

Abstract
Background
Active surveillance (AS) is increasingly accepted for managing low-risk prostate cancer, yet there is no consensus about implementation. The lack of consensus is due in part to uncertainty about risks of disease progression, which have not been systematically compared or integrated across AS studies with variable surveillance protocols and dropout to treatment.
Objective
To (1) compare risks of upgrading from Gleason score (GS) ≤6 to ≥7 across AS studies after accounting for differences in surveillance intervals and competing treatments and (2) evaluate tradeoffs of more vs less frequent biopsies.
Design
Joint statistical model of longitudinal prostate-specific antigen (PSA) levels and risks of biopsy upgrading.
Setting
Johns Hopkins University (JHU), Canary Prostate Active Surveillance Study (PASS), University of California San Francisco (UCSF), and University of Toronto (UT) AS studies.
Patients
2,576 men aged 40–79 years diagnosed with GS ≤6 and clinical stage ≤T2 prostate cancer enrolled in 1995–2014.
Measurements
PSA levels and biopsy GS.
Results
After accounting for variable surveillance intervals and competing treatment, estimated risks of biopsy upgrading in PASS and UT were similar, but UCSF was higher and JHU lower. All cohorts produced a delay of 3–5 months in detecting upgrading under biennial biopsies starting after a first confirmatory biopsy relative to annual biopsies.
Limitations
The model does not account for possible biopsy GS misclassification.
Conclusions
Men in different AS studies exhibit different risks of biopsy upgrading after accounting for variable surveillance protocols and competing treatments. Nonetheless, despite these differences, the consequences of more versus less frequent biopsies appear to be similar across cohorts. Biennial biopsies appear to be an acceptable alternative to annual biopsies.
Funding Source
National Cancer Institute and others.
Keywords: active surveillance, biopsy upgrading, Gleason score, joint model, prostate-specific antigen, prostatic neoplasms
Introduction
Active surveillance (AS) is now the preferred approach for managing newly diagnosed, low-risk prostate cancer (PC) (1, 2). A recent guideline from the American Society of Clinical Oncology (ASCO) (2) supports the use of AS for low-risk PC and provides recommendations regarding target population and surveillance protocol. However, the recommendations lack specifics about how AS should be implemented.
Several studies are ongoing in North America (3–6) and Europe (7) to learn about the outcomes of AS, but they involve different populations, follow-up durations, inclusion criteria, surveillance protocols, and definitions of progression that lead to treatment referral.
The accumulation of data across multiple AS cohorts provides an opportunity to learn more about disease progression on AS. Indeed, recent analyses within specific cohorts (6, 8, 9) have pointed to prostate-specific antigen (PSA) level, number of prior stable biopsies, PSA density, and history of any negative biopsies as important predictors of progression. However, differences in AS implementation and compliance across cohorts preclude straightforward comparison of progression risks and prevent direct integration of results to inform best practices (10).
This article brings together individual-level data from four of the largest North American AS studies to compare and integrate their information about PC progression on AS. We evaluate whether progression rates are consistent across cohorts after standardizing inclusion criteria and the definition of progression and after controlling for variable surveillance intervals and risks of competing treatments. In addition, we examine the expected consequences of more versus less frequent biopsies across AS studies. This cross-cohort analysis is critical to assessing representativeness of individual studies and to developing sound AS guidelines that balance timely intervention against the morbidity from intensive surveillance.
Methods
Data sources
De-identified, individual-level data were obtained from four AS cohorts following institutional review board approval. Records included patient age and year of diagnosis, clinical and pathologic information at diagnosis, and dates and results of all surveillance tests, including PSA values and biopsy results, dates of curative treatment, and vital status. Cohort inclusion criteria, surveillance strategies, and conditions for referral to treatment are summarized in Table 1.
Table 1.
Eligibility criteria, surveillance protocol, and definition of progression in four active surveillance studies.
| Institution, start year, sample size* | Eligibility criteria | Follow-up schedule | Definition of progression according to protocol |
|---|---|---|---|
| Johns Hopkins University, 1995, N = 1,298 |
Younger men: T1c stage; < 0.15 μg/L/cc PSA density; ≤ 6 Gleason score; ≤ 2 positive cores; ≤ 50% core involvement Older men: ≤ T2a stage; < 10 μg/L PSA; ≤ 6 Gleason score |
~ 6 months: PSA, DRE ~ 12 months: biopsy |
Any adverse change on prostate biopsy |
| Canary Prostate Active Surveillance Study, 2008, N = 1,067 | ≤ T2 stage; 10-core biopsy ≤ 1 year or ≥ 2 biopsies ≥ 1 year |
~ 3 months: PSA; ~ 6 months: DRE; 6–12, 24, 48, and 72 months: biopsy |
Increase in biopsy Gleason score or in volume from < 34% to ≥ 34% of cores positive |
| University of Toronto, 1995, N = 1,104 |
Before 1999: For age <70 years: ≤ 6 Gleason score; ≤ 10 μg/L PSA; For age ≥70 years: ≤ 15 μg/L PSA or ≤ 3+4 Gleason score After 1999: Additionally, ≤ 20 μg/L PSA and ≤ 3+4 Gleason score in men with clinically significant comorbidities or < 10 year life expectancy |
First year: ~ 3 months: PSA; ~ 12 months: biopsy Second year: ~ 3 months: PSA Third year and beyond: ~ 6 months: PSA ~ 36–48 months: biopsy |
Histologic upgrading or clinical progression between biopsies; until 2009 also < 3 years PSA doubling time |
| University of California San Francisco, 1990, N = 1,319 | Has evolved over time Currently: ≤ T2 Stage; ≤ 10 μg/L PSA; ≤ 6 Gleason score; ≤ 33% positive cores; ≤ 50% core involvement; Exceptions can be made |
~ 3 months: PSA; ~ 6 months: Transrectal ultrasound ~ 12–24 months: biopsy depending on risk |
Increase to ≥ 3+4 Gleason score, > 33% positive cores, or > 50% involvement |
Sample size used in this study before exclusions, which may be different from previous published papers.
means “every”.
