

Abstract
Camptothecin (CPT) was discovered from plant extracts more than 60 years ago. Since then, only two CPT analogues (irinotecan and topotecan) have been approved for cancer treatment, although several thousand CPT derivatives have been synthesized and many of them were actively studied in our research community over the past 6+ decades. In this review article, we briefly summarize: (1) the discovery and early development of CPTs, (2) the recognized CPT mechanism of action (MOA), (3) the synthesis of CPT and CPT analogues, and (4) the structure-activity relationship (SAR) of CPT and its analogues. Next, we provide evidence that certain CPT analogues can exert improved efficacy with low toxicity independently of topoisomerase I (Top1) inhibition; instead, these CPT analogues use novel MOAs by targeting important cancer survival-associated oncogenic proteins and/or by bypassing various treatment-resistant mechanisms. We then present a comprehensive review of the most advanced CPT analogues in clinical development, with the goal of resolving why no new CPTs have been FDA approved for cancer treatment, beyond irinotecan and topotecan. We argue that new CPT Top1 inhibitor drugs are unlikely being found to be significantly better than irinotecan and/or topotecan in terms of the overall antitumor activity and toxicity. The significance of CPT analogues that possess novel MOAs has not been sufficiently recognized so far. In our opinion, this is a research area with great potential to make a breakthrough for development of the next generation of CPT analogues that possess high efficacy (due to novel targets) and low toxicity (due to low inhibition of Top1 activity/function) for effective treatment of human disease, including cancer.
Keywords: Camptothecin (CPT), topoisomerase I (Top1), analogue/derivative, FL118, novel mechanism of action, survivin, Mcl-1, XIAP, cIAP2, clinical trials
Camptothecin discovery and early development
Camptothecin (CPT) (Figure 1A) is a pentacyclic alkaloid that was first isolated from stem wood of Camptotheca acuminata by botanists working in the USDA’s Plant Introduction Division in the mid-1950s [1]. Camptotheca acuminata is a tree native to China and its bark is a recognized Chinese traditional medicine. The process of CPT discovery was well reviewed by Drs. Monroe Wall and Mansukh Wani [2], the co-discovers of CPT and Taxol. Chemical synthesis of CPT in laboratories, and follow-up preclinical and clinical studies were actively conducted in the late 1950s and mid to late 1960s [3]. CPT was investigated in the United States in cancer patients in both Phase I [4,5] and Phase II [6] clinical trials. Clinical use of CPT for the treatment of stomach and bladder cancer and certain types of leukemia, often in combination with corticosteroids, continued into the mid-70s in China [7]. Those early studies indicated that the water-soluble carboxylate form of CPT (Figure 1B) possesses much less antitumor activity than the water-insoluble lactone form of CPT (Figure 1A). Clinical trials for about one thousand patients with colorectal, head-&-neck or bladder cancer in China using carboxylate form of CPT (CPT sodium salt) showed some positive results [8]. However, results from US trials with the carboxylate form of CPT appeared to be not as promising [4-6]. This inconsistency could be attributed to the fact that the US clinical trials included only patients that had already shown resistance to other treatment. Nevertheless, the lack of consistent efficacy of using the carboxylate form of CPT in clinical trials drove researchers to focus on the CPT lactone form for development. However, clinical trials with CPTs were essentially discontinued in the 1970s due to the inability to resolve the water-insoluble property of CPT in the lactone form (the form used in references throughout this article), low response rates [4-6] and high toxicity (e.g. myelosuppression, gastrointestinal toxicities, and hemorrhagic cystitis) [9,10], as well as an unclear CPT mechanism of action.
Figure 1.

The chemical structure of CPT analogues and non-CPT compounds.
Discovery of mechanism of action (MOA) for CPT
Although CPT clinical trials ended in the 1970s, its mechanism of action studies continued to be an area of interest. The husband-and-wife team of Drs. Marshall and Susan Horwitz at Albert Einstein College of Medicine, as well as others, made the early findings related to the CPT mechanism of action. Their studies revealed that CPT inhibits DNA and RNA (including ribosomal RNA) synthesis and induces DNA damage [11-15]. These scientists observed that CPT is most potent during the S-phase of the cell cycle and predicted that the DNA replication fork must play a role in CPT-induced cell death [15]. Later studies indicated that CPT arrested cell cycle at both S and G2 phases, which were needed for CPT cytotoxicity [16,17].
