

Abstract
The tripeptide glutathione (GSH) is a crucial intracellular reductant and radical scavenger, but it may also coordinate the soft Cu(I) cation and thereby yield pro-oxidant species. The GSH-Cu(I) interaction is thus a key consideration for both redox and copper homeostasis in cells. However, even after nearly four decades of investigation, the nature and stability of the GSH-Cu(I) complexes formed under biologically relevant conditions remain controversial. Here, we revealed the unexpected predominance of a tetranuclear [Cu4(GS)6] cluster that is sufficiently stable to limit the effective free aquacopper(I) concentration to the sub-femtomolar regime. Combined spectrophotometric–potentiometric titrations at biologically realistic GSH/Cu(I) ratios, enabled by our recently developed Cu(I) affinity standards and corroborated by low-temperature phosphorescence studies, established cooperative assembly of [Cu4(GS)6] as the dominant species over a wide pH range, from 5.5 to 7.5. Our robust model for the glutathione-Cu(I) equilibrium system sets a firm upper limit on the thermodynamic availability of intracellular copper that is 3 orders of magnitude lower than previously estimated. Taking into account their ability to catalyze the production of deleterious superoxide, the formation of Cu(I)-glutathione complexes might be avoided under normal physiological conditions. The actual intracellular Cu(I) availability may thus be regulated a further 3 orders of magnitude below the GSH/Cu(I) affinity limit, consistent with the most recent affinity determinations of Cu(I) chaperones.
Keywords: copper, metal homeostasis, peptides, spectroscopy, thermodynamics, phosphorescence, potentiometry
Introduction
Glutathione (GSH)2 is a ubiquitous thiol-containing non-ribosomal tripeptide that plays a central role in cellular redox homeostasis (1–3), detoxification (4), and signaling pathways (5). Present at millimolar concentrations, GSH serves as a cytosolic redox buffer (6) and is essential for combating oxidative stress. By virtue of its sulfhydryl group, GSH functions not only as an intracellular reductant and radical scavenger but may also serve as a ligand for soft transition metal cations. The formation of complexes with copper ions is of particular interest as GSH plays a critical role in cellular copper homeostasis (7). For example, depletion of GSH in HEK293 cells resulted in a 50% decrease in the initial rate of copper uptake (8). At the same time, recent in vitro experiments demonstrated that such complexes may catalyze the production of superoxide from dioxygen (9), thus challenging the presumed role of GSH as an immediate copper ligand. Because Cu(II) is rapidly converted to Cu(I) within the reducing cellular environment, the coordination chemistry of GSH with Cu(I) is key toward understanding its potential role as a ligand in cellular copper homeostasis. Despite the importance of the GSH-Cu(I) interaction, studies spanning the past four decades have yet to reach a consensus on the nature and stability of the complexes produced under physiological conditions (10–16). Previously reported stability constants, derived from equilibrium titrations using stoichiometric quantities of Cu(I) and GSH, imply predominance of species with 1:1 and 1:2 stoichiometry (10–14), which is contradicted by X-ray absorption data showing a trigonal CuS3 coordination environment (15, 16). Although these discrepancies might be attributed to differences in experimental conditions, the usefulness of complexation studies is contingent on their applicability to biological systems, where GSH is present in large excess compared with copper. Standard potentiometric techniques are not applicable at biologically realistic GSH/copper ratios, and the redox instability of free aquacopper(I) at typical analytical concentrations poses an additional challenge for determining reliable formation constants.
We recently reported a set of three water-soluble monovalent copper ligands, MCL-1, MCL-2, and MCL-3 (Scheme 1), that form discrete air-stable Cu(I) complexes with well-defined 1:1 metal-ligand binding stoichiometry (17). By offering complementary buffer windows stretching from 10−10 to 10−17 m, this ensemble of ligands is well suited to determine Cu(I)-complex dissociation constants in the pico- to sub-femtomolar range by means of ligand competition titrations. Taking advantage of these properties, we employed the monovalent copper ligand reagents as affinity standards to elucidate the nature of the Cu(I)-GSH equilibrium system at biologically relevant concentrations, where GSH is present at large excess compared with Cu(I).
Scheme 1.

Results
Competition titration at constant pH
The three MCL-affinity standards form complexes with apparent dissociation constants ranging from 10 pm to 100 am at pH 7 (17, 18), thus covering the full range of Cu(I) affinities previously reported for GSH. Both the free ligands and their Cu(I) complexes are transparent across the visible and near-UV range, permitting spectroscopic observation of Cu(I) complexation equilibria in their presence. Monitored by UV absorption spectroscopy under anaerobic conditions (Fig. 1A, red trace), addition of 100 μm aqueous Cu(II)aq to 4 mm GSH in pH 7 buffer produced a strong charge-transfer band with a shoulder near 300 nm. Consistent with quantitative reduction to Cu(I), an identical spectrum was observed after addition of [Cu(I)-(MCL-2)]PF6. (Fig. 1B). Back titration with the high-affinity ligand MCL-1 (17), which offers a logK of 16.0 at pH 7, yielded a steady decrease in absorbance (Fig. 1A), indicating competitive chelation of GSH-bound Cu(I). Although evolving factor analysis of these data supported only formation of a single new absorbing species, non-linear least-squares fitting using equilibrium models with either 1:1 or 1:2 Cu(I)-GSH stoichiometries resulted in trending residuals (Fig. 1C).
Figure 1.

Spectrophotometric titration data and non-linear least-squares fitting for the Cu(I)-glutathione system at constant pH. A, UV spectra of in situ formed GSH-Cu(I) complex titrated with MCL-1. Red trace: 100 μm CuSO4, 4 mm GSH, 10 mm PIPES, 0.1 m KCl, pH 7.0 (25 °C). Black traces: addition of MCL-1 up to 356 μm over 40 aliquots (odd-numbered aliquots were omitted for clarity; the arrow indicates the direction of spectral changes with increasing MCL-1 content). Inset: absorbance at 300 nm versus total MCL-1 concentration. B, absorption spectrum of [Cu(I)-(MCL-2)]PF6 (100 μm) in the presence of GSH (4 mm) at pH 7.0 (10 mm PIPES, 0.1 m KCl, 25 °C). For comparison, the spectrum after addition of Cu(II)SO4 (100 μm) to 4 mm GSH is overlaid as a green dashed trace. C, residual absorbances (observed − calculated) at 300 nm for a single titration dataset (A) modeled with three different complex stoichiometries. The raw absorbance spectra were factored to reduce random noise for improved visualization of model-dependent fitting error. D, mean absolute error (absorbance units) for a single titration dataset (A) as a function of model complex nuclearity. All models assumed a 1:1 Cu(I)/glutathione ratio for comparison purposes.
