

Abstract
Atypical fibroxanthomas (AFX) and pleomorphic dermal sarcomas (PDS) are frequent cutaneous sarcomas typically arising on sun-exposed skin in elderly patients. In contrast to AFX, which generally do not recur after complete excision, PDS locally recur in up to 50% and metastasize in up to 20%.
We recently detected characteristic UV-induced TP53 mutations as potential driver mutation in almost all PDS investigated as well as activating PIK3CA and RAS gene mutations in around one third of our tumors representing targets for personalized treatments in patients with unresectable or metastasized PDS.
In the present study, we identified amplifications and deletions in a small part of the PDS (6 of 27 cases) but not in AFX suggesting that copy number variations (CNV) might not be an initial event in tumor development but rather important during tumor progression. In addition to BRAF, KNSTRN, IDH1 and PDGFRA amplification, CNV analyses revealed deletions in the CDKN2A, KIT and PDGFRA genes. In cases where an appropriate FISH assay was established, the CNV results could be verified by FISH analysis.
Amplification of BRAF, KIT or PDGFRA and/or losses of CDKN2A might represent bad prognostic markers, although larger studies are needed to clarify their association with prognosis or progression in PDS.
Keywords: atypical fibroxanthoma, BRAF, copy number variation, pleomorphic dermal sarcoma
INTRODUCTION
Atypical fibroxanthomas (AFX) and pleomorphic dermal sarcomas (PDS) represent frequent subtypes of cutaneous sarcomas typically arising on sun-exposed skin of the head and neck in elderly patients. AFX generally do not recur after complete excision whereas PDS have a high potential for local recurrence, metastasis and disease-specific death in case of unresectable tumors [1, 2].
Using next generation sequencing (NGS)-based mutation analyses, we recently detected characteristic UV-induced TP53 mutations as potential driver mutation in almost all our PDS investigated. Besides, we detected CCND1/CDK4 alterations, PIK3CA and different RAS gene mutations as well as an ALK translocation as additional underlying genetic alterations representing targets for personalized treatments in patients with unresectable or metastasized PDS [2].
In addition to somatic mutations, copy number variations (CNV) by gain of specific chromosomal segments containing relevant oncogenes or loss of chromosomal segments harboring critical tumor suppressor genes have been shown to be highly characteristic of other UV-induced skin tumors such as malignant melanoma [3], cutaneous squamous cell carcinoma (SCC) [4] and basal cell carcinoma (BCC) [5]. The aim of the present study was to analyze CNV in a large sample cohort of AFX and PDS to get further insights into their evolutionary process as well as to detect additional diagnostic or therapeutic target structures.
RESULTS
Detection of mutations by next generation sequencing
The results of all NGS analyses are summarized in Table 1 and are partly published by Helbig et al. [2].
Table 1. Results of the NGS, CNV and FISH analyses.
| Case no. | NGS result | CNV gain | CNV loss | FISH result |
|---|---|---|---|---|
| A1 | TP53: c.658T>C p.Y220H | none | none | |
| A2 | KRAS: c.35G>A p.G12D | none | none | |
| A51 |
BRAF: c.1799T>A p.V600E; PIK3CA: c.1624G>A p.E542K; TP53: c.672+1G>C; TP53: c.818G>A p.R273H |
none | none | |
| P1 |
KNSTRN: c.28G>A p.D10N; TP53: c.585_586delinsTT p.R196*; TP53: c.748_749CC>TT p.P250F |
none | none | |
| P2 | TP53: c.[734G>A];[741C>T] p.[G245D];R248W] | none | none | |
