level of background fluorescence for each marker (e.g., H3Cit) using standard software.
- To define MET expression, count the cells with the typical phenotype (e.g., extracellular chromatin with co-expression of H3Cit and MMP9) and for each MET, measure the level of fluorescence of markers (e.g., H3Cit and MMP9). Exclude cells producing METs if their staining was not above background and isotype control.
- For human BAL samples, analyze a minimum of 100 cells for each sample for the percentage of macrophages that make METs. For murine samples, as the cells are smaller and METs are harder to define, analyze a larger number of cells for each sample (e.g., 200 macrophages).
- Analysis of lung tissue samples
- To determine the presence of METs in lung tissue, use specific macrophage marker such as F4/80.
- Define the presence of METs in lung tissue by using the co-localization of F4/80 in cells that express extracellular chromatin with other parameters as listed in section 6.1.
- Determine the levels of background fluorescence in samples with secondary antibody and the isotype controls. Exclude METs from analysis that are not above background.
Representative Results
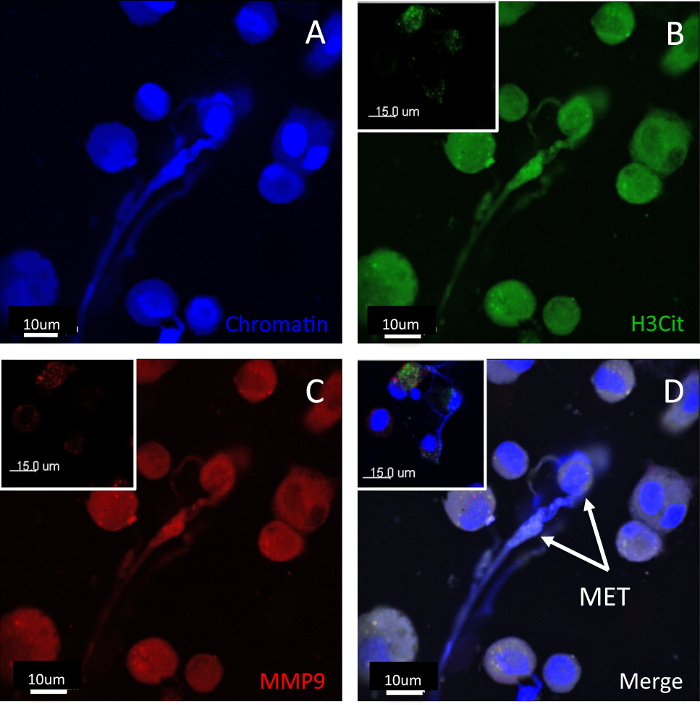
METs may be visualized from BAL samples, in lung tissue and in thicker lung sections with 3-D images. An example of METs visualized in a BAL sample is shown in Figure 1. The morphology of the METs will vary according to their stage of maturation. The first detectable feature on microscopy is the movement of the nucleus to the edge of the cell. This is followed by extracellular chromatin with other co-expressed mediators, such as H3Cit and granule proteases. In the earlier stages, the cell still has a roughly spherical shape with the extracellular trap expressed. At later stages, the MET formation is characterized by elongation of the body of the cell.
Lung tissue METs are shown in Figure 2. As the lungs are inflated with fluid to define the lung architecture, the METs will generally be pushed against the alveolar walls. The morphology of the METs will also vary depending on which plane the tissue was cut in. The standard sections used for lung tissue samples are 4-5 µm in thickness. Thicker tissue sections enable multiple images to be taken and the METs can then be viewed in 3-D. The antibodies will only penetrate so far into the lung tissue and a section thickness of 25-50 µm is optimal. An example of a 3-D image is shown in Figure 3 and in Video 1.
