

Abstract
Ceramides are bioactive lipids that mediate cell death in cancer cells and ceramide-based therapy is now being tested in dose-escalating phase 1 clinical trials as a cancer treatment. Multiple nanoscale delivery systems for ceramide have been proposed to overcome the inherent toxicities, poor pharmacokinetics and difficult biophysics associated with ceramide. Using the ceramide nanoliposomes (CNL) we now investigate the therapeutic efficacy and signaling mechanisms of this nanoscale delivery platform in refractory ovarian cancer. Treatment of ovarian cancer cells with CNL decreased the number of living cells through necroptosis but not apoptosis. Mechanistically, dying SKOV3 ovarian cancer cells exhibit activation of pseudokinase mixed lineage kinase domain-like (MLKL) as evidenced by oligomerization and relocalization to the blebbing membranes, showing necroptotic characteristics. Knock-down of MLKL, but not its upstream protein kinases such as receptor-interacting protein kinases, with siRNA significantly abolished CNL-induced cell death. Monomeric MLKL protein expression inversely correlated with the IC50 values of CNL in distinct ovarian cancer cell lines, suggesting MLKL as a possible determinant for CNL-induced cell death. Finally, systemic CNL administration suppressed metastatic growth in an ovarian cancer cell xenograft model. Taken together, these results suggest that MLKL is a novel pro-necroptotic target for ceramide in ovarian cancer models.
Keywords: ceramide, ceramide nanoliposomes, necroptosis, ovarian cancer, pseudokinase mixed lineage kinase domain-like
Introduction
Despite advances in ovarian cancer diagnosis and treatment, more than half of patients relapse and develop a multifactorial drug resistance (1, 2). There is an urgent need to develop novel treatment strategy for refractory ovarian cancer.
Ceramide, a sphingolipid metabolite, is a bioactive lipid that plays a critical role in activating cell death signaling initiated by chemotherapeutic reagents and ionizing radiation (3–6). Accumulating evidence indicates that ceramide species are reduced in carcinoma tissues and chemo-resistance transformed cell lines (7–9), providing the rationale to investigate ceramide-based therapeutic strategies as cancer treatments (10, 11).
Regardless of the therapeutic potential of ceramides, the delivery of bioactive lipids into cancer cells has been hampered due to the inherent hydrophobicity and insolubility. For this reason, there is a critical need for improved delivery systems to optimize the delivery of ceramides to cells. Nanoscale formulations have been shown to dramatically improve the pharmacokinetic and toxicological profile of ceramide delivery to cancer cells. Therefore, several laboratories, including our own, are now employing nanotechnologies to improve therapeutic efficacy of ceramides (11, 12). Ceramides have been successfully intercalated within nanoscale liposomal bilayers generating pegylated (11) and transferrin-conjugated delivery vehicles (13, 14). Even though Torchilin’s studies have shown that ceramide delivery vehicles suppress tumor growth in ovarian cancer cells (13), the molecular mechanisms underlying cell death were not fully determined.
Ceramide nanoliposomes (CNL) (11, 15, 16) have been engineered and validated by our laboratory, and published preclinical studies support an FDA phase 1 first-in-man-dose escalation study under IND# 109571. Our formulation contains 30 molar percent C6-ceramide in a 12 molar percent pegylated 80 nm, −7mV vehicle that extends the T1/2 of ceramide from 15 min to >15 h (11). Multiple studies demonstrated that CNL is selectively anti-proliferative in numerous cancer cell types in cell-based assay as well as animal models. For instance, CNL significantly inhibited proliferation and induced cell death in human breast adenocarcinoma cells (17) and melanoma cells (18). In addition to solid tumor models, CNL was also effective in nonsolid leukemia models (19). Mechanistically, ceramides inhibit prosurvival protein kinase Cζ-dependent AKT and ERK signaling cascades (20). Despite validating the effects of CNL upon these pro-survival regulatory pathways, the mechanisms underlying cell death of cancer cells are relatively controversial with both apoptotic and necrotic mechanisms proposed. In addition, the mechanisms responsible for the selectivity of the cell death response to ceramide to transformed cell lines are also relatively undefined, even though the selectivity of CNL has been shown to be dependent upon elevated glycolytic metabolism (Warburg effect) in transformed cells (15).
Necrosis is often considered an accidental and unregulated event. However, growing evidence suggests that necrosis, like apoptosis, can be executed by regulated mechanisms. Necroptosis is the best-characterized form of regulated necrosis (21–23). Necroptosis is mediated with regulatory and/or effector proteins such as protein kinase receptor-interacting protein kinase (RIPK) 1, RIPK3 and pseudokinase mixed lineage kinase domain-like (MLKL) (24, 25). In the present study, we demonstrate a novel mechanism of action for CNL in ovarian cancer models, identifying MLKL, but not RIPK1/3, as a target for CNL-induced necroptotic cell death.
Materials and Methods
Antibodies and reagents
PARP antibody (sc7150) and horseradish peroxidase-conjugated antibodies for mouse (sc2005) and rabbit IgG (sc2004) were from Santa Cruz Biotechnology (Dallas, TX, USA). MLKL antibody (ab184718) was obtained from Abcam (Cambridge, MA, USA). GAPDH antibody (#A01622) was from GenScript (Tokyo, Japan). Antibodies for LC3B (#2775), RIPK1 (#4926) and RIPK3 (#13526) were from Cell Signaling Technology (Massachusetts, USA). RNAiMax, Lipofectamine2000, siRNAs for control (4390846) and MLKL (s47088 and s47089), and mouse monoclonal V5 antibody (R96025) were from Life Technologies (Carlsbad, California, USA). Tetramethylrhodamine isothiocyanate (TRITC)-conjugated phalloidin, biotin and β-actin antibodies (A5441) were obtained from Sigma (St Louis, MO, USA). Hoechst 33342 was from Dojindo (Kumamoto, Japan). CellTiter-Glo luminescent cell viability assay kit was obtained from Promega (Fitchburg, Wisconsin, USA). Annexin V/7-AAD kit and matrigels were from BD Biosciences (San Jose, CA, USA).