During the early 1980s, a number of unrelated DNA damaging agents were being explored clinically for the treatment of both cancer and bacterial infection. Studies revealed two different classes of DNA damaging drugs: the quinolone antibiotics (e.g. cinoxacin, nalidixic acid, ciprofloxacin) and the podophyllotoxin derivatives (etoposide, teniposide). Both classes of drugs shared the same mechanism of action: inhibition of topoisomerase II (Top2), an enzyme active during S-phase that assists with DNA replication (reviewed in [18]). Noting that CPT is also most active during the S-phase and that the DNA replication fork was believed to be necessary for CPT-induced cell death, Dr. Leroy F. Liu’s team at Johns Hopkins, in collaboration with Smith Kline & French Laboratories in Philadelphia, set out to test whether CPT could be an inhibitor of Top2 [19]. To their surprise, even 125 µM CPT failed to inhibit Top2-dependent DNA cleavage [19]. However, when they tested other enzymes associated with DNA replication, they observed potent and dose-dependent induction of DNA damage in the presence of topoisomerase I (Top1) [19].
Top1 orthologues are found in all eukaryotes, and appear to be an essential enzyme during development in a wide variety of animals. For example, knocking out TOP1 is embryonically lethal in both Mus musculus [20] and Drosophila melanogaster [21]. During the process of DNA replication and transcription, Top1 is responsible for relaxing supercoiled DNA. Specifically, Top1 first cut supercoiled DNA to introduce a single-strand break, or “nick”, into the DNA and covalently binds to the nicked 3’-end DNA and allows the 5-nicked strand to rotate around the intact strand in a controlled manner; after rotation Top1 re-ligates the nicked strand [22]. This Top1-DNA complex during DNA replication is commonly referred to as the “Top1 covalent complex”, owing to the covalent bond between Top1 and the nicked strand (reviewed in [23]).
CPT and CPT analogues function by inhibition of Top1 activity [24,25]. In the cell, CPT integrates itself into the Top1/DNA covalent complex, forming a ternary complex. Both Top1 and DNA are required for CPT binding, and CPT does not have a significant binding to either in the absence of the other [26]. CPT binds to both the Top1 enzyme and the intact DNA strand through hydrogen bonding, and prevents both the re-ligation of the nicked DNA and dissociation of Top1 from the DNA. During replication, this CPT-involved ternary complex acts as a roadblock for the replication fork. Collision between the ternary complex and the replication fork results in shear stress upon the intact DNA strand, resulting in breakage, DNA double-strand breaks, and cell death. Interestingly, the known target for CPT and its analogues is the Top1-DNA complex. However, as mentioned above it was demonstrated that CPT affects cellular protein, RNA and DNA synthesis [11-15], which may suggest that CPT could have other targets. Yeast cells with deleted TOP1 become functionally immune to CPT and its analogues [24,25], and mammalian/human cancer cells become resistant to CPTs when TOP1 is mutated [27-32] or overexpression of a mutant Top1 [33], while events that can increase Top1 activity enhance CPT sensitivity [34]. While Top1 activity inhibition is a well-documented MOA for CPTs and its analogues, we present evidence below for CPT and CPT-derived analogues that have different molecular targets, and importantly, these targets (but not Top1 expression) are involved in their anticancer activity.
Synthesis of CPT and its derived analogues
The discovery of Top1 being the molecular target of CPT [19] further stimulated the research interest to synthesize new CPT analogues with a hope that new CPT analogues may overcome the weakness of CPT (e.g. improved water solubility, better Top1 activity inhibition) and thus, enhance antitumor activity. Since the CPT structure was available [1], early chemistry efforts developed a number of ways to synthesize CPT (reviewed in [3]). However, these methods are not useful for synthesizing CPT analogues. Drs. Wani and Wall’s research team at RTI (North Carolina, US) employed a Friedländer condensation reaction and developed a much more flexible approach for generating CPT or CPT analogues by coupling the tricycle CDE compound (Figure 1A) to the A ring-relevant compound to make the pentacycle CPT or CPT analogues [35-39], in which these authors resolved the separation of 20 (S) and 20 (R) configuration. This is important since the CPT or CPT analogues in the 20 (R) configuration are found to be functionally inactive [38]. Based on the current development of CTP medicinal chemistry, it is clear that multiple approaches have been developed for the synthesis of CPT and its analogues. These CPT synthetic methods have been optimized over time. For example, the broadly used method of coupling the tricycle CDE compound to the A ring-relevant compound to make CPT analogues through Friedländer condensation reaction introduced by Drs. Wani and Wall for synthesis of various CPT analogues [35-39], based on the early studies [40-42], were further developed and optimized by Henegar et al in 1997 to fit a versatile and large scale of CPT analogue synthesis [43]. This approach was further developed specifically for enantiopure 20 (S)-CPT by Tang et al in 2006 [44], and we believe that this could also be applied to various 20 (S)-CPT analogue syntheses. Li et al summarized various CPT and its analogue synthetic methods in a review article [45]. These methods or their modified methods, especially the Friedländer reaction-based approach [44,45], are practical for the efficient synthesis of various CPT analogues.