Consistent with the large excess of unbound GSH, changes to the GSH stoichiometry or inclusion of multiple mononuclear species had no discernible effect on the fit quality. In contrast, varying the nuclearity revealed a sharp minimum in fitting error for a tetranuclear model (Fig. 1D). This behavior could be explained by cooperative assembly of an adamantane-like {Cu4S6} cluster (Fig. 2A), which is the most common structural motif for crystalline Cu(I)-thiolate complexes (19). With macroscopic pKa values of 1.98, 3.49, 8.64, and 9.36 (20), corresponding to sequential deprotonation of the Glu-α-COOH, Gly-COOH, –SH, and Glu-NH3+ groups, GSH exists primarily as a monoanion at neutral pH. A species distribution diagram for the stepwise protonation of glutathione as a function of pH is provided in Fig. S1. As Cu(I)-thiolate cluster formation entails loss of the –SH proton, the corresponding pKa should be taken into account when extracting the stability constant of the cluster from titration data collected at pH 7. Employing a thiol pKa of 8.75, derived by correcting the macroscopic pKa of 8.64 from a proton concentration to a proton activity scale (17, 21), a Cu4(GS)6 complexation model indeed yielded small, random residuals (Fig. 1C) with a reproducible stability constant of logβ46 = 85.0.
Figure 2.

Low-temperature phosphorescence spectra indicating Cu(I)-thiolate clusters. A, structures of the copper-sulfur cores of model Cu(I)-thiolate clusters from literature X-ray crystallographic data. Only copper, sulfur, and carbon directly attached to sulfur are shown for clarity. Cu4S6, [Me4N+]2[Cu4(SMe)6]2−; Cu5S7, [Ph4P+]2[Cu5(SMe)7]2− ·(CH2OH)2; Cu5S6, [Et4N+][Cu5(SCMe3)6]−. B, normalized emission spectra (λex = 310 nm) of 1 μm and 1 mm Cu(I) in the presence of 4 mm GSH in pH 7 PIPES-KCl buffer. C, normalized emission spectra (λex = 310 nm) of 100 μm Cu(I) in the presence of 1 or 10 mm GSH in variable pH universal buffer (see under “Experimental procedures”). D and E, comparison of the long-wavelength region of the emission spectrum (normalized at the respective maxima) at two different pH values and excitation wavelengths (100 μm Cu(I), 1 mm GSH, universal buffer). F, emission spectra of solutions containing 100 μm Cu(I) and 1, 4, or 10 mm GSH at pH 5 (universal buffer, λex = 310 nm, normalized at 423 nm). All phosphorescence spectra were recorded at 77 K using vitrified samples containing 33% propylene glycol.
Low-temperature phosphorescence
Because many Cu(I)-thiolate clusters are phosphorescent, we turned to luminescence spectroscopy to further probe the nature of the multinuclear Cu(I)-glutathione complex. When employing conditions identical to above complexation studies, no emission was observed at ambient temperature; however, at 77 K a vitrified sample revealed a strong and unusually narrow phosphorescence band at 423 nm (Fig. 2B) with a luminescence lifetime of τ = 42 μs (Fig. S2). Because propylene glycol (33% v/v) was added to achieve vitrification, we also acquired the phosphorescence spectrum of a glycol-free frozen solution as control. These measurements revealed the same band, albeit slightly broadened. Furthermore, glycol addition had no significant effect on the UV spectrum at room temperature.
The distinctive signal at 423 nm can be attributed to a cluster-centered emission previously reported for complexes containing an adamantane-type {Cu4S6} core, including crystalline [Et4N+]2[Cu4(SPh)62−] (22) and certain copper-loaded metallothioneins (23, 24), whereas no other Cu(I)-thiolate cluster arrangement is known to emit below 550 nm (22, 23, 25). Although the 423-nm band remains the dominant spectral feature over total Cu(I) concentrations spanning at least 3 orders of magnitude from 1 μm to 1 mm (Fig. 2B), close examination of the baseline revealed a second, much weaker emission at ∼590 nm (Fig. 2B, inset).
To determine whether the long-wavelength band is due to a change in protonation state or a different cluster stoichiometry, we explored the effect of GSH concentration and pH on the phosphorescence spectrum. As noted above, free GSH exists primarily in monoanionic form at pH 7, whereas cluster formation presumably entails S-deprotonation to the corresponding dianion, which we represent as GS−. An equilibrium between clusters differing in Cu(I)/glutathione ratio would therefore depend on [GS−], which is influenced by both [GSH] and pH. Once the latter is significantly below the thiol pKa of 8.75, each further unit drop in pH will decrease the GS−/GSH ratio by 10-fold. We therefore measured pairs of solutions containing either 1 mm GSH or 10 mm GSH at 1 unit lower pH.
As evident from Fig. 2C, the long-wavelength band steadily increases relative to the 423-nm emission upon acidification from pH 8 to 5; however, each spectrum is essentially superimposable with its matched [GS−] counterpart. These data unequivocally demonstrate that the long-wavelength band originates from a distinct Cu(I)-thiolate cluster with a lower GS/Cu(I) ratio than the predominant {Cu4S6} species. Each order of magnitude decrease in [GS−] increased the abundance of the minor species by only 3-fold, or 0.5 orders of magnitude, implying that the equilibrium between {Cu4S6} and the minor complex yields only half an equivalent of GS− per eq of the latter. Of all previously observed Cu(I)-thiolate cluster stoichiometries, only {Cu5S7} is consistent with this result (see below).
Although the emission maximum of the long-wavelength band appears unchanged at the scale presented in Fig. 2C, normalization of the 1 mm GSH spectra at 590 nm revealed a subtle shift from 588 nm at pH 7 and above to 594 nm at pH 6 (Fig. 2, D and E). Increasing the excitation wavelength to 350 nm had no effect at pH 7 but further shifted the long wavelength band to 611 nm at pH 6, indicating that the onset of the shift corresponds to the appearance of a third coordination species. This was verified by decreasing the total glutathione concentration under otherwise identical conditions (100 μm Cu(I), pH 5, λex 310 nm), which shifted the long-wavelength emission from 593 nm at 10 mm GSH to 605 and 617 nm at 4 and 1 mm, respectively (Fig. 2F). At pH 5 and 1 mm GSH, a fourth emission is apparent as a shoulder on the 423-nm band but is already greatly diminished at 4 mm GSH. Precipitation occurs upon further acidification as the pH approaches 4, suggesting formation of a coordination polymer. Taking the 423 nm {Cu4S6} emission as a reference point, the band in the longest wavelength region (>630 nm) increases 21-fold in response to a 10-fold reduction in GSH concentration. This implies the presence of a third species whose net formation from Cu4(GS)6 yields more than 1 molar eq of GS−. The most likely candidate is a {Cu5S6} cluster, given the previously reported long-wavelength phosphorescence at 618 nm of Et4N+[Cu5(SAd)6−] (Ad = 1-adamantyl) (25). The symmetrical {Cu5S6} cage is the second most frequently crystallized Cu(I)-thiolate motif after {Cu4S6} and is structurally related to the {Cu5S7} by a missing bridging thiolate (Fig. 2A).