Cell culture
Ovarian cancer cell lines including SKOV3, TOV112D, A2780, A2780CP, PE01, and PE04 were kindly provided by Dr. Carla Grandori (Fred Hutchinson Cancer Research Center, Seattle, WA, USA). All cell lines were free of mycoplasma contamination, determined by using the MycoAlert™ mycoplasma detection kit (LONZA). Cell lines SKOV3 and A2780 were authenticated by JCRB Cell Bank (Osaka, Japan) in 2013. Ovarian cancer cells were cultured (passage number 5–20) in Dulbecco’s Modified Eagle’s Medium (DMEM), supplemented with 10% fetal bovine serum (FBS) at 37°C in a humidified incubator containing 5% CO2. Cells were maintained at <80% confluence.
Preparation of nanoliposomal ceramide
The physiochemical characterization and validation of CNL as well as the preclinical evaluation of the CNL platform were previously described by Kester et al. (11).
Trypan blue exclusion assay
Briefly, cells were plated into 6-well plates (1 × 105 cells/well) and pre-incubated with growth medium overnight. The following day, the cells were treated with CNL or ghost nanoliposomes up to 48 h. The number of trypan blue-excluding living cells was counted.
Transfection with siRNAs
Cells were transfected with 5 nM of siRNAs using Lipofectamine RNAiMAX transfection reagent (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Preparation of MLKL vector constructs
Human MLKL cDNAs were amplified by PCR using human MLKL pFN21A HaloTag®CMV Flexi®Vector (Kazusa DNA Research Institute, Kisarazu, Japan). The PCR products and pcDNA3.1/V5-His A (ThermoFisher Scientific) were digested with EcoRI/XhoI and each fragment was ligated, forming human MLKL pcDNA3.1/V5-His A vectors.
Transfection with vectors
Cells were transfected with empty or MLKL-V5 vector (2 μg/dish) using Lipofectamine 2000 transfection reagent (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Immunoblotting
Cells (2 × 105 cells/60 mm dish) were treated with CNL, ghost nanoliposomes or staurosporine for 24 h. Cells were harvested, washed with ice-cold PBS and lysed in RIPA lysis buffer supplemented with Halt Phosphatase Inhibitor Cocktail. After brief centrifugation, the protein content of the samples was measured using the BCA protein assay reagent. Extracted proteins were submitted to SDS-PAGE (4–20% gradient gels). Proteins were electrophoretically transferred on to nitrocellulose membranes. Nonspecific binding to the membrane was blocked for 30 min at room temperature with PBS/0.1% Tween 20 (PBS-T) containing 5% nonfat dried milk. The membranes were incubated overnight at 4°C with the following primary antibodies: GAPDH (1:1,000), LC3B (1:1,000), MLKL (1:1,000), PARP (1:2,000), RIPK1 (1:1,000), RIPK3 (1:500), V5 (1:5,000). β-actin (1:200,000) was used as loading control. After an overnight incubation, the membranes were washed with PBS-T and incubated with a secondary antibody conjugated with horseradish peroxidase in PBS-T containing 5% nonfat dried milk for 1 h at 4°C. The labeled proteins were visualized using enhanced chemiluminescence reagents, and quantification of the chemiluminescent signals was performed with a digital imaging system.
Immunofluorescence
Cells (2 × 104 cells) were seeded in 35 mm glass-bottom dishes. After exposure to the CNL, cells were washed in PBS and then fixed in 4% formaldehyde for 10 min at room temperature. Fixed cells were treated with 0.1% Triton X-100 for 10 min, and then blocked for 1 h with PBS containing 20% human serum. Cells were incubated with primary antibody against MLKL in PBS containing 20% human serum overnight. After three times washed with PBS, cells were incubated with Alexa488-conjugated anti-rabbit IgG antibody for 1 h. Nuclear staining was performed with Hoechst 33342 in the dark and all images were captured using Zeiss LSM710 confocal microscope (Carl Zeiss, Thornwood, NY, USA).
Cell viability assay
Ovarian cancer cells (1 × 103 cells/well) were transfected with siRNA following treatment with nanoliposomes. The cell viability was determined using a CellTiter-Glo luminescent cell viability assay according to the manufacturer’s protocol.
Apoptosis assay (Annexin V/7-AAD)
To determine apoptotic cells, Annexin V apoptosis detection kit was used according to the manufacturer’s protocol. Cells were exposed to CNL, ghost nanoliposomes or staurosporine for 18 h. Cells were trypsinized, collected by centrifugation. After removing the supernatant, cells were washed with PBS and resuspended in Annexin V binding buffer. Cells were labeled with 2.5 μl of Annexin V-FITC and 1 μl of 7-AAD in 50 μl Annexin V binding buffer, and incubated in the dark at room temperature for 30 min. Apoptotic cells were then detected by flow cytometer. Cells are considered necrotic, if they allow the penetration of 7-AAD without annexin V staining.
Analysis of pseudopodia formation
Cells (2 × 104 cells) were seeded in 35 mm glass bottom dishes. After treatment with CNL, cells were washed with PBS twice and fixed with 4% formaldehyde for 10 min at room temperature. Fixed cells were further treated with 0.1% TritonX-100 for 10 min followed by staining with Hoechst 33342 and TRITC-conjugated phalloidin for 5 min in the dark. Fluorescence microscopy was performed with the Zeiss LSM 780 laser scanning confocal microscope with a 60× oil objective. The percentages of cells forming lamellipodia were determined by counting more than 300 cells.
