

Abstract
We aimed to assess the utility of a novel, non-invasive method of detecting genomic alterations in patients with gastrointestinal malignancies i.e., the use of liquid biopsies to obtain blood-derived circulating tumor DNA (ctDNA) through an analysis of the genomic landscape of ctDNA (68 genes) from 213 patients with advanced gastrointestinal cancers. The most common cancer types were colorectal adenocarcinoma (N=55 (26%)), appendiceal adenocarcinoma (N=46 (22%)), hepatocellular carcinoma (N=31 (15%)), and pancreatic ductal adenocarcinoma (N=25 (12%)). The majority of patients (58%) had ≥1 characterized alteration (excluded variants of unknown significance). The median number of characterized alterations was 1 (range, 0–13). The number of detected alterations per patient varied between different cancer types: in hepatocellular carcinoma, 74% of patients (23/31) had ≥1 characterized alteration(s) versus 24% of appendiceal adenocarcinoma patients (11/46). The median percent ctDNA among characterized alterations was 2.50% (interquartile range, 0.76–8.96%). Overall, 95% of patients (117/123) had distinct molecular portfolios with 143 unique characterized alterations within 56 genes. Overall concordance rates of 96%, 94%, 95%, and 91%, respectively, were found between ctDNA and tissue-biopsy (N = 105 patients) in the four most common alterations (KRAS amplification, MYC amplification, KRAS G12V, and EGFR amplification). Of 123 patients with characterized alterations, >99% (122/123) (57% of entire population tested (122/213)) had one or more alterations potentially actionable by experimental or approved drugs. These observations suggest that many patients with gastrointestinal tumors, including difficult-to-biopsy malignancies like hepatocellular cancers, frequently have discernible and theoretically pharmacologically tractable ctDNA alterations that merit further studies in prospective trials.
Keywords: circulating tumor DNA, gastrointestinal tumors, sequencing
INTRODUCTION
Advances in tumor sequencing have made possible unprecedented data-driven decision-making in clinical and academic oncology, using panels of markers for the development and implementation of matched targeted therapies (1–4) and new research paradigms such as multi-histology “basket trials” (5). For example, the discovery of KRAS mutations predictive of primary resistance to cetuximab in colorectal cancer (CRC) (6) has led to changes in clinical use of this and other EGFR targeting agents. Indeed, KRAS, BRAF, NRAS, ERBB2, and MET (amongst other) mutations or amplifications all confer secondary resistance to EGFR blockade (7), and suggest the need for combination therapies (8). However, the implementation of this knowledge in the clinical setting relies on a robust sequencing protocol with a panel of actionable genes and alterations, and the ability to biopsy tumors, perhaps repeatedly, prior to the selection of each new treatment regimen. Traditional tissue biopsies are invasive and sometimes costly procedures that carry their own risks. Furthermore, in breast cancer, for instance, genetic characterization of primary tumors, lymph node metastases, and distant metastases found a great deal of heterogeneity, not only between metastases in general and the primary tumor, but also (and especially pronounced) between distant metastases, likely both because of random evolutionary forces and tissue-specific selective pressures (9,10). This diversity may not be appreciable with biopsy of a single solid tumor. More importantly, exclusively using data from biopsy of a single tumor in treatment decisions may be suboptimal, both because of missed opportunities for targeted therapy and because of inadvertent selection of drugs for which distinct resistant clones are already present. For example, a study of circulating tumor cells (CTCs) in breast cancer found that 54% of patient with estrogen-receptor (ER-) negative primary tumors expressed ER-positive circulating tumor cells (CTCs) (11).
The need for a method of easily repeatable, minimally invasive, and spatially unbiased tumor characterization makes technology such as next generation sequencing (NGS) of circulating tumor cell DNA (ctDNA) especially appealing (12). Cancer cells, either through direct secretion, or secretion secondary to immune cell phagocytosis of necrotic or apoptotic cell fragments, release ctDNA (13). This DNA can then be detected in various body compartments (e.g. blood), and sequenced. Because of the minimally invasive nature of a blood test (as opposed to tissue biopsy), the reproducibility of liquid biopsies has been leveraged for several proof-of-concept studies predicting response and resistance (14,15), as well as prognosis and recurrence (16,17). Here we examined the ctDNA mutational landscape of 213 patients with a variety of gastrointestinal malignancies.
METHODS
Patients
We performed a retrospective analysis of ctDNA liquid biopsies collected between October 2014 through December 2015 from 213 patients with gastrointestinal cancers at the University of California San Diego Moores Cancer Center. All patients in the study either had metastatic or locally advanced unresectable disease. Data was collected from a de-identified database with minimal clinical information present. Data analysis and consent acquisition was performed in accordance with UCSD IRB guidelines (NCT02478931). All authors had access to study data, and contributed to reviewing and approving the final manuscript.
Next Generation Sequencing
Next generation sequencing (NGS) of plasma ctDNA (liquid biopsies) was done by Guardant Health (www.guardanthealth.com/guardant360/), a College of American Pathologists (CAP)-accredited and Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory. This is a highly analytically/clinically sensitive and specific test, able to detect single molecules of tumor DNA in 10mL blood samples, >85% of single nucleotide polymorphisms (SNPs) found in tumors of patients with advanced cancers, with analytic specificity of >99.9999%. At the time of this study, this test detected alterations (e.g. mutations, fusions, copy number change, etc.) in a panel of 68 genes (Supplemental Table 1). Sequencing covered all cell-free DNA, including germline found in the bloodstream (e.g. as a result of immune lysis), as well as the ctDNA. The percent ctDNA was quantified by dividing the number of ctDNA SNP variants by the number of wild type DNA fragments at the respective nucleotide. Gene amplifications were reported by absolute copy number detected in plasma, as compared to normal controls from healthy patients included in each run (18).