Johns Hopkins University (JHU) (4)
The JHU study began enrollment in 1995. Eligibility criteria include PSA density <0.15 μg/L/cc, clinical stage ≤T1c, Gleason score (GS) ≤6, ≤2 positive biopsy cores, and ≤50% involvement of any biopsy core with cancer. Men are monitored with PSA and digital rectal exam (DRE) every 6 months and with annual biopsies. Curative intervention is recommended for disease progression, defined as any adverse change on prostate biopsy.
Canary Prostate Active Surveillance Study (PASS) (3)
The PASS study began enrollment in 2008. Eligibility criteria include clinical stage ≤T2 disease and either a 10-core biopsy ≤1 year before enrollment or ≥2 biopsies with at least one ≤1 year before enrollment (3). Men are monitored with PSA tests every 3 months, DRE every 6 months, and biopsies 6–12, 24, 48, and 72 months after enrollment. Curative intervention is recommended if there is an increase in either biopsy GS or volume (from ≤33% to >33% positive cores).
University of Toronto (UT) (5)
The UT study began enrollment in 1995. Between 1995 and 1999, eligibility criteria included PSA ≤10 μg/L and GS ≤6 for men age < 70 years and PSA ≤15 μg/L and GS ≤3+4 for men age ≥70 years. In January 2000, eligibility criteria were expanded to include PSA ≤20 μg/L and GS ≤3+4 in men with substantial comorbidities or life expectancy <10 years. Men are monitored with PSA tests every 3 months for 2 years and then every 6 months. A confirmatory biopsy is performed within 12 months of the initial biopsy and then every 3–4 years until the patient reaches age 80 years. Curative intervention is recommended in case of histologic upgrading on repeat biopsy or clinical progression between biopsies (or PSA kinetics before 2009).
University of California San Francisco (UCSF) (6)
The UCSF study began enrollment in 1990. Eligibility criteria evolved over time and currently include PSA ≤10 μg/L, clinical stage ≤T2, biopsy GS ≤6, ≤33% positive cores, and ≤50% tumor in any single core. Selected patients who do not satisfy these criteria may be enrolled, and these comprise >30% of the cohort (6). Men are monitored with a confirmatory biopsy within 12 months of the initial biopsy and every 12–24 months thereafter. Curative intervention is recommended for any biopsy reclassification.
Data Synthesis and Analysis
Exclusion criteria and endpoint definitions
Patients diagnosed before 1995, age >80 years at enrollment, or with GS ≥7 at diagnosis were excluded from the analysis to obtain a more homogeneous population (Appendix Table 1). Further, we standardized the definition of disease progression to focus exclusively on biopsy upgrading, i.e., the first point at which a biopsy GS ≥7 is reached. We also defined competing treatments as initiation of treatment in the absence of biopsy upgrading, for example, in response to an increase in biopsy volume or an increase in PSA growth. Accounting for differences in risks of competing treatments is important because cohorts with a high frequency of competing treatments may appear to have a lower risk of biopsy upgrading than comparable cohorts with a low frequency of competing treatments even if their underlying risk of biopsy upgrading is similar.
Estimating underlying risks of upgrading
The empirical risk of upgrading is affected by both the surveillance protocol and the frequency of competing treatment. Our first objective is to compare underlying risks of upgrading across surveillance cohorts, i.e., risks that would be observed in the absence of competing treatments. A standard approach for obtaining underlying risks—a Kaplan-Meier (K-M) curve—is only valid if the competing event is independent of the event of interest. In the AS setting, upgrading and treatment initiation may be dependent. For example, if patients with higher PSA level or PSA velocity tend to initiate treatment more frequently, this could induce a dependence between treatment initiation and upgrading risk. In the case of a dependent competing risk, the K-M approach is biased (11, 12). However, a commonly-used alternative—the cumulative incidence estimate—captures the risk of the event of interest in the presence of the competing event and can therefore be sensitive to the competing risk. For example, two cohorts could have the same underlying risk of upgrading, but one with a higher incidence of competing treatment would appear to have a lower incidence of upgrading.
To overcome this problem, we first evaluate the dependence of the two events using a regression model that allows both events to depend on patient age and PSA kinetics. Allowing risks of both upgrading and competing treatment to depend on these common patient variables allows us to capture their potential dependence on each other. For example, if the risks of upgrading and treatment initiation both increase with PSA velocity, the two risks will be positively correlated. In practice, we estimate a so-called joint model for the evolution of the patient variables and the risks of upgrading and treatment initiation (13–15). After fitting the joint model, we extract the risk of upgrading in the absence of competing treatments using standard statistical formulas for obtaining marginal from conditional data summaries. This avoids the limitations of the K-M and cumulative incidence approaches.
The joint model has three components.
The PSA model. A linear mixed-effects model for log PSA captures heterogeneity in patient PSA kinetics, i.e., baseline PSA and PSA velocity, which is defined as the annual percent change in the PSA level.
The model for time to upgrading. A Weibull regression models the risk of upgrading given patient age and PSA kinetics. This model assumes that biopsy GS has no misclassification error. Thus, patients with all biopsy GS ≤6 biopsy are right censored for the event of upgrading, i.e., their upgrading event can only occur after the end of their follow-up, and patients with an observed biopsy GS ≥7 must have upgraded after the prior biopsy but before this biopsy.