Cell migration assay
The migration of treated cells was assessed by the ability of cells to migrate through a porous (8 μm) polycarbonate membrane of a transwell device towards a chemotactic cue. Thereafter, equal numbers of cells (1 × 105) were plated onto the upper chamber in serum-free medium. The chamber was lodged into the lower chamber (10% FBS medium). After incubating at 37°C for 6 h, non-migrated cells on the top of the filter membrane were removed by a cotton swab, while migratory cells on the bottom of the filter membrane were fixed in 100% methanol and stained using Hoechst 33342. The imaging was performed by fluorescence microscopy. Cell migration was assessed by counting migrated cellc in fluorescence microscopy pictures (at least ten fields for each determination).
Invasion assay
Cells (1 × 105) in serum-free medium were seeded onto the upper chamber with 8 μm pores that were coated with 5% matrigel. The lower chamber was filled with 10% FBS-containing DMEM. After 18 h incubation with CNL or ghost nanoliposomes, non-invasive cells were removed from the upper surface of the filter membrane. The invading cells attached to the bottom surface of the filter membrane were stained with Hoechst 33342 in the dark. Cell invasiveness was determined by counting invading cell number. The number of invading cells was counted in at least ten randomly selected regions of interest.
Ethics statement
All animal experiments were performed according to the protocols approved by the Institutional Animal Care and Use Committee of Tohoku University, Japan.
Human ovarian cancer cell xenograft studies
Nude mice (BALB/c; Charles River Japan) were housed under pathogen-free conditions with unlimited access to food and water. Cells (5 × 106 cells/mouse) in RPMI-1640 serum-free medium were injected intraperitoneally into 4 weeks-old female mice. Mice were randomly allocated to two groups (n = 5). CNL or ghost nanoliposomes were intraperitoneally injected daily for 3 consecutive days. After 4 weeks, the mice were euthanized and the number and extent of overt metastases (>1 mm) were quantified. Mice that died of unrelated causes were excluded from analyses.
Statistical analysis
Data represent the mean ± SD of at least three independent experiments. Statistical analyses were performed using GraphPad Prism 6.0. p values lower than 0.05 are considered significant.
Results
To characterize anti-tumor effects of CNL in ovarian cancer cells, we initially determined cytotoxicity of CNL in ovarian cancer cells. Treatment of SKOV3 ovarian cancer cells with 30 μM CNL was revealed to show cytotoxic effects (Figure 1A) and most of cells were dead within 48 h after treatment with 100 μM CNL (Figure 1B). The decrease in cell viability with CNL treatment was both dose and time responsive in SKOV3 cells (Figure 1B and 1C). Moreover, we determined the CNL IC50 values using GraphPad prism in various ovarian cancer cell lines. CNL exhibited cytotoxic effects in six ovarian cancer cell lines and IC50 values ranged between 2 and 57 μM (Figure 1D, and Supplementary Figure 1). Ghost nanoliposomes, consisting of the exact amount of bilayer lipids, but without ceramide, had no cytotoxic effect upon ovarian cancer cell lines. Of interest, the IC50 values were higher for cisplatin-resistant cell lines (PE04 and A2780CP) and lower for cisplatin-sensitive cell lines (TOV112D and PE01).
Figure 1. Cytotoxic effects of ceramide nanoliposomes in ovarian cancer cells.
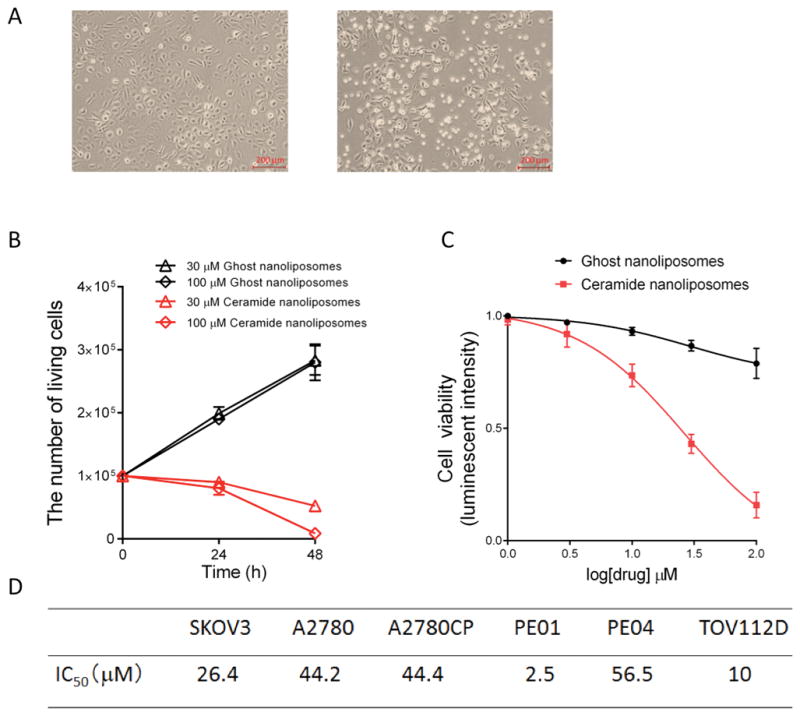
A, SKOV3 cells (1 × 105/well) were treated with 30 μM ceramide or ghost nanoliposomes for 24 h and then imaged by a phase-contrast microscopy. B, SKOV3 cells were treated with 30 or 100 μM nanoliposomes up to 48 h. The number of living cells was counted. The data represent the mean ± SD (n = 3). C, SKOV3 cells were treated with 1, 3, 10, 30, or 100 μM nanoliposomes. The cell viability was determined using a CellTiter-Glo luminescent cell viability assay according to the manufacturer’s protocol. The results are expressed as the percentages of 1 μM ghost nanoliposomes and the data represent the mean ± SD (n = 3). D, IC50 values were determined by GraphPad prism. The IC50 values of ceramide nanoliposomes in six kinds of ovarian cancer cell lines were shown.
