

Abstract
This phase I study evaluates the safety, maximum tolerated dose (MTD), pharmacokinetics (PK), pharmacodynamics, and preliminary anticancer activity of enavatuzumab, a humanized IgG1 antibody to the TWEAK receptor, in patients with advanced solid malignancies. Patients received escalating doses of enavatuzumab given intravenously over 60 minutes every 2 weeks. Blood was obtained for PK and biomarker assessment. Three patients were enrolled per dose level in a standard 3+3 design with response assessment by RECIST version 1.0, every 8 weeks. Thirty patients were enrolled at 6 dose levels ranging from 0.1 to 1.5 mg/kg. Dose limiting toxicities (DLT) included grade 4 (G4) lipase, G3 bilirubin, and G4 amylase elevations. There was no apparent correlation of liver or pancreatic enzyme elevation with drug exposure or the presence of liver metastases. Enavatuzumab exhibited a two-compartment linear PK model. Estimated systemic clearance was 23–33 mL/h with an elimination half-life of 7–18 days. The predicted target efficacious peak and trough concentrations occurred at 1.0 mg/kg following the second dose. There were no objective responses; 4 patients had stable disease. The maximum tolerated dose of enavatuzumab is 1.0 mg/kg IV every 2 weeks. Higher doses were not tolerated due to hepatopancreatic lab abnormalities. Further evaluation of the mechanisms of the liver and pancreatic enzyme toxicities are needed before embarking on further single agent or combination strategies.
Keywords: Enavatuzumab, PDL192, ABT-361, phase 1, Tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK), TweakR, advanced solid cancer
INTRODUCTION
Tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK, aka TNFSF12, APO3L, or CD255) and its receptor TweakR (Fn14, TNFRSF12A, CD266) are members of the TNF and TNF receptor superfamilies, respectively (1). TWEAK is a multifunctional cytokine that affects cell proliferation, migration and survival, cytokine induction, cytotoxicity and apoptosis (2). In situations of acute injury, TWEAK and TweakR normally interact to coordinate inflammatory and progenitor cell responses in order to facilitate acute tissue repair. However, in chronic inflammatory disease, this ligand-receptor pair is persistently activated and pathologically amplifies inflammation and promotes tissue damage (3). TweakR is overexpressed in numerous cancers, including melanoma, breast, brain, non-small cell lung, pancreas, esophageal, colorectal, renal, ovarian, and prostate cancers (4–7). The pro-tumorigenic effects of TWEAK are postulated to occur via multiple mechanisms: protecting cancer cells from apoptosis by inducing Bcl-2, promoting migration and invasion, inducing cell proliferation, promoting angiogenesis of tumor vasculature, and repressing tumor surveillance (3). TWEAK also has anti-cancer properties, including the induction of apoptosis and/or growth inhibition of cancer cells through activation of the non-canonical NFκB pathway, which underscores the pleiotropic nature of this signalling pathway (8–10).
Enavatuzumab (PDL192, ABT-361) is a first-in-class humanized IgG1 recombinant monoclonal antibody to TweakR. It is a moderate agonist of TweakR that mediates anti-tumor activity in preclinical models both by signalling through TweakR and by inducing antibody-dependent cellular cytotoxicity (ADCC) (4). Enavatuzumab and its murine parental antibody, 19.2.1, exert some of the biological functions of TWEAK by binding to TweakR. However, enavatuzumab significantly differs from TWEAK in that it exhibits weaker effects on the stimulation of cytokine expression and has no pro-angiogenic activity. Enavatuzumab and 19.2.1 both have been shown to inhibit growth of tumor cell lines in vitro and of TweakR-expressing xenograft models representing a range of solid tumor types (4,11). In some xenograft models, antibody-dependent cell-mediated cytotoxicity (ADCC) appears to play a major role in the anti-tumor activity, while in other models the direct signalling function of enavatuzumab appears to dominate (4).
In preclinical toxicity studies in cynomolgus monkeys, administration of enavatuzumab at doses up to 100 mg/kg (every other week for 13 weeks or weekly doses for 1 month) was generally well tolerated. The toxicity observed was generally reversible and observed at the high range of doses tested, with the primary targets of toxicity being the kidney (tubular degeneration/regeneration), liver (bile duct hyperplasia), and pancreas (fibrotic replacement of acini, mononuclear cell infiltration, decreased zymogen granules in acinar cells). Based on a no-observed-adverse-effect-level (NOAEL) of < 3 mg/kg observed in the good laboratory practice (GLP) 13-week toxicity study in cynomolgus monkeys, a 0.1 mg/kg clinical starting dose was selected for this phase 1 study.
This was a first-in-class, phase 1, multi-center study designed to evaluate the safety, pharmacokinetics, pharmacodynamics, and preliminary effectiveness of enavatuzumab in patients with advanced solid malignancies.