Definition of actionability
An alteration was considered actionable if an FDA-approved drug or an investigational drug in a clinical trial could specifically target the resulting protein product, either directly (e.g. EGFR inhibition) or through a downstream pathway (e.g. mTOR inhibitor in the context of a PIK3CA mutation). For small molecule inhibitors, this classification was met if the drug directly impacted the aberration with low nanomolar 50% inhibitory concentration (IC50). Antibody therapeutics had to have primary selectivity against the altered protein. If an important downstream effector of protein products of a gene anomaly could be targeted, that anomaly was considered druggable. Finally, if a drug could recognize a protein that was differentially expressed on tumor versus normal cells, the gene responsible for that protein was considered potentially actionable regardless of whether or not the drug affected protein function.
Definition of Variant of Unknown Significance (VUS)
A non-synonymous change in the gene sequence for which the risk of malignancy was unclear or unknown was designated as a VUS. Characterization of variants (known versus unknown significance) was performed by N-of-One, Inc. (https://n-of-one.com/), using the published literature as well as publicly available databases, including COSMIC, cBioPortal, EVS, ExAC, and ClinVar. Novel variants are assessed for their incidence in somatic cancers vs. incidence in the germline and evaluated based on the literature for evidence of molecular effect based on cellular or biochemical assays, or clinical evidence of association with disease or predictive, prognostic, or diagnostic value. Variants with little available evidence may also be evaluated based on their location at a conserved amino acid or an amino acid that is a hotspot for mutation if the specific mutation has not been characterized. Protein prediction algorithms may be used to contribute to the analysis, but are not used as the sole basis for classification of an alteration. Synonymous alterations were not included in our analysis.
Data extraction and analysis
Data examined included the date(s) at which samples were collected for sequencing, gene alterations detected, the type and details of the alteration, the interpretation of the alteration, demographic information that was part of the deideintified report (gender, age, and histology of cancer), as well as the corresponding actionability classification for each alteration. Analysis was predominantly descriptive, but when appropriate the median and confidence intervals or (interquartile) range were reported. There were 213 individual patients who collectively underwent a total of 386 ctDNA liquid biopsies. When counting alterations in the patients who underwent multiple tests, an alteration was considered present if it appeared in at least one of the tests. When describing percent ctDNA for patients with multiple tests, we selected the larger value.
Assessment of concordance with solid tissue sequencing
A separate analysis compared the concordance between solid and liquid biopsy sequences in the four most common alterations (KRAS amplification, MYC amplification, KRAS G12V, and EGFR amplification) in patients who underwent both tests. Tissue biopsy genotyping was performed using a 315 gene next generation sequencing panel (https://www.foundationmedicine.com/). An overall concordance matrix was tabulated by examining detection of alteration(s) within the 68 genes included in both the liquid and tissue sequencing panels and iterating this across the 105 patients who had undergone both types of studies. The same process was repeated for the 16 genes that are evaluated for amplification in both liquid and tissue biopsies to yield the amplification concordance statistics.
Results
Patient Characteristics
Data from 213 patients’ gastrointestinal cancers were analyzed. The most common malignancies were colorectal adenocarcinoma (N=55 (26%)), appendiceal adenocarcinoma (N=46 (22%)), hepatocellular carcinoma (N=31 (15%)), and pancreatic ductal adenocarcinoma (N=25 (12%)). Within the overall population, the majority were men (N=124 (58%)) and the median age was 61 years (range, 25 to 92 years) (Table 1).
Table 1.
Patient characteristics
| Number of patients with ≥1 alteration(s) (includes VUS) | 149 (70.0%) | ||
|---|---|---|---|
|
| |||
| Number of patients with ≥1 characterizeda alteration(s) (excludes VUS) | 123 (58%) | ||
|
| |||
| Median number of characterizeda alterations per patient (range) | 1 (0–13) | ||
|
| |||
| Women | Men | Overall | |
| Number | 89 (41.8%) | 124 (58.2%) | 213 |
| Age (median, range) | 60 (25–92) | 62 (29–88) | 61 (25–92) |
| Cancer Type (N, %)* | |||
| Colorectal Adenocarcinoma | 27 (49.1%) | 28 (50.9%) | 55 (25.8%) |
| Appendix Adenocarcinoma | 23 (50.0%) | 23 (50.0%) | 46 (21.6) |
| Hepatocellular Carcinoma | 8 (25.8%) | 23 (74.2%) | 31(14.6%) |
| Pancreatic Ductal Adenocarcinoma | 15 (60.0%) | 10 (40.0%) | 25 (11.7%) |
| Cholangiocarcinoma | 4 (30.8%) | 9 (69.2%) | 13 (6.1%) |
| Gastric Adenocarcinoma | 6 (54.5%) | 5 (45.5%) | 11 (5.2%) |
| Esophageal Adenocarcinoma | 0 | 8 (100.0%) | 8 (3.8%) |
| Gastrointestinal Stromal Tumor (GIST) | 0 | 7 (100.0%) | 7 (3.3%) |
| Pancreatic Neuroendocrine Tumor | 0 | 5 (100.0%) | 5 (2.3%) |
| Anal Carcinoma | 3 (75.0%) | 1 (25.0%) | 4 (1.9%) |
| Gallbladder Carcinoma | 2 (66.7%) | 1 (33.3%) | 3 (1.4%) |
| Duodenum Adenocarcinoma | 0 | 2 (100.0%) | 2 (0.9%) |
| GI Carcinoma Small Cell | 0 | 1 (100.0%) | 1 (0.5%) |
| Small bowel adenocarcinoma (small intestine) | 0 | 1 (100.0%) | 1 (0.5%) |
| Stomach carcinoma (Signet ring cell carcinoma) | 1 (100.0%) | 0 | 1 (0.5%) |
Abbreviations: VUS = Variant of unknown significance
Characterized alterations do not, by definition, include VUSs
Percentages for women and men are the percent of cases within a given cancer type.