We next investigated the underlying molecular mechanism of cell death induced by CNL. Cell death was initially assessed by an Annexin V/7-AAD apoptosis assay. Upon treatment with staurosporine known as an apoptosis-inducing reagent, Annexin V-positive/7-ADD-negative cell population was increased, confirming apoptosis induction. Surprisingly, CNL treatment predominantly induced necrosis, but not apoptosis (Figure 2A). This phenomenon was not unique to SKOV3 cells, because we observed the same tendency in other multiple ovarian cancer cells (Supplementary Figure 2A). Furthermore, CNL had minimal effects on DNA fragmentation (Supplementary Figure 2B) and PARP cleavage (Figure 2B). Interestingly, procaspase 3 was cleaved upon 100 μM CNL treatment, although obvious cleavage of PARP was not coincidently observed. In addition, knockdown of apoptosis-inducing factor (AIF) (Supplementary Figure 3A and 3B) or enforced expression of anti-apoptotic protein Mcl-1 (Supplementary Figure 3C and 3D) had no protective effects against CNL-induced cytotoxicities. These results suggest that CNL promotes specifically necrosis induction in ovarian cancer cells. In contrast to non-solid including chronic lymphocytic leukemia cell lines (15), GAPDH expression remained unchanged after CNL treatment of SKOV3 cells (Figure 2B).
Figure 2. Effects of ceramide nanoliposomes on cell death.

A, SKOV3 cells were treated with 30 μM ceramide or ghost nanoliposomes, DMSO or 1 μM staurosporine for 18 h. Annexin V/7-AAD assay was performed for determining apoptotic cell death. B, SKOV3 cells were treated with ceramide nanoliposomes, ghost nanoliposomes or staurosporine for 24 h. Extracted cellular proteins were subjected to immunoblot analysis with antibodies for β-actin, GAPDH, LC3B, PARP, and procaspase 3 (apoptosis detection antibodies cocktail). Equal amounts of proteins were loaded in each lane. Three independent experiments were performed and representative images are shown. C, SKOV3 cells were incubated with the indicated concentrations of 3-methyladenine for 1 h and then treated with 30 μM ceramide or ghost nanoliposomes for 48 h. Cell viability was determined using a CellTiter-Glo luminescent assay according to the manufacturer’s protocol. The results are expressed as the percentages of 30 μM ghost nanoliposomes in non-3-methyladenine treatment group and the data represent the mean ± SD (n = 3).
Ceramide is known to induce autophagy. CNL treatment promoted the formation of LC3B-II representing biochemical characteristics of autophagy (Figure 2B), whereas the loss of cell viability induced by CNL was not significantly restored by inhibition of autophagy by 3-methyladenine (an autophagy inhibitor targeting class III phosphatidylinositol 3-kinase) (Figure 2C). Therefore, autophagy appears to have minimal effects on the CNL-induced cell death.
To evaluate possible involvement of necroptosis in cell death induced by CNL, we evaluated MLKL activation as evidenced by MLKL oligomerization and relocalization to the blebbing membranes. MLKL protein size is approximately 50 kDa as a monomer (26) and MLKL is oligomerized as a tetramer or an octamer upon necroptosis (27). Importantly, high molecular-mass complexes of V5-tagged MLKL (~100 kDa), possibly multiple forms of SDS-resistant oligomers, were detected with CNL treatment in V5-tagged MLKL overexpressing SKOV3 cells and the formation of oligomeric MLKLs was CNL-dose dependent (Figure 3A). Moreover, SDS-resistant octamer-like oligomers of endogenous MLKL were also formed in response to CNL treatment (Supplementary Figure 4). To further confirm MLKL activation, we next investigated intracellular localization with immunofluorescence using specific antibodies. MLKL was localized in cytoplasm and lamellipodia in ghost nanoliposomes-treated cells. Treatment with CNL promoted the relocalization of MLKL to the blebbing membranes (Figure 3B). MLKL oligomerization and relocalization to the plasma membranes are consistent with the late breakage of plasma membranes observed in necroptosis.
Figure 3. Activation of MLKL by ceramide nanoliposomes.

A, SKOV3 cells were transfected with MLKL-V5 vectors for 24 h followed by treatment with the indicated concentrations of ceramide or ghost nanoliposomes for 24 h. Cellular proteins extracted without reducing reagents were subjected to SDS-PAGE. Immunoblotting was performed using antibodies against V5 and β-actin. Three independent experiments were performed. Representative images are shown. B, SKOV3 cells were incubated with 30 μM ceramide or ghost nanoliposomes for 6 h and then were fixed followed by staining with TRITC-conjugated phalloidin (red), Hoechst 33342 (blue), MLKL (green). Imaging was performed by confocal microscopy, and representative images are shown. Arrows show the blebbing membranes.
We next utilized a molecular approach to further confirm the importance of MLKL in necroptotic cell death. Two individual siRNAs for MLKL were employed and confirmed to efficiently knockdown MLKL protein expression in SKOV3 cells (Figure 4A). The reduction in cell viability induced by CNL was significantly restored by MLKL siRNA compared to control siRNA (Figure 4B). Similar results were observed in TOV112D ovarian cancer cells (Supplementary Figure 5A). These results indicate that MLKL is required for CNL-induced necroptosis.
Figure 4. MLKL-dependent cell death.