| P3 |
PIK3CA: c.1600A>G p.I534V; TP53: c.464_465CC>TT p.T155I; TP53: c.853G>A p.E285K; TP53: c.783-2A>T |
none | none | |
| P4 |
OXA1L: c.169C>T p.L57F; TP53: c.773A>C p.E258A; TP53: c.808T>A p.F270I |
none | none | |
| P5 |
TP53: c.574C>T p.Q192*; TP53: c.742C>T p.R248W |
BRAF (CN=4.5, p=1.4e-23) | none | BRAF ratio 4.3 |
| P6 | TP53: c.949C>T p.Q317* | none | none |
BRAF ratio 1.2; CDK4 ratio 1.9; KIT ratio 1.8; PDGFRA ratio 1.4 |
| P7 |
TP53: c.530C>T p.P177L; TP53: c.742C>T p.R248W |
none | none | |
| P8 | wildtype | none | none |
BRAF ratio 1.0; CDK4 ratio 0.85; KIT ratio 0.7; PDGFRA ratio 1.0 |
| P9 |
RAC1: c.85C>T p.P29S; TP53: c.380C>T p.S127F; TP53: c.585C>T p.R196* |
KNSTRN (CN=3.7, p=2.7e-14) | none | |
| P10 |
KRAS: c.38G>A p.G13D; TP53: c.406C>T p.Q136*; TP53: c.949C>T p.Q317* |
none | none | |
| P11 |
KNSTRN: c.13G>A p.E5K; PIK3CA: c.1633G>A p.E545K; TP53: c.644G>C p.S215T; TP53: c.730G>A p.G244S; TP53: c.833C>T p.P278L |
none | none |
BRAF ratio 1.2; CDK4 ratio 1.0; KIT ratio 1.9; PDGFRA ratio 1.3 |
| P13 |
HRAS: c.38G>A p.G13D; TP53: c.747G>T p.R249S |
none | none | |
| P14 |
TP53: c.856G>A p.E286K; TP53: c.841G>A p.D281N |
none | none | |
| P15 |
KNSTRN: c.13G>A p.E5K; TP53: c.581T>G p.L194R |
none | CDKN2A (CN=0.5, p=0.037) | |
| P16 |
TP53: c.528C>G p.C176W; TP53: c.580C>T p.L194F |
none | none | |
| P18 |
NRAS: c.181C>A p.Q61K; PIK3CA: c.3145G>A p.G1049S; TP53: c.422G>T p.C141F; TP53: c.655C>T p.P219S; TP53: c.743_744GG>AA p.R248Q |
IDH1 (CN=2.9, p=7.7e-06) | PDGFRA (CN=1, p=0.0031); KIT (CN=0.8, p=3.5e-05) |
KIT ratio 0.6; PDGFRA NA |
| P19 |
RAC1: c.85C>T p.P29S; TP53: c.535C>T p.H179Y; TP53: c.586C>T p.R196* |
none | none | |
| P20 | TP53: c.859G>T p.E287* | none | none | |
| P21 |
PIK3CA: c.1624G>A p.E542K; TP53: c.584_585TC>GT p.I195S; TP53: c.716A>G p.N239S |
none | none | |
| P221 |
PIK3CA: c.1624G>A p.E542K; TP53: c.672+1G>C; TP53: c.818G>A p.R273H |
none | none | |
| P23 |
IDH1: c.379C>T p.P127S; TP53: c.637C>T p.R213* |
none | none | |
| P24 |
TP53: c.585_586CC>TT p.R196*; TP53: c.712T>A p.C238S |
none | PDGFRA (CN=1.4, p=0.0034) | PDGFRA ratio 0.4 |
| P25 |
TP53: c.490A>T p.K164*; TP53: c.586C>T p.R196* |
PDGFRA (CN=4.2, p=7.1e-25) | none | PDGFRA ratio 3.8 |
| P261 |
PIK3CA: c.1624G>A p.E542K; TP53: c.672+1G>C; TP53: c.818G>A p.R273H |
none | none |
1 1 AFX (Case A5) and two recurring PDS samples (Case P22 and P26) came from the same patient at the age of 83, 86 and 87 years.
NA=Not applicable, Cases A3, A4 and P12, P17 were not analyzable due to insufficient tumor material.
Copy number variations detected by ioncopy
One PDS harbored a BRAF amplification (CN=4.5, p=1.4e-23, ndetected=3/5, mean_CN=4.1; see materials and methods for details, case P5) (Figure 1). In addition, a KNSTRN amplification (CN=3.7, p=2.7e-14, ndetected=2/3, mean_CN=3.2, case P9), an IDH1 amplification (CN=2.9, p=7.7e-06, ndetected=1/2, mean_CN=2.5, case P18) and a PDGFRA amplification PDGFRA (CN=4.2, p=7.1e-25, ndetected=4/8, mean_CN=3.6, case P25) was detected.
Figure 1. Copy number variation (CNV) in pleomorphic dermal sarcomas (PDS).
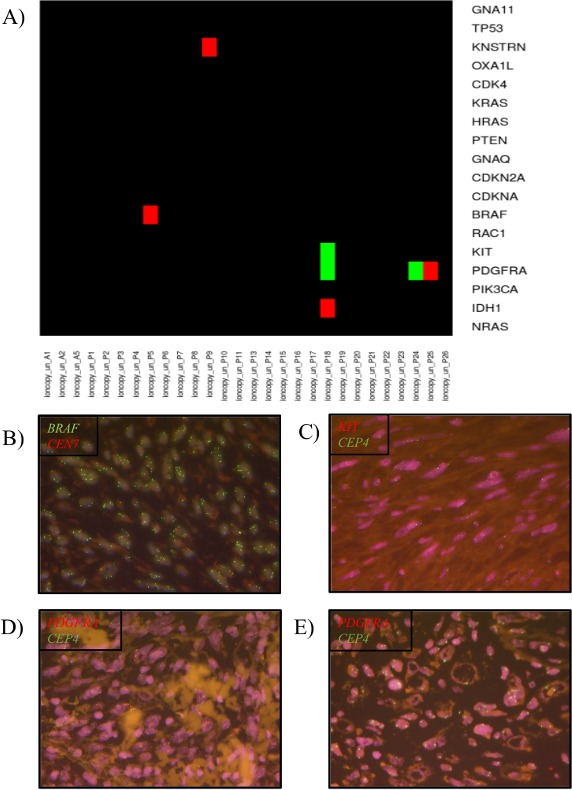
(A) Heatmap diagram of the CNVs in a melanoma specific gene panel detected by the software tool Ioncopy. Red indicates an amplification and green a deletion. (B) Verification of BRAF amplification by FISH analysis. (C) Verification of KIT deletion by FISH analysis. (D) Verification of PDGFRA amplification by FISH analysis. (E) Verification of PDGFRA deletion by FISH analysis.