The model for time to competing treatment. Another Weibull regression models the risk of treatment initiation given patient age and PSA kinetics.
We use Bayesian methods to estimate the three models simultaneously (Appendix 1). We do not attempt to model pathologic GS because previous work encountered substantial difficulties in doing so using serial biopsies among men on AS (16).
Predicting consequences under more versus less intensive surveillance protocols
Using the fitted joint model, we simulated times to upgrading in each cohort in the absence of competing treatments. Then, we superimposed surveillance protocols that involved regular biopsies every 1, 2, 3, and 4 years to determine the earliest point that biopsy would detect the upgrade. To acknowledge the clinical value of a confirmatory biopsy, we also consider biopsies every 2 years starting after the first confirmatory biopsy (i.e., 1 year after enrollment). The model predictions consist of the average number of biopsies performed and the average time delay between detection under each protocol relative to annual biopsies. To project the consequences of delaying the detection of biopsy upgrade, we harnessed a previously developed algorithm (17) that translates delays in diagnosis and treatment into relative risks of PC death. Specifically, the algorithm applies an established nomogram for PC death given the predicted change in PSA due to the delay since the true point of biopsy upgrade while keeping other prognostic covariates (e.g., volume) constant. Our projections are for clinically localized disease with GS ≥7.
Role of the funding source
The funding sources had no role in the design, conduct, or reporting of this study.
Results
Empirical risks of upgrading
Descriptive statistics for the four cohorts are shown in Table 2. Appendix Table 1 reports the number of patients omitted by our exclusion criteria to obtain more comparable patient populations; the resulting sample consists of 699, 613, 421, and 843 patients from JHU, PASS, UT, and UCSF cohorts, respectively. Figure 1 presents the empirical cumulative incidence (CI) of upgrading across cohorts after applying the exclusions. (Appendix Figure 1 shows corresponding curves before exclusions.) The empirical CI represents the cumulative risk of upgrading in the presence of competing treatment; i.e., if treatment is initiated for a given patient, that patient is no longer at risk of upgrading. Figure 1 shows that UCSF has the highest CI of disease upgrading; JHU has the lowest; and PASS and UT are intermediate between UCSF and JHU. In contrast, JHU has the highest risk of competing treatment, possibly due to the relatively high incidence of reclassification by volume-only in this cohort, which accounts for about half the reclassified patients (4), whereas the other cohorts have similar, lower risks of competing treatments.
Table 2.
Descriptive statistics for four active surveillance cohorts.
| JHU N = 913 |
PASS N = 1,067 |
UT N = 1,104 |
UCSF N = 1,319 |
|
|---|---|---|---|---|
| Year of Diagnosis | ||||
| ≤ 1995 | 8 (0.9%) | 0 | 9 (0.8%) | 8 (0.6%) |
| 1995–2000 | 91 (10.0%) | 0 | 176 (15.9%) | 57 (4.3%) |
| 2000–2005 | 273 (30.0%) | 30 (2.8%) | 235 (21.3%) | 221 (16.8%) |
| 2005–2010 | 458 (50.2%) | 416 (39.0%) | 389 (35.2%) | 473 (35.9%) |
| 2010–2015 | 65 (7.1%) | 621 (58.2%) | 263 (23.8%) | 552 (41.8%) |
| 2015+ | 0 | 0 | 6 (0.5%) | 8 (0.6%) |
| Missing | 18 (2.0%) | 0 | 26 (2.4%) | 8 (0.6%) |
|
| ||||
| Age at diagnosis (years) | ||||
| ≤ 40 | 0 | 1 (0.09%) | 0 | 0 |
| 40–50 | 12 (1.3%) | 44 (4.1%) | 33 (3.0%) | 66 (5.0%) |
| 50–60 | 143 (15.7%) | 296 (27.7%) | 197 (17.8%) | 412 (31.2%) |
| 60–70 | 548 (60.0%) | 566 (53.0%) | 464 (42.0%) | 618 (46.8%) |
| 70–80 | 204 (22.3%) | 158 (14.8%) | 337 (30.5%) | 202 (15.3%) |
| 80+ | 6 (0.6%) | 2 (0.2%) | 47 (4.2%) | 21 (1.6%) |
| Missing | 0 | 0 | 26 (2.3%) | 0 |
|
| ||||
| Gleason Score at Diagnosis | ||||
| 4 | 3 (0.3%) | 1 (0.09%) | 7 (0.6%) | 9 (0.7%) |
| 5 | 13 (1.4%) | 3 (0.3%) | 40 (3.6%) | 16 (1.2%) |
| 6 | 876 (95.9%) | 984 (92.2%) | 887 (80.3%) | 1126 (85.4%) |
| 7+ | 2 (0.2%) | 79 (7.4%) | 148 (13.4%) | 154 (11.7%) |
| Missing | 19 (2.1%) | 0 | 22 (2.0%) | 15 (1.1%) |