Overall mutational landscape
Number of alterations
The majority of patients (70%) had one or more detected alterations, of which 83% had ≥1 characterized alteration(s) (excluding VUS). Overall, 58% of patients had ≥1 characterized alterations (Table 1; Figure 1A). However, the number of patients with a given number of detected alterations decreased proportionally to the number of alterations, with only 15% (N=32) of our patients having more than three alterations of known significance (Figures 2A and 2B). The median number of characterized alterations per patient was 1 (range, 0–13); the median number of alterations that included VUS (but still excluded synonymous alterations) was 2 (range, 0 to 26). When examining the number of alterations by cancer histology, we found significant differences (p<.00001 (Kruskall-Wallis test)). Within the more common histologies in this study, this was best illustrated by the difference between hepatocellular carcinoma, where almost 74% of patients (23/31) had one or more characterized alterations, and appendiceal adenocarcinoma, in which 76% of patients (35/46) presented with no detectable alterations of known significance (Figure 2B).
Figure 1. Most frequently altered genes and percent ctDNA by gene.
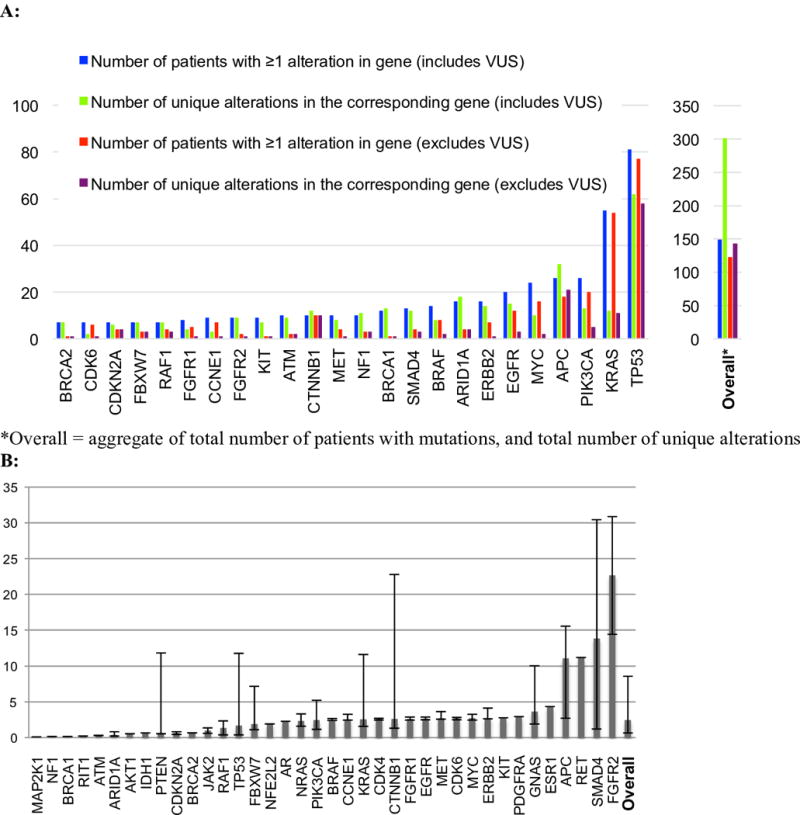
Abbreviations: VUS = variant of unknown significance
A: Bar plot representing the number of patients with an alteration(s) in a given gene, and the number of unique alterations in each gene. Unique alterations are defined as the number of specific alterations (point mutation, amplification, etc.) occurring in a given gene across the population. This plot only includes genes for which there were >5 patients with an alteration in the given gene; for others, refer to Figure S1. B: Bar plot representing median percent ctDNA (see Methods for definition) and 1st and 3rd quartiles. Only patients who had a characterized alteration in the specified gene were included in the calculation. Variants of unknown significance were excluded. In patients who had multiple tests that detected the same alteration, only the greatest value was included.
Figure 2. Number of patients with a given number of alterations and percent breakdown by cancer type (excludes VUS).
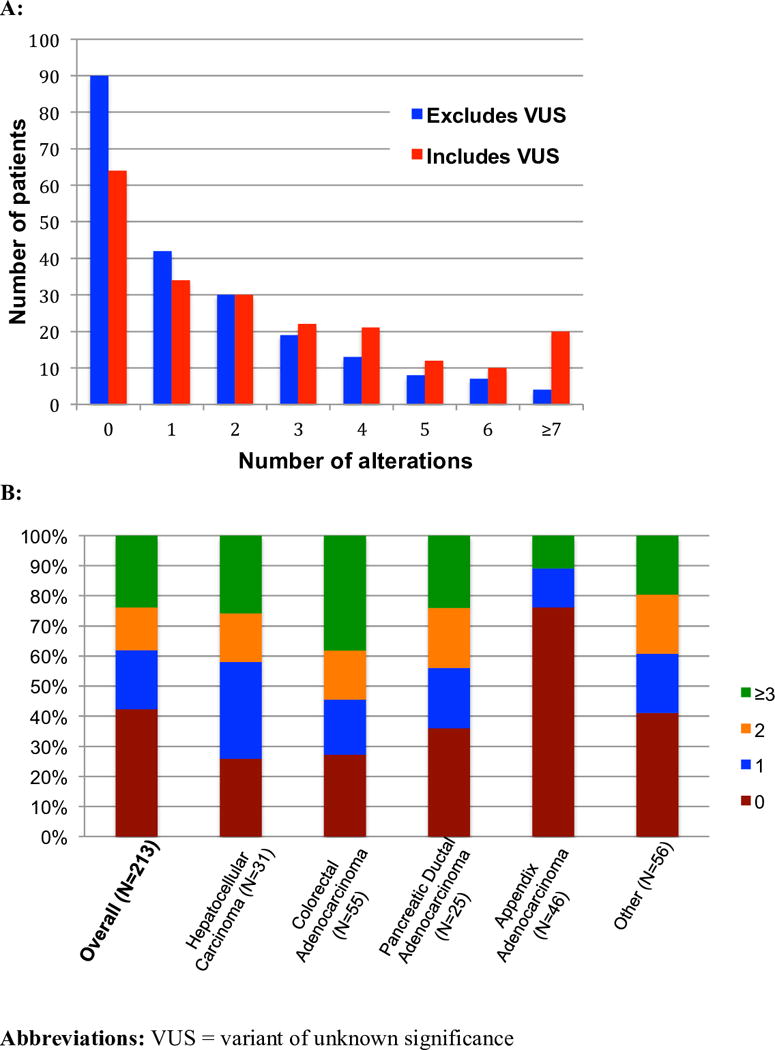
A: Histogram representing the number of patients with each given number of detected alterations. There were 123 patients with characterized alteration(s) (excluding VUSs), and 149 patients with detected alteration(s) (including VUSs). B: Percentage of patients with given number(s) of characterized alterations, separated by most common cancers in data set. All 213 patients were included. Patients for whom all detected alterations were VUS, as well as patients with no alterations were counted as having zero characterized alterations. In some cancers such as hepatocellular carcinoma, the majority of patients (74%) harbor one or more characterized alterations; in contrast, the vast majority (76%) of the patients with appendiceal adenocarcinoma patients had no characterized alterations (either no detected alterations, or all detected were VUS).