Deletions could be detected in 2 tumors (Figure 1). Simultaneous PDGFRA (CN=1, p=0.0031, ndetected=4/8, mean_CN=1.5) and KIT (CN=0.8, p=3.5e-05, ndetected=7/14, mean_CN=1.5) deletions were detected in case P18. Single gene copy number deletions of PDGFRA (CN=1.4, p=0.0034, ndetected=5/8, mean_CN=1.4, case P24) was identified in one PDS.
In all other tumors including all AFX and the recurring case (AFX= case A5, PDS= case P22 and P26) no CNV could be detected. For details see Table 1. In the whole cohort no simultaneous mutation and CNV was detected in the same gene. BRAF CNV gain was detected with TP53 mutations, KNSTRN CNV gain with RAC1 and TP53 mutations, IDH1 with NRAS, PIK3CA and TP53 mutations and simultaneous PDGFRA and KIT CNV loss, PDGFRA with TP53 mutations. Single CNV loss in PDGFRA was accompanied with TP53 mutations.
Verification of the copy number variations by fluorescence in-situ hybridization
To verify our Ioncopy results we compared the bioinformatically obtained CNV results with those determined by the current gold standard fluorescent in-situ hybridization (FISH). All amplified or deleted samples were analyzed by FISH if an appropriate FISH assay was established in our institute. To exclude false negative samples FISH was performed exemplarily in three cases.
The BRAF amplification in the PDS case P5 could be confirmed by FISH analysis (Figure 1B). The case showed a ratio of 4.3 and a gene copy number of ≥ 6.0. The PDS with PDGFRA amplification (case P25) was also positive by FISH analysis showing a ratio of 3.8.
Case P18, with a simultaneous PDGFRA and KIT deletion, showed for KIT a ratio of 0.6 in the FISH analysis confirming the deletion detected by Ioncopy (Figure 1C). The PDGFRA FISH could not be analyzed due to low signal intensity even after repetition.
The CNV loss of PDGFRA detected by Ioncopy in case P24 could be confirmed by FISH analysis showing a ratio of 0.4.
The single copy number variations detected in KNSTRN (case P9), IDH1 (case P18) were not verified due to the lack of an appropriate FISH assay.
Four bioinformatically negative cases were also analyzed by FISH for BRAF, CDK4, KIT and PDGFRA copy number variations to exclude false negative samples. The negative results could be confirmed in FISH assays in all samples. Figure 2 shows exemplarily the negative FISH results of case P6. This case showed a ratio of 1.2 for BRAF, one of 1.9 for CDK4, one of 1.8 for KIT and one of 1.4 for PDGFRA FISH analysis.
Figure 2. Verification of negative case P6.
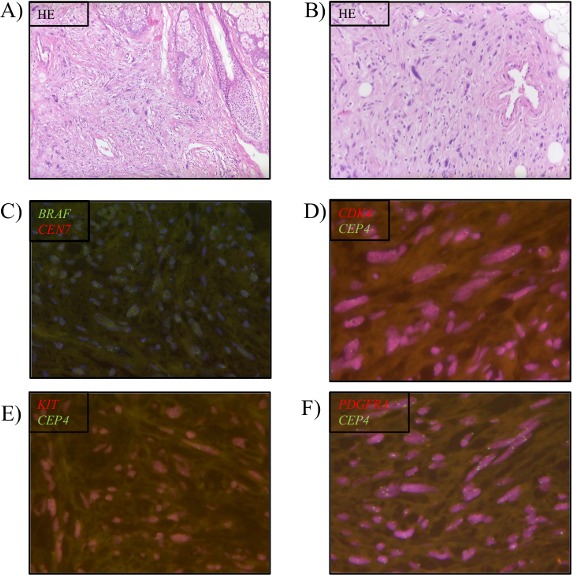
Negative results by Ioncopy were verified by FISH analysis if an appropriate assay was available. (A, B) HE staining, (C) BRAF FISH, (D) CDK4 FISH, (E) KIT FISH, and (F) PDGFRA FISH.
DISCUSSION
In the present study, we used amplicon sequencing data generated on a MiSeq Benchtop sequencer to perform CNV analysis by the software tool Ioncopy. As described by Budczies et al. the detection of amplifications is possible with one amplicon covering the region of interest and without corresponding normal tissue [6]. However, confidences increase when the region of interest is covered by at least two amplicons. Furthermore, it is essential that the raw data is carefully inspected before analysis to exclude bad quality cases. A minimum coverage of 100x for all amplicons is recommended but the reliability increases with the coverage. Ioncopy is a good tool to analyze the CNVs without normal control tissue with a minimum of 20 samples as shown in the present study. All CNVs detected by Ioncopy could be verified by FISH analysis if an appropriate FISH assay was established.