|
| ||||
| Clinical Stage | ||||
| T1 | 913 (100.0%) | 947 (77.8%) | 859 (77.8%) | 901 (68.3%) |
| T2 (not otherwise specified) | 0 | 0 | 4 (0.4%) | 46 (3.5%) |
| T2a | 0 | 113 (12.4%) | 133 (12.0%) | 284 (21.5%) |
| T2b or higher | 0 | 7 (0.6%) | 42 (3.8%) | 75 (5.7%) |
| Missing | 0 | 0 | 66 (6.0%) | 13 (1.0%) |
|
| ||||
| Biopsies per person | ||||
| 0 | 2 (0.2%) | 0 | 26 (2.4%) | 0 |
| 1 | 289 (31.6%) | 363 (34.0%) | 318 (28.8%) | 288 (21.8%) |
| 2 | 336 (36.8%) | 431 (40.4%) | 472 (42.8%) | 511 (38.7%) |
| 3 | 145 (15.9%) | 179 (16.8%) | 224 (20.3%) | 250 (19.0%) |
| 4 | 77 (8.4%) | 70 (6.5%) | 48 (4.3%) | 133 (10.1%) |
| 5 | 30 (3.3%) | 18 (1.7%) | 13 (1.2%) | 62 (4.7%) |
| 6+ | 34 (3.7%) | 6 (0.5%) | 3 (0.3%) | 62 (4.7%) |
|
| ||||
| PSA at enrollment (μg/L) (mean, sd)* | ||||
| ≤ 4 | 259 (2.4, 1.1) | 269 (2.5, 1.0) | 219 (2.6, 10.1) | 270 (2.6, 1.0)) |
| 4–8.0 | 491 (5.4, 1.1) | 561 (5.5, 1.0) | 426 (5.9, 1.1) | 777 (5.6, 1.1)) |
| 8.0–12.0 | 79 (9.5, 1.1) | 109 (9.6, 1.1) | 156 (9.6, 1.1) | 202 (9.6, 1.1)) |
| 12.0+ | 21 (15.3, 2.6) | 39 (16.3, 3.4) | 57 (15.9, 5.2) | 70 (17.4, 7.2) |
| Missing | 63 | 89 | 246 | 0 |
|
| ||||
| PSA tests per person | ||||
| 0 | 0 | 0 | 17 (1.5%) | 0 |
| ≤ 5 | 308 (33.7%) | 170 (15.9%) | 150 (13.6%) | 102 (7.7%) |
| 5–10 | 336 (36.8%) | 387 (36.3%) | 222 (20.1%) | 331 (25.1%) |
| 10–20 | 240 (26.2%) | 372 (34.9%) | 435 (39.4%) | 477 (36.2%) |
| 20–30 | 26 (2.8%) | 127 (11.9%) | 198 (17.9%) | 267 (20.2%) |
| 30+ | 3 (0.3%) | 11 (1.0%) | 82 (7.4%) | 142 (10.8%) |
|
| ||||
| Number with disease reclassification | ||||
| Biopsy upgrade | 178 (19.5%) | 240 (22.5%) | 364 (33.0%) | 599 (45.4% |
| Volume change without upgrade | 234 (25.6%) | 169 (15.8%) | 238 (21.6%) | 240 (18.2%) |
At enrollment or at first PSA on active surveillance 3 months prior or up to 6 months after enrollment.
JHU – Johns Hopkins University; PASS – Canary Prostate Active Surveillance Study; UT – University of Toronto; UCSF – University of California San Francisco.
Figure 1.
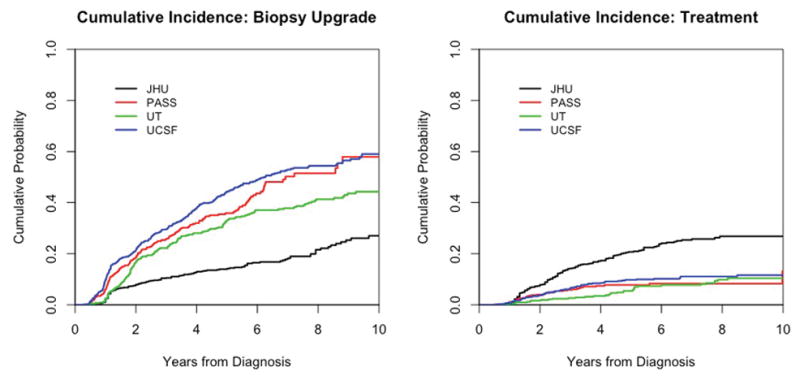
Empirical cumulative incidence of biopsy upgrading and competing treatments across active surveillance studies after applying exclusions to yield more comparable patient characteristics across cohorts (Appendix 1).
Risks of upgrading and associations with baseline PSA and PSA velocity
Exploratory plots show a range of PSA profiles across 3 selected patients from each cohort, illustrating that the PSA models capture the observed diversity (Appendix Figure 2). Mean PSA velocity is similar across the cohorts, with an estimated 5% (95% confidence interval (4%, 6%)) annual increase for JHU, 6% (4%, 7%) for PASS, 8% (6%, 9%) for UT, and 7% (6%,8%) for UCSF. Across cohorts, men with higher PSA levels tend to have higher risks of upgrading as indicated by the baseline PSA (intercept) and/or PSA velocity (slope) significantly associated with the risk of upgrading in every cohort (Appendix Table 2). Older men tend to have higher risks of upgrading, most notably in JHU and UT, and lower risks of treatment, most notably in JHU.
Annual risks of upgrading
Figure 2 shows the estimated cumulative risks of upgrading by year in the absence of competing treatments as derived from the joint model, where disease upgrading is considered to occur in the interval between a last negative (no cancer or GS ≤6) biopsy and a first positive (GS ≥7) biopsy. (Appendix Figure 3 shows empirical K-M curves in which upgrading occurs at the first observed biopsy GS ≥7.) While the upgrading risk is still highest in UCSF and lowest in JHU, the risk in UT closely matches the risk in PASS, in contrast to the empirical results in Figure 1. This convergence is due to the interval censoring, which impacts the estimates from UT more than other cohorts due its longer surveillance intervals.
Figure 2.

Model-based cumulative probabilities of biopsy upgrading and competing treatments (each in the absence of the other) by cohort. For each curve, we assume baseline PSA and PSA velocity fixed at cohort-specific mean values. Estimates accommodate interval censoring for biopsy upgrading between the last negative and first positive biopsy.