Types of alterations
There were 143 unique characterized alterations, and if VUSs were included, there were 301 unique alterations. Overall, 56 genes were altered in at least one patient. The three genes that most commonly had characterized alterations (excluded VUS) in patients across the different cancers were TP53 (N = 77: 36% of patients), KRAS (N = 54; 25%), and PIK3CA (N = 20; 9%) (Figure 1A).
Quantification of alterations
The median percent ctDNA (including VUS and repeat biopsies in the same patients) was 2.42% (interquartile range (IQR) 1.76–6.13%), and, when excluding VUS and repeats, 2.51% (IQR 0.76–8.96%) (Figure 1B). Prior work has found that a percent ctDNA threshold at ≥5% versus <5% is a prognostic marker for overall survival l (19). In our cohort, of the 123 patients with one or more characterized alterations, 57 (46%) had one or more alterations for which the percent ctDNA was ≥ 5%.
Inimitability of the alterations
Most patients with characterized alterations had unique genomic portfolios. Of the 123 patients with one or more characterized alterations, there were 13 sets of patients with identical genomic alterations (involving 60 patients) (Table 2). The most common such pairings were single-mutations in either TP53 (N = 19 patients) or KRAS (N=10 patients). Of these 13 shared portfolios, nine were comprised of only two patients. Identical genetic portfolios have been reported to arise from different primary tissues-of-origin (20). Our study found the majority (N=8 of 13 portfolios) of the shared gene portfolios to be comprised of different histologies.
Table 2.
Frequency and tissues of origin of patients sharing gene or alteration profiles* (excludes VUS)
| Shared gene profile (N = 60) | Number of patients | Represented histologies in liquid biopsy |
|---|---|---|
| TP53 | 19 | Hepatocellular carcinoma (N = 7), colorectal adenocarcinoma (N = 4), pancreatic ductal adenocarcinoma (N=3), esophageal adenocarcinoma (N = 2), cholangiocarcinoma (N = 1), appendix adenocarcinoma (N = 1), gallbladder carcinoma (N = 1) |
| KRAS | 10 | Colorectal adenocarcinoma (N=4), pancreatic ductal adenocarcinoma (N=2), appendix adenocarcinoma (N=2), cholangiocarcinoma (N=1), pancreatic neuroendocrine tumor (N=1) |
| KRAS, TP53 | 9 | Colorectal adenocarcinoma (N=5), pancreatic ductal adenocarcinoma (N=2), esophageal adenocarcinoma (N=1), GIST (N=1) |
| TP53, KRAS, MYC | 4 | Appendix adenocarcinoma (N=1), colorectal adenocarcinoma (N=1), gastric adenocarcinoma (N=1), pancreatic ductal adenocarcinoma (N=1) |
| RAF1 | 2 | Duodenum adenocarcinoma (N=1), gastric adenocarcinoma (N=1) |
| APC | 2 | Colorectal adenocarcinoma (N=2) |
| GNAS | 2 | Appendix adenocarcinoma (N=1), colorectal adenocarcinoma (N=1) |
| CTNNB1 | 2 | Hepatocellular carcinoma (N=2) |
| KRAS, MYC | 2 | Colorectal adenocarcinoma (N=1), pancreatic ductal adenocarcinoma (N=1) |
| CTNNB1, TP53 | 2 | Hepatocellular carcinoma (N=2) |
| APC, TP53 | 2 | Colorectal adenocarcinoma (N=2) |
| PIK3CA, TP53 | 2 | Colorectal adenocarcinoma (N=1), hepatocellular carcinoma (N=1) |
| APC, KRAS, TP53 | 2 | Colorectal adenocarcinoma (N=2) |
| Shared molecular alteration profile (N = 6) | Number of patients | Represented histologies in liquid biopsy |
| KRAS G12D | 2 | Pancreatic ductal adenocarcinoma (N=1), colorectal adenocarcinoma (N=1) |
| GNAS R201H | 2 | Appendix adenocarcinoma (N=1), colorectal adenocarcinoma (N=1) |
| KRAS G12V | 2 | Appendix adenocarcinoma (N=2) |
| Shared molecular alteration profile (N = 2) | Number of patients | Represented histologies in tissue biopsy |
| KRAS G12R | 2 | Pancreatic ductal adenocarcinoma (N=1), colorectal adenocarcinoma (N=1) |
Abbreviations: VUS = variant of unknown significance, GIST = gastrointestinal stromal tumor
Patients sharing gene profiles had the same sets of genes altered, while shared alteration profiles were patients who had the same sets of specific alterations at the same gene.
Unsurprisingly, identical molecular signatures (identical alteration(s) at the respective genes altered) were much less common than portfolios considering only the affected genes (N=6 versus N=60, respectively). There were a total of six patients who had a molecular signature that was identical to at least one other patient, and each of the three such molecular portfolios were single-gene alterations shared between two patients (Table 2). Therefore, 95% of patients (117 of 123) with detected characterized alteration(s) had distinct molecular portfolios. This uniqueness was even more marked within the 105 patients who had undergone the larger panel tissue biopsy NGS sequencing: only two patients shared an identical molecular signature (KRAS G12R single-mutation portfolio, in colorectal and pancreatic cancers).