A, SKOV3 cells were transfected with 5 nM control siRNA or two individual MLKL siRNA (sequence-1 and -2) for 24 h. The knockdown efficiency was confirmed by immunoblotting. B, Cells treated with siRNAs were further incubated with ceramide or ghost nanoliposomes for 48 h. Cell viability was determined using a CellTiter-Glo luminescent assay according to the manufacturer’s protocol. The results are expressed as the percentages of 10 μM ghost nanoliposomes in control siRNA group and the data represent the mean ± SD (n = 3). Statistical analyses were performed by unpaired, Student t-test. * p < 0.05 compared with control siRNA group. C, SKOV3 cells were incubated with the indicated concentrations of necrostatin-1 for 1 h and then treated with 30 μM ceramide or ghost nanoliposomes for 48 h. Cell viability was determined using a CellTiter-Glo luminescent assay according to the manufacturer’s protocol. The results are expressed as the percentages of 30 μM ghost nanoliposomes in non-necrostatin-1 treatment group and the data represent the mean ± SD (n = 3). D, SKOV3 cells were transfected with 5 nM control or RIPK3 siRNA for 24 h followed by treatment with ceramide or ghost nanoliposomes for 48 h. Cell viability was determined using a CellTiter-Glo luminescent assay according to the manufacturer’s protocol. The results are expressed as the percentages of 10 μM ghost nanoliposomes in control siRNA group and the data represent the mean ± SD (n = 3). E, proposed action mechanisms of ceramide nanoliposomes.
Necrosulfonamide has been identified as a necroptosis inhibitor that specifically targets the Cys86 of MLKL protein (28). Its treatment had no significant effects on the loss of cell viability and the oligomerization of MLKL induced by CNL (Supplementary Figure 6). CNL is unlikey to target the Cys86 of human MLKL.
We next examined whether the MLKL upstream kinases RIPK1 and RIPK3 are also required for CNL-induced cell death. SKOV3 and TOV112D ovarian cancer cells were treated with a RIPK1 inhibitor necrostatin-1 (29) or RIPK3 siRNA in the presence of CNL or ghost nanoliposomes. Inhibition of MLKL upstream molecules by necrostatin-1 or RIPK3 siRNA had no effect on CNL-induced cytotoxicities in SKOV3 cells (Figure 4C and 4D) and TOV112D cells (Supplementary Figure 5). Moreover, inhibition of RIPK1/3 had no effects on the expression of monomeric MLKL in SKOV3 cells (Supplementary Figure 7). Taken together, CNL is suggested to regulate necroptosis in a MLKL-dependent but not RIPK-dependent mechanism (Figure 4E).
RIPK3 is known to phosphorylate MLKL at Thr357/Ser358 within the MLKL activation loop (28). Upon CNL treatment, the amount of phosphorylated MLKL at Ser358 did not change in TOV112D cells (X. Zhang; unpublished observations).
To investigate MLKL expression with therapeutic efficacy of CNL, we quantified monomeric MLKL expression. Immunoblotting analysis showed that MLKL expression varied in the six ovarian cancer cell lines (Figure 5A). Quantified MLKL expression was plotted against the IC50 values to CNL. Interestingly, monomeric MLKL expression inversely correlated with the IC50 values for CNL (Figure 5B), suggesting that monomeric MLKL protein expression possibly determines susceptibilities of ovarian cancer cells to CNL-induced cell death. In contrast, there was no correlation of RIPK1/RIPK3 expression with sensitivities to CNL-induced cell death (Figure 5C and 5D).
Figure 5. Significant correlation of MLKL expression with susceptibilities of ovarian cancer cells to ceramide nanoliposomes-induced cell death.

A, Extracted cellular proteins from the indicated ovarian cancer cell lines were subjected to immunoblot analysis with antibodies for MLKL, RIPK-1, RIPK-3 and β-actin. Equal amounts of protein were loaded in each lane. Three independent experiments were performed and representative images are shown. The intensity of MLKL protein expression was quantified using Image Lab. IC50 values for ceramide nanoliposomes are plotted with MLKL protein expression (B), RIPK1 protein expression (C) and RIPK3 protein expression (D). R2 values were determined by GraphPad prism.
Ovarian cancer usually metastasizes widely, and metastatic growth is the leading cause of death for patients (30, 31). Thus, cell motility and invasiveness are critical therapeutic targets for metastatic ovarian cancer. Considering that ceramide serves as a metastasis-suppressor lipid in ovarian cancer (14), it is important to determine effects of CNL on metastatic growth as evidenced by cell motility corresponding to lamellipodia formation and cell migration. Confocal microscopy demonstrated that CNL significantly decreased the formation of lamellipodia in SKOV3 cells (Figure 6A). Treatment with CNL for 18 h also showed a significant decrease in cell motility as assessed by transwell assay as compared with ghost nanoliposomes in SKOV3 cells (Figure 6B) and other ovarian cancer cells (A2780, A2780CP, and TOV112D cells; Supplementary Figure 8). Similar to inhibitory effects on cell motility, CNL also significantly reduced cell invasion by 50% relative to ghost nanoliposomes group (Figure 6C). Therefore, CNL is suggested to suppress the metastatic potential.
Figure 6. Effects of ceramide nanoliposomes on metastatic growth.

A, SKOV3 cells were treated with 30 μM ceramide or ghost nanoliposomes for 6 h. Cells were fixed and counterstained with TRITC-conjugated phalloidin (red) and Hoechst 33342 (blue). Imaging was performed by confocal microscopy and the cell number forming lamellipodia were determined by counting more than 300 cells. The values represent the percentage of cells forming lamellipodia relative to total cells and the data represent the mean ± SD (n = 4). Four independent experiments were performed and yellow arrows show lamellipodia. Statistical analyses were performed by unpaired, student t-test. B and C, SKOV3 cells were treated with 30 μM ceramide nanoliposomes or ghost nanoliposomes for 18 h and then the assay for migration (B) and invasion (C) was performed as described in “Materials and Methods”. Data are represented as the percentage compared with ghost nanoliposomes’ group. The data represent are the means ± SD (n = 9). Statistical analyses were performed by unpaired, Student t-test. D, SKOV3 cells (5 × 106 / mouse) were injected intraperitoneally into peritoneal cavity of 4 weeks-old female nude mice. One day after implantation of cells, mice were intraperitoneally treated with ceramide or ghost nanoliposomes (40 mg/kg/day) continuously for 3 days. Four weeks later after inoculation, mice were euthanized to determine metastatic growth in the mesentery. The number of metastatic nodules was determined and mouse body weight was measured (n = 5). The data represent the mean ± SD and the representative images of specimens from mice were shown. White arrowheads indicate metastatic nodules. Statistical analyses were performed by unpaired, student t-test.