Characteristic CNV have been identified in other UV-induced skin tumors such as malignant melanomas [3], cutaneous SCC [4] and BCC [5]. In only one previous study comparative genomic hybridization (CGH) revealed many shared CNVs in AFX and PDS (most frequently deletions on chromosomes 9p and 13q). Overall, the number of CNVs detected in PDS was higher than in AFX (8.8 ± 1.1 versus 3.3 ± 0.7) [7]. CNVs on single gene level have not been analyzed yet in AFX and PDS.
In the present study, we identified amplifications and deletions in a small part of the PDS suggesting that CNVs might not be an initial event in tumor development but rather are important during tumor progression. The analyzed AFX did not show any CNVs.
The BRAF amplification detected in one PDS may play an essential role in this tumor. The BRAF gene encodes the serine/threonine-protein kinase BRAF, which regulates normal cell growth and proliferation. It is frequently mutated in cancer with highest mutation rates in hairy cell leukemia, malignant melanoma, and papillary thyroid cancer [8–10]. Overall, the most frequent BRAF mutation is V600E which leads to a constitutive activation of the BRAF kinase and its downstream signaling through the mitogen-activated protein kinase (MAPK) pathway [8, 11]. Two BRAF inhibitors (vemurafenib and dabrafenib) and one MEK inhibitor (trametinib) are recently approved for the treatment of metastatic BRAF V600E mutated melanoma [12–18]. Following this, a clinically relevant antitumor activity of these BRAF inhibitors could also be demonstrated in patients with advanced V600E-mutant non–small-cell lung cancer, thyroid cancer, and hairy cell leukemia [19, 20]. Despite a prolonged disease free and patient’s overall survival under those BRAF inhibitors, the combination with a MEK inhibitor is useful to delay acquired drug resistances [21, 22]. BRAF amplifications have been described in various frequencies in melanomas (5.6-66%) and seemed to be associated with decreased progression free survival (PFS), independent of the underlying BRAF mutation [23]. More frequently, BRAF amplifications have been identified as a key regulator for treatment resistance to BRAF and MEK inhibitors, either alone or in combination [23–27]. In colorectal carcinoma, BRAF amplification induced resistance to RAF/EGFR or RAF/MEK combinations through sustained MAPK pathway activity. Interestingly, an ERK inhibitor was able to overcome this resistance [28]. As a BRAF amplification is associated with decreased response to BRAF and or MEK inhibition and poor clinical outcome, this specific genetic alteration may serve as a predictive biomarker. Furthermore, treatment regimens and combinations should be adapted to this specific molecular alteration to prevent resistance.
In addition to this BRAF amplification, our CNV analyses revealed amplifications in PDGFRA as well as deletions in KIT and PDGFRA. KIT (CD117) and PDGFRA (both localized on chromosome 4q) encode for two homologous receptor tyrosine kinases. Oncogenic mutations in KIT occur in 75-80% of gastrointestinal stromal tumors (GIST) and in 20-25% in PDGFRA [29]. GIST with a mutation in either KIT or PDGFRA can be treated with selective small inhibitors such as Imatinib, Sunitinib or Regorafenib [30–32]. KIT copy number variations are described in melanoma, especially in acral (27.3%) and mucosal (26.3%) melanoma [33–35], in glioblastoma multiforme [36] and in intimal sarcoma [37]. They are detected together with BRAF V600E, NRAS or KIT mutations or even alone [38]. In the study of Minor et al., a patient with a KIT amplification and NRAS mutation, as shown in the present study, showed progressive disease under Sunitinib treatment [38]. In the whole cohort, only one of the six detected patients with KIT amplification had a partial response. In the study of Hodi et al., none of the patients with KIT wildtype amplification responded to Imatinib treatment [39]. In a multicenter phase II study with Nilotinib, a novel phenylaminopyrimidine derivate with potent activity against KIT, KIT amplification alone was detected in 35.7% of melanoma and combined with KIT mutations in 4.8%. Seven patients responded to Nilotinib treatment and one out of these seven harbored a KIT amplification only [40]. Therefore, the presence of not only a KIT mutation but also a KIT amplification seems to be a poor prognostic marker [41, 42].
PDGFRA amplifications are also described as a bad prognostic marker in adult IDH1 mutated high-grade glioma [43] and adult anaplastic astrocytoma [44]. In vitro studies suggest Dasatinib as a good therapeutic option for PDGFRA amplified pediatric high-grade glioma [45].
Beside this, we could detect CNV gain in KNSTRN and IDH1. KNSTRN (kinetochore-localized astrin-binding protein) encodes a kinetochore-associated protein that regulates anaphase onset and chromosomal segregation during mitosis. Point mutations in KNSTRN are described in cutaneous squamous cell carcinoma. Here, the hotspot mutation in codon 24 leads to a disrupted sister chromatin cohesion, an increased tumor aneuploidy and tumor growth in vivo [40]. To our knowledge, KNSTRN CNV gain not yet described in the literature so that the clinical significance of this aberration is not known.