Potential actionability of alterations (Figure 3 and 4, and Supplemental Table 2)
Figure 3. Overall potential actionability of characterized alterations based on available agents.
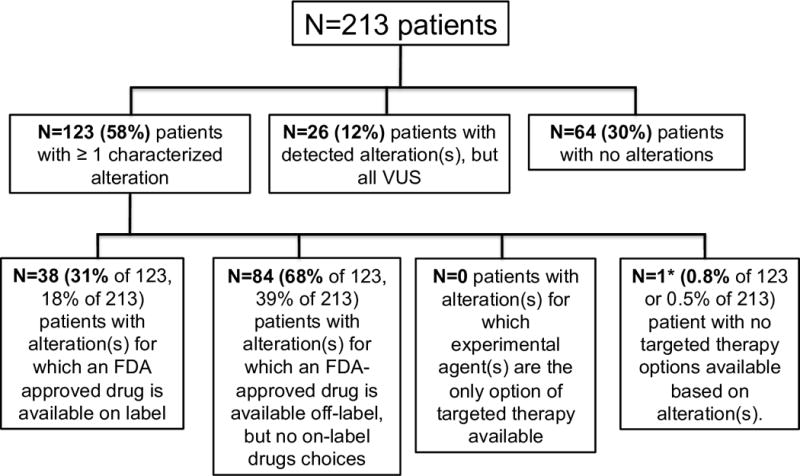
Almost all patients (122/123) with characterized alterations(s) had one or more targeted therapy(ies) available, either through an on-label drug, an off-label drug, or a clinical trial. For patients who had multiple therapies available, FDA approved agents were prioritized over experimental drugs. Amongst FDA approved agents, on-label drugs were prioritized over off-label drugs.
Figure 4. Theoretical treatment options for characterized alterations across diverse gastrointestinal cancers.
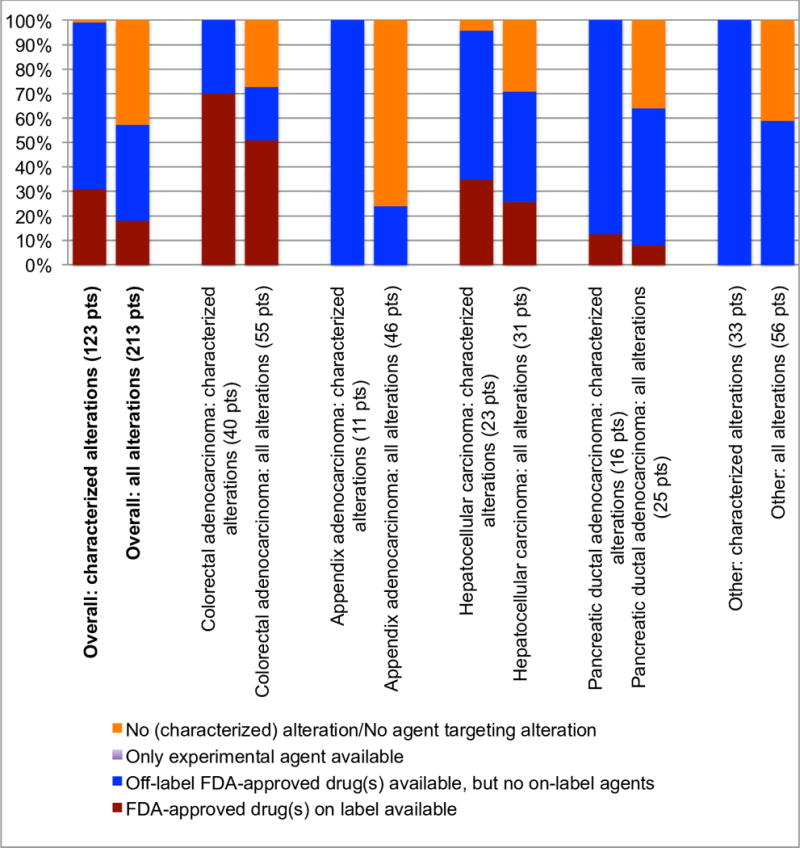
Abbreviations: FDA = Food and Drug Administration; pts = patients
Excluding variants of unknown significance, the percent breakdown of available treatment options for patients with the most common cancer-types in the cohort. FDA approved agents were prioritized over experimental drugs. Amongst FDA approved agents, on-label drugs were prioritized over off-label drugs. In our population, all of the patients who were candidates for an experimental agent also had targeted FDA-approved drugs available, either on or off-label (see Figure 3).
Of the 123 patients with characterized alterations, >99% (122 of 123) had one or more hypothetical (experimental or approved) treatment options available (Figure 3). Overall, of the 213 patients in the study, 122 (57%) had potential targetable alteration(s). This number varied by cancer type. For instance, in hepatocellular carcinoma, 74% of patients (22 of 31) had one or more characterized alterations for which there is theoretically an FDA-approved drug available (on- or off-label). In contrast, that number is approximately three-fold lower (24%) for appendiceal adenocarcinoma (11 of 46) (Figure 4).
Concordance between tissue next generation sequencing and ctDNA analysis (Supplemental Table 3)
The median time between specimen acquisition for tissue NGS and ctDNA in 105 patients who had both performed was 53 days (range −359 days to 3544 days). We assessed tissue versus ctDNA concordance, using the four most commonly found alterations in the liquid biopsy data set (KRAS amplification, MYC amplification, KRAS G12V, and EGFR amplification). The concordance rates were 96%, 94%, 95%, and 91%, respectively. With only the last of these alterations (EGFR amplification) was there a significant difference between tissue and ctDNA NGS (p = 0.04). This difference was mainly due to the detection of EGFR amplification in ctDNA in patients in which it was not found in tissue NGS. An analysis of overall concordance at the gene level for the 68 genes included in both tissue and liquid biopsy panels was performed, and found a concordance rate of 96%, a kappa correlation of 0.42, and a p-value by McNemar’s exact test of 0.68. (kappa p value indicates that there is moderate concordance and the p value indicates that the two tests (ctDNA versus tissue NGS) did not perform significantly differently). Finally, analysis of concordance across the 16 genes evaluable for amplifications in both ctDNA and tissue found overall concordance of 97%, kappa correlation of 0.251, and a p-value by McNemar’s exact test of 0.0066, indicating that there was a difference between ctDNA and tissue in detection of amplification. The positive predictive value for tissue biopsy was 37.5% and for ctDNA was 20%.