To test the effects of CNL on ovarian cancer metastatic growth in vivo, we utilized a human ovarian SKOV3 cancer xenograft model (14). CNL or ghost nanoliposomes were administered intraperitoneally for 3 consecutive days after inoculation of SKOV3 cells into peritoneal cavity. After additional 4 weeks of no treatment, metastatic tumor growth in the peritoneal cavity was determined. The number of metastatic nodules in CNL-treated mice was reduced by approximately 50% without signs of systemic toxicities (Figure 6D). Taken together, these data suggest that CNL can limit the metastatic growth of ovarian cancer through both pro-death (necroptosis) and anti-metastatic mechanisms.
Discussion
In this study, we demonstrated that CNL activates MLKL, leading to necroptosis in ovarian cancer cell lines. CNL displayed cytotoxicity in multiple ovarian cancer cell lines. The IC50 values of growth inhibition for CNL (2 to 57 μM, Figure 1D and Supplementary Figure 1) are similar to those of pancreatic cancer cells (IC50 > 20 μM) (32) and breast cancer cells (IC50 > 4 μM) (33). Though the cytotoxic effects of CNL seem to be conserved across cancer types, the necroptotic mechanism of action seems to be unique to ovarian cancer and thus offers additional therapeutic options, especially to drug resistant ovarian cancers. Our data (Figure 1 and Supplementary Figure 1) indicated that CNL is able to significantly induce cell death in multiple ovarian cancer cell lines, including several cisplatin-resistant cell lines (A2780cp and PE04). Our studies are consistent with recent findings documenting dysfunctional ceramide metabolism in multidrug resistant cancer models (8, 10) and argues that targeting necroptotic cell death with ceramide-based therapy could be an attractive treatment strategy for overcoming chemoresistance.
Recent studies demonstrated that necroptosis is mediated through the RIPK-MLKL pathway (34–37). Intramolecular autophosphorylation of RIPK results in the phosphorylation and subsequent oligomerization and recruitment of MLKL to the plasma membrane (28). This translocation is believed to increase sodium influx, leading to water influx and ultimately plasma membrane rupture (38). It was reported that exogenous C2-ceramide caused programmed necrosis (39), however the molecular mechanism remains unknown. In the present study, our findings strongly implicate that CNL targets MLKL activation independently of RIPK1/RIPK3–regulated pathway. Recently, Chen et al. showed that MLKL is translocated to lipid rafts in the plasma membrane during necroptosis (40). Considering that the lipid rafts are enriched with several sphingolipids, including ceramides, and serve as a signaling platform (41, 42), we argue that CNL forms lipid rafts-mimetics on plasma membranes or promotes MLKL relocalization to the lipid rafts. Even though our molecular and pharmacological experimentation suggests that MLKL is a critical protein for executing CNL-induced cell death, we have not demonstrated if MLKL is directly targeted by nanoliposomal ceramide. Ongoing studies are aimed at elucidating the specific molecular association of ceramide with MLKL in a cell-free system.
Given our data demonstrating an inverse relationship between monomeric MLKL expression and CNL efficacy, it can be suggested that MLKL serves as a possible biomarker of predicting therapeutic efficiency of CNL-based therapy. A corollary to this finding is that necroptotic signaling molecules such as MLKL can serve as precision medicine biomarkers for all chemotherapeutic agents that selectively induce necroptosis.
Our major finding demonstrating that CNL exhibits a cytotoxic effect by inducing MLKL-dependent necroptosis in ovarian cancer supports the therapeutic utility of CNL due to this unique mechanism of action. Moreover, our studies demonstrating the dual effect of CNL to induce necroptosis as well as reduce metastases in ovarian cancer models may offer additional opportunities for combinatorial cancer therapies (43, 44). For example, CNL may synergize with many of the recently validated necroptotic reagents including FTY720, β-lapachone and kuguaglycoside c (45–48). In addition, validating MLKL as a CNL-mediated necroptotic target suggests the utility of rationally designing more specific MLKL therapeutics as necroptosis-inducing drugs. Nevertheless, more preclinical studies are needed to evaluate the potential for necroptosis-targeted cancer therapies to further augment the efficacy of anti-metastatic or cytotoxic therapies.
Supplementary Material
Acknowledgments
Financial support: JSPS KAKENHI Grants (16K11125 to K.K., 26462509 to M.T., 16K15697 to N.Y.), NIH NCI P01 (to M.K.), Mizutani Foundation for Glycoscience (to K.K.) and Takeda Science Foundation (to K.K.).
We express our gratitude to the laboratory members of the Biomedical Research Unit of Tohoku University Hospital and Department of Obstetrics and Gynecology (Tohoku University, Sendai, Japan) for critical discussion.
Footnotes
Materials and Methods for Supplementary Figures are available (Supplementary Materials and Methods).