IDH1 (Isocitrate dehydrogenase 1) is located on chromosome 2q33.3 and encodes for a protein that catalyzes the reversible oxidative decarboxylation of isocitrate to alpha-ketoglutarate (αKG) while reducing NADP+ to NADPH. Mutations in the IDH1 gene (mainly in codon 132) are described in solid tumors as well as in haematologic malignancies such as glioma, acute myeloid leukemia and chondrosarcoma [46]. In low grade gliomas IDH1 mutations are the earliest genetic alteration whereas higher grade gliomas are associated with IDH1 copy number variations [47]. Therefore, IDH1 copy number variation seems to be a worse prognostic biomarker.
Based on these results and our findings, an amplification of BRAF and/or losses of PDGFRA as well as KIT could represent markers for worse clinical outcome, although larger studies are needed to clarify their association with prognosis or progression in PDS.
As we already suggested earlier, a subset of patients (showing gene mutations in PIK3CA, HRAS, KRAS, NRAS and BRAF or ALK translocation) could benefit from a systemic treatment with a PI3K, MEK, BRAF or ALK inhibitor in the case of unresectable or metastasized tumor stage [2].
MATERIALS AND METHODS
Patient characteristics and tumor material
3 AFX and 24 PDS, initially diagnosed in different dermatopathology centers (Department of Dermatology, University Hospital Cologne, Germany; Institute of Pathology Weger Emberger OG, Salzburg, Austria; Department of Dermatology, University Hospital Würzburg, Germany) were included. Cases A3, A4 and P12, P17 were not analyzable due to insufficient tumor material. One patient developed an AFX (case A5) which recurred twice as PDS (case P22 and P26) 3 and 4 years after the initial diagnosis. All tumors were selected and reevaluated by D.H. and A.Q. according to our definitions and part of the cohort has been already described before [2]. For all patient and tumor details see Table 2.
Table 2. Patient and tumor characteristics.
| PDS (n= 261) | AFX (n=51) | |
|---|---|---|
| Male | 23 | 5 |
| Female | 3 | |
| Age (Years) | ||
| - range | 58-94 | 63-83 |
| - median | 80.5 | 76 |
| - mean | 79 | 76 |
| Tumor localization | ||
| - Capillitium/Face | 25 | 4 |
| - Shoulder | 1 | |
| - Not known | 1 |
1 1 AFX and 2 recurring PDS samples came from the same patient at the age of 83, 86 and 87 years.
Next-generation-sequencing (NGS)
NGS analysis has been described earlier [2].
Copy number variations (CNV)
CNVs were detected with the freely available software tool Ioncopy [6]. This method allows the detection of CNVs based on amplicon sequencing data. The advantage of this method is that no normal controls are needed and that somatic mutations and CNVs (amplifications and deletions) can be detected from the same raw data. Coverage files were loaded into Ioncopy to detect CNVs. They were generated from the same BAM files as for mutational analysis generated by a MiSeq benchtop sequencer (Illumina, [48]). Copy numbers were estimated after sample and amplicon normalization. Significance of CNVs was assessed separately for each amplicon including only amplicons with mean coverage ≥ 100 in the analysis. Similar to Budczies et al. an amplification was called, when detected by a single amplicon and a significance (p<0.05) after Bonferroni correction for the total number of samples and amplicons [6]. A deletion was called, when detected by at least four amplicons (p<0.05 without multiple testing correction for each of them). For each gene called, CN and p-value of the most significant amplicon, the number of significant amplicons compared to the total number of the amplicons interrogating the gene (ndetected) and average CN over all amplicons (mean_CV) were reported.
Quality criteria were a mean coverage ≥ 100 and a significance of copy number variation of p = 0.05. For each CNV the copy number (CN) with significance (p=0.05) is described. Ndetected gives the number of amplicons with a significant amplification compared to the total number of the amplicons covering a gene. Mean_CV describes the mean of copy numbers of the total amplicons of one gene.
Fluorescence in-situ hybridization (FISH)
Four μm thick sections from formalin-fixed and paraffin embedded (FFPE) tissue blocks were mounted on sialinized slides for FISH analysis.
Briefly, pretreatments and washes were performed on the half-automated VP2000 processor system (Abbott Molecular, Wiesbaden, Germany). The slides were then denatured for five minutes at 75°C and incubated with the appropriate probes at 37°C overnight: ZytoLight® Spec BRAF/CEN7 dual color probe (purchased from ZytoVision GmbH, Bremerhaven, Germany) for BRAF copy number variation, SureFISH 4q12 KIT 155 kb, orange-red probe combined with SureFISH Chr4 CEP 613 kb, green probe (both purchased from Agilent Technologies, Cedar Creek, USA) for KIT copy number variations and in-house made PDGFRA or CDK4 probes (Bacterial artificial chromosome clone: RP11-XY labeled with Spectrum Orange or bacterial artificial chromosome clone: RP11-571M6 labeled with Spectrum Orange both combined with a centromere specific Spectrum Green probe) for PDGFRA or CDK4 CNV`s.
Unbound probes were washed away with 2x post-hybridization SSC buffer at 72°C and nuclei were counterstained with DAPI (Sigma-Aldrich GmbH, Taufkirchen, Germany). Fluorescent signals of twenty non-overlapping contiguous tumor cells were analyzed in random areas with a DM5500B fluorescent microscope (Leica, Wetzlar, Germany). The ratio of BRAF/CEN7, KIT/CEN4, PDGFRA/CEN4 or CDK4/CEN12 signals and the average gene copy number were estimated for each sample. Criteria for an amplification were either a ratio ≥ 2.0 or an average gene copy number per cell of ≥ 6.0, criteria for a deletion was a ratio < 0.8.