Discussion
As molecularly targeted therapeutics are becoming a mainstay of contemporary oncology, accounting for over $10 billion in annual spending in the U.S. alone, there is a growing need for effective new molecular tests (21). The specific advantages of liquid biopsies (e.g. repeatable, relatively inexpensive, and minimally invasive), has led to research in real-time prognostics and drug cycling applications, such as the use of KRAS mutations detected by liquid biopsy for prognosis in advanced pancreatic cancer (22), the use of serial ctDNA biopsies to track treatment-conditional clonal evolution to cycle EGFR inhibitors in colorectal cancer (23), and the use of cell-free plasma exome sequencing to identify new pathways of acquired resistance to targeted therapeutics in a variety of cancers (14).
In our study, we explored the mutational landscape of various gastrointestinal malignancies, as detected by ctDNA derived from liquid biopsy of blood. We found the genomic portfolio of the malignancies to be very diverse. The three genes most commonly found to have characterized alterations in patients across the different cancers were TP53 (N = 77: 36% of patients), KRAS (N = 54; 25%), and PIK3CA (N = 20; 9%) (Figure 1A). The median percent ctDNA in alterations of known significance was 2.42%, but the interquartile range was wide (0.6% to 8.5%). While common alterations like TP53 and/or KRAS were identified in 69% of patients with detected alteration(s), we also discerned aberrations in 56 different genes, only 8 of which were found in more than 10% of the population with detected alteration(s). Altogether, there were 143 unique characterized alterations and, if VUSs were included, there were 301 unique alterations. Certain specific alterations were more common within detected genes.
We examined how frequently shared portfolios of genes or specific alterations arose as detected by ctDNA liquid biopsies. When considering only shared portfolios at the level of genes (rather than specific alterations), there were thirteen sets (60 patients in total) of identical genomic portfolios (the genes involved were identical though the specific alteration within the genes may have been different). Analyzing data at the level of genes rather than alterations has limitations since different defects/gains of function in the same gene may have a vastly distinct biologic impact. At the alteration level, we found only three pairs of tumors to have identical profiles. These data suggest that, even with the limited panel of genes used here, the vast majority of patients have unique or near unique molecular portfolios, a finding also noted in tissue NGS for several tumors in the existing literature (20,21,24–26). Larger ctDNA panels may reveal further inter-tumor heterogeneity. Indeed, tissue NGS (with 315 genes) showed only one pair of patients with an identical molecular portfolio among the 105 patients tested (both had a KRAS G12R alteration) (Table 2). The complexity of cancers and their uniqueness has significant therapeutic implications, and suggests that customized combination therapy may be needed for many patients.
We also found that the current utility of liquid biopsy sequencing technology may be dependent on the tumor of origin. While on aggregate the majority of patients (58%) had characterized alteration(s), some tumor types were exceptions. For instance, less than a quarter of patients with appendiceal adenocarcinoma harbored alterations of known significance. As the gene panel was constructed, at least in part, based on potentially actionable alterations, the number of detected alterations in a patient’s blood directly correlated with the number of available cognate targeted agents. Indeed, only one of the 123 patients with detected alteration(s) of known significance had no FDA-approved (on or off-label) or experimental agents available (Figure 3).
In addition to potentially helping to select therapy, ctDNA results might also provide prognostic data. For instance, it was previously shown that percent ctDNA greater than or equal to 5% is associated with poorer survival (19). Furthermore, specific alterations, such as BRAF mutations in colorectal cancer, are associated with a poor prognosis (27).
One hundred and five patients had both ctDNA and tissue NGS assessment. The median time between the two forms of biopsies was 53 days (range, −359 to 3544 days). Of interest, the concordance rates between tissue NGS and liquid biopsy ctDNA were 96%, 94%, 95%, and 91%, respectively, for the four most commonly detected alterations in the liquid biopsy dataset (KRAS amplification, MYC amplification, KRAS G12V, and EGFR amplification). Concordance data for these genes also suggests that there may be alteration-specific shortcomings/advantages of tissue NGS versus liquid biopsy. For example, NGS of ctDNA detected EGFR amplifications in eight patients for whom tissue biopsies did not find the alteration (p < 0.05), and NGS of tissue biopsies for KRAS G12V detected the mutation in five patients in whom ctDNA sequencing did not (p = 0.06) (Supplement Table 3). Though this latter finding was not statistically significant at the 0.05 threshold, it warrants further study, as the presence/absence of activating KRAS alterations guides ERGR-targeting treatment decisions. Alongside the alteration-specific concordance, we also surveyed overall concordance of the two assays at the gene level (not alteration-specific) for all 105 patients for the 68 genes covered by both the tissue and liquid biopsy panels, and found a concordance rate of 96%. Overall, 119 of 263 (45%) of altered genes detected by ctDNA liquid biopsy were also detected by tissue NGS, and 119 of 271 (44%) of altered genes detected by tissue NGS were also detected in ctDNA liquid biopsy. The kappa correlation of 0.42 suggested moderate correlation between the two tests, and a p-value by McNemar’s exact test of 0.68 indicated that differences in detection rate at the gene level between the two assays overall was not statistically significant (Supplemental Table 3). An analysis of the 16 genes (Supplemental Table 1) assessable for amplifications by both assays found that tissue biopsy detected alterations significantly more often (p < 0.01) than tissue biopsy. Overall concordance was 97%, but of the 44 patients in whom amplifications were detected by ctDNA liquid biopsy, only 9 of these were also found by tissue NGS (20%); reciprocally, 9 of 24 (37.5%) genes detected by tissue NGS were also detected by ctDNA liquid biopsy (Supplemental Table 3).”