MATERIALS AND METHODS
Patients
Eligible patients had advanced solid tumors refractory to standard therapies, age ≥ 18 years, Eastern Cooperative Oncology Group (ECOG) performance status 0–1, adequate hematologic function (hemoglobin ≥ 9 g/dL, absolute neutrophil count ≥ 1500/mm3, platelet count ≥ 100,000/mm3), and adequate kidney, liver, and pancreatic function [serum creatinine ≤ 1.5 × upper limit of normal (ULN), aspartate aminotransferase (AST) < 2.5 × ULN, alanine aminotransferase (ALT) < 2.5 × ULN, bilirubin ≤ ULN, amylase < 1.5 × ULN, lipase < 1.5 × ULN]. Exclusion criteria included symptomatic or progressive central nervous system metastases or leptomeningeal disease, diagnosis of glioblastoma, known chronic viral hepatitis, history of cirrhotic liver disease, history of pancreatitis, acute cholecystitis within 6 months of study drug dosing, proteinuria > 1 g/24 hours, ongoing ≥ grade 2 toxicities (according to the National Cancer Institute Common Toxicity Criteria version 3.0, NCI-CTC v3.0) (12) from prior therapies, systemic steroid therapy > 10 mg/day of prednisone or its equivalent, immunosuppressive medications, pregnancy or breastfeeding, and any uncontrolled medical problems. Concomitant anti-cancer therapy was not allowed. The institutional review boards at both participating institutions approved the study and written informed consent was obtained from each patient prior to study entry.
Study Design
The primary objective of the study was to determine the maximum tolerated dose (MTD) of enavatuzumab, defined as the highest dose level with 0/3 or ≤ 1/6 patients experiencing a dose-limiting toxicity (DLT) during the first treatment cycle. Secondary objectives were to evaluate the safety, pharmacokinetic profiles, and anti-tumor activity of enavatuzumab. Exploratory objectives were to explore the relationship between pharmacodynamic markers (biomarkers) and pharmacokinetic profile, clinical response, and toxicity.
Enavatuzumab was administered as an intravenous (IV) infusion over 60 minutes every 2 weeks, on days 1 and 15 of each 4-week treatment cycle. The starting dose was 0.1 mg/kg with planned escalation dose levels of 0.3, 0.7, 1.5, 3.0, 5.0, and 7.5 mg/kg. Three patients were enrolled per dose level in a standard 3+3 design. The first patient in each cohort was observed for at least 7 days prior to initiation of treatment for subsequent patients in that cohort. The DLT window was one treatment cycle (28 days). If none of 3 patients in a cohort experienced DLT in the first treatment cycle, then the subsequent cohort was to receive the next highest dose level of enavatuzumab. If 1 of 3 patients experienced a DLT, the cohort was expanded to evaluate 6 patients. If 2 or more patients experienced a DLT, then dose escalation was to stop. The MTD cohort was to be expanded to 6 patients unless previously enrolled.
Additionally, upon review of cohort safety data from the first treatment cycle, there were options to de-escalate to an intermediate dose level and to re-evaluate a dose level by incorporating steroid premedication in additional patients if 2 or more patients experienced pancreatic or hepatobiliary DLTs, or further evaluate the dose level below the one at which DLT occurred for additional safety evaluation. If a dose level was re-evaluated using steroid premedication, a new cohort of patients was enrolled. Dose escalation using premedication could resume if 0/3 or ≤ 1/6 patients experienced a DLT during the first treatment cycle and all subsequent patients would also receive premedication before every dose of enavatuzumab.
During the course of this study, the DLT language was revised to provide clarity and enable accurate assessments of DLT. The final DLT definition included any of the following drug-related toxicities occurring during the first cycle of treatment: any ≥ grade 3 hematologic toxicity, grade 3 AST/ALT elevation persisting for > 14 days, grade 3 AST/ALT elevations associated with clinical hepatitis, any grade 4 AST/ALT elevation, ≥ grade 2 bilirubin elevation with concomitant ≥ grade 3 AST/ALT elevation, ≥ grade 3 bilirubin elevation, any other ≥ grade 3 hepatic toxicity [excluding isolated ≥ grade 3 gamma-glutamyltransferase (GGT) abnormalities] according to Hy’s law (13), grade 3 lipase or amylase lasting > 14 days, grade 4 lipase or amylase, acute pancreatitis meeting Banks and Freeman criteria (≥ 2 of the following: symptoms of abdominal pain consistent with acute pancreatitis, lipase or amylase ≥ 3 times ULN, and characteristic findings of acute pancreatitis on CT scan) (14), any ≥ grade 3 gastrointestinal toxicity, grade 4 cytokine release syndrome (CRS) or infusion reaction, CRS or reactions requiring treatment with epinephrine or other vasopressors, recurrent grade 3 CRS or infusion reaction occurring despite premedication, grade 3 CRS requiring hospitalization or persisting >4 hours, and any other ≥ grade 3 toxicity.
Drug administration
PDL192 was supplied in sterile, 10-mL, single-use vials containing 10 mg/mL PDL192, 20 mM citrate, 120 mM sodium chloride, and 0.01% polysorbate 80, pH 6.0. Pre-medications for infusion reactions and prophylactic anti-emetics or growth factors were not permitted prior to the first study drug infusion.
Patient evaluation
All patients underwent scheduled safety assessments including physical examinations, vital signs, and chemistry and hematology laboratory evaluations. Toxicities were graded according to NCI-CTC v3.0 (12). Patients were evaluable for DLT if they completed the DLT observation period (28 days) or experienced a DLT prior to completing the first cycle of 2 doses. Patients were evaluated for safety if they received at least one dose of enavatuzumab. Anti-tumor activity was evaluated according to Response Evaluation Criteria in Solid Tumors (RECIST 1.0) (15) using computed tomography/magnetic resonance imaging (CT/MRI) at 8-week intervals throughout the study. Patients with stable disease (SD) or better at the completion of 8 weeks (2 cycles) of treatment were eligible to continue treatment with enavatuzumab. Patients were followed, when feasible, for 90 days after the last dose of study drug.