Structure-activity relationship (SAR) of CPTs
The findings from the earlier studies on CPT structure-activity relationship (SAR) can be summarized as: 1) the E-ring in a lactone form is much more potent than the E-ring in a carboxylate form (Figure 1A versus 1B); 2) the chiral center located at position 20 of the E-ring with an S-configuration is absolutely required for CPT compound activity and the R-configuration is inactive [38]; and 3) CPT without A and B rings (de-AB-CPT) shows no discernible inhibition of DNA and RNA synthesis at a µM concentration where CPT reached 50% inhibition. Indeed, de-AB-CPT reaching a 20% inhibition of DNA and RNA synthesis needs 50 µM concentration [46] and, furthermore, no meaningful activity in L1210 carcinoma screen assay at a concentration where CPT is quite active [46]. This suggests that the A and B rings are important for CPT antitumor potential. Together, these early findings on CPT SAR studies lay a foundation for further chemistry modulation of the CPT structure in hopes of discovering CPT analogues with better Top1 activity inhibition.
Novel MOAs for CPT and CPT analogues
CPTs’ regulation of gene expression independent of Top1
Since Top1 has important functions in gene transcription control [23], a critical question in the CPT and CPT analogue research field is whether CPT or CPT analogues could modulate gene expression (e.g. modulate key drug targets in cancer) independent of Top1 activity inhibition by CPT or CPT analogues. In May 2016, Mabb, et al published an interesting study in PLOS ONE [47]. In this study, the authors used multiple approaches to knock down or delete the Top1 gene (TOP1) in neurons to determine the role of Top1 in topotecan-mediated gene modulation. These authors found that in the presence of Top1, topotecan modulates much more gene expression than in the absence of Top1 through both Top1/DNA cleavage complex-dependent and -independent mechanisms [47]. We analyzed the raw data provided in the Table S1 from Mabb, et al.’s publication for the topotecan-induced 38-downregulated genes and 4 upregulated genes in the neurons presented with conditional knockout (cKO) of TOP1 [47]. We wanted to know whether the inhibition or induction of these genes by topotecan is Top1-independent or due to the incomplete TOP1 cKO. The result from the analysis of these topotecan-modulated genes was described in detail in our recent publication [48]. Based on the analysis, our conclusion was that the topotecan-downregulated 38 genes and topotecan-upregulated 4 genes are true Top1-independent events [48]. The study clearly indicated that certain CPTs (topotecan used in this study) could modulate gene expression independent of Top1 function [47]. The key point that we want to emphasize here is that certain CPTs can inhibit or induce gene expression independent of Top1 activity inhibition by CPTs.
We propose that certain novel CPT analogues that possess high efficacy and low toxicity in treatment of cancer (e.g. FL118, which will be reviewed in detail below) may mainly use Top1-independent mechanisms to deliver their antitumor activity and cancer cell killing [48], while inhibition of Top1 activity may mainly be involved in toxicity to the host as suggested in our recent studies [48]. In this regard, it is known that Top1 is a ubiquitously expressed gene that is essential for mammalian cell proliferation during embryo development, as well as human normal tissue and cell renewal over a lifetime. Top1 plays a critical role in cellular DNA replication, and thus blocking of Top1 function will result in early embryo lethality during development [49] or induces serious toxicity in children and adults in various renewal tissues (e.g. hematopoietic toxicity). Due to the high hematopoietic toxicity of irinotecan and topotecan, during the use of irinotecan or topotecan for cancer patient chemotherapy, peripheral-blood stem cell infusion or bone marrow transplantation was also used in parallel in order to alleviate the intensity of hematopoietic toxicity [50-52]. In summary, Top1 is not an ideal target for cancer therapeutics. Development of novel CPT analogues that do not use Top1 as a major target, but use other cancer proliferation and survival-associated oncogenes as major targets for anticancer activity would be a promising direction for future efforts to generate novel CPT analogues with low toxicity (due to low inhibition of Top1 activity) and high efficacy (due to targeting cancer-associated key genes/proteins) for treatment of cancer.