Combined potentiometric and spectrophotometric titrations
To fully characterize the GSH-Cu(I) solution equilibrium system, including the two additional clusters detected by phosphorescence, we performed further ligand competition titrations using pH as the independent variable. Because the thiol group deprotonates upon Cu(I) coordination, the position of the GSH-Cu(I) equilibrium system is sensitive toward pH changes. Increasing the pH in the presence of a non-protic competitor ligand should therefore produce a distinct binding isotherm, reflective of the complex stoichiometry, for each coordination species. Simulated titrations predicted that MCL-3, a non-protic Cu(I) chelator with a logK of 13.8 (17), should yield titration midpoints in the acidic range (Fig. 3A), where all three Cu(I)-glutathione species can be detected according to the phosphorescence data. With variable MCL-3 concentrations as a second titration dimension, discrimination between species of similar Cu/GS ratio such as Cu4(GS)6 and Cu5(GS)7 can be further improved (Fig. 3B).
Figure 3.
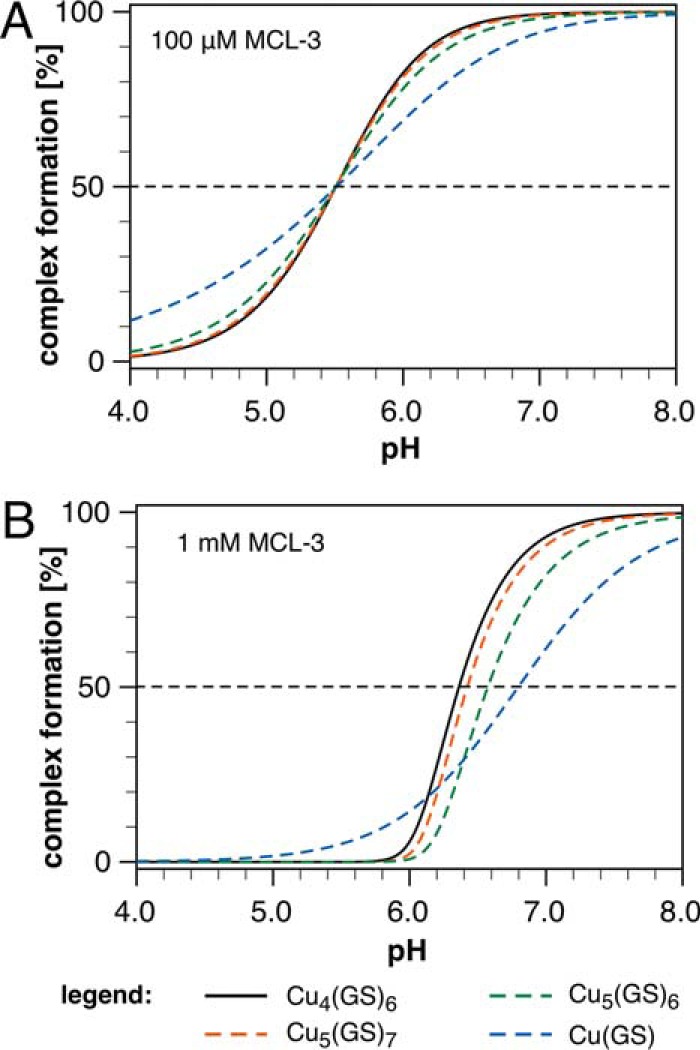
Computer simulations of the Cu(I)-GSH equilibrium system as a function of pH in the presence of MCL-3 as competing Cu(I) chelator. A, binding isotherms illustrating the degree of complex formation for selected Cu(I)-GSH cluster stoichiometries in the presence of 100 μm MCL-3 (logK = 13.80, I = 0.1, 25 °C). The experimental logβ46 value of 85.0 was assumed for the formation constant of the Cu4(GS)6 cluster. For all other cluster stoichiometries, the formation constants were chosen such that the inflection point at 50% complex formation occurs at the same pH value as for the Cu4(GS)6 cluster, thus yielding logβ11 = 15.3, logβ56 = 98.6, and logβ57 = 104.4. Simulation conditions: 100 μm MCL-3, 100 μm Cu(I), 4 mm GSH (pKa = 8.64, adjusted to 8.75 to account for activity at I = 0.1). B, simulated binding isotherms for selected Cu(I)-GSH cluster stoichiometries assuming the same stability constants and conditions as in A except for the MCL-3 concentration, which was set to 1 mm. Note that the isotherms for the 4:6 and 5:7 cluster stoichiometries are almost identical at 100 μm MCL-3 (A) but can be clearly distinguished at 1 mm MCL-3 (B).
We thus performed a series of spectrophotometric pH titrations using 4 mm GSH, 100 μm Cu(I), and 16 different MCL-3 concentrations ranging from 0.1 to 1.7 mm (Fig. 4). Each titration was conducted in duplicate and entailed 50 absorption traces for which the pH was varied between 4.5 and 7.5 by addition of KOH in the presence of a non-coordinating universal buffer (Fig. 4A; see “Experimental procedures” for details). An additional pair of titrations was conducted in the absence of MCL-3 to aid in spectral discrimination of the minor species (Fig. 4B). The cumulative data set, which entails a total of 900 spectra or 86,400 individual data points, was subjected to a global least-squares fitting procedure implemented in BeerOz (26). As evident from the absorption profiles at 308 nm, the previously proposed models with 1:1 or 1:2 Cu(I)-GSH stoichiometries yielded either a poor fit (Fig. 4C) or a close fit only at intermediate MCL-3 concentrations (Fig. 4D). In contrast, an equilibrium system comprising Cu4(GS)6, Cu5(GS)7, and Cu5(GS)6 produces an excellent fit across the entire data set, including the titrations devoid of MCL-3 (Fig. 4E). The corresponding stability constants for each of the three species are compiled in Table 1, and detailed residual maps for the global fitting results are provided in Fig. S3. It is noteworthy that the stability constant of logβ46 = 84.9 for Cu4(GS)6 closely mirrors the value of 85.0 determined using MCL-1 under an orthogonal titration mode with a single-component fit at pH 7 (Fig. 1A).
Figure 4.

Spectrophotometric pH titration data and global non-linear least-squares fitting results for the Cu(I)-glutathione equilibrium system using MCL-3 as competitor ligand (100 μm Cu(I), 4 mm GSH, and universal buffer, 0. 1 m KCl, 25 °C). A, representative UV absorption spectra for a pH titration in the presence of 290 μm MCL-3. The red trace was acquired at pH 4.5 where Cu(I) is complexed to MCL-3 only. Inset, non-linear least squares fit shown for the pH-dependent absorption change at 308 nm. B, UV-visible absorption spectra for a pH titration in the absence of MCL-3. The arrows indicate the direction of spectral changes with increasing pH. C–F, data (solid circles) and global non-linear least square fit (continuous lines) for 18 independent pH titrations in the presence of 0 to 17 molar eq of MCL-3 relative to total Cu(I). For clarity, absorption data and fitted traces are shown only at 308 nm as a function of pH (for comprehensive residual plots see Fig. S3). Equilibrium models and fitted parameters are as follows: [Cu(GS)] with logβ11 = 15.42 (χ2 = 7.885) (C); [Cu(GS)2] with logβ12 = 20.97 (χ2 = 2.705) (D); [Cu4(GS)6], [Cu5(GS)7], and [Cu5(GS)6], with logβ46 = 84.92, logβ56 = 97.99, logβ57 = 103.92, (χ2 = 0.424) (E); and [Cu4(GS)6] with logβ46 = 84.97 (χ2 = 1.029) (F).