Blood sampling and pharmacokinetic analysis
Samples were collected for pharmacokinetics (PK) at pre-specified time intervals during treatment (pre-treatment, then 1, 5, and 23 hours post-infusion, days 8 and 15 during cycle 1; days 1 and 15 of subsequent cycles and at follow-up (days 30, 45, 60 and 90 after study completion). Serum concentrations of enavatuzumab were assessed by a validated enzyme-linked immunosorbent assay (ELISA) method and used to calculate the values of PK parameters, including maximum serum concentration (Cmax), time of Cmax (Tmax), the area under the concentration-time curve (AUC), systemic clearance (CL), and elimination half-life (t1/2).
Pharmacodynamics measurements and analysis
Pharmacodynamic sampling was obtained at pre-specified time points (same as those for PK analysis) to determine the potential relationship between exposure and response. Serum samples were evaluated by Luminex® for levels of MCP-1, IP-10, IL-1α, IL-8, bFGF, fractalkine (CX3CL1), GM-CSF, IFNγ, IL-6, MIP-1α, TNFα, and VEGF. For each analyte, the measured concentration at each post-treatment time point was divided by the concentration measured in the pre-treatment sample. The largest-fold increase from baseline is reported. Peripheral blood was evaluated for absolute levels of T-, B-, and NK-cell counts and analysis of NK cell activation.
Statistical considerations
Descriptive statistics were used for baseline characteristics, safety assessment, pharmacokinetic, and pharmacodynamic data. The best overall response (complete response [CR], partial response [PR], SD, or progressive disease [PD]) was summarized by cohort and pooled across cohorts. The objective response rate (CR + PR) and disease control rate (CR + PR + SD) were calculated for all evaluable patients.
RESULTS
Patient population
Between July 2008 and October 2011, 30 patients were enrolled and treated at 2 centers. Baseline patient characteristics are depicted in Table 1. Overall, the mean age of the study participants was 64.5 years, and 60% of the patients were female. Most patients (77%) had received ≥ 3 prior chemotherapy regimens, with a range of 1–10 for prior chemotherapy regimens. Thirteen percent of patients had received prior immunotherapy and 37% of patients had prior surgery. The majority of patients had gastrointestinal or hepatobiliary cancers (15 patients had colorectal cancer, 4 had pancreas cancer, and 1 patient had hepatocellular carcinoma. Fifty-three percent of all patients had liver metastases.
Table 1.
Patient Characteristics
| Characteristic | No. (%) |
|---|---|
| Total no. of patients | 30 |
| Male | 12 (40) |
| Female | 18 (60) |
| Age (years) | |
| Median | 64.5 |
| Range | 36–82 |
| ECOG performance status | |
| 0 | 5 (17) |
| 1 | 25 (83) |
| Time since diagnosis (years) | |
| Median | 2.93 |
| Range | 0.6–7.3 |
| Prior chemotherapy regimens | |
| 1 | 0 |
| 2 | 7 (23) |
| ≥3 | 23 (77) |
| Median | 3 |
| Range | 1–10 |
| Prior immunotherapy | 4 (13) |
| Prior radiotherapy | 11 (37) |
| Prior surgery | 30 (100) |
| Tumor type | |
| Colorectal | 15 (50) |
| Pancreas | 4 (13) |
| Ovarian | 4 (13) |
| Head and Neck (tongue) | 1 (3) |
| Breast | 1 (3) |
| Prostate | 1 (3) |
| Cervical | 1 (3) |
| Endometrial | 1 (3) |
| Hepatocellular | 1 (3) |
| Thyroid | 1 (3) |
| Liver metastases | 16 (53) |
Treatment
Patients were enrolled at 6 dose levels: 0.1 (n=7), 0.3 (n=7), 0.5 (n=5), 0.7 (n=3), 1.0 (n=7), and 1.5 mg/kg (n=1). The median number of treatment cycles was 2 (range 1–6), and the median number of infusions was 3 (range 1–11). Only two patients were treated beyond cycle 2: one patient received six cycles at the 0.1 mg/kg dose and a second patient received 4 cycles at the 0.5mg/kg dose level. Reasons for discontinuation included disease progression (n=20), adverse event (n=7), investigator decision, (n=1), patient decision (n=1), and delay in study drug administration greater than 2 weeks (n=1).
Safety and dose-limiting toxicities
Two patients experienced DLTs based on the final DLT definition. One patient at the 1.0-mg/kg dose level developed grade 3 bilirubin and grade 4 lipase elevations. One patient at the 1.5- mg/kg dose level had grade 4 lipase and amylase increases, along with ALT and AST values > 3 × ULN and total bilirubin values > 2 × ULN, meeting Hy’s Law criteria for drug-induced liver injury. While there was intolerable toxicity at the 1.5-mg/kg dose level, only 1 of 7 patients experienced a DLT at the intermediate 1.0-mg/kg dose level, leading to declaration of the MTD at 1.0 mg/kg.