Discovery of the novel CPT analogue FL118
We recently discovered a novel antitumor compound (named as FL118, Figure 1C) using the survivin gene expression as a biomarker [53] via high throughput screening (HTS) of small molecule libraries, followed by hit-to-lead-to-analogue characterization in vitro and in vivo [54]. The logic of using the antiapoptotic survivin gene expression as a biomarker for drug discovery and leading to the finding of FL118 is that studies have revealed that survivin is a pivotal molecule at the junction of cancer cell survival, division and apoptosis control [55,56]. Survivin is also a critical factor in the inherent and induced drug/radiation resistance for cancer during treatment and is involved in cancer metastasis [57-63]. This is consistent with a potential role of survivin in the latent cancer stem cells (CSCs) [64-71]. A role for survivin in CSCs is independently revealed by computer analysis of the death-from-cancer signature genes. The study showed that cancer cells with stem cell-like expression profiles possess three characteristics: increased expression of inhibitor of apoptosis (IAP) proteins, activated mitotic spindle checkpoint proteins, and elevated cell cycle control proteins [72]. Accordingly, survivin is a key member in the IAP family and possesses all three characteristics: apoptosis inhibition, mitotic/cell division control, and cell cycle regulation [55,56,73-76]. Therefore, survivin is considered as a critical cancer target and is important for both highly proliferative cancer cells and for latent CSCs. Inhibition of survivin expression or function would result in both bulk tumor regression and latent CSC elimination; thus, avoiding tumor metastasis and/or relapse. FL118 shows exceptional antitumor activity, is safe, and works through a MOA of downregulation of multiple cancer-associated oncogenic proteins including survivin regardless of the presence or absence of Top1 expression in cancer cells, as summarized below.
Coincidently, FL118 is a novel CPT analogue with a unique chemical structure identical to 10, 11-methylenedioxy-20 (S)-CPT. The racemic mixture of FL118 (10,11-OCH2O-20(RS)-CPT) was synthesized and tested in mouse L1210 leukemia assays by Drs. Wani and Wall’s research group in 1980s [36,37]. Together with other CPT analogues, they demonstrated that CPT analogues in the “R” configuration are at least 10 to 100-fold less active than the corresponding CPT analogues in the “S” configuration either in mouse leukemia assays or in the test of Top1 inhibition by cleavable complex formation [38,39]. Consistent with our finding that FL118 possesses exceptional antitumor activity, their mouse L1210 leukemia assay indicated that 10, 11-methylenedioxy (MD)-20 (RS)-CPT exhibited a good life prolongation, although it was not among the most effective CPT analogues [39]. Due to water-insolubility and the relative lower efficacy in a mouse tumor model, FL118 was never pursued as an anticancer agent toward clinical trials. In our view, the 10, 11-MD-20 (RS)-CPT did not stand out from other CPT analogues tested then in their mouse leukemia life prolongation assay studies for two reasons. First, they used a racemic “RS” mixture, thus decreasing the apparent efficacy. Second, 10, 11-MD-20 (RS)-CPT is extremely water-insoluble and thus, poor formulation of 10, 11-MD-20 (RS)-CPT would have a poor bioavailability in the in vivo mouse L1210 leukemia life prolongation test. However, we screened FL118 along with other compounds against human tumors in vitro and in vivo; we found that FL118 was very active against human tumors. In fact, FL118 showed inferior antitumor activity to YM155 (Astellas, Japan) to inhibit mouse E0771 breast cancer cell line-established tumor, but FL118 exhibited superiority to YM155 in anti-human tumors (the Li Lab unpublished observation). This suggests that FL118 prefers to inhibit human tumors but not mouse tumors. For testing in vivo, our research group at Roswell Park Cancer Institute developed a novel formulation for FL118 and other linear/arched highly water-insoluble compounds [77]. Our in vivo studies demonstrated that FL118 possesses exceptional antitumor activity against colorectal and head-&-neck cancer in human tumor animal models [54] and can effectively overcome human xenograft tumor resistance to irinotecan (CPT-11, Figure 1D) and topotecan (Figure 1E) [78], two FDA-approved CPT analogues used in the clinic. Given that FL118 is a CPT analogue with high antitumor efficacy, we thought that FL118 might be an effective Top1 activity inhibitor. However, our Top1-DNA complex biochemical cleavage assay showed that even at a 1 µM concentration, FL118 was less effective at inhibiting Top1 activity than SN-38 (active metabolite of irinotecan, Figure 1F) [54]. In contrast, FL118 can effectively inhibit cancer cell growth at or below nM levels, depending on cancer cell types [54].