Table 1.
Experimental formation constants for Cu(I)-glutathione clusters at 25 °C (0.1 m ionic strength)
Data were determined based on global non-linear least-squares fitting of spectrophotometric titrations data covering the pH range from 4.5 to 7.5 (see text for details).
a GS refers to monoprotonated glutathione, a net dianion.
b Apparent formation constants valid within the pH window of 4.5 to 7.5.
Speciation of the Cu(I)-GSH equilibrium system
Consistent with this remarkable agreement, the fitted stability constants for all three species (Table 1) indicate a distribution dominated by Cu4(GS)6 above pH 5 (Fig. 5A). Accordingly, fitting the entire titration dataset with this component alone yields an essentially unchanged logβ46 of 85.0, albeit with a reduced fit quality most apparent in the titrations without MCL-3 (Fig. 4F).
Figure 5.

Speciation for the Cu(I)-GSH equilibrium system as a function of pH and total GSH concentration. A, species distribution diagram as a function of pH at constant total Cu(I) (100 μm) and GSH (4 mm) concentrations (derived based on the stability constants compiled in Table 1). B, linear correlation between the calculated mole fraction of Cu4(GS)6 and the fraction of the emission intensity corresponding to the {Cu4S6} cluster observed in the phosphorescence spectrum, (I400–440)/(I515–750 + I400–440), at 100 μm Cu(I) and select pH and total GSH concentrations. Data points from left to right: 1 mm GSH, pH 5; 4 mm GSH, pH 5; 4 mm GSH, pH 6; 10 mm GSH, pH 7. C, contour plot of the calculated pCu (−log[Cu(I)aq]) as a function of total Cu(I) and GSH concentrations at constant pH 7.2.
Because such direct, independent corroborating evidence is not available for the minor species apparent at low pH, we verified our equilibrium model by comparing the species distribution calculated from the titration-derived stability constants to the relative intensities of the phosphorescence bands over conditions selected to cover a broad range of different speciations (Fig. 5B). The close agreement between the two independent techniques supports the assignment of all major components of the Cu(I)-GSH equilibrium system within the concentration and pH ranges investigated and reaffirms the predominance of Cu4(GS)6 under biologically relevant conditions. It should be noted that the observed 1:1 correspondence is expected only if the product of molar absorptivity and phosphorescence quantum yield is comparable for all three clusters. This condition is apparently met given the similar molar absorptivities at 310 nm indicated by the deconvoluted absorption spectra and the suppression of most non-radiative excited-state deactivation pathways at 77 K. The actual phosphorescence quantum yields could not be measured due to strong and variable scattering of the excitation beam by the samples.
Discussion
Cluster speciation
Both the nuclearity analysis of the MCL-1 titration dataset (Fig. 1D) and the predominance of the 423-nm phosphorescence emission (Fig. 2B) suggest that the glutathione-Cu(I) solution equilibrium is dominated by formation of an adamantane-like {Cu4S6} cluster, which remains the predominant coordination species over a Cu(I) concentration range spanning at least 3 orders of magnitude. This result is also fully consistent with previous X-ray absorption studies, which indicate a trigonal coordination environment (15, 16). Each face of the {Cu4S6} cluster consists of four trigonal CuS3 faces (Fig. 2A), and the Cu–S and Cu–Cu bond distances of 2.26 and 2.69 Å, which were determined for the Cu(I)-GSH system at pH 8 (16), match the values of 2.28 and 2.72 Å observed in the crystal structure of [Me4N+]2[Cu4(SMe)62−] (27).
Although the literature extended X-ray absorption fine structure data and our titration results at pH 7 can be fully accounted for by {Cu4S6} alone, the phosphorescence data reveal the presence of a minor coordination species characterized by a broad emission at 590 nm. Although several Cu(I) clusters with various stoichiometries have been reported to exhibit low-temperature phosphorescence within this spectral region, we deduced that the GSH-derived species is {Cu5S7} based on the observed dependence of the phosphorescence spectrum on the concentration of S-deprotonated glutathione (GS−, Fig. 2C). The fact that the spectrum does not respond directly to pH but rather to [GS−] indicates both that –SH is the only protic group linked to the equilibrium between the two clusters and that GS− appears directly in the equilibrium expression. Within the experimental pH window, GSH can therefore be modeled as a mono-protic thiol RSH and the tetranuclear cluster as Cu4(RS)62−. Thus, the relative ratio of Cu4(RS)62− and a minor species with stoichiometry Cux(RS)yz− is related to pH-dependent [GS−] according to Equation 1,
| (Eq. 1) |
with nominal cluster charge z = y − x and stoichiometric coefficients A = 4/x and B = 6 − Ay. The corresponding equilibrium constant for this reaction can be expressed as in Equation 2,
| (Eq. 2) |
and solving for the concentration of the minor cluster yields Equation 3,
| (Eq. 3) |
with ratio B/A = (3x − 2y)/2.
Given the large excess of glutathione over Cu(I), the concentration of [GS−] remains essentially unaltered for a given pH regardless of the cluster stoichiometry. Furthermore, over the range of RS− concentrations where Cu4(RS)62− remains the predominant Cu(I) species, its concentration can be approximated as constant, making the concentration of the minor species Cux(RS)yz− inversely proportional to [RS−]B/A. Thus, an order of magnitude reduction in [RS−] will increase the abundance of the minor species by a factor of 10B/A. Concluding from the B/A ratios calculated for all stoichiometric combinations up to 12 Cu(I) and 12 thiolates (Table S1), only formation of species with 1:1, 3:4, 5:7, or 7:10 Cu(I)-glutathione stoichiometry could account for the observed 0.5 order of magnitude increase in relative abundance per 10-fold reduction in [RS−]. Within these possibilities, only Cu5(RS)72− has been experimentally observed and structurally characterized. Furthermore, {Cu5S7} cages have been synthesized under similar conditions or even crystallized from the same solution as the corresponding {Cu4S6} species, implying that the two species coexist in equilibrium (28, 29).
Because the accuracy of the preceding approximate treatment is expected to diminish once the minor cluster represents a significant fraction of total copper, we confirmed the results by calculating the actual change in species ratio versus Cu4(GS)6 upon lowering the pH from 7 to 6 for all crystallographically observed Cu(I)-thiolate cluster ratios where B/A is in the range of 0–2 (Table S2), as well as hypothetical species with B/A = 0.5 for comparison. Using the program HySS (30) and an activity-corrected thiol pKa of 8.75 for GSH, we adjusted the formation constant of each minor species such that its abundance is 10% relative to Cu4(GS)6 (logβ46 = 84.9) at pH 7, 100 μm Cu(I), and 1 mm GSH, consistent with the relative areas of the integrated long-wavelength and short-wavelength phosphorescence bands observed under these conditions. The results in Table S2 confirm that Cu5(GS)7 is the only stoichiometry that is both consistent with the phosphorescence behavior and corresponds to a crystallographically observed Cu(I)-thiolate cluster structure. Furthermore, the predicted 22-fold increase in [Cu5(GS)6] per unit reduction in pH is consistent with the 21-fold increase in long-wavelength phosphorescence observed upon reduction of [GSH] from 10 to 1 mm at pH 5, where {Cu5S6} is expected to overtake {Cu5S7} as the most abundant pentanuclear species. The excellent correlation between the phosphorescence spectra and the calculated species distributions derived from the independent competition titration dataset (Fig. 5B) suggests that both pentanuclear species have been correctly assigned.