The most common adverse events (in ≥10% patients) were fatigue (47%), nausea (37%) and vomiting (33%). Grade 3 or higher adverse events in ≥5% of patients included neutropenia, abdominal pain, fatigue, pneumonia, hyponatremia, hypoxia, dyspnea, and elevated AST, ALT, GGT, lipase, and amylase. Table 2 summarizes treatment-related adverse events (occurring in ≥ 2% of patients overall) by dose level. Thirteen patients (43%) experienced an adverse event grade ≥ 3 related to enavatuzumab; the most frequent were elevations in lipase (n = 7, 23%), blood amylase (n = 5, 17%), GGT (n = 5, 17%), ALT (n = 4, 13%), and AST (n = 4, 13%). All cases of grade 3 or higher hepatic or pancreatic toxicities occurred during cycle 1 of treatment. In most cases, the hepatopancreatic lab abnormalities decreased to a lower grade or resolved with discontinuing of study treatment. There was no apparent correlation of liver or pancreatic enzyme elevation with tumor type or presence of liver metastases. Five patients died during the study due to disease progression. The date of death ranged from 1.2 to 2.6 months after the last dose of study drug. Seven patients (23%) discontinued treatment due to an adverse event. Four of these patients experienced adverse events that were related to study drug. Two of these 4 patients were discontinued due to grade 4 lipase elevations that met DLT criteria (described above), but did not meet Banks’ clinical criteria for acute pancreatitis. Adverse events of particular interest for this study were defined as allergic/infusion-type reactions, clinical signs/symptoms of pancreatic toxicity, and clinical signs/symptoms of liver toxicity. Twelve patients experienced at least one of these adverse events. In this study, one patient received premedication with dexamethasone 10 mg intravenously during cycle 2 of treatment; however, this did not avert the development of hepatic or pancreatic toxicity.
Table 2.
Treatment-Related Adverse Events (In ≥ 2% Patients Overall) by Dose Level of Enavatuzumab (n [%])
| Cohort 1 | Cohort 2 | Cohort 3 | Cohort 4 | Cohort 5 | Cohort 6 | Total | |
|---|---|---|---|---|---|---|---|
| 0.1 mg/kg | 0.3 mg/kg | 0.5 mg/kg | 0.7 mg/kg | 1.0 mg/kg | 1.5 mg/kg | ||
| n = 7 | n = 7 | n = 5 | n = 3 | n = 7 | n = 1 | N = 30 | |
| Gastrointestinal disorders | |||||||
| Nausea | 1 (14) | 0 | 1 (20) | 0 | 1 (14) | 0 | 3 (10) |
| Vomiting | 1 (14) | 1 (14) | 0 | 0 | 0 | 0 | 2 (7) |
| General disorders and administration site conditions | |||||||
| Fever | 2 (29) | 2 (29) | 0 | 1 (33) | 2 (29) | 0 | 7 (23) |
| Chills | 2 (29) | 0 | 0 | 0 | 2 (29) | 0 | 4 (13) |
| Fatigue | 2 (29) | 1 (14) | 1 (20) | 0 | 0 | 0 | 4 (13) |
| Investigations | |||||||
| ALT increased | 1 (14) | 0 | 2 (40) | 1 (33) | 3 (43) | 1 (100) | 8 (27) |
| Amylase increased | 0 | 0 | 1 (20) | 1 (33) | 5 (71) | 1 (100) | 8 (27) |
| AST increased | 1 (14) | 0 | 2 (40) | 0 | 3 (43) | 1 (100) | 7 (23) |
| Lipase increased | 0 | 0 | 1 (20) | 0 | 5 (71) | 1 (100) | 7 (23) |
| GGT increased | 2 (29) | 2 (29) | 2 (40) | 0 | 0 | 0 | 6 (20) |
| Alkaline phosphatase increased | 2 (29) | 0 | 1 (20 | 0 | 0 | 0 | 3 (10) |
| Bilirubin increased | 0 | 0 | 0 | 0 | 2 (29) | 1 (100) | 3 (10) |
| Metabolism and nutrition disorders | |||||||
| Decreased appetite | 0 | 0 | 2 (40) | 0 | 0 | 0 | 2 (7) |
| Musculoskeletal disorders | |||||||
| Pain in extremity | 1 (14) | 0 | 0 | 1 (33) | 0 | 0 | 2 (7%) |
| Nervous system disorders | |||||||
| Headache | 2 (29) | 1 (14) | 0 | 0 | 0 | 0 | 3 (10%) |
| Skin disorders | |||||||
| Pruritus | 1 (14) | 0 | 0 | 1 (33) | 0 | 0 | 2 (7%) |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; GGT = gamma glutamyl transferase.
Anti-tumor activity
Among 20 evaluable patients, there were no objective RECIST responses. Four patients (20%) had stable disease (2–4 months duration), 3 at the 0.1-mg/kg dose level and 1 in the 0.5-mg/kg cohort. The remaining 16 patients (80%) had progressive disease.
Pharmacokinetics
Due to the lack of clinical efficacy and DLTs, most patients received only 2 to 4 doses (1 to 2 cycles) of enavatuzumab and collection of washout samples was inconsistent. As a result of the limited available data, no formal PK modeling was performed for this study. Instead, the observed enavatuzumab maximum serum concentrations following the first dose (C1max) and the AUC of enavatuzumab for the first cycle are summarized (Table 3).
Table 3.