Issues for CPTs to use Top1 as a target and how to avoid them
A problem with CPTs Top1 inhibitors is the CPT resistance resulted from the proclivity of CPTs to downregulate the expression of Top1 protein targets by which the CPTs exert their MOA [79-82]. Mechanistically, downregulation of Top1 proteins by CPTs is through ubiquitin/26S proteasome-mediated degradation of Top1 in cancer cells [83,84]. Interestingly, Top1 inhibition by CPTs is usually associated with a Top2 activity increase [81,85], suggesting that Top2 increase could be a CPTs resistant factor. Thus, Top2 may partially do the Top1 work in cancer cells, since CPTs are Top1 activity inhibitors but not Top2 activity inhibitors, this phenomenon would be expected to contribute to resistance to CPTs. In fact, the intensity of CPT-induced downregulation of Top1 expression is positively associated with the intensity of cell resistance to CPTs. For example, CPT effectively inhibits Top1 expression in the CPT-resistant breast cancer cell line BT474, while CPT is unable to inhibit Top1 expression in the CPT-sensitive breast cancer cell line ZR75-1 [83]. Consistent with these observations, it was reported that reduced Top1 expression and/or Top1 catalytic activity in cancer cells is associated with increased resistance to CPTs [86,87], while increased Top1 expression in cancer cells sensitizes CPTs [88-90]. Similarly, previous studies also revealed that cancer cells become resistant to CPTs when the Top1 gene is mutated [27-32]. In this regard, using the Du145 parental prostate cancer cells (wild type Top1) in parallel with Du145-derived two sublines, RC0.1 and RC1, with Top1 R364H mutations [91], we demonstrated that FL118 IC50 is 10-50-fold less affected by Top1 mutation in comparison with the affected degree in IC50 for CPT, SN-38 and topotecan [92]. Furthermore, our recent studies revealed that the sensitivity of human colorectal cancer xenograft tumors to FL118 is independent of Top1 expression levels [48]. Human xenograft tumors with high Top1 expression can be resistant to FL118 treatment, while tumors with low/negative Top1 expression can be sensitive to FL118 treatment (Figure 2) [48], which is distinct from other CPTs. These observations are consistent with the fact that FL118 exhibits high in vivo antitumor efficacy and effectively overcomes topotecan and irinotecan-resistant human tumors [78].
Figure 2.
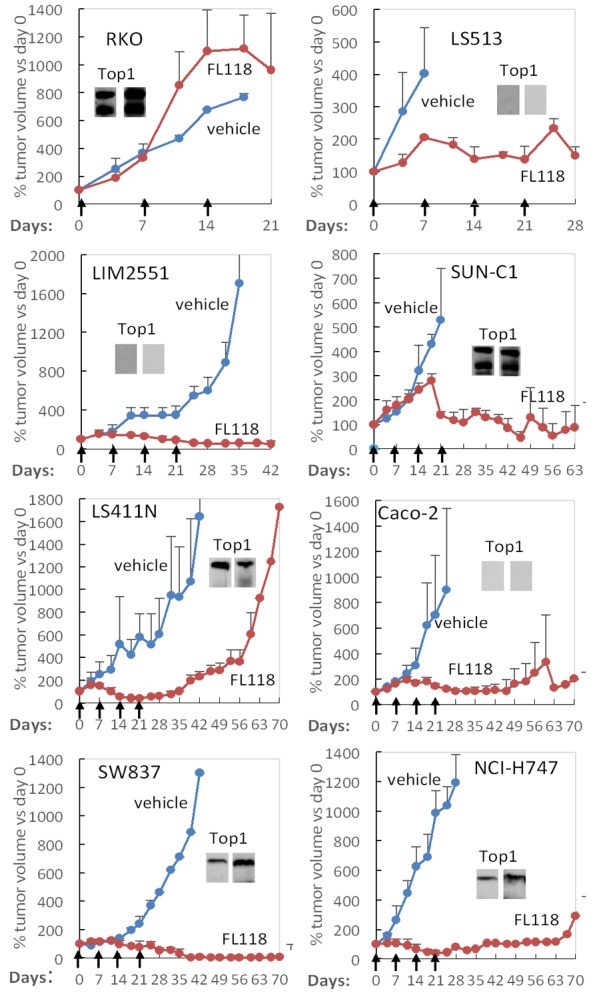
The sensitivity of colorectalcancer (CRC) xenograft tumors to FL118 treatment is not associated with the expression level of Top1: The small image insert within each xenograft tumor histogram curve was the expression of Top1 proteins measured using western blots with Top1 antibodies from two independent commercial sources. Individual xenograft tumors were first established from their corresponding CRC cell lines (RKO, LS513, LIM2551, SUN-C1, LS411N, Caco-2, SW837, NCI-H747,) by subcutaneous injection of 2 million cells at the flank area of SCID mice, respectively. Then the established tumors were inoculated into SCID mice at the flank area for testing FL118 sensitivity. FL118 treatment was initiated at the time when the inoculated individual xenograft tumors reached 100-200 mm3 (designated day 0). FL118 was administered with the schedule of weekly × 4 (arrowed) via po (per oral) routes at a dose of 10 mg/kg (MTD: maximum tolerated dose). Individual tumor curves were derived from the mean tumor sizes ± SD from up to five mice. These in vivo experimental studies were performed following the mouse protocol that was approved by the Institutional Animal Care and Use Committee (IACUC) at Roswell Park Cancer Institute. (Data were adapted from our previous publication: Li et al Am J Cancer Res 2017; 7: 370-382).