Role of GSH in copper homeostasis
The equilibria unraveled in this work have important implications for intracellular copper transport. Remarkably, GSH alone is sufficient to hold the thermodynamically available Cu(I)aq concentration below 1 fm under cytosolic conditions, even in a scenario where the entire cellular copper content would be available for GSH complexation (Fig. 5C). Furthermore, the cooperative assembly of a tetranuclear cluster provides a clamping effect, insulating −log[Cu(I)aq] (pCu) 4-fold more effectively from increases in total Cu(I) than a 1:1 binding stoichiometry would imply. This is best illustrated by the simplified pCu Equation 4, which was derived from the formation constant expression of Cu4(GS)6 under the approximation that [GS−] ≪ [GSH] ≫ [total Cu(I)] (see supporting information). The corresponding estimates for pCu match within 0.1 logarithmic units those of the full model under biologically relevant conditions with [GSH]/[Cu(I)] ratios of greater than 20 and a pH window of 5.5 to 7.5 (at 25 °C).
| (Eq. 4) |
Although our data indicate that GSH has the ability to clamp cytosolic Cu(I)aq to the sub-femtomolar range, it remains debatable whether this mechanism serves a physiological role under ordinary conditions. Because the GSH-Cu(I)-O2 system can produce deleterious superoxide (9), it would appear reasonable that intracellular copper is regulated below the range where assembly of Cu(I)-glutathione clusters can occur. This would be consistent with the most recent affinity determinations of Cu(I) chaperones, which yielded dissociation constants in the attomolar range (31–34). Perhaps the remarkable Cu(I) cluster-forming tendency of glutathione, and potentially other cysteine-derived monothiols as well, is an underlying reason why these chaperones have evolved such large Cu(I) affinities.
Implications for cellular copper stores
It is important to note that the high affinity of proteins involved in cellular copper transport does not necessarily imply a slow metal-exchange kinetics. Below the picomolar range, metal exchange may still proceed rapidly through associative mechanisms without intermediacy of the free aquated Cu(I) cation. In fact, single molecule FRET studies of the copper metallochaperone Atox1 and its binding partners imply a highly dynamic exchange mechanism involving formation of multiple interaction complexes characterized by short lifetimes (35–37). Although the transfer of Cu(I) could not be observed directly based on these measurements, the short lifetime of the underlying protein–protein interaction complexes is consistent with rapid Cu(I) transfer through an associative exchange as originally proposed by O'Halloran and co-workers (38).
An associative metal-transfer mechanism is likely also responsible for shuttling Cu(I) in and out of proteins maintaining cellular copper stores. Although specific proteins that contribute to this labile pool may vary between the cytosol, different compartments, and organelles, or across various organisms, there is increasing evidence that, as a unifying theme, the underlying Cu(I)-binding sites are rich in thiols and characterized by sub-femtomolar dissociation constants. For example, Pushkar et al. (39) reported copper accumulation in the glia of the sub-ventricular zone in rodent brain. Quantified by X-ray fluorescence microscopy, the copper levels were thousands of times higher compared with cells in the surrounding tissue. Microprobe K-edge X-ray absorption spectra of copper-rich locations were consistent with reduced Cu(I) and resembled the spectrum of copper-metallothionein, suggesting that metallothionein might in fact serve as a ligand for copper storage. Given the high Cu(I) affinity of metallothionein with reported dissociation constants between 10−19 and 10−20 (40, 41), GSH would not be able to compete for Cu(I) binding, even when present at tens of millimolar concentrations. A similar accumulation of copper in the form of intracellular deposits was also observed in the brain tissue of Ctr2 knock-out mice (42), although the speciation of this pool was not further investigated. The utilization of dedicated high-affinity ligands for maintaining safe copper stores appears to apply also to prokaryotes as underscored by the recent discovery of a novel Cu(I) storage protein (Csp1) isolated from a methane-oxidizing bacteria requiring large quantities of copper (43). Composed of a cysteine-rich four-helix bundle, the tetrameric protein is capable of binding up to 52 Cu(I) ions with an average dissociation constant of 10−17.
In summary, based on the presented data we established a robust model for the GSH-Cu(I) equilibrium system under physiologically relevant conditions. Anchored by low-temperature phosphorescence studies, our model is consistent with cooperative assembly of a tetranuclear cluster that sets a firm upper limit of ∼1 fm on the thermodynamic availability of intracellular copper. Conversely, the existence of intracellular Cu(I) pools that are buffered to the upper femtomolar or picomolar range, as implied from studies with synthetic fluorescent probes (44–46), is precluded by normal physiological levels of GSH, both in the cytosol as well as in GSH-rich compartments and organelles. Further studies of intracellular Cu(I) fluxes thus will require analytical tools capable of detecting Cu(I) at sub-femtomolar buffered concentrations. Although GSH occupies a prominent role as cellular redox buffer, the interaction of Cu(I) with less abundant low-molecular weight thiols, including cysteine, coenzyme A, or lipoic acid, might prove important toward a comprehensive understanding of cellular copper homeostasis and copper-induced stress response. Likewise, the interaction of Cu(I) with prokaryotic counterparts to GSH such as mycothiol (47) or bacillithiol (48), might have important implications for organisms that do not utilize GSH for maintaining the cellular redox balance. Although we can only speculate regarding the speciation of these systems, the formation of high-affinity tetranuclear clusters might take shape as a unifying concept, especially given the prevalence of tetranuclear clusters in inorganic Cu(I)-thiolate coordination chemistry (19).
Experimental procedures
Reagents
Glutathione (≥98%) and [Cu(MeCN)4]BF4 (97%) were purchased from Sigma. Copper(II) sulfate stock solutions were prepared from ACS grade CuSO4·5H2O (Thermo Fisher Scientific). The universal buffer was prepared from 1,4-diethylpiperazine (DEPP, Alfa Aesar, 98%, purified as described below), PIPPS (GFS Chemicals), MES, ultrapure (VWR®), aq. KOH (0.1 and 1 m, standardized, Fluka), and aq. HCl (0.1 and 1 m, standardized, Mallinckrodt Baker). PIPES pH 7 buffer was prepared from PIPES, free acid (GFS Chemicals), and standardized aq. KOH. Inert gas used was argon (ultra-high purity, Airgas).
Instrumentation
Automated spectrophotometric titrations were performed using a Varian Cary Bio50 spectrophotometer equipped with a temperature-controlled cuvette holder (Quantum Northwest), a computer-controlled syringe pump (J-KEM Scientific), and a pH meter (Orion 3 Star) equipped with a double junction glass electrode (Orion 9110DJWP). The electrode was calibrated with standard pH buffers (VWR®) at 25 °C (water bath). Titrations were conducted under a continuous stream of argon (passed through a 5-μm filter and pre-humidified by bubbling through deionized water).