Appendix Figure 4 shows the estimated cumulative risks of upgrading in the absence of competing treatments for a man age 60 years at entry and specified values for baseline PSA and PSA velocity. Conditional on his PSA kinetics, comparisons across cohorts may shift. Upgrading risks are closer in PASS, UCSF, and UT for lower PSA levels (3 μg/L) at entry, but the curves fan out for men with higher PSA levels (6 μg/L) at entry, with UCSF showing a marked increase in the risk of upgrading with baseline PSA level. This finding is consistent with Appendix Table 2, which shows a stronger positive association between PSA at entry and the risk of upgrading in UCSF than in the other cohorts. Even when age and PSA kinetics are held constant, the JHU cohort still has a lower risk of upgrading than the other cohorts.
More versus less intensive surveillance protocols
Table 3 shows model-predicted outcomes over a range of surveillance intervals. For example, on average, biennial biopsies starting at enrollment reduces the number of biopsies relative to annual biopsies by 42–48% but delays detection of upgrading by 6–8 months. Biennial biopsies starting after the first confirmatory biopsy (1 year after enrollment) reduces the number of biopsies relative to annual biopsies by 32–38% but delays detection of upgrading by only 3–5 months.
Table 3.
Predicted outcomes under surveillance protocols that biopsy every 1, 2, 3, and 4 years for up to 12 years for men age 60 years at enrollment.
| Cohort | Number of years between biopsies | Time to upgrade detection | Number of biopsies | Delay in upgrade detection relative to annual biopsies | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||||
| Mean | SD | Median | Q1 | Q3 | Mean | SD | Median | Q1 | Q3 | Mean | SD | Median | Q1 | Q3 | ||
| JHU | 1 | 5.18 | 3.22 | 5 | 2 | 8 | 5.18 | 3.22 | 5 | 2 | 8 | (reference) | ||||
| 2 | 5.75 | 3.20 | 6 | 2 | 8 | 2.87 | 1.60 | 3 | 1 | 4 | 0.56 | 0.50 | 1 | 0 | 1 | |
| 3 | 6.32 | 3.20 | 6 | 3 | 9 | 2.11 | 1.07 | 2 | 1 | 3 | 1.14 | 0.80 | 1 | 0 | 2 | |
| 4 | 6.94 | 3.10 | 8 | 4 | 8 | 1.74 | 0.78 | 2 | 1 | 2 | 1.76 | 1.08 | 2 | 1 | 3 | |
| 2* | 5.58 | 3.41 | 6 | 2 | 8 | 3.71 | 1.82 | 4 | 2 | 5 | 0.40 | 0.49 | 0 | 0 | 1 | |
|
| ||||||||||||||||
| PASS | 1 | 3.86 | 3.07 | 3 | 1 | 6 | 3.86 | 3.07 | 3 | 1 | 6 | (reference) | ||||
| 2 | 4.49 | 3.00 | 4 | 2 | 6 | 2.24 | 1.5 | 2 | 1 | 3 | 0.63 | 0.48 | 1 | 0 | 1 | |
| 3 | 5.15 | 2.89 | 3 | 3 | 6 | 1.72 | 0.96 | 1 | 1 | 2 | 1.29 | 0.78 | 1 | 1 | 2 | |
| 4 | 5.85 | 2.75 | 4 | 4 | 8 | 1.46 | 0.69 | 1 | 1 | 2 | 2.00 | 1.08 | 2 | 1 | 3 | |
| 2* | 4.15 | 3.30 | 4 | 1 | 6 | 2.91 | 1.82 | 3 | 1 | 4 | 0.3 | 0.46 | 0 | 0 | 1 | |
|
| ||||||||||||||||
| UT | 1 | 3.29 | 2.94 | 2 | 1 | 5 | 3.29 | 2.94 | 2 | 1 | 5 | (reference) | ||||
| 2 | 3.99 | 2.83 | 2 | 2 | 6 | 1.99 | 1.42 | 1 | 1 | 3 | 0.69 | 0.46 | 1 | 0 | 1 | |
| 3 | 4.74 | 2.72 | 3 | 3 | 6 | 1.58 | 0.91 | 1 | 1 | 2 | 1.45 | 0.75 | 2 | 1 | 2 | |
| 4 | 5.48 | 2.57 | 4 | 4 | 8 | 1.37 | 0.64 | 1 | 1 | 2 | 2.18 | 1.05 | 3 | 1 | 3 | |
| 2* | 3.53 | 3.17 | 2 | 1 | 6 | 2.54 | 1.78 | 2 | 1 | 4 | 0.24 | 0.43 | 0 | 0 | 0 | |
|
| ||||||||||||||||
| UCSF | 1 | 3.64 | 2.92 | 3 | 1 | 5 | 3.64 | 2.92 | 3 | 1 | 5 | (reference) | ||||
| 2 | 4.27 | 2.84 | 4 | 2 | 6 | 2.14 | 1.42 | 2 | 1 | 3 | 0.63 | 0.48 | 1 | 0 | 1 | |
| 3 | 4.97 | 2.75 | 3 | 3 | 6 | 1.66 | 0.92 | 1 | 1 | 2 | 1.33 | 0.78 | 2 | 1 | 2 | |
| 4 | 5.67 | 2.63 | 4 | 4 | 8 | 1.42 | 0.66 | 1 | 1 | 2 | 2.03 | 1.08 | 2 | 1 | 3 | |
| 2* | 3.92 | 3.14 | 4 | 1 | 6 | 2.79 | 1.74 | 3 | 1 | 4 | 0.28 | 0.45 | 0 | 0 | 1 | |
SD – standard deviation; Q1 – first quartile; Q3 – first quartile; JHU – Johns Hopkins University; PASS – Canary Prostate Active Surveillance Study; UT – University of Toronto; UCSF – University of California San Francisco.