In summary, these observations suggest that FL118 does not exert its antitumor effects through inhibition of Top1 activity; instead, FL118-mediated inhibition of Top1 activity may mainly be involved in FL118-induced hematopoietic toxicity as suggested in our recent studies [48].
Unique mechanism of actions for certain CPTs such as FL118
If FL118 does not use Top1 as a major target for its anticancer activity, then what target(s) does FL118 use for its antitumor efficacy? Our studies revealed that FL118 selectively inhibits the expression of multiple antiapoptotic proteins (survivin, Mcl-1, XIAP, cIAP2). We found that the inhibition of these proteins by FL118 is independent of the tumor suppressor p53 status (wild type, mutant or null) [54]. This is another important feature of FL118, because most (if not all) DNA damaging drugs are ineffective when p53 is mutated or lost (null). Next, we asked whether these gene products (survivin, Mcl-1, XIAP, cIAP2) are involved in FL118’s inhibition of cancer cell growth and induction of apoptosis. Our studies demonstrated that when we genetically overexpress or silence these proteins individually, each of these proteins plays a role in FL118-mediated cancer cell growth inhibition and apoptosis induction [54,93]. Furthermore, in p53 wild type colorectal cancer cells, FL118 induces p53-dependent senescence by promoting MdmX (also called Mdm4) ubiquitination and degradation [94]. Intriguingly, in the absence of p53, FL118 exhibits an even stronger ability to inhibit colorectal cancer (CRC) cell growth and induce apoptosis [94]. We further demonstrated that forced expression of exogenous MdmX in HCT116 colon cancer cells further enhances FL118 ability to inhibit cell growth and induce apoptosis [94]. This suggests that the oncogenic protein MdmX is a unique biomarker and target for FL118, as well. Mechanistically, the inhibition of MdmX expression by FL118 is through FL118 switching Mdm2-mediated ubiquitination and degradation of the tumor suppressor p53 (oncogenic effects) to Mdm2-mediated ubiquitination and degradation of MdmX (tumor suppression effects) [94]. Intriguingly, the degradation of oncogenic protein MdmX by Mdm2 is independent of the DNA damage signaling regulator ATM and the status of p53 and p21 [94]. Furthermore, our recent studies indicated that in addition to its inhibition of survivin, Mcl-1, XIAP, cIAP2 and MdmX, FL118 can effectively inhibit the expression of ERCC6 (The Li Lab unpublished observations), a critical DNA repair regulator that is involved in active gene repair [95], correcting transcription-coupled DNA repair defects [96] and drug resistance [97]. This finding supports the idea that FL118 in combination with other DNA damaging drugs have the potential to even treat the most difficult-to-treat cancers. Additionally, different from irinotecan, SN-38 and topotecan, which are substrates of the efflux pump proteins ABCG2/BCRP [98-103] and P-gp/MDR1 [104-109], FL118 is not a substrate of ABCG2 and P-gp, and can overcome treatment resistance resulting from the expression of ABCG2 [110] or P-gp [78]. This might be one of the reasons that FL118 can effectively overcome irinotecan and topotecan resistance [78] and can be orally administered with high antitumor activity [48]. Actually, development of non-efflux pump (e.g. ABCG2) substrate drug instead of inhibition of them is a new trend in the field for anticancer drug development [111].