Acknowledgments
We thank Wiebke Jeske for technical assistance.
Author contributions
D.H., A.Q. and M.A.I designed the study, selected cases, conceived and carried out all experiments, analyzed and interpreted results, generated figures and tables and performed literature research, and writing of the manuscript. M.A.I., J.B. and J.R. conceived CNV and analyzed the CNV data. V.R., K.P. and E.B. performed the FISH analyses and analyzed the results. M.E. and M. W. provided cases. C.M., S.M.B and R.B. were involved in designing the project. All authors were involved in writing the paper and had final approval of the submitted and published version.
CONFLICTS OF INTEREST
There is no conflict of interest. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki as reflected by the approval of the institution’s human research review committee (registration no. 15-307). Informed consent has been obtained.
FUNDING
This work was supported by the Deutsche Forschungsgemeinschaft through the SFB829 (Z4 to D.M. and C.M.) and Köln Fortune (Project Number 191/2016 to D.H. and A.Q.).
REFERENCES
- 1.Tardio JC, Pinedo F, Aramburu JA, Suarez-Massa D, Pampin A, Requena L, Santonja C. Pleomorphic dermal sarcoma: a more aggressive neoplasm than previously estimated. J Cutan Pathol. 2016;43:101–112. doi: 10.1111/cup.12603. [DOI] [PubMed] [Google Scholar]
- 2.Helbig D, Ihle MA, Putz K, Tantcheva-Poor I, Mauch C, Buttner R, Quaas A. Oncogene and therapeutic target analyses in atypical fibroxanthomas and pleomorphic dermal sarcomas. Oncotarget. 2016;7:21763–21774. doi: 10.18632/oncotarget.7845. https://doi.org/10.18632/oncotarget.7845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bastian BC, LeBoit PE, Hamm H, Brocker EB, Pinkel D. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridization. Cancer Res. 1998;58:2170–2175. [PubMed] [Google Scholar]
- 4.Jacobs MS, Persons DL, Fraga GR. EGFR and MYC gene copy number aberrations are more common in squamous cell carcinoma than keratoacanthoma: a FISH study. J Cutan Pathol. 2013;40:447–454. doi: 10.1111/cup.12117. [DOI] [PubMed] [Google Scholar]
- 5.Pesz KA, Bieniek A, Makowska I, Sasiadek MM. Basal cell carcinoma of the skin: whole genome screening by comparative genome hybridization revisited. J Cutan Pathol. 2013;40:25–29. doi: 10.1111/cup.12033. [DOI] [PubMed] [Google Scholar]
- 6.Budczies J, Pfarr N, Stenzinger A, Treue D, Endris V, Ismaeel F, Bangemann N, Blohmer JU, Dietel M, Loibl S, Klauschen F, Weichert W, Denkert C. Ioncopy: a novel method for calling copy number alterations in amplicon sequencing data including significance assessment. Oncotarget. 2016;7:13236–13247. doi: 10.18632/oncotarget.7451. https://doi.org/10.18632/oncotarget.7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mihic-Probst D, Zhao J, Saremaslani P, Baer A, Oehlschlegel C, Paredes B, Komminoth P, Heitz PU. CGH analysis shows genetic similarities and differences in atypical fibroxanthoma and undifferentiated high grade pleomorphic sarcoma. Anticancer Res. 2004;24:19–26. [PubMed] [Google Scholar]
- 8.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 9.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R, Cancer Genome Project Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 10.Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer. 2014;14:455–467. doi: 10.1038/nrc3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Brocker EB, LeBoit PE, Pinkel D, Bastian BC. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 12.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH, Jr, Kaempgen E, Martin-Algarra S, Karaszewska B, Mauch C, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 14.Paik PK, Arcila ME, Fara M, Sima CS, Miller VA, Kris MG, Ladanyi M, Riely GJ. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol. 2011;29:2046–2051. doi: 10.1200/JCO.2010.33.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cardarella S, Ogino A, Nishino M, Butaney M, Shen J, Lydon C, Yeap BY, Sholl LM, Johnson BE, Janne PA. Clinical, pathologic, and biologic features associated with BRAF mutations in non-small cell lung cancer. Clin Cancer Res. 2013;19:4532–4540. doi: 10.1158/1078-0432.CCR-13-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kinno T, Tsuta K, Shiraishi K, Mizukami T, Suzuki M, Yoshida A, Suzuki K, Asamura H, Furuta K, Kohno T, Kushima R. Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann Oncol. 2014;25:138–142. doi: 10.1093/annonc/mdt495. [DOI] [PubMed] [Google Scholar]