Biopsy every 2 years starting after one year.
Table 4 provides more detail about annual versus biennial surveillance protocols. Except for year 1, annual probabilities of upgrades are reasonably constant. Thus, while both biennial strategies materially reduce the number of biopsies, biennial surveillance starting after a first confirmatory biopsy may represent a more acceptable balance. To assess the clinical consequences of detection delays, Appendix Figure 5 shows that the hazard of PC death increases with delayed detection relative to the point of true upgrading. However, for the range of delays assessed (1–4 years), this increase is <3% across cohorts. While this long-term prediction provides a useful benchmark, it assumes other prognostic covariates (e.g., volume) remain constant during the delay.
Table 4.
Predicted outcomes under surveillance protocols that biopsy every 1 or 2 years.
| Cohort | Year | Incidence | No. upgrades | Annual | Biennial1 | Biennial2* | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||||||
| Annual | Cum. | No. biopsies | Cum. biopsies | No. biopsies | Cum. biopsies | Biopsies prevented† | No. biopsies | Cum. biopsies | Biopsies prevented† | |||
| JHU | 1 | 4% | 4% | 26 | 699 | 699 | 699 | 699 | 0 | |||
| 2 | 3% | 7% | 21 | 673 | 1372 | 699 | 699 | 673 | 673 | 1372 | 0 | |
| 3 | 3% | 9% | 19 | 651 | 2023 | |||||||
| 4 | 2% | 12% | 16 | 633 | 2656 | 673 | 1372 | 1284 | 651 | 2023 | 633 | |
| 5 | 2% | 14% | 14 | 617 | 3272 | |||||||
| 6 | 2% | 16% | 15 | 602 | 3875 | 633 | 2004 | 1870 | 617 | 2640 | 1235 | |
| 7 | 2% | 18% | 15 | 588 | 4463 | |||||||
| 8 | 2% | 20% | 15 | 573 | 5036 | 602 | 2607 | 2429 | 588 | 3228 | 1808 | |
| 9 | 2% | 22% | 13 | 558 | 5594 | |||||||
| 10 | 2% | 24% | 11 | 545 | 6139 | 573 | 3180 | 2959 | 558 | 3786 | 2353 | |
|
| ||||||||||||
| PASS | 1 | 20% | 20% | 121 | 613 | 613 | 613 | 613 | 0 | |||
| 2 | 8% | 28% | 50 | 492 | 1105 | 613 | 613 | 492 | 492 | 1105 | 0 | |
| 3 | 6% | 34% | 35 | 442 | 1547 | |||||||
| 4 | 5% | 38% | 29 | 407 | 1954 | 492 | 1105 | 849 | 442 | 1547 | 407 | |
| 5 | 4% | 42% | 25 | 379 | 2333 | |||||||
| 6 | 3% | 46% | 21 | 354 | 2687 | 407 | 1512 | 1175 | 379 | 1926 | 761 | |
| 7 | 3% | 49% | 19 | 333 | 3020 | |||||||
| 8 | 3% | 51% | 16 | 314 | 3334 | 354 | 1866 | 1468 | 333 | 2259 | 1075 | |
| 9 | 3% | 54% | 16 | 298 | 3631 | |||||||
| 10 | 2% | 56% | 13 | 282 | 3913 | 314 | 2180 | 1733 | 298 | 2556 | 1357 | |
|
| ||||||||||||
| UT | 1 | 22% | 22% | 91 | 421 | 421 | 421 | 421 | 0 | |||
| 2 | 6% | 28% | 25 | 330 | 751 | 421 | 421 | 330 | 330 | 751 | 0 | |
| 3 | 4% | 32% | 17 | 305 | 1056 | |||||||
| 4 | 3% | 35% | 13 | 288 | 1344 | 330 | 751 | 593 | 305 | 1056 | 288 | |
| 5 | 3% | 37% | 12 | 275 | 1619 | |||||||
| 6 | 2% | 40% | 10 | 263 | 1882 | 288 | 1039 | 843 | 275 | 1331 | 552 | |
| 7 | 2% | 42% | 8 | 253 | 2136 | |||||||
| 8 | 2% | 44% | 8 | 246 | 2381 | 263 | 1302 | 1079 | 253 | 1584 | 797 | |
| 9 | 2% | 45% | 7 | 237 | 2618 | |||||||
| 10 | 1% | 47% | 6 | 230 | 2848 | 246 | 1548 | 1300 | 237 | 1821 | 1027 | |
|
| ||||||||||||
| UCSF | 1 | 25% | 25% | 214 | 843 | 843 | 843 | 843 | 0 | |||
| 2 | 10% | 36% | 88 | 629 | 1472 | 843 | 843 | 629 | 629 | 1472 | 0 | |
| 3 | 8% | 43% | 64 | 542 | 2014 | |||||||
| 4 | 6% | 49% | 47 | 478 | 2492 | 629 | 1472 | 1020 | 542 | 2014 | 478 | |
| 5 | 5% | 54% | 39 | 431 | 2923 | |||||||
| 6 | 4% | 57% | 33 | 392 | 3314 | 478 | 1950 | 1364 | 431 | 2445 | 870 | |
| 7 | 3% | 61% | 29 | 359 | 3674 | |||||||
| 8 | 3% | 64% | 26 | 330 | 4004 | 392 | 2342 | 1662 | 359 | 2804 | 1200 | |
| 9 | 2% | 66% | 19 | 304 | 4307 | |||||||
| 10 | 2% | 69% | 20 | 284 | 4592 | 330 | 2672 | 1920 | 304 | 3108 | 1484 | |
JHU – Johns Hopkins University; PASS – Canary Prostate Active Surveillance Study; UT – University of Toronto; UCSF – University of California San Francisco.