Summary of Enavatuzumab Pharmacokinetic Parameters (Mean [± SD])
| Enavatuzumab Dose | ||||||
|---|---|---|---|---|---|---|
| Pharmacokinetic Parameter Mean (SD) | 0.1 mg/kg (n = 5) |
0.3 mg/kg (n = 4) |
0.5 mg/kg (n = 1) |
0.7 mg/kg (n = 3) |
mg/kg (n = 5) |
1.5 mg/kg (n = 1) |
| Cycle 1 (28 days) | ||||||
| Tmax (h) | 1.0 (0.00) |
1.57 (1.51) |
1.80 (1.79) |
2.33 (2.31) |
3.86 (1.95) |
1.0 (NA) |
| C1max (μg/mL) | 2.32 (40.71) |
7.56 (3.19) |
12.74 (4.02) |
13.03 (3.11) |
28.09 (5.87) |
26.54 (NA) |
| C1max/D (μg/mL/mg/kg) | 23.16 (7.14) |
25.19 (10.63) |
25.48 (8.04) |
18.61 (4.45) |
28.09 (5.87) |
17.69 (NA) |
| AUCcycle1 (mg•h/mL) | 0.70 (0.11) |
2.33 (0.66) |
1.85 (NA) |
4.32 (0.25) |
10.59 (0.48) |
NA |
|
AUCcycle1/2xD (mg•h/mL/mg/kg) |
3.48 (0.54) |
3.89 (1.09) |
1.85 (NA) |
3.08 (0.18) |
5.29 (0.24) |
NA |
| Troughcycle1 (mcg/mL) | 0.32 | 0.79 | 1.81 | 2.56 | 4.21 | N/A |
| Troughcycle2-6 (mcg/mL) | 0.27 | 1.26–1.57 | 0.46–0.48* | 1.98–2.34 | 6.50–7.49 | N/A |
AUCcycle 1 = area under the drug concentration-time curve for Cycle 1; C1max = maximum serum concentration following the first dose; D = dose of enavatuzumab; h = hour; NA = not applicable; SD = standard deviation; Tmax = time to maximum serum concentration;
represents results from a single patient.
The average serum exposure of enavatuzumab measured by AUCcycle1 was 0.7 mg•h/mL for the 0.1-mg/kg dose group, and approximately 10.6 mg•h/mL for the 1.0-mg/kg dose group. Upon every other week IV infusion administration, both C1max and AUC values of enavatuzumab increased approximately dose proportionally with increasing dose, indicating linear pharmacokinetics in the dose range from 0.1 to 1.5 mg/kg. Enavatuzumab followed a two-compartment linear PK model. The estimated clearance was 23–33 mL/h and the elimination half-life was 7–18 days. The mean Cmax after the first dose was 2.3 mcg/mL and 28.1 mcg/mL for the 0.1-mg/kg and 1.0-mg/kg cohorts, respectively. Based on preclinical studies in xenograft models, the target efficacious trough and peak levels were 1.6 mcg/mL and 44 mcg/mL, respectively. The target peak level was not reached; however, the target trough level was achieved following the second dose for the 1.0-mg/kg cohort (Figure 1, Table 3).
Figure 1. Serum drug concentrations for all patients treated in the 1.0-mg/kg cohort.
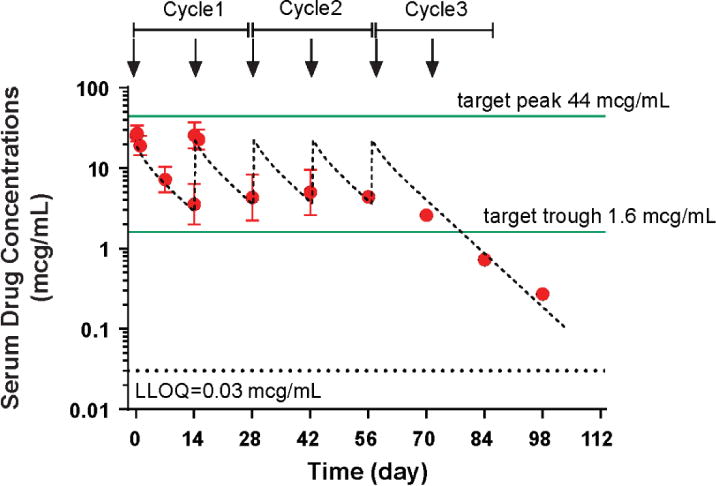
Figure 1 contains the serum concentration-time profiles of enavatuzumab (1.0 mg/kg) intravenously every 2 weeks (1 cycle = 4 weeks). The observed average maximum concentration after the 1st dose was 28.1 mcg/mL. Steady state serum concentrations appeared to be reached following the second dose, with average minimum concentrations of 4.3 mcg/mL.
Pharmacodynamics
A subset of patients exhibited elevations of circulating cytokines/chemokines > 2-fold over baseline. Of the time points tested, elevations were only observed at 5hr or 23hr after the first dose, suggesting that the cytokine/chemokine elevations were transient. The most frequently observed elevations were for interferon-inducible protein 10 (IP-10) (15 patients), IL-1α (14 patients), monocyte chemo-attractant protein-1 (MCP-1) (13 patients), and interleukin-8 (IL-8) (10 patients). MCP-1 elevations were found to correlate loosely with increases in ALT and AST levels (R2 = 0.158 and 0.192, respectively; Figure 2). Immunophenotyping of peripheral blood revealed no consistent changes in T, B, or NK cell counts after enavatuzumab dosing, nor was evidence of NK cell activation observed in patients.
Figure 2. Correlation between monocyte chemo-attractant protein-1 (MCP-1) elevations with increases in ALT and AST levels.