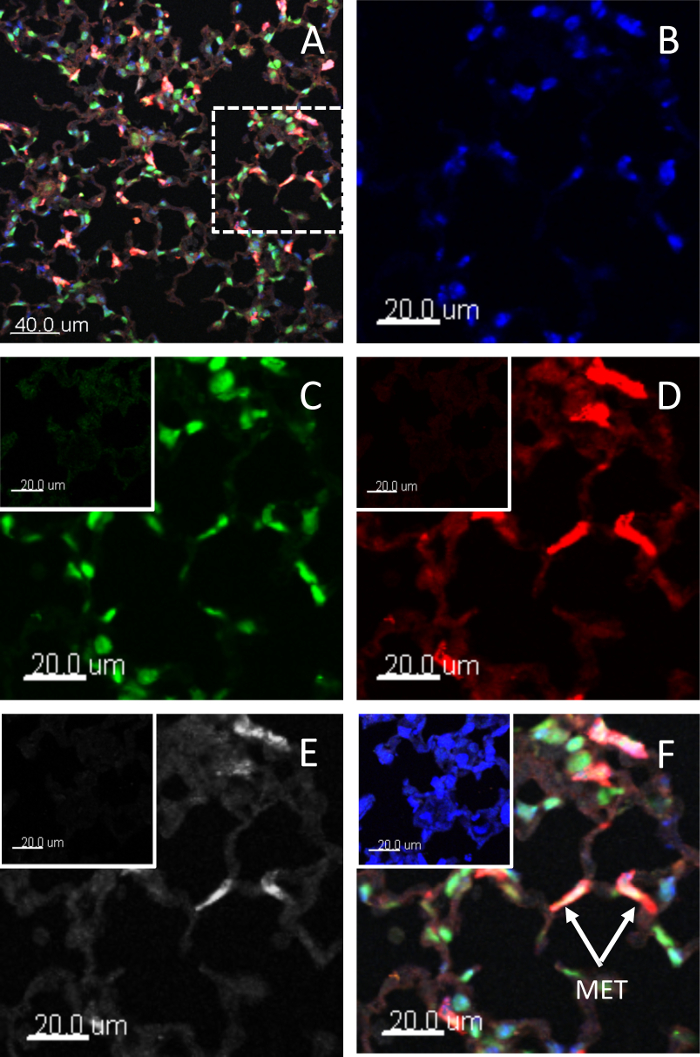
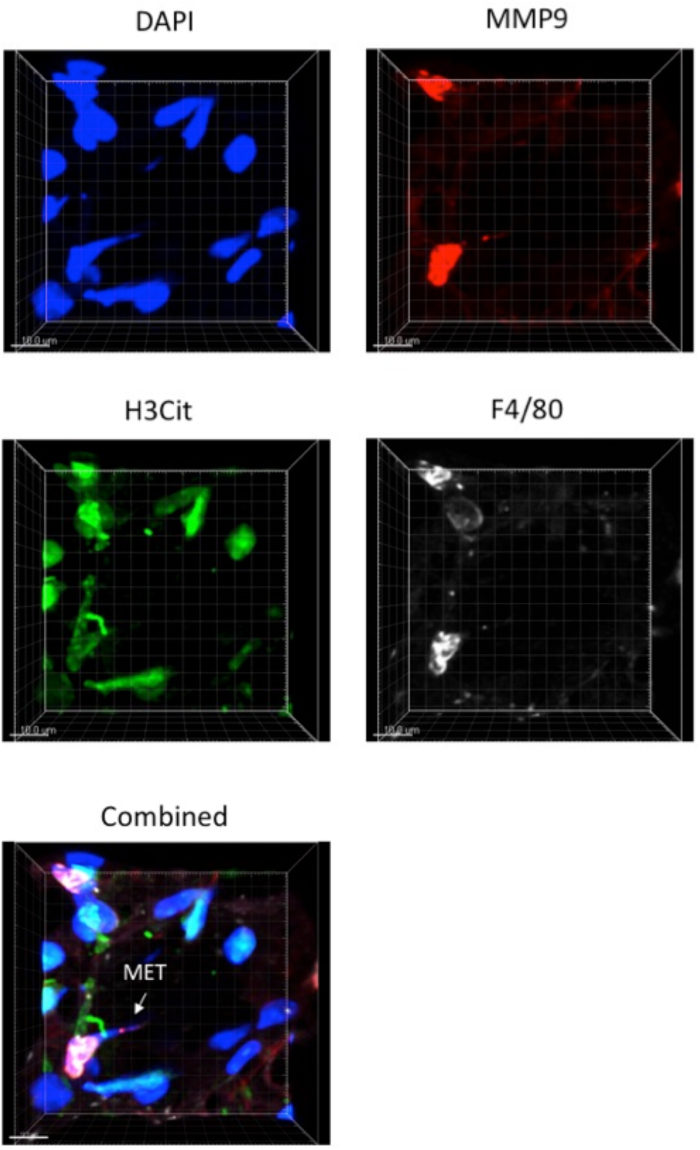
Figure 1: BAL METs. Human BAL METs formed after stimulation at different stages of development. METs were characterized by extracellular chromatin with co-expression of H3Cit and MMP9. Staining for (A) chromatin, (B) H3Cit, (C) MMP9, and (D) the merged image. Panel inserts are isotype controls. Scale bars = 10 µm (15 µm for isotype). An earlier stage MET is shown in the top right corner and has a roughly spherical shape. A more mature MET is shown in the middle of the field. Images were taken using a laser scanning confocal microscope and a 40x 1.0 NA oil objective. Please click here to view a larger version of this figure.