Biopsy every 2 years starting after one year.
Relative to annual biopsies.
Discussion
Active surveillance continues to evolve and gain acceptance as a strategy for managing low-risk PC, but consensus on an optimal protocol is lacking (10). To inform any consensus, we need a representative assessment of the extent to which PC patients on AS face risks of disease progression and adverse downstream outcomes. This study is the first to bring together individual-level data across AS studies for such an assessment.
Integrating evidence across AS studies means confronting major differences in inclusion criteria, protocols, and endpoints. It also means addressing differences in competing treatment. All these differences materially affect published risks of progression. However, even when we level the playing field in terms of inclusion criteria, surveillance intervals, and endpoints and present results in the absence of competing treatment, there appear to be fundamental differences in underlying risks of biopsy upgrading across cohorts. Indeed, the estimated 10-year cumulative risk of upgrading in the absence of competing treatments ranges from 25% (JHU) to 65% (UCSF).
How can we explain these persistent differences? They may be partly explained by minor differences in biopsy cores sampled but are more likely due to variations in the profiles of entering patients that are not captured by the characteristics measured in this study. For example, most JHU patients were very low risk with PSA density <0.15 μg/L/cc. Since low PSA density may result from a higher prostate volume, the chance of identifying high-grade foci could have been reduced. This is supported by the results of imposing this condition on PASS patients (the only other cohort that recorded this information), where approximately 75% of patients had PSA density <0.15 μg/L/cc. While the estimated risk of upgrading in this subset is modestly reduced relative to the risk in the whole cohort (Appendix Figure 6), it does not become comparable to the risk in the JHU cohort. The low risk of upgrading in JHU may also reflect other factors associated with selection into this cohort, such as the biopsy volume. JHU restricted enrollment to ≤2 cores with cancer and <50% involvement of every core, while UCSF allowed up to 33% of cores and included some patients with higher volumes.
Our results indicate that a single AS study may not reflect the risks of PC progression in another population. Nonetheless, our analysis suggests that consequences of varying the intensity of surveillance are highly robust across levels of underlying risk. For example, biennial biopsies starting at enrollment will detect upgrading on average only 6–8 months later than annual biopsies, a time delay that is unlikely to confer a substantially increased risk of adverse downstream outcomes based on our assessment and on published studies of treatment delays in low- to moderate-risk patients (18, 19). Biennial biopsies starting after a confirmatory biopsy (1 year after enrollment) imply even shorter expected delays.
A crucial assumption of our analysis is that the risks of upgrading and competing treatment are independent given patient age and PSA kinetics and that no other factors correlate with these events.
A key limitation of this work is that we only consider biopsy upgrading rather than true, pathologic upgrading. Because of potential misclassification, biopsy grade may not capture the true pathologic grade, which is a better surrogate for aggressiveness. We previously showed (16) that we cannot identify the true grade trajectory from serial biopsies on AS even if we know the misclassification probabilities. Thus, grade progression is the first biopsy with GS ≥7, and our analysis does not allow grade to have progressed before the last biopsy with GS ≤6. Our analysis also does not allow for false positive biopsies or address other questions of interest, such as inclusion of GS 3+4 patients, optimal ages of enrollment, or the appropriateness of AS for African-American men. Finally, we do not address questions about magnetic resonance imaging or novel biomarkers for predicting disease progression or aggressiveness; further research is needed to tailor surveillance protocols to underlying or evolving risk of progression.
In conclusion, our analysis reveals practical challenges to medical decision making and guideline development around AS. In particular, risks of cancer upgrading do not generalize from one cohort to another even after controlling for differences in eligibility criteria, surveillance protocols, and competing treatments. However, expected delays to detecting upgrading under more versus less intensive protocols are robust across cohorts. Our analysis suggests that biennial rather than annual biopsies may be justified, particularly given the invasiveness and potential morbidity of annual biopsies. This finding provides quantitative justification for the ASCO clinical practice guideline (2), which also recommends less frequent biopsies after a confirmatory biopsy within a year of entering AS. Both that guideline and our analysis support less frequent biopsies for men on AS, which should reduce morbidity and complications of this conservative approach as it becomes further established as a preferred method for managing low-risk prostate cancer.
Supplementary Material
Acknowledgments
Funding/Support This work was supported by the National Cancer Institute Award Number R01 CA183570 for the Prostate Modeling to Identify Surveillance Strategies (PROMISS) consortium (all authors), R01 CA160239 for statistical methods to study cancer recurrence using longitudinal multi-state models (LYTI), P50 CA097186 as part of the Pacific Northwest Prostate Cancer Specialized Program in Research Excellence (SPORE) (ASL), and U01 CA199338 as part of the Cancer Intervention and Surveillance Modeling Network (CISNET) (RG, RE); the Canary Foundation (DWL, LFN); Genomic Health Inc. and the US Department of Defense Translational Impact Award for Prostate Cancer Award Number W81XWH-13-2-0074 (PRC, MRC, JEC); and Prostate Cancer Canada (LHK).
Role of the Funders/Sponsors: The funders/sponsors had no role in the design and conduct of the study; collection, management, analysis, or interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit the manuscript for publication.
Footnotes
Disclaimer: The contents are solely the responsibility of the authors and do not necessarily represent the official views of the funders/sponsors.
Reproducible Research Statement: Study protocol: Not available. Statistical code: Available from Dr. Inoue (linoue@u.washington.edu). Data set: Not available.