Examples of certain CPT compounds that are not dependent on the inhibition of Top1 activity were suggested in previous studies. Pommier and his team previously showed that while most of the CPTs tested have a well association of Top1 activity inhibition with antitumor activity, two CPT analogues, 10-NH2-(RS)-CPT and 11-CN-(RS)-CPT, showing very poor Top1 activity inhibition, extended mouse survival time much longer than other CPTs (which have strong Top1 activity inhibition) in the L1210 leukemia metastatic survival mouse model [112]. The disagreement between antitumor efficacy and the potential inhibition of Top1 enzyme activity suggests that 10-NH2-(RS)-CPT and 11-CN-(RS)-CPT may use alternative targets instead of Top1 for their anti-leukemia activity. Furthermore, it was recently reported that a CPT analogue O2-16 inactive against Top1 activity showed broadly antiviral HIV-1 activity through a Top1-independent mechanism [113]. These observations indicate that CPT analogues can show antitumor and antiviral activity independently of Top1 activity inhibition. Similarly, recent studies revealed that suppression of methyltransferase KMT1A by CPT in alveolar rhabdomyosarcoma tumor cells to induce cell differentiation is independent of CPT-mediated Top1 inhibition (Wolff, et al in press). Additional examples for novel antitumor mechanism of CPT analogues can be found in the sub-section of “Other CPT analogues or CPT conjugates” under the section of “Update of the outcomes of clinically developing CPT analogues” below.
It is now clear that CPTs can exert therapeutic effects independently of Top1 activity inhibition. However, those CPT analogues may still have Top1 inhibitory activity. This remaining Top1 activity inhibition may contribute to the drug side effects (e.g. hematopoietic toxicity), as suggested by our recent studies [48]. In this regard, avoiding or reducing a CPT analogue’s inhibition of Top1 activity would be one way to generate the next generation of low toxicity and high efficacious CPT structure-based anticancer therapeutic drugs.
Update of the outcomes of clinically developing CPT analogues
There are many reviews in the CPT and CPT analogue research field that summarized various aspects of the studies. In this regard, we found three of those review articles having comprehensive coverage largely without being redundant. One is from Legarza K and Yang LX in 2005 [114]; this article reviewed preclinical and clinical studies of individual CPT analogues, and the article format is very good for overviewing individual CPT analogue clinical development status then. The second article is from Venditto VJ and Simanek EE in 2010 [115]. This article used a similar review format but put more emphasis on the pharmacokinetics (PK) data-driven evaluation of various CPT analogues and clinical potential when the data was available. These two publications focused on distinct emphases on the available CPT analogues, and largely cited different publications. The third one is from Liu YQ et al in 2015. [116]. This article comprehensively reviewed the biological property of various CPT derivatives for potential treatment of cancer and other human disease. In this section of our review, the major goal is to review up-to-date CPT-based analogues that have been moved into clinical trials for cancer treatment and recent research progression in the CPT analogue research area.
Rubitecan (Orathecin, RFS2000/RFS-2000) (Figure 1G)
During irinotecan (CPT-11) development in the late 1980s, many CPT analogues were generated by adding a chemical group on the A-ring of CPT. Two of these compounds are 9-nitrocamptothecin (9-NC, rubitecan) and 9-aminocamptothecin (9-AC) [36]. Rubitecan/9-NC can be converted to 9-AC in vivo [117]. Studies indicated that 9-AC but not 9-NC is a substrate of ABCG2/BCRP [118]. So the advantage of 9-NC is that it was found to have a better overall PK profile via oral routes versus intravenous routes shown in rats [119], and thus oral administration of 9-NC may clinically be more effective, although its poor absorption in the given formulation is still an issue [119]. Generally speaking, at least some of the preclinical studies on 9-NC or 9-AC in animal models obtained promising results when used alone [114,115] or in combination with radiation [120]. However, most (if not all) clinical Phases I and II trials with 9-AC obtained disappointing results indicating the lack of antitumor activity for various human cancers with severe toxicity (Table 1). Interestingly, although there was a lack of promising clinical trial data in place in the mid-to-late 1990s, multiple clinical trials with 9-AC were continued until 2009, and later studies were also unable to obtain strong positive results for arguing further development (Table 1). The general conclusion is that further development of 9-AC for clinical application is not warranted in any cancer type that was clinically tested. Meanwhile, clinical trials with 9-NC were also continuously conducted throughout the mid 1990’s until 2011. Most of these clinical trials with 9-NC did not result in promising data to warrant further commercialization of 9-NC with the exception of pancreatic cancer with or without radiation/drug combination (Table 1), which seemingly provided a hope for commercialization of 9-NC for treatment of advanced pancreatic cancer. Two Phase III clinical studies on advanced pancreatic cancer patients reported in ASCO Annual Meeting in 2004 and 2005 (Table 1), though no full papers were followed for detailed evaluation. Nevertheless, based on the information on rubitecan/9-NC at the Astex Pharmaceuticals website (of note, the rubitecan’s sponsor SuperGen merged with Astex in 2011), the FDA officially accepted the rubitecan capsules’ New Drug Application (NDA) filed by SuperGen as a treatment for pancreatic cancer patients who have failed at least one prior chemotherapy in 2004 [121]. The news release indicated that the NDA filing contained data on more than 1,000 pancreatic cancer patients who failed at least one prior chemotherapy [121]. Of this population, more than 600 patients received Orathecin/rubitecan/9-NC capsules and the other ~400 patients were given control therapies [121]. However, based on an unclear source (likely SuperGen website then) provided in the Clark’s 2006 rubitecan review article, the NDA was withdrawn in 2005 by SuperGen, when the FDA informed SuperGen that the data at that point did not support approval of the drug for patients with advanced pancreatic cancer who had progressed on prior therapy [122]. Now after another decade has passed, rubitecan/9-NC may likely become an example that has provided us with many lessons for making a go or no/go decision much earlier in order to avoid the need of over 60 clinical trials on 9-NC and 9-AC (Table 1).