References
- 1.Lengyel E. Ovarian cancer development and metastasis. Am J Pathol. 2010;177:1053–1064. doi: 10.2353/ajpath.2010.100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banerjee S, Kaye SB. New strategies in the treatment of ovarian cancer: current clinical perspectives and future potential. Clin Cancer Res. 2013;19:961–968. doi: 10.1158/1078-0432.CCR-12-2243. [DOI] [PubMed] [Google Scholar]
- 3.Kolesnick RN, Kronke M. Regulation of ceramide production and apoptosis. Annu Rev Physiol. 1998;60:643–665. doi: 10.1146/annurev.physiol.60.1.643. [DOI] [PubMed] [Google Scholar]
- 4.Hannun YA. Functions of ceramide in coordinating cellular responses to stress. Science. 1996;274:1855–1859. doi: 10.1126/science.274.5294.1855. [DOI] [PubMed] [Google Scholar]
- 5.Kitatani K, Idkowiak-Baldys J, Hannun YA. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal. 2008;20:1010–1018. doi: 10.1016/j.cellsig.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kitatani K, Taniguchi M, Okazaki T. Role of Sphingolipids and Metabolizing Enzymes in Hematological Malignancies. Molecules and cells. 2015;38:482–495. doi: 10.14348/molcells.2015.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krautbauer S, Meier EM, Rein-Fischboeck L, Pohl R, Weiss TS, Sigruener A, Aslanidis C, Liebisch G, Buechler C. Ceramide and polyunsaturated phospholipids are strongly reduced in human hepatocellular carcinoma. Biochim Biophys Acta. 2016;1861:1767–1774. doi: 10.1016/j.bbalip.2016.08.014. [DOI] [PubMed] [Google Scholar]
- 8.Senchenkov A, Litvak DA, Cabot MC. Targeting ceramide metabolism--a strategy for overcoming drug resistance. J Natl Cancer Inst. 2001;93:347–357. doi: 10.1093/jnci/93.5.347. [DOI] [PubMed] [Google Scholar]
- 9.Karahatay S, Thomas K, Koybasi S, Senkal CE, Elojeimy S, Liu X, Bielawski J, Day TA, Gillespie MB, Sinha D, Norris JS, Hannun YA, Ogretmen B. Clinical relevance of ceramide metabolism in the pathogenesis of human head and neck squamous cell carcinoma (HNSCC): attenuation of C(18)-ceramide in HNSCC tumors correlates with lymphovascular invasion and nodal metastasis. Cancer Lett. 2007;256:101–111. doi: 10.1016/j.canlet.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morad SA, Cabot MC. Ceramide-orchestrated signalling in cancer cells. Nat Rev Cancer. 2013;13:51–65. doi: 10.1038/nrc3398. [DOI] [PubMed] [Google Scholar]
- 11.Kester M, Bassler J, Fox TE, Carter CJ, Davidson JA, Parette MR. Preclinical development of a C6-ceramide NanoLiposome, a novel sphingolipid therapeutic. Biol Chem. 2015;396:737–747. doi: 10.1515/hsz-2015-0129. [DOI] [PubMed] [Google Scholar]
- 12.Torchilin VP. Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nat Rev Drug Discov. 2014;13:813–827. doi: 10.1038/nrd4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koshkaryev A, Piroyan A, Torchilin VP. Increased apoptosis in cancer cells in vitro and in vivo by ceramides in transferrin-modified liposomes. Cancer biology & therapy. 2012;13:50–60. doi: 10.4161/cbt.13.1.18871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kitatani K, Usui T, Sriraman SK, Toyoshima M, Ishibashi M, Shigeta S, Nagase S, Sakamoto M, Ogiso H, Okazaki T, Hannun YA, Torchilin VP, Yaegashi N. Ceramide limits phosphatidylinositol-3-kinase C2beta-controlled cell motility in ovarian cancer: potential of ceramide as a metastasis-suppressor lipid. Oncogene. 2016;35:2801–2812. doi: 10.1038/onc.2015.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ryland LK, Doshi UA, Shanmugavelandy SS, Fox TE, Aliaga C, Broeg K, Baab KT, Young M, Khan O, Haakenson JK, Jarbadan NR, Liao J, Wang HG, Feith DJ, Loughran TP, Jr, Liu X, Kester M. C6-ceramide nanoliposomes target the Warburg effect in chronic lymphocytic leukemia. PLoS One. 2013;8:e84648. doi: 10.1371/journal.pone.0084648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tagaram HR, Divittore NA, Barth BM, Kaiser JM, Avella D, Kimchi ET, Jiang Y, Isom HC, Kester M, Staveley-O’Carroll KF. Nanoliposomal ceramide prevents in vivo growth of hepatocellular carcinoma. Gut. 2011;60:695–701. doi: 10.1136/gut.2010.216671. [DOI] [PubMed] [Google Scholar]
- 17.Stover T, Kester M. Liposomal delivery enhances short-chain ceramide-induced apoptosis of breast cancer cells. J Pharmacol Exp Ther. 2003;307:468–475. doi: 10.1124/jpet.103.054056. [DOI] [PubMed] [Google Scholar]
- 18.Tran MA, Smith CD, Kester M, Robertson GP. Combining nanoliposomal ceramide with sorafenib synergistically inhibits melanoma and breast cancer cell survival to decrease tumor development. Clin Cancer Res. 2008;14:3571–3581. doi: 10.1158/1078-0432.CCR-07-4881. [DOI] [PubMed] [Google Scholar]
- 19.Liu X, Ryland L, Yang J, Liao A, Aliaga C, Watts R, Tan SF, Kaiser J, Shanmugavelandy SS, Rogers A, Loughran K, Petersen B, Yuen J, Meng F, Baab KT, Jarbadan NR, Broeg K, Zhang R, Liao J, Sayers TJ, Kester M, Loughran TP., Jr Targeting of survivin by nanoliposomal ceramide induces complete remission in a rat model of NK-LGL leukemia. Blood. 2010;116:4192–4201. doi: 10.1182/blood-2010-02-271080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang P, Fu C, Hu Y, Dong C, Song Y, Song E. C6-ceramide nanoliposome suppresses tumor metastasis by eliciting PI3K and PKCzeta tumor-suppressive activities and regulating integrin affinity modulation. Sci Rep. 2015;5:9275. doi: 10.1038/srep09275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 22.Galluzzi L, Kepp O, Kroemer G. RIP kinases initiate programmed necrosis. J Mol Cell Biol. 2009;1:8–10. doi: 10.1093/jmcb/mjp007. [DOI] [PubMed] [Google Scholar]