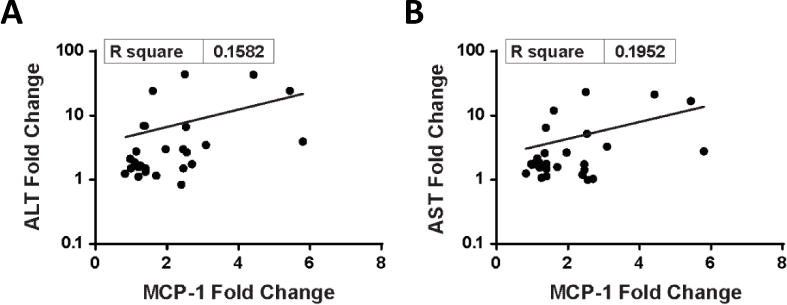
Figure 2 shows the relationship between change in plasma MCP-1 levels and ALT (A) and AST (B), respectively, when comparing pre-study and post-dosing samples. Plasma MCP-1 elevations correlate with increases in ALT (R2 = 0.158) and AST levels (R2 = 0.192).
TweakR expression
TweakR expression data were available only for 12 patients, of which 7 had positive staining for TweakR. Elevated TweakR expression was observed in baseline liver biopsies of tumors obtained from 3 patients in this study, one of whom developed hepatic DLT after enavatuzumab was administered. Investigative studies on pancreatic cells or tissues were not performed due to the lack of available samples. Due to the limited dataset, the immunohistochemistry data are inconclusive and insufficient to correlate TweakR expression with either response or toxicity.
DISCUSSION
Preclinical evidence demonstrating both the pro-tumorigenic and anti-tumor effects of TWEAK, as well as the overexpression of TweakR in variety of tumor types makes targeting TWEAK-TweakR signalling an attractive therapeutic approach. Lassen and colleagues have recently reported on a phase 1 study of RG7212, a monoclonal antibody targeting TWEAK, reporting good tolerability (no DLTs and no significant hepatopancreatic toxicities), expected pharmacodynamic changes, and some evidence of antitumor activity (16). RG7212 was shown to reduce tumor TWEAK-TweakR signaling in an exposure-dependent manner (17). To our knowledge, our study is the first phase 1 clinical trial of a humanized IgG1 antibody to TweakR. The MTD of enavatuzumab was determined to be 1 mg/kg administered intravenous every 14 days. Observed DLTs included grade 4 lipase, grade 4 amylase, and grade 3 bilirubin elevations.
Over 40% of patients in this study experienced a severe (grade ≥ 3) drug-related adverse event, most commonly consisting of elevations in serum lipase, amylase, GGT, ALT, and AST. In most cases, these abnormalities improved or resolved upon discontinuation of enavatuzumab. There was no apparent correlation of liver or pancreatic enzyme elevation to duration of drug exposure or presence of liver metastases. There were insufficient data in this study to correlate TweakR expression with either response or toxicity. Preselection based on TweakR and other biomarkers should be considered in future studies with drugs targeting TWEAK-TweakR signalling.
In preclinical studies in non-human primates, mild elevations in liver enzyme levels were observed only at the highest doses tested (100 mg/kg). Preclinical studies utilizing cultured hepatocytes have identified a potential mechanism for the liver enzyme elevations observed following enavatuzumab dosing. While enavatuzumab had little effect on cultured hepatocytes alone, exposure of hepatocytes to enavatuzumab in the presence of immune cells resulted in elevated concentrations of secreted ALT, AST, cytokines and chemokines. These elevations were largely suppressed by pre-treatment with dexamethasone. Together, these findings suggest that the presence of immune cells and/or increased TweakR expression may sensitize tissues to enavatuzumab and that the toxicity may be associated with cytokine release induced by enavatuzumab.
The elevated transaminase, lipase, and bilirubin levels observed in our study of enavatuzumab which targets TweakR contrasts with lack of these laboratory adverse events in the phase 1 study of RG7212, which targets the TWEAK ligand (16). This suggests that the liver and pancreatic toxicities may be mediated by signalling through the TWEAK receptor. The role of the TWEAK/TweakR pathway in human liver injury may also shed light on the mechanism of hepatotoxicity by enavatuzumab. Jakubowski and colleagues have demonstrated that liver progenitor cell expansion was significantly reduced in TweakR-null mice, as well as in adult wild-type mice treated with a blocking anti-TWEAK monoclonal antibody and that TWEAK-stimulated proliferation of the progenitor cells in vitro, suggesting that TWEAK has a selective mitogenic effect on liver progenitor cells (18). In a follow-up study, these investigators demonstrated that TWEAK stimulates liver progenitor cell mitosis via a TweakR and NFĸB-dependent fashion (19). In studies after acute partial hepatectomies in mice, rapid proliferation of TweakR-positive cells were observed, indicating that TWEAK-TweakR signalling was required for the healthy liver to regenerate. When that signalling was disrupted, induction of pro-regenerative cytokines and proliferation of hepatocytes is inhibited and post-hepatectomy liver damage and elevated bilirubin levels were observed (20).
The effect of dexamethasone on enavatuzumab anti-tumor activity was assessed in two xenograft models. In both the ADCC-dependent and ADCC-independent models, dexamethasone did not decrease the anti-tumor activity of enavatuzumab, suggesting that dexamethasone may be considered as a means to decrease enavatuzumab-stimulated cytokine secretion without eliminating potency. However, as noted above, in our study, only one patient received dexamethasone premedication (prior to cycle 2) and this did not appear to prevent hepatic or pancreatic toxicity. The data are insufficient to draw any conclusions, and future clinical investigation is needed to determine whether dexamethasone can mitigate the hepatopancreatic toxicity of enavatuzumab.