Figure 2: Lung tissue METs. Murine METs in lung tissue after stimulation. The METs have been characterized by extracellular chromatin with co-expression of H3Cit, MMP9, and F4/80 (as a macrophage marker). Panel (A) shows a typical high-power field of view used to measure the number of METs; Panels (B-F) show the dashed square area under higher magnification. Staining for (B) chromatin, (C) H3Cit, (D) MMP9, (E) F4/80, and (F) the merged image. Insert panel is the isotype control. Scale bars = 20 µm (20 µm for isotype). METs are generally pushed against the alveolar walls. Images were taken using a laser scanning confocal microscope and a 40x 1.3 NA oil objective. Please click here to view a larger version of this figure.
Figure 3: Three-dimensional view of lung section. Mouse lung tissue was fixed and cut into 25-35 µm thick sections. Tissue was labeled with markers for METs and imaged with sections at 1 µm thickness. Images were combined with a z-stack to create a 3-dimensional picture of a MET. Images were taken using a laser scanning confocal microscope and a 40x 1.3 NA oil objective. Please click here to view a larger version of this figure.
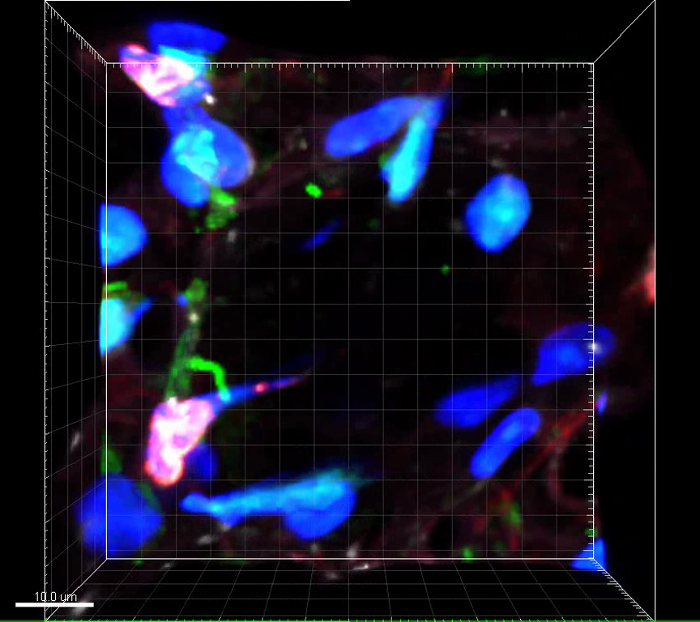 Video 1: Three-dimensional view of lung section. Video corresponds to samples presented in Figure 3. Axis ticks = 5 µm. Please click here to view this video. (Right-click to download.)
Video 1: Three-dimensional view of lung section. Video corresponds to samples presented in Figure 3. Axis ticks = 5 µm. Please click here to view this video. (Right-click to download.)
Discussion
The method described in this review is based on that used for defining the presence of NETs14. Macrophages are by far the dominant cell type in BAL samples and this method of collection is particularly suited for studying METs. If red blood cells are present in the BAL, these cells should be lysed by using ammonium chloride. The BAL procedure will generally activate the macrophages and therefore it is expected that there will be METs present in un-stimulated samples. The BAL macrophages are usually very adherent. The BAL macrophages will generally not need a specific marker as these cells are the dominant cell type and can also be distinguished by their morphology. Usually in a BAL, greater than 80% of the cells are macrophages (and neutrophils are less than 5%), and after multiple washes, generally only the adherent leukocytes (i.e., the macrophages) remain6. If there is doubt about the ability to distinguish macrophages from neutrophils in humans, a neutrophil marker such as neutrophil elastase may be used. In lung tissue, a specific macrophage marker (e.g., F4/80) is required as macrophages are difficult to distinguish from other cell types. Macrophages have auto-fluorescence so the use of background and isotype controls is important; this is particularly the case with stimulated samples.
The best way to define the presence of METs remains to be determined. Specific pathways/processes, which are common to both neutrophils and macrophages are: (1) granular proteases, (2) citrullination of histones, and (3) PAD. PAD4 is more specific for neutrophils, whilst PAD2 is more prominent in monocytes/macrophages. The use of F4/80 is a well-accepted marker for murine macrophages, but human macrophages do not have such well-defined markers and this may pose issues when assessing human lung tissue for MET expression. Analyzing MET expression in lung tissue may be more difficult as there are many other cell types and the METs tend to be pushed onto the alveolar walls. When analyzing tissue sections, METs are best visualized when they are flat in cross-section; if the METs are in a different plane it may be difficult to determine if a MET is present. This issue can be addressed to some extent by making z-stacks to enable deeper tissue (3-D) planes for examining METs and the potential to visualize the METs in different aspects.