- 23.Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, Chan FK, Wu H. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339–350. doi: 10.1016/j.cell.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370:455–465. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoon S, Bogdanov K, Kovalenko A, Wallach D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ. 2016;23:253–260. doi: 10.1038/cdd.2015.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanzer MC, Tripaydonis A, Webb AI, Young SN, Varghese LN, Hall C, Alexander WS, Hildebrand JM, Silke J, Murphy JM. Necroptosis signalling is tuned by phosphorylation of MLKL residues outside the pseudokinase domain activation loop. Biochem J. 2015;471:255–265. doi: 10.1042/BJ20150678. [DOI] [PubMed] [Google Scholar]
- 27.Huang D, Zheng X, Wang ZA, Chen X, He WT, Zhang Y, Xu JG, Zhao H, Shi W, Wang X, Zhu Y, Han J. The MLKL Channel in Necroptosis Is an Octamer Formed by Tetramers in a Dyadic Process. Mol Cell Biol. 2017:37. doi: 10.1128/MCB.00497-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 29.Su Z, Yang Z, Xie L, DeWitt JP, Chen Y. Cancer therapy in the necroptosis era. Cell Death Differ. 2016;23:748–756. doi: 10.1038/cdd.2016.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naora H, Montell DJ. Ovarian cancer metastasis: integrating insights from disparate model organisms. Nat Rev Cancer. 2005;5:355–366. doi: 10.1038/nrc1611. [DOI] [PubMed] [Google Scholar]
- 31.Tan DS, Agarwal R, Kaye SB. Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol. 2006;7:925–934. doi: 10.1016/S1470-2045(06)70939-1. [DOI] [PubMed] [Google Scholar]
- 32.Jiang Y, DiVittore NA, Kaiser JM, Shanmugavelandy SS, Fritz JL, Heakal Y, Tagaram HR, Cheng H, Cabot MC, Staveley-O’Carroll KF, Tran MA, Fox TE, Barth BM, Kester M. Combinatorial therapies improve the therapeutic efficacy of nanoliposomal ceramide for pancreatic cancer. Cancer biology & therapy. 2011;12:574–585. doi: 10.4161/cbt.12.7.15971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stover TC, Sharma A, Robertson GP, Kester M. Systemic delivery of liposomal short-chain ceramide limits solid tumor growth in murine models of breast adenocarcinoma. Clin Cancer Res. 2005;11:3465–3474. doi: 10.1158/1078-0432.CCR-04-1770. [DOI] [PubMed] [Google Scholar]
- 34.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 37.Vandenabeele P, Declercq W, Van Herreweghe F, Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal. 2010;3:re4. doi: 10.1126/scisignal.3115re4. [DOI] [PubMed] [Google Scholar]
- 38.Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 39.Zhu W, Wang X, Zhou Y, Wang H. C2-ceramide induces cell death and protective autophagy in head and neck squamous cell carcinoma cells. Int J Mol Sci. 2014;15:3336–3355. doi: 10.3390/ijms15023336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen X, Li W, Ren J, Huang D, He WT, Song Y, Yang C, Li W, Zheng X, Chen P, Han J. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24:105–121. doi: 10.1038/cr.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 42.Gulbins E, Kolesnick R. Raft ceramide in molecular medicine. Oncogene. 2003;22:7070–7077. doi: 10.1038/sj.onc.1207146. [DOI] [PubMed] [Google Scholar]
- 43.Liu P, Xu B, Shen W, Zhu H, Wu W, Fu Y, Chen H, Dong H, Zhu Y, Miao K, Xu W, Li J. Dysregulation of TNFalpha-induced necroptotic signaling in chronic lymphocytic leukemia: suppression of CYLD gene by LEF1. Leukemia. 2012;26:1293–1300. doi: 10.1038/leu.2011.357. [DOI] [PubMed] [Google Scholar]
- 44.Meng MB, Wang HH, Cui YL, Wu ZQ, Shi YY, Zaorsky NG, Deng L, Yuan ZY, Lu Y, Wang P. Necroptosis in tumorigenesis, activation of anti-tumor immunity, and cancer therapy. Oncotarget. 2016;7:57391–57413. doi: 10.18632/oncotarget.10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tabata K, Hamano A, Akihisa T, Suzuki T. Kuguaglycoside C, a constituent of Momordica charantia, induces caspase-independent cell death of neuroblastoma cells. Cancer Sci. 2012;103:2153–2158. doi: 10.1111/cas.12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Voigt S, Philipp S, Davarnia P, Winoto-Morbach S, Roder C, Arenz C, Trauzold A, Kabelitz D, Schutze S, Kalthoff H, Adam D. TRAIL-induced programmed necrosis as a novel approach to eliminate tumor cells. BMC Cancer. 2014;14:74. doi: 10.1186/1471-2407-14-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park EJ, Min KJ, Lee TJ, Yoo YH, Kim YS, Kwon TK. beta-Lapachone induces programmed necrosis through the RIP1-PARP-AIF-dependent pathway in human hepatocellular carcinoma SK-Hep1 cells. Cell Death Dis. 2014;5:e1230. doi: 10.1038/cddis.2014.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saddoughi SA, Gencer S, Peterson YK, Ward KE, Mukhopadhyay A, Oaks J, Bielawski J, Szulc ZM, Thomas RJ, Selvam SP, Senkal CE, Garrett-Mayer E, De Palma RM, Fedarovich D, Liu A, Habib AA, Stahelin RV, Perrotti D, Ogretmen B. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol Med. 2013;5:105–121. doi: 10.1002/emmm.201201283. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.