There was no preliminary evidence of clinical activity of enavatuzumab, given the absence of objective responses or prolonged disease stability. However, due to the presence of DLTs, the overall drug exposure for most patients was low. Although the drug exposure following the second dose at 1.0 mg/kg was within the target efficacious trough concentration, the target efficacious peak concentration was not met, and the overall duration of enavatuzumab exposure may have been inadequate to induce tumor responses. The phase 1 study of RG7212, in which all 54 patients enrolled had TweakR positivity, (at least 10% or IHC 1+) also did not show any objective response. Among the 54 patients treated, 23 (43%) had stable disease including 15 (28%) who had prolonged stable disease lasting 16 weeks or more; 1 patient with BRAF wild-type melanoma has tumor regression and pharmacodynamic changes consistent with antitumor effects (16).
In vitro studies have established that NFkB drives the growth inhibitory activity of enavatuzumab, suggesting that targeting TweakR may still be a plausible cancer treatment strategy (8). In vivo studies in patient-derived breast cancer xenografts found an 8-gene signature of predictive of response to PDL192 among genes evaluated in the following signalling pathways: TweakR pathway, apoptosis, the NFkB pathway, proliferation, migration/invasion, vascularization, and epithelial-mesenchymal transition (21). Given the lack of single agent activity and the hepatopancreatic toxicities that limit the evaluation of enavatuzumab at higher doses in this study, one strategy may be to combine lower doses of enavatuzumab with chemotherapy to promote synergistic efficacy. For example, in preclinical studies of pancreatic cancer models, the combination of enavatuzumab and gemcitabine exhibited more potent activity than either agent alone (22).
There are other clinical trials of monoclonal antibodies against the TWEAK ligand in cancer and non-cancer conditions. A phase I study of RO5458640, a TWEAK antagonist, in patients with advanced solid tumors has completed accrual, and results are yet to be presented (NCT01383733). A phase I study of another anti-TWEAK antagonist, BIIB023, as monotherapy and in combination with TNF blockers in patients with rheumatoid arthritis reported no hepatic enzyme abnormalities in the monotherapy cohorts and an 8% incidence (1/12 patients) of hepatic enzyme elevation in the combination arm (23). There is an ongoing study of BIIB023 in patients with lupus nephritis (NCT01499355). The toxicity results from these trials may also inform future development of anti-TweakR agents such as enavatuzumab.
TweakR is a potential oncologic target, given the differential expression patterns and preclinical efficacy observed with TweakR antibodies. Although preclinical safety evaluations confirmed a safe starting dose, this work did not predict the severity of hepatopancreatic enzyme toxicities that were observed in the clinic. The human pharmacokinetic results demonstrate that the concentrations of enavatuzumab in this study were near or within range of those required for monotherapy efficacy in xenograft models. Although the MTD was defined at 1.0 mg/kg, this might not result in a recommended phase 2 dose, given the high rate of grade ≥ 2 toxicities noted in the patients treated at this dose level. Thus, it is not recommended to continue the development of enavatuzumab as monotherapy at this dose and schedule. However, further evaluation of enavatuzumab may be reasonable if altering the pharmacokinetics in preclinical models with a lower dose at more frequent dosing intervals demonstrates similar anti-tumor effects while avoiding some of the hepatopancreatic toxicities. Additionally, clinical evaluation of lower doses of enavatuzumab in combination with chemotherapy such as gemcitabine in pre-selected patients with positive tumor TweakR expression may also be reasonable. Future studies of TWEAK/Tweak R signalling agents, and studies with agents known to stimulate inflammatory responses, should include careful evaluation of delayed dose-limiting toxicity.
Acknowledgments
Financial Information: Elaine Lam receives funding support from the NIH/NCI Paul Calabresi Award in Clinical Oncology Research (K12CA086913).
References
- 1.Wiley SR, Cassiano L, Lofton T, Davis-Smith T, Winkles JA, Lindner V, et al. A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity. 2001;15:837–846. doi: 10.1016/s1074-7613(01)00232-1. [DOI] [PubMed] [Google Scholar]
- 2.Wiley SR, Winkles JA. TWEAK, a member of the TNF superfamily, is a multifunctional cytokine that binds the TweakR/Fn14 receptor. Cytokine Growth Factor Rev. 2003;14:241–249. doi: 10.1016/s1359-6101(03)00019-4. [DOI] [PubMed] [Google Scholar]
- 3.Burkly LC, Michaelson JS, Hahm K, Jakubowski A, Zheng TS. TWEAKing tissue remodeling by a multifunctional cytokine: role of TWEAK/Fn14 pathway in health and disease. Cytokine. 2007;40:1–16. doi: 10.1016/j.cyto.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 4.Culp PA, Choi D, Zhang Y, Yin J, Seto P, Ybarra SE, et al. Antibodies to TWEAK receptor inhibit human tumor growth through dual mechanisms. Clin Cancer Res. 2010;16:497–508. doi: 10.1158/1078-0432.CCR-09-1929. [DOI] [PubMed] [Google Scholar]