There is not a well-accepted method for defining the presence of METs in lung samples. The described methods are based on those used for visualizing NETs. Chow et al. described that the mitogen PMA induced METs in a macrophage cell line, as assessed by the presence of extracellular chromatin15. A subsequent study demonstrated that Mycobacterium tuberculosis induced METs in vitro, as defined by the presence of extracellular chromatin and H3Cit16. We have shown in vivo in a model of glomerulonephritis, that METs are present in the kidney as defined by co-localized chromatin, histone, and myeloperoxidase7. Finally, we have demonstrated the presence of METs in vitro in human lung macrophage by the combination of extracellular chromatin and MMP126. Our method can be used by other investigators to research this important facet of the inflammatory response.
It is likely that METs will be recognized to have a major role in chronic inflammatory disease. The processes that lead to NETosis are still not well-defined. The study of METs may provide significant insights into the mechanisms of trap formation. The development of protocols with more markers and functional studies of METs will further define the role of METs in disease.
There are critical steps that we have identified in this protocol. The human BAL samples are likely to have secondary bacterial contamination and the use of antibiotics in culture medium is important for limiting contamination. The primary and secondary antibodies can vary in their fluorescence, even with the same manufacturer. Finally, the analysis of lung tissue samples is more complex than BAL samples and requires time to develop a standardized approach.
Disclosures
The authors have no disclosures to make in relation to this work.
Acknowledgments
This work was funded by grants from Monash University, the National Health and Medical Research Council, and the Monash Lung and Sleep Institute. The authors would like to thank the staff of Clinical Immunology at Monash Health, Judy Callaghan, Alex Fulcher, Kirstin Elgass, and Camden Lo of Monash Micro Imaging (MMI) for help with the confocal microscopy imaging, and the authors acknowledge the MMI facilities of Monash University. The authors acknowledge the facilities, and scientific and technical assistance of the Monash Histology Platform.
References
- Brinkmann V, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Zychlinsky A. Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol. 2012;198(5):773–783. doi: 10.1083/jcb.201203170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. 2017;23(3):279–287. doi: 10.1038/nm.4294. [DOI] [PubMed] [Google Scholar]
- Cheng OZ, Palaniyar N. NET balancing: a problem in inflammatory lung diseases. Frontiers immunol. 2013;4:1. doi: 10.3389/fimmu.2013.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boe DM, Curtis BJ, Chen MM, Ippolito JA, Kovacs EJ. Extracellular traps and macrophages: new roles for the versatile phagocyte. J Leukoc Biol. 2015;97(6):1023–1035. doi: 10.1189/jlb.4RI1014-521R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King PT, et al. Nontypeable Haemophilus influenzae induces sustained lung oxidative stress and protease expression. PloS one. 2015;10(3):e0120371. doi: 10.1371/journal.pone.0120371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan KM, et al. Renal participation of myeloperoxidase in antineutrophil cytoplasmic antibody (ANCA)-associated glomerulonephritis. Kidney Int. 2015;88(5):1030–1046. doi: 10.1038/ki.2015.202. [DOI] [PubMed] [Google Scholar]
- Fuchs TA, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2):231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184(2):205–213. doi: 10.1083/jcb.200806072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191(3):677–691. doi: 10.1083/jcb.201006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbach AS, Slade DJ, Thompson PR, Mowen KA. Activation of PAD4 in NET formation. Front Immunol. 2012;3:360. doi: 10.3389/fimmu.2012.00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis HD, et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat Chem Biol. 2015;11(3):189–191. doi: 10.1038/nchembio.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruwanpura SM, McLeod L, Dousha LF, et al. Therapeutic Targeting of the IL-6 Trans-Signaling/Mechanistic Target of Rapamycin Complex 1 Axis in Pulmonary Emphysema. Am J Respir Crit Care Med. 2016;194(12):1494–1505. doi: 10.1164/rccm.201512-2368OC. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Laube B, Abu Abed U, Goosmann C, Zychlinsky A. Neutrophil extracellular traps: how to generate and visualize them. Journal of visualized experiments : JoVE. 2010. [DOI] [PMC free article] [PubMed]
- Chow OA, von Kockritz-Blickwede M, Bright AT, et al. Statins enhance formation of phagocyte extracellular traps. Cell host & microbe. 2010;8(5):445–454. doi: 10.1016/j.chom.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KW, Jacobs WR., Jr Mycobacterium tuberculosis exploits human interferon gamma to stimulate macrophage extracellular trap formation and necrosis. J Infect Dis. 2013;208(1):109–119. doi: 10.1093/infdis/jit097. [DOI] [PMC free article] [PubMed] [Google Scholar]