- 17.Villaruz LC, Socinski MA, Abberbock S, Berry LD, Johnson BE, Kwiatkowski DJ, Iafrate AJ, Varella-Garcia M, Franklin WA, Camidge DR, Sequist LV, Haura EB, Ladanyi M, et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer. 2015;121:448–456. doi: 10.1002/cncr.29042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Labocha MK, Jung SK, Aleman-Meza B, Liu Z, Zhong W. WormGender - open-source software for automatic caenorhabditis elegans sex ratio measurement. PLoS One. 2015;10:e0139724. doi: 10.1371/journal.pone.0139724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mazieres J, Peters S, Lepage B, Cortot AB, Barlesi F, Beau-Faller M, Besse B, Blons H, Mansuet-Lupo A, Urban T, Moro-Sibilot D, Dansin E, Chouaid C, et al. Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol. 2013;31:1997–2003. doi: 10.1200/JCO.2012.45.6095. [DOI] [PubMed] [Google Scholar]
- 20.Mazieres J, Zalcman G, Crino L, Biondani P, Barlesi F, Filleron T, Dingemans AM, Lena H, Monnet I, Rothschild SI, Cappuzzo F, Besse B, Thiberville L, et al. Crizotinib therapy for advanced lung adenocarcinoma and a ROS1 rearrangement: results from the EUROS1 cohort. J Clin Oncol. 2015;33:992–999. doi: 10.1200/JCO.2014.58.3302. [DOI] [PubMed] [Google Scholar]
- 21.Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, Mandala M, Demidov L, Stroyakovskiy D, Thomas L, de la Cruz-Merino L, Dutriaux C, Garbe C, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371:1867–1876. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- 22.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, Chiarion-Sileni V, Drucis K, Krajsova I, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30–39. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 23.Wilson MA, Zhao F, Khare S, Roszik J, Woodman SE, D'Andrea K, Wubbenhorst B, Rimm DL, Kirkwood JM, Kluger HM, Schuchter LM, Lee SJ, Flaherty KT, Nathanson KL. Copy number changes are associated with response to treatment with carboplatin, paclitaxel, and sorafenib in melanoma. Clin Cancer Res. 2016;22:374–382. doi: 10.1158/1078-0432.CCR-15-1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, Kelley MC, Kefford RF, Chmielowski B, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, Hodis E, Rosenberg M, McKenna A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4:94–109. doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi H, Moriceau G, Kong X, Lee MK, Lee H, Koya RC, Ng C, Chodon T, Scolyer RA, Dahlman KB, Sosman JA, Kefford RF, Long GV, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. doi: 10.1038/ncomms1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Helias-Rodzewicz Z, Funck-Brentano E, Baudoux L, Jung CK, Zimmermann U, Marin C, Clerici T, Le Gall C, Peschaud F, Taly V, Saiag P, Emile JF. Variations of BRAF mutant allele percentage in melanomas. BMC Cancer. 2015;15:497. doi: 10.1186/s12885-015-1515-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahronian LG, Sennott EM, Van Allen EM, Wagle N, Kwak EL, Faris JE, Godfrey JT, Nishimura K, Lynch KD, Mermel CH, Lockerman EL, Kalsy A, Gurski JM, Jr, et al. Clinical acquired resistance to RAF inhibitor combinations in BRAF-mutant colorectal cancer through MAPK pathway alterations. Cancer Discov. 2015;5:358–367. doi: 10.1158/2159-8290.CD-14-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad Tunio G, Matsuzawa Y, Kanakura Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–580. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 30.Frolov A, Chahwan S, Ochs M, Arnoletti JP, Pan ZZ, Favorova O, Fletcher J, von Mehren M, Eisenberg B, Godwin AK. Response markers and the molecular mechanisms of action of Gleevec in gastrointestinal stromal tumors. Mol Cancer Ther. 2003;2:699–709. [PubMed] [Google Scholar]
- 31.Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368:1329–1338. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 32.Nishida T, Doi T, Naito Y. Tyrosine kinase inhibitors in the treatment of unresectable or metastatic gastrointestinal stromal tumors. Expert Opin Pharmacother. 2014;15:1979–1989. doi: 10.1517/14656566.2014.937707. [DOI] [PubMed] [Google Scholar]
- 33.Beadling C, Jacobson-Dunlop E, Hodi FS, Le C, Warrick A, Patterson J, Town A, Harlow A, Cruz F, 3rd, Azar S, Rubin BP, Muller S, West R, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. 2008;14:6821–6828. doi: 10.1158/1078-0432.CCR-08-0575. [DOI] [PubMed] [Google Scholar]
- 34.Antonescu CR, Busam KJ, Francone TD, Wong GC, Guo T, Agaram NP, Besmer P, Jungbluth A, Gimbel M, Chen CT, Veach D, Clarkson BD, Paty PB, Weiser MR. L576P KIT mutation in anal melanomas correlates with KIT protein expression and is sensitive to specific kinase inhibition. Int J Cancer. 2007;121:257–264. doi: 10.1002/ijc.22681. [DOI] [PubMed] [Google Scholar]