- 5.Tran NL, McDonough WS, Savitch BA, Fortin SP, Winkles JA, Symons M, et al. Increased fibroblast growth factor-inducible 14 expression levels promote glioma cell invasion via Rac1 and nuclear factor-kappaB and correlate with poor patient outcome. Cancer Res. 2006;66:9535–9542. doi: 10.1158/0008-5472.CAN-06-0418. [DOI] [PubMed] [Google Scholar]
- 6.Zhou H, Ekmekcioglu S, Marks JW, Mohamedali KA, Asrani K, Phillips KK, et al. The TWEAK receptor Fn14 is a therapeutic target in melanoma: immunotoxins targeting Fn14 receptor for malignant melanoma treatment. J Invest Dermatol. 2013;133:1052–1062. doi: 10.1038/jid.2012.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang M, Narita S, Tsuchiya N, Numakura K, Obara T, Tsuruta H, et al. Overexpression of Fn14 promotes androgen-independent prostate cancer progression through MMP-9 and correlates with poor treatment outcome. Carcinogenesis. 2011;32:1589–1596. doi: 10.1093/carcin/bgr182. [DOI] [PubMed] [Google Scholar]
- 8.Purcell JW, Kim HK, Tanlimco SG, Doan M, Fox M, Lambert P, et al. Nuclear Factor kappaB is Required for Tumor Growth Inhibition Mediated by Enavatuzumab (PDL192), a Humanized Monoclonal Antibody to TweakR. Front Immunol. 2014;4:505. doi: 10.3389/fimmu.2013.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakayama M, Ishidoh K, Kayagaki N, Kojima Y, Yamaguchi N, Nakano H, et al. Multiple pathways of TWEAK-induced cell death. J Immunol. 2002;168:734–743. doi: 10.4049/jimmunol.168.2.734. [DOI] [PubMed] [Google Scholar]
- 10.Armstrong CL, Galisteo R, Brown SA, Winkles JA. TWEAK activation of the non-canonical NF-kappaB signaling pathway differentially regulates melanoma and prostate cancer cell invasion. Oncotarget. 2016;49:81474–81492. doi: 10.18632/oncotarget.13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chao DT, Su M, Tanlimco S, Sho M, Choi D, Fox M, et al. Expression of TweakR in breast cancer and preclinical activity of enavatuzumab, a humanized anti-TweakR mAb. J Cancer Res Clin Oncol. 2013;139:315–325. doi: 10.1007/s00432-012-1332-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rivera E, Lee J, Davies A. Clinical Development of Ixabepilone and Other Epothilones in Patients with Advanced Solid Tumors. Oncologist. 2008;13:1207–1223. doi: 10.1634/theoncologist.2008-0143. [DOI] [PubMed] [Google Scholar]
- 13.Reuben A. Hy’s law. Hepatology. 2004;39:574–578. doi: 10.1002/hep.20081. [DOI] [PubMed] [Google Scholar]
- 14.Banks PA, Freeman ML. Practice guidelines in acute pancreatitis. Am J Gastroenterol. 2006;101:2379–2400. doi: 10.1111/j.1572-0241.2006.00856.x. [DOI] [PubMed] [Google Scholar]
- 15.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 16.Lassen UN, Meulendijks D, Siu LL, Karanikas V, Mau-Sorensen M, Schellens JH, et al. A Phase I Monotherapy Study of RG7212, a First-in-Class Monoclonal Antibody Targeting TWEAK Signaling in Patients with Advanced Cancers. Clin Cancer Res. 2015;21:258–266. doi: 10.1158/1078-0432.CCR-14-1334. [DOI] [PubMed] [Google Scholar]
- 17.Meulendijks D, Lassen UN, Siu LL, Huitema AD, Karanikas V, Mau-Sorensen M, et al. Exposure and Tumor Fn14 Expression as Determinants of Pharmacodynamics of the Anti-TWEAK Monoclonal Antibody RG7212 in Patients with Fn14-Positive Solid Tumors. Clin Cancer Res. 2016;22:858–867. doi: 10.1158/1078-0432.CCR-15-1506. [DOI] [PubMed] [Google Scholar]
- 18.Jakubowski A, Ambrose C, Parr M, Lincecum JM, Wang MZ, Zheng TS, et al. TWEAK induces liver progenitor cell proliferation. J Clin Invest. 2005;115:2330–2340. doi: 10.1172/JCI23486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tirnitz-Parker JE, Viebahn CS, Jakubowski A, Klopcic BR, Olynyk JK, Yeoh GC, et al. Tumor necrosis factor-like weak inducer of apoptosis is a mitogen for liver progenitor cells. Hepatology. 2010;52:291–302. doi: 10.1002/hep.23663. [DOI] [PubMed] [Google Scholar]
- 20.Karaca G, Swiderska-Syn M, Xie G, Syn WK, Kruger L, Machado MV, et al. TWEAK/Fn14 signaling is required for liver regeneration after partial hepatectomy in mice. PLoS ONE. 2014;9:e83987. doi: 10.1371/journal.pone.0083987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Plater L, Vincent-Salomon A, Berger F, Nicolas A, Vacher S, Gravier E, et al. Predictive gene signature of response to the anti-TweakR mAb PDL192 in patient-derived breast cancer xenografts. PLoS ONE. 2014;9:e104227. doi: 10.1371/journal.pone.0104227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hylander BL, Choi D, Sho M, Chao DT, Starling GC, Culp PA, et al. PDL192, a humanized antibody to TweakR exhibits potent antitumor activity in pancreatic cancer models. Clin Cancer Res. 2010;16:B17. [Google Scholar]
- 23.Wisniacki N, Amaravadi L, Galluppi GR, Zheng TS, Zhang R, Kong J, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of anti-TWEAK monoclonal antibody in patients with rheumatoid arthritis. Clin Ther. 2013;35:1137–1149. doi: 10.1016/j.clinthera.2013.06.008. [DOI] [PubMed] [Google Scholar]