Steady-state phosphorescence spectra were acquired on a PTI fluorimeter with cold finger Dewar accessory. Interference from second-order diffraction of the scattered excitation beam was eliminated using a 400-nm long-pass filter (Thorlabs FEL0400). Spectra were corrected for the intensity of the excitation source, the spectral response of the detector, and the transmittance of the filter element. Phosphorescence decay profiles were recorded on a Horiba Fluorolog-3 instrument equipped with a 75-watt xenon flash lamp (3-μs full width at half-maximum) employing the same cold finger Dewar and long-pass filter as described above.
Preparation and handling of Cu(I)-GSH solutions
All experiments were conducted under exclusion of oxygen using the Schlenk technique. To avoid contamination with UV-absorbing impurities, rubber septa were washed with two portions of acetonitrile followed by two portions of water prior to use. For molar ratio titrations and phosphorescence measurements, the appropriate amount of reduced glutathione was dissolved in 10 ml of aqueous buffer and purged with argon for 15 min. Addition of CuSO4 or the specified Cu(I) complex followed by pH adjustment was carried out under argon. The precise copper concentrations of the prepared stock solutions were determined by inductively coupled plasma atomic emission spectroscopy.
Low-temperature phosphorescence measurements
To account for dilution by propylene glycol, GSH-Cu(I) solutions were prepared using 1.5 times the target concentrations. Solutions with final concentrations of 1–100 μm Cu(I) were obtained by means of in situ reduction of CuSO4 supplied from 3 to 60 mm stock solutions. The initial GSH concentration was adjusted to compensate for stoichiometric oxidation to GSSG upon Cu(II) reduction. For each sample, 1.0 ml of aqueous GSH-Cu(I) solution and 500 μl of deoxygenated of propylene glycol were combined, and a 200-μl aliquot of the resulting solution was transferred into a quartz NMR tube and vitrified by immersion in liquid nitrogen. For variable pH experiments, a single bulk GSH-Cu(I) solution was prepared in universal buffer at pH 5 for each GSH concentration, and the pH was adjusted progressively by addition of 1 m aq. KOH. Each solution was allowed to equilibrate for at least 10 min following pH adjustment before vitrification.
Universal buffer
Full-range buffering was achieved using only non-coordinating reagents with the exception of biologically relevant chloride. The buffer consisted of 10 mm MES, 10 mm PIPPS, 5 mm DEPP dihydrochloride, and 5 mm KOH in 90 mm aq. KCl for an initial pH near 3.7 and a total chloride concentration of 100 mm. The calculated pH response to added alkali is nearly linear (R2 =0.9986) up to a pH of 9 with a slope of 0.17 pH-units per mm of added hydroxide. Commercial-grade DEPP was purified as the dihydrochloride salt. To this end, 12 ml of aq. HCl (37%) were added to a solution of 10.1 g (70.7 mmol) of DEPP in 200 ml of ethanol, and the slurry was concentrated under reduced pressure. After taking up the thick paste in boiling ethanol (200 ml), methanol was added to the point of dissolution, and the mixture was boiled down to the onset of crystallization. The mixture was diluted with ethanol, allowed to cool, and filtered under argon. After washing with ethanol, the colorless crystalline product was dried under vacuum. Yield was 13.2 g (87%).
Spectrophotometric competition titrations at constant pH
A 1-cm path length screw-top quartz cuvette was fitted with a magnetic stir bar, sealed with a PTFE-faced silicone septum, placed in a thermostatted holder at 25 °C, and purged with humidified argon for 15 min. Glutathione was dissolved in pH 7 buffer (10 mm PIPES, 0.1 m KCl) at a concentration of 4.0 mm, and the pH was adjusted to 7.00 with 1 m KOH. A 3.0-ml aliquot of the resulting solution was delivered to the sealed cuvette via syringe, and CuSO4 was added from a stock solution (5 μl, 60 mm). A solution of MCL-1 was prepared in 0.1 m KCl, adjusted to pH 7.0 with HCl, deoxygenated, and loaded into the syringe pump. Argon was introduced through a pipette tip (0.3-mm inner diameter) and exited through a 25-gauge needle to a water-filled bubbler. A total of 400 μl of titrant was injected into 10-μl aliquots with a 10-min equilibration time between spectral acquisitions.
The UV-absorption traces were analyzed by evolving factor analysis and non-linear least-squares fitting with the SPECFIT software package (49) to yield the corresponding apparent stability constant for pH 7.0 at 0.1 m ionic strength (KCl) and 25 °C. The published protonation constant of GSH (pKa = 8.64) (20) was adjusted upward by 0.11 to correct for the proton activity at 0.1 m ionic strength (17, 21).
Spectrophotometric pH titrations
Titrations were conducted in a modified quartz cuvette with a 3-cm extended neck, a sidearm fitted with a rubber septum for introduction of argon and titrant, and an internally threaded top port with an O-ring compression seal holding the pH electrode. Humidified argon was introduced at the bottom of the cuvette neck through a polyethylene tube (1.5-mm outer diameter) and vented at the top of the neck to a water bubbler.
For variable pH titrations in the presence of MCL-3, stock solutions of 100 μm [Cu(I)MCL-3]PF6 were prepared in 100-ml batches using the above universal buffer. Each batch was purged with argon for 15 min and stored at 4 °C under 1.3 atm of argon in a flask sealed with a rubber septum. For each set of titrations, 40 μmol (12.29 mg) of solid glutathione were placed in a 25-ml round-bottom flask, which was sealed with a rubber septum, evacuated, and back-filled with argon. Using a syringe, 10.0 ml of [Cu(I)MCL-3]PF6 stock solution were added followed by the corresponding amount of free MCL-3 supplied from a 100 mm aqueous stock solution. A 3.5-ml aliquot of the resulting working solution was transferred to the cuvette under a blanket of argon. The solution was titrated at 25 °C with 4-μl aliquots of 0.3 m KOH up to a total volume of 400 μl with 10-min equilibration time between aliquots.
For variable pH titrations in the absence of MCL-3, a 4.1 mm glutathione solution was prepared in universal buffer and deoxygenated by bubbling with argon. A 3.5-ml aliquot was transferred by syringe to the cuvette under a blanket of argon and adjusted to approximately pH 4.5 with KOH (28 μl, 1.0 m) before addition of CuSO4 stock solution (5.83 μl, 60 mm). The titration was then carried out as described above.
The proton titration data were analyzed by fitting the cumulative UV absorption traces to a single equilibrium model using the global non-linear least-squares algorithm implemented in BeerOz (26). The data set included a total of 18 independent titrations with MCL-3 concentrations varying between 0 and 1.7 mm, whereas the initial GSH and Cu(I) concentrations were kept constant at 4 mm and 100 μm, respectively. Specifically, duplicate spectrophotometric pH titrations were carried out at total MCL-3 concentrations of 0, 100, 120, 150, 200, 300, 500, 1000, and 1700 μm. For each titration, a total of 50 absorption traces were acquired over the range of pH 4.5 to 7.5. For the global fitting procedure, spectral data between 285 and 380 nm were utilized, corresponding to a total of 86,400 independent data points. To account for proton activity, the protonation constant of GSH (pKa = 8.64) (20) was corrected upward by 0.11 pH units, whereas all other species were expressed in terms of their concentrations (17). Species distributions and free Cu(I) concentrations were calculated based on the fitted stability constants with the program HySS (30).