Although time elapsed between the tissue and liquid biopsies and technical problems in either tissue or ctDNA NGS cannot be ruled out as explanations for discrepancies, there are good biologic hypotheses for these differences. For instance, ctDNA NGS reflects genomic alterations from shed DNA derived from multiple metastatic sites while tissue NGS demonstrates only the molecular findings in the small piece of tissue assessed. Conversely, not all tumors may shed DNA into the bloodstream at an equivalent rate. Further, ctDNA may be suppressed if assessed while a patient is on therapy. Finally, there is an inverse correlation between time interval between tissue and blood-based NGS and concordance of findings (19).
There are several limitations to our analysis. First, the genomics data were obtained from a de-identified database and, hence, only limited clinical information was available. Second, only 105 patients also had tissue NGS performed, and these biopsies were rarely collected at the same time. Thus, (targeted) treatment courses could have arisen in the time between the two biopsy collections, and the patients could have had changes in their clinical/disease status. Furthermore, without access to pathology or full sequencing reports, it was not possible to adjust for possible confounders like tumor purity. Even so, concordance rates between tissue and ctDNA NGS was high (91% to 96%) for the four most frequently identified alterations in the liquid biopsy data. While there were occasional patients with similar molecular portfolios from different histologies, the numbers were too small to draw conclusions about the implications of this observation. Furthermore, more extensive genomic sequencing might reveal that the molecular portfolios in these patients differed. Finally, while the majority of patients had theoretically actionable alterations, it is important to point out that the clinical significance of these alterations will need to be evaluated in additional studies to confirm the true potential of liquid biopsies to discover drivers that are worth acting upon.
In conclusion, liquid biopsy of ctDNA in gastrointestinal malignancies is a promising new technique for obtaining genomic information in a minimally invasive, repeatable fashion. Its potential utility for selecting targeted therapeutic agents or estimating prognoses warrants further investigation.
Supplementary Material
Acknowledgments
Grant Support:
Razelle Kurzrock a recipient of the Joan and Irwin Jacobs Fund. Paul Riviere is a recipient of the National Institutes of Health Grant TL1TR001443. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH
Abbreviations
- ctDNA
circulating tumor DNA
- VUS
Variant of unknown significance
Footnotes
Paul J Riviere: medical student, Paul Fanta: associate clinical professor, Sadakatsu Ikeda: assistant professor of medicine, Joel Baumgartner: assistant professor of surgery, Gregory Heestand: clinical assistant professor, Razelle Kurzrock: Director of Center for Personalized Cancer Therapy and Clinical Trials Office, Senior Deputy Director of Clinical Science at Moores Cancer Center
References
- 1.Schwaederle M, Husain H, Fanta PT, Piccioni DE, Kesari S, Schwab RB, et al. Detection rate of actionable mutations in diverse cancers using a biopsy-free (blood) circulating tumor cell DNA assay. Oncotarget Impact Journals. 2016;7:9707–17. doi: 10.18632/oncotarget.7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wheler JJ, Janku F, Naing A, Li Y, Stephen B, Zinner R, et al. Cancer Therapy Directed by Comprehensive Genomic Profiling: A Single Center Study. Cancer Res [Internet] 2016 doi: 10.1158/0008-5472.CAN-15-3043. 76:3690–701. Available from: http://cancerres.aacrjournals.org/cgi/doi/10.1158/0008-5472.CAN-15-3043. [DOI] [PubMed]
- 3.Tsimberidou A-M, Iskander NG, Hong DS, Wheler JJ, Falchook GS, Fu S, et al. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clin Cancer Res [Internet] Clinical Cancer Research. 2012 doi: 10.1158/1078-0432.CCR-12-1627. [cited 2016 Aug 11];18:6373–83. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22966018. [DOI] [PMC free article] [PubMed]
- 4.Von Hoff DD, Stephenson JJ, Rosen P, Loesch DM, Borad MJ, Anthony S, et al. J Clin Oncol [Internet] American Society of Clinical Oncology; 2010. Pilot Study Using Molecular Profiling of Patients’ Tumors to Find Potential Targets and Select Treatments for Their Refractory Cancers. [cited 2016 Aug 11];28:4877–83. Available from: http://jco.ascopubs.org/cgi/doi/10.1200/JCO.2009.26.5983. [DOI] [PubMed] [Google Scholar]
- 5.Hyman DM, Solit DB, Arcila ME, Cheng DT, Sabbatini P, Baselga J, et al. Precision medicine at Memorial Sloan Kettering Cancer Center: clinical next-generation sequencing enabling next-generation targeted therapy trials. Drug Discov Today. 2015;20:1422–8. doi: 10.1016/j.drudis.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lièvre A, Bachet J-BB, Le Corre D, Boige V, Landi B, Emile J-FF, et al. Cancer Res [Internet] American Association for Cancer Research; 2006. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. [cited 2016 Jul 21];66:3992–5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16618717. [DOI] [PubMed] [Google Scholar]
- 7.Misale S, Di Nicolantonio F, Sartore-Bianchi A, Siena S, Bardelli A. Cancer Discov [Internet] American Association for Cancer Research; 2014. Resistance to Anti-EGFR Therapy in Colorectal Cancer: From Heterogeneity to Convergent Evolution. [cited 2016 Jul 7];4:1269–80. Available from: http://cancerdiscovery.aacrjournals.org/cgi/doi/10.1158/2159-8290.CD-14-0462. [DOI] [PubMed] [Google Scholar]
- 8.Misale S, Arena S, Lamba S, Siravegna G, Lallo A, Hobor S, et al. Sci Transl Med [Internet] American Association for the Advancement of Science; 2014. Blockade of EGFR and MEK intercepts heterogeneous mechanisms of acquired resistance to anti-EGFR therapies in colorectal cancer. [cited 2016 Jul 7];6:224ra26. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24553387. [DOI] [PubMed] [Google Scholar]