References
- 1.Tosoian JJ, Carter HB, Lepor A, Loeb S. Active surveillance for prostate cancer: current evidence and contemporary state of practice. Nat Rev Urol. 2016;13(4):205–15. doi: 10.1038/nrurol.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen RC, Rumble RB, Jain S. Active Surveillance for the Management of Localized Prostate Cancer (Cancer Care Ontario guideline): American Society of Clinical Oncology Clinical Practice Guideline Endorsement Summary. J Oncol Pract. 2016;12(3):267–9. doi: 10.1200/JOP.2015.010017. [DOI] [PubMed] [Google Scholar]
- 3.Newcomb LF, Thompson IM, Jr, Boyer HD, Brooks JD, Carroll PR, Cooperberg MR, et al. Outcomes of Active Surveillance for Clinically Localized Prostate Cancer in the Prospective, Multi-Institutional Canary PASS Cohort. J Urol. 2016;195(2):313–20. doi: 10.1016/j.juro.2015.08.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tosoian JJ, Mamawala M, Epstein JI, Landis P, Wolf S, Trock BJ, et al. Intermediate and Longer-Term Outcomes From a Prospective Active-Surveillance Program for Favorable-Risk Prostate Cancer. J Clin Oncol. 2015;33(30):3379–85. doi: 10.1200/JCO.2015.62.5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klotz L, Vesprini D, Sethukavalan P, Jethava V, Zhang L, Jain S, et al. Long-term follow-up of a large active surveillance cohort of patients with prostate cancer. J Clin Oncol. 2015;33(3):272–7. doi: 10.1200/JCO.2014.55.1192. [DOI] [PubMed] [Google Scholar]
- 6.Welty CJ, Cowan JE, Nguyen H, Shinohara K, Perez N, Greene KL, et al. Extended followup and risk factors for disease reclassification in a large active surveillance cohort for localized prostate cancer. J Urol. 2015;193(3):807–11. doi: 10.1016/j.juro.2014.09.094. [DOI] [PubMed] [Google Scholar]
- 7.Bul M, Zhu X, Valdagni R, Pickles T, Kakehi Y, Rannikko A, et al. Active surveillance for low-risk prostate cancer worldwide: the PRIAS study. Eur Urol. 2013;63(4):597–603. doi: 10.1016/j.eururo.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 8.Alam R, Carter HB, Landis P, Epstein JI, Mamawala M. Conditional probability of reclassification in an active surveillance program for prostate cancer. J Urol. 2015;193(6):1950–5. doi: 10.1016/j.juro.2014.12.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ankerst DP, Xia J, Thompson IM, Jr, Hoefler J, Newcomb LF, Brooks JD, et al. Precision Medicine in Active Surveillance for Prostate Cancer: Development of the Canary-Early Detection Research Network Active Surveillance Biopsy Risk Calculator. Eur Urol. 2015;68(6):1083–8. doi: 10.1016/j.eururo.2015.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bruinsma SM, Bangma CH, Carroll PR, Leapman MS, Rannikko A, Petrides N, et al. Active surveillance for prostate cancer: a narrative review of clinical guidelines. Nat Rev Urol. 2016;13(3):151–67. doi: 10.1038/nrurol.2015.313. [DOI] [PubMed] [Google Scholar]
- 11.Pepe MS, Mori M. Kaplan-Meier, marginal or conditional probability curves in summarizing competing risks failure time data? Statistics in Medicine. 1993;12(8):737–51. doi: 10.1002/sim.4780120803. [DOI] [PubMed] [Google Scholar]
- 12.Gooley TA, Leisenring W, Crowley J, Storer BE. Estimation of failure probabilities in the presence of competing risks: new representations of old estimators. Statistics in Medicine. 1999;18(6):695–706. doi: 10.1002/(sici)1097-0258(19990330)18:6<695::aid-sim60>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 13.Taylor JM, Park Y, Ankerst DP, Proust-Lima C, Williams S, Kestin L, et al. Real-time individual predictions of prostate cancer recurrence using joint models. Biometrics. 2013;69(1):206–13. doi: 10.1111/j.1541-0420.2012.01823.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiang AJ, Chen J, Chung YC, Huang HJ, Liou WS, Chang C. A longitudinal analysis with CA-125 to predict overall survival in patients with ovarian cancer. J Gynecol Oncol. 2014;25(1):51–7. doi: 10.3802/jgo.2014.25.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ibrahim JG, Chu H, Chen LM. Basic concepts and methods for joint models of longitudinal and survival data. J Clin Oncol. 2010;28(16):2796–801. doi: 10.1200/JCO.2009.25.0654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inoue LY, Trock BJ, Partin AW, Carter HB, Etzioni R. Modeling grade progression in an active surveillance study. Statistics in Medicine. 2014;33(6):930–9. doi: 10.1002/sim.6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inoue LY, Gulati R, Yu C, Kattan MW, Etzioni R. Deriving benefit of early detection from biomarker-based prognostic models. Biostatistics. 2013;14(1):15–27. doi: 10.1093/biostatistics/kxs018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boorjian SA, Bianco FJ, Jr, Scardino PT, Eastham JA. Does the time from biopsy to surgery affect biochemical recurrence after radical prostatectomy? BJU Int. 2005;96(6):773–6. doi: 10.1111/j.1464-410X.2005.05763.x. [DOI] [PubMed] [Google Scholar]
- 19.Fossati N, Rossi MS, Cucchiara V, Gandaglia G, Dell’Oglio P, Moschini M, et al. Evaluating the effect of time from prostate cancer diagnosis to radical prostatectomy on cancer control: Can surgery be postponed safely? Urol Oncol. 2017;35(4):150.e9–e15. doi: 10.1016/j.urolonc.2016.11.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.