- 35.Willmore-Payne C, Holden JA, Tripp S, Layfield LJ. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol. 2005;36:486–493. doi: 10.1016/j.humpath.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 36.Kumar A, Boyle EA, Tokita M, Mikheev AM, Sanger MC, Girard E, Silber JR, Gonzalez-Cuyar LF, Hiatt JB, Adey A, Lee C, Kitzman JO, Born DE, et al. Deep sequencing of multiple regions of glial tumors reveals spatial heterogeneity for mutations in clinically relevant genes. Genome Biol. 2014;15:530. doi: 10.1186/s13059-014-0530-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tajima S, Takanashi Y, Takahashi T, Neyatani H. Intimal sarcoma of the abdominal aorta with platelet-derived growth factor receptor alpha overexpression and amplification in mural invasive cells and pulmonary metastatic cells but not in intimal spreading cells. Pathol Int. 2015;65:426–431. doi: 10.1111/pin.12308. [DOI] [PubMed] [Google Scholar]
- 38.Minor DR, Kashani-Sabet M, Garrido M, O'Day SJ, Hamid O, Bastian BC. Sunitinib therapy for melanoma patients with KIT mutations. Clin Cancer Res. 2012;18:1457–1463. doi: 10.1158/1078-0432.CCR-11-1987. [DOI] [PubMed] [Google Scholar]
- 39.Hodi FS, Corless CL, Giobbie-Hurder A, Fletcher JA, Zhu M, Marino-Enriquez A, Friedlander P, Gonzalez R, Weber JS, Gajewski TF, O'Day SJ, Kim KB, Lawrence D, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol. 2013;31:3182–3190. doi: 10.1200/JCO.2012.47.7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee SJ, Kim TM, Kim YJ, Jang KT, Lee HJ, Lee SN, Ahn MS, Hwang IG, Lee S, Lee MH, Lee J. Phase II trial of nilotinib in patients with metastatic malignant melanoma harboring KIT gene aberration: a multicenter trial of Korean Cancer Study Group (UN10-06) Oncologist. 2015;20:1312–1319. doi: 10.1634/theoncologist.2015-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kong Y, Si L, Zhu Y, Xu X, Corless CL, Flaherty KT, Li L, Li H, Sheng X, Cui C, Chi Z, Li S, Han M, et al. Large-scale analysis of KIT aberrations in Chinese patients with melanoma. Clin Cancer Res. 2011;17:1684–1691. doi: 10.1158/1078-0432.CCR-10-2346. [DOI] [PubMed] [Google Scholar]
- 42.Bastian BC, Esteve-Puig R. Targeting activated KIT signaling for melanoma therapy. J Clin Oncol. 2013;31:3288–3290. doi: 10.1200/JCO.2013.50.3227. [DOI] [PubMed] [Google Scholar]
- 43.Phillips JJ, Aranda D, Ellison DW, Judkins AR, Croul SE, Brat DJ, Ligon KL, Horbinski C, Venneti S, Zadeh G, Santi M, Zhou S, Appin CL, et al. PDGFRA amplification is common in pediatric and adult high-grade astrocytomas and identifies a poor prognostic group in IDH1 mutant glioblastoma. Brain Pathol. 2013;23:565–573. doi: 10.1111/bpa.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alentorn A, Marie Y, Carpentier C, Boisselier B, Giry M, Labussiere M, Mokhtari K, Hoang-Xuan K, Sanson M, Delattre JY, Idbaih A. Prevalence, clinico-pathological value, and co-occurrence of PDGFRA abnormalities in diffuse gliomas. Neuro Oncol. 2012;14:1393–1403. doi: 10.1093/neuonc/nos217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koschmann C, Zamler D, MacKay A, Robinson D, Wu YM, Doherty R, Marini B, Tran D, Garton H, Muraszko K, Robertson P, Leonard M, Zhao L, et al. Characterizing and targeting PDGFRA alterations in pediatric high-grade glioma. Oncotarget. 2016;7:65696–65706. doi: 10.18632/oncotarget.11602. https://doi.org/10.18632/oncotarget.11602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oki K, Takita J, Hiwatari M, Nishimura R, Sanada M, Okubo J, Adachi M, Sotomatsu M, Kikuchi A, Igarashi T, Hayashi Y, Ogawa S. IDH1 and IDH2 mutations are rare in pediatric myeloid malignancies. Leukemia. 2011;25:382–384. doi: 10.1038/leu.2010.307. [DOI] [PubMed] [Google Scholar]
- 47.Mazor T, Chesnelong C, Pankov A, Jalbert LE, Hong C, Hayes J, Smirnov IV, Marshall R, Souza CF, Shen Y, Viswanath P, Noushmehr H, Ronen SM, et al. Clonal expansion and epigenetic reprogramming following deletion or amplification of mutant IDH1. Proc Natl Acad Sci U S A. 2017;114:10743–10748. doi: 10.1073/pnas.1708914114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Konig K, Peifer M, Fassunke J, Ihle MA, Kunstlinger H, Heydt C, Stamm K, Ueckeroth F, Vollbrecht C, Bos M, Gardizi M, Scheffler M, Nogova L, et al. Implementation of amplicon parallel sequencing leads to improvement of diagnosis and therapy of lung cancer patients. J Thorac Oncol. 2015;10:1049–1057. doi: 10.1097/JTO.0000000000000570. [DOI] [PubMed] [Google Scholar]