Author contributions
All authors contributed to the design of the experiments and data interpretation; experiments were performed by M. T. M. (Figs. 1, 2, and 5B, Fig. S2), L. A. H. N. (Fig. 4 and Fig. S3), and H. L. H. (Fig. 1A). The manuscript was written by C. J. F. and M. T. M. All authors discussed the results and commented on the manuscript.
Supplementary Material
This work was supported by National Institutes of Health Grant GM067169. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1-S3, Tables S1 and S2, and a derivation of Equation 4.
- GSH
- glutathione
- aq
- aqueous
- DEPP
- 1,4-diethylpiperazine
- PIPPS
- piperazine-N,N′-bis(3-propanesulfonic acid).
References
- 1. Menon S. G., and Goswami P. C. (2007) A redox cycle within the cell cycle: ring in the old with the new. Oncogene 26, 1101–1109 [DOI] [PubMed] [Google Scholar]
- 2. Cotgreave I. A., and Gerdes R. G. (1998) Recent trends in glutathione biochemistry-glutathione-protein interactions: a molecular link between oxidative stress and cell proliferation? Biochem. Biophys. Res. Commun. 242, 1–9 [DOI] [PubMed] [Google Scholar]
- 3. Meyer A. J., and Hell R. (2005) Glutathione homeostasis and redox-regulation by sulfhydryl groups. Photosynth. Res. 86, 435–457 [DOI] [PubMed] [Google Scholar]
- 4. Pastore A., Federici G., Bertini E., and Piemonte F. (2003) Analysis of glutathione: implication in redox and detoxification. Clin. Chim. Acta 333, 19–39 [DOI] [PubMed] [Google Scholar]
- 5. Coppola S., and Ghibelli L. (2000) GSH extrusion and the mitochondrial pathway of apoptotic signalling. Biochem. Soc. Trans. 28, 56–61 [DOI] [PubMed] [Google Scholar]
- 6. Schafer F. Q., and Buettner G. R. (2001) Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 30, 1191–1212 [DOI] [PubMed] [Google Scholar]
- 7. Hatori Y., and Lutsenko S. (2016) The role of copper chaperone Atox1 in coupling redox homeostasis to intracellular copper distribution. Antioxidants 5, E25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maryon E. B., Molloy S. A., and Kaplan J. H. (2013) Cellular glutathione plays a key role in copper uptake mediated by human copper transporter 1. Am. J. Physiol. Cell Physiol. 304, C768–C779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Speisky H., Gómez M., Carrasco-Pozo C., Pastene E., Lopez-Alarcón C., and Olea-Azar C. (2008) Cu(I)-glutathione complex: a potential source of superoxide radicals generation. Bioorg. Med. Chem. 16, 6568–6574 [DOI] [PubMed] [Google Scholar]
- 10. Österberg R., Ligaarden R., and Persson D. (1979) Copper(I) complexes of penicillamine and glutathione. J. Inorg. Biochem. 10, 341–355 [DOI] [PubMed] [Google Scholar]
- 11. Königsberger L. C., Königsberger E., Hefter G., and May P. M. (2015) Formation constants of copper(I) complexes with cysteine, penicillamine and glutathione: implications for copper speciation in the human eye. Dalton Trans. 44, 20413–20425 [DOI] [PubMed] [Google Scholar]
- 12. Walsh M. J., and Ahner B. A. (2013) Determination of stability constants of Cu(I), Cd(II) and Zn(II) complexes with thiols using fluorescent probes. J. Inorg. Biochem. 128, 112–123 [DOI] [PubMed] [Google Scholar]
- 13. Banci L., Bertini I., Ciofi-Baffoni S., Kozyreva T., Zovo K., and Palumaa P. (2010) Affinity gradients drive copper to cellular destinations. Nature 465, 645–648 [DOI] [PubMed] [Google Scholar]
- 14. Johnson D. K., Stevenson M. J., Almadidy Z. A., Jenkins S. E., Wilcox D. E., and Grossoehme N. E. (2015) Stabilization of Cu(I) for binding and calorimetric measurements in aqueous solution. Dalton Trans. 44, 16494–16505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Corazza A., Harvey I., and Sadler P. J. (1996) 1H, 13C-NMR and X-ray absorption studies of copper(I) glutathione complexes. Eur. J. Biochem. 236, 697–705 [DOI] [PubMed] [Google Scholar]
- 16. Poger D., Fillaux C., Miras R., Crouzy S., Delangle P., Mintz E., Den Auwer C., and Ferrand M. (2008) Interplay between glutathione, Atx1 and copper: X-ray absorption spectroscopy determination of Cu(I) environment in an Atx1 dimer. J. Biol. Inorg. Chem. 13, 1239–1248 [DOI] [PubMed] [Google Scholar]
- 17. Bagchi P., Morgan M. T., Bacsa J., and Fahrni C. J. (2013) Robust affinity standards for Cu(I) biochemistry. J. Am. Chem. Soc. 135, 18549–18559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Morgan M. T., McCallum A., and Fahrni C. J. (2016) Rational design of a water-soluble, lipid-compatible fluorescent probe for Cu(I) with sub-part-per-trillion sensitivity. Chem. Sci. 7, 1468–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Henkel G., and Krebs B. (2004) Metallothioneins: zinc, cadmium, mercury, and copper thiolates and selenolates mimicking protein active site features-structural aspects and biological implications. Chem. Rev. 104, 801–824 [DOI] [PubMed] [Google Scholar]
- 20. Martell A. E., Smith R. M., and Motekaitis R. J. (2004) NIST Standard Reference Database 46 Version 8.0: NIST Critically Selected Stability Constants of Metal Complexes. National Institute of Standards and Technology, Texas A&M University College Station, TX [Google Scholar]
- 21. Martell A. E., and Smith R. M. (1974) Critical Stability Constants, Plenum Press, New York [Google Scholar]
- 22. Maiti B. K., Pal K., and Sarkar S. (2007) Flexible CuI-thiolate clusters with relevance to metallothioneins. Eur. J. Inorg. Chem. 2007, 5548–5555 [Google Scholar]
- 23. Pountney D. L., Schauwecker I., Zarn J., and Vašák M. (1994) Formation of mammalian Cu8-metallothionein in vitro: evidence for the existence of two Cu(I)4-thiolate clusters. Biochemistry 33, 9699–9705 [DOI] [PubMed] [Google Scholar]
- 24. Roschitzki B., and Vašák M. (2002) A distinct Cu4-thiolate cluster of human metallothionein-3 is located in the N-terminal domain. J. Biol. Inorg. Chem. 7, 611–616 [DOI] [PubMed] [Google Scholar]
- 25. Fujisawa K., Imai S., Kitajima N., and Moro-oka Y. (1998) Preparation, spectroscopic characterization, and molecular structure of copper(I) aliphatic thiolate complexes. Inorg. Chem. 37, 168–169 [Google Scholar]