- 9.Almendro V, Kim HJ, Cheng Y-K, Gönen M, Itzkovitz S, Argani P, et al. Cancer Res [Internet] American Association for Cancer Research; 2014. Genetic and phenotypic diversity in breast tumor metastases. [cited 2016 Jul 7]74:1338–48. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24448237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. N Engl J Med [Internet] Massachusetts Medical Society; 2012. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. [cited 2016 Jul 7];366:883–92. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22397650%5Cnhttp://www.nejm.org/doi/abs/10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Onstenk W, Sieuwerts AM, Weekhout M, Mostert B, Reijm EA, van Deurzen CHM, et al. Gene expression profiles of circulating tumor cells versus primary tumors in metastatic breast cancer. Cancer Lett. 2015;362:36–44. doi: 10.1016/j.canlet.2015.03.020. [DOI] [PubMed] [Google Scholar]
- 12.De Mattos-Arruda L, Weigelt B, Cortes J, Won HH, Ng CKY, Nuciforo P, et al. Ann Oncol [Internet] Oxford University Press; 2014. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. [cited 2016 Jul 7];25:1729–35. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25009010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park BH, Parsons HA, Beaver JA, Parsons HA, Beaver JA, Park BH. Circulating Plasma Tumor DNA. Springer International Publishing; 2016. [cited 2016 Jul 5];259–76. Available from: http://link.springer.com/10.1007/978-3-319-22909-6_11. [Google Scholar]
- 14.Murtaza M, Dawson S-J, Tsui DWY, Gale D, Forshew T, Piskorz AM, et al. Nature [Internet] Nature Publishing Group; 2013. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. [cited 2016 Jul 7];497:108–12. Available from: http://www.nature.com/doifinder/10.1038/nature12065. [DOI] [PubMed] [Google Scholar]
- 15.Xia S, Kohli M, Du M, Dittmar RL, Lee A, Nandy D, et al. Plasma genetic and genomic abnormalities predict treatment response and clinical outcome in advanced prostate cancer. Oncotarget Impact Journals. 2015;6:16411–21. doi: 10.18632/oncotarget.3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dawson S-J, Tsui DWY, Murtaza M, Biggs H, Rueda OM, Chin S-F, et al. Analysis of Circulating Tumor DNA to Monitor Metastatic Breast Cancer. Massachusetts Medical Society; 2013. http://dx.doi.org/101056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- 17.Shaw JA, Page K, Blighe K, Hava N, Guttery D, Ward B, et al. Genomic analysis of circulating cell-free DNA infers breast cancer dormancy Genome Res [Internet] Cold Spring Harbor Laboratory Press; 2012. [cited 2016 Jul 7];22:220–31. Available from: http://genome.cshlp.org/cgi/doi/10.1101/gr.123497.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lanman RB, Mortimer SA, Zill OA, Sebisanovic D, Lopez R, Blau S, et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. In: Hoheisel JD, editor. PLoS One [Internet] Public Library of Science; 2015. [cited 2017 Jul 8];10:e0140712. Available from: http://dx.plos.org/10.1371/journal.pone.0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwaederle M, Husain H, Fanta PT, Piccioni DE, Kesari S, Schwab RB, et al. Use of Liquid Biopsies in Clinical Oncology: Pilot Experience in 168 Patients. Clin Cancer Res [Internet] Clinical Cancer Research. 2016 doi: 10.1158/1078-0432.CCR-16-0318. [cited 2016 Jul 26];clincanres.0318.2016. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27185373. [DOI] [PubMed]
- 20.Patel SP, Schwaederle M, Daniels GA, Fanta PT, Schwab RB, Shimabukuro KA, et al. Oncotarget [Internet] Impact Journals, LLC; 2015. Molecular inimitability amongst tumors: implications for precision cancer medicine in the age of personalized oncology. [cited 2016 Jul 7];6:32602–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26418953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lyman GH, Moses HL. Biomarker Tests for Molecularly Targeted Therapies — The Key to Unlocking Precision Medicine. Massachusetts Medical Society; 2016. http://dx.doi.org/101056/NEJMp1604033. [DOI] [PubMed] [Google Scholar]
- 22.Tjensvoll K, Lapin M, Buhl T, Oltedal S, Steen-Ottosen Berry K, Gilje B, et al. Clinical relevance of circulating KRAS mutated DNA in plasma from patients with advanced pancreatic cancer. Mol Oncol. 2016;10:635–43. doi: 10.1016/j.molonc.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, et al. Nat Med [Internet] Nature Publishing Group; 2015. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. [cited 2016 Jul 7];21:795–801. Available from: http://www.nature.com/doifinder/10.1038/nm.3870. [DOI] [PubMed] [Google Scholar]
- 24.Kurzrock R, Giles FJ. Precision oncology for patients with advanced cancer: the challenges of malignant snowflakes. Cell Cycle [Internet] 2015 doi: 10.1080/15384101.2015.1041695. [cited 2016 Nov 28];14:2219–21. Available from: http://www.tandfonline.com/doi/full/10.1080/15384101.2015.1041695. [DOI] [PMC free article] [PubMed]
- 25.Wheler J, Lee JJ, Kurzrock R. Unique Molecular Landscapes in Cancer: Implications for Individualized, Curated Drug Combinations. Cancer Res. 2014;74 doi: 10.1158/0008-5472.CAN-14-2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wheler JJ, Parker BA, Lee JJ, Atkins JT, Janku F, Tsimberidou AM, et al. Unique molecular signatures as a hallmark of patients with metastatic breast cancer: Implications for current treatment paradigms. Oncotarget [Internet] 2014 doi: 10.18632/oncotarget.1946. Impact Journals. [cited 2016 Nov 28];5:2349–54. Available from: http://oncotarget.com/abstract/1946. [DOI] [PMC free article] [PubMed]
- 27.Chen K-H, Lin Y-L, Liau J-Y, Tsai J-H, Tseng L-H, Lin L-I, et al. Med Oncol [Internet] Springer; US: 2016. BRAF mutation may have different prognostic implications in early- and late-stage colorectal cancer. [cited 2017 Jan 12];33:39. Available from: http://link.springer.com/10.1007/s12032-016-0756-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.