- 26. Brugger J. (2007) BeerOz, a set of Matlab routines for the quantitative interpretation of spectrophotometric measurements of metal speciation in solution. Comp. Geosci. 33, 248–261 [Google Scholar]
- 27. Dance I. G., Bowmaker G. A., Clark G. R., and Seadon J. K. (1983) The formation and crystal and molecular structures of hexa(μ-organothiolato)tetracuprate(I) cage dianions: bis-(tetramethylammonium)hexa-(μ-methanethiolato)tetracuprate(I) and two polymorphs of bis(tetramethylammonium)hexa-(μ-benzenethiolato)-tetracuprate(I). Polyhedron 2, 1031–1043 [Google Scholar]
- 28. Baumgartner M., Schmalle H., and Baerlocher C. (1993) Synthesis, characterization, and crystal structure of three homoleptic copper(I) thiolates:(Cu(CH3S−))∞, [(C6H5)4P+]2[Cu5(CH3S−)7]·C2H6O2, and [(C3H7)4N+]2[Cu4(CH3S−)6·CH4O. J. Solid State Chem. 107, 63–75 [Google Scholar]
- 29. Dance I. G. (1978) The hepta(μ-benzenethiolato)pentametallate(I) dianions of copper and silver: formation and crystal structures. Aust. J. Chem. 31, 2195–2206 [Google Scholar]
- 30. Alderighi L., Gans P., Ienco A., Peters D., Sabatini A., and Vacca A. (1999) Hyperquad simulation and speciation (HySS): a utility program for the investigation of equilibria involving soluble and partially soluble species. Coord. Chem. Rev. 184, 311–318 [Google Scholar]
- 31. Xiao Z., Brose J., Schimo S., Ackland S. M., La Fontaine S., and Wedd A. G. (2011) Unification of the copper(I) binding affinities of the metallo-chaperones Atx1, Atox1 and related proteins: detection probes and affinity standards. J. Biol. Chem. 286, 11047–11055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Allen S., Badarau A., and Dennison C. (2012) Cu(I) affinities of the domain 1 and 3 sites in the human metallochaperone for Cu,Zn-superoxide dismutase. Biochemistry 51, 1439–1448 [DOI] [PubMed] [Google Scholar]
- 33. Liu T., Ramesh A., Ma Z., Ward S. K., Zhang L., George G. N., Talaat A. M., Sacchettini J. C., and Giedroc D. P. (2007) CsoR is a novel Mycobacterium tuberculosis copper-sensing transcriptional regulator. Nat. Chem. Biol. 3, 60–68 [DOI] [PubMed] [Google Scholar]
- 34. Wegner S. V., Sun F., Hernandez N., and He C. (2011) The tightly regulated copper window in yeast. Chem. Comm. 47, 2571–2573 [DOI] [PubMed] [Google Scholar]
- 35. Benítez J. J., Keller A. M., Huffman D. L., Yatsunyk L. A., Rosenzweig A. C., and Chen P. (2011) Relating dynamic protein interactions of metallochaperones with metal transfer at the single-molecule level. Faraday Discuss. 148, 71–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Benítez J. J., Keller A. M., Ochieng P., Yatsunyk L. A., Huffman D. L., Rosenzweig A. C., and Chen P. (2008) Probing transient copper chaperone-Wilson disease protein interactions at the single-molecule level with nanovesicle trapping. J. Am. Chem. Soc. 130, 2446–2447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Keller A. M., Benítez J. J., Klarin D., Zhong L., Goldfogel M., Yang F., Chen T.-Y., and Chen P. (2012) Dynamic multibody protein interactions suggest versatile pathways for copper trafficking. J. Am. Chem. Soc. 134, 8934–8943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pufahl R. A., Singer C. P., Peariso K. L., Lin S. J., Schmidt P. J., Fahrni C. J., Culotta V. C., Penner-Hahn J. E., and O'Halloran T. V. (1997) Metal ion chaperone function of the soluble Cu(I) receptor Atx1. Science 278, 853–856 [DOI] [PubMed] [Google Scholar]
- 39. Pushkar Y., Robison G., Sullivan B., Fu S. X., Kohne M., Jiang W., Rohr S., Lai B., Marcus M. A., Zakharova T., and Zheng W. (2013) Aging results in copper accumulations in glial fibrillary acidic protein-positive cells in the subventricular zone. Aging Cell 12, 823–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Faller P. (2010) Neuronal growth-inhibitory factor (metallothionein-3): reactivity and structure of metal-thiolate clusters. FEBS J. 277, 2921–2930 [DOI] [PubMed] [Google Scholar]
- 41. Scheller J. S., Irvine G. W., Wong D. L., Hartwig A., and Stillman M. J. (2017) Stepwise copper(I) binding to metallothionein: a mixed cooperative and non-cooperative mechanism for all 20 copper ions. Metallomics 9, 447–462 [DOI] [PubMed] [Google Scholar]
- 42. Öhrvik H., Nose Y., Wood L. K., Kim B.-E., Gleber S.-C., and Ralle M., and Thiele D. J. (2013) Ctr2 regulates biogenesis of a cleaved form of mammalian Ctr1 metal transporter lacking the copper- and cisplatin-binding ecto-domain. Proc. Natl. Acad. Sci. U.S.A. 110, E4279–E4288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vita N., Platsaki S., Baslé A., Allen S. J., Paterson N. G., Crombie A. T., Murrell J. C., Waldron K. J., and Dennison C. (2015) A four-helix bundle stores copper for methane oxidation. Nature 525, 140–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fahrni C. J. (2013) Synthetic fluorescent probes for monovalent copper. Curr. Opin. Chem. Biol. 17, 656–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shen C., and New E. J. (2015) What has fluorescent sensing told us about copper and brain malfunction? Metallomics 7, 56–65 [DOI] [PubMed] [Google Scholar]
- 46. Cotruvo J. A. Jr., Aron A. T., Ramos-Torres K. M., and Chang C. J. (2015) Synthetic fluorescent probes for studying copper in biological systems. Chem. Soc. Rev. 44, 4400–4414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Newton G. L., Arnold K., Price M. S., Sherrill C., Delcardayre S. B., Aharonowitz Y., Cohen G., Davies J., Fahey R. C., and Davis C. (1996) Distribution of thiols in microorganisms: mycothiol is a major thiol in most actinomycetes. J. Bacteriol. 178, 1990–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Newton G. L., Rawat M., La Clair J. J., Jothivasan V. K., Budiarto T., Hamilton C. J., Claiborne A., Helmann J. D., and Fahey R. C. (2009) Bacillithiol is an antioxidant thiol produced in bacilli. Nat. Chem. Biol. 5, 625–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Binstead R. A., and Zuberbühler A. D. (2001) SPECFIT Global Analysis System. Spectrum Software Associates, Marlborough, MA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.