Table 1.
Clinical trials of 9-aminocamptothecin (9-AC) and 9-nitrocamptothecin (9-NC, rubitecan)
| Key | Route & dose | Formulation | Cancer type and key clinical trial outcome | Refs |
|---|---|---|---|---|
| 1995 9-AC Phase I | 72 h iv 3 wk/c*; MTD: 45 µg/m2/h | Dissolve in DMA, PEG400, phosphoric acid (see No 2) | 31 pts w resistant solid tumors; DLT: NP, TCP; minimal responses were seen in pts w gastric, colon, and NSCLC; try alternative schedules. | No 1 [123] |
| 1996 9-AC Phase I | 72 h iv at 0.1 mg/ml; 2 wk/c*; | 2% DMA, 98% PEG400 w 10 mM phosphoric acid diluted w saline | 48 pts w progressive solid tumor, DLT: NP; PR: 1 pt (finally progressed); 1 pt w 49% shrinkage in pulmonary nodules; 1 NSCLC pt w a 29% decrease in his lung metastases. MTD: 35 µg/m2/h or 47 µg/m2/h w G-CSF | No 2 [124] |
| 1996 9-NC Phase I | po: CPT at 0.3 mg/m2/d x 21 4 wk/c,; 9-NC at 1 mg/m2/d, 5/wk* | Drug powder is encapsulated in gelatin capsules (see No 7 and No 34) | 52 (CPT) and 29 (9-NC) pts w refractory solid & liquid cancer; DLT: CPT, diarrhea; 9-NC, NP, anemia and TCP; favorable responses (11% CPT, 24% 9-NC). | No 3 [125] |
| 1997 9-AC Phase I | 72 h iv 2 wk/c*; 5-74 µg/m2/h | Same as No 5 (also see No 2) | 48 pts w malignant solid tumor; total 9-AC circulating in plasma as the active lactone was less than 10%, no antitumor activity reported. | No 4 [126] |
| 1997 9-AC Phase II | 72 h iv at 35 µg/m2/h in 2 wk, 4 wk/c* | 0.1 mg/ml in 2% DMA, 49% PEG400 w 5 mM phosphoric acid (see No 2) | 17 naïve pts w metastatic colorectal carcinoma; No CR or PR; toxicity: neutropenia, nausea, vomiting, stomatitis, fatigue, and anemia but tolerated; lack of antitumor activity | No 5 [127] |
| 1997 9-AC Phase II | 72 h iv at 59 then 50 µg/m2/h w G-CSF in 2 wks* | Same as No 5 (also see No 2) | 16 pts w metastatic colorectal cancer; no OR; SD: 8 pts; DLT: myelosuppression; conclusion: no promising for this regimen | No 6 [128] |
| 1998 9-AC Phase I | po, 1.5 on d1 & iv, 1 mg/m2 on d8 or vice versa | Gelatin capsule containing 9-AC-PEG1000 molten mix | 12 pts w solid tumors; active lactone accounting for < 10% of total drug at the terminal disposition phase; the study is not activity focused | No 7 [129] |
| 1998 9-AC Phase I | 72 h iv at ≥ 37.5 µg/m2/h, 3 wk/c*; | Lyophilized CD in DMPC, DMPG & mannitol in 20% dextrose/saline | 25 pts w primary solid tumors; DLT: NP; OR: 0 pt; SD: 9 pts for 2-6 months; Phase II: 54.2 µg/m2/h, 72 h iv infusion every 3 weeks. | No 8 [130] |
| 1998 9-AC Phase I | 72 h iv at 36 to 62 µg/m2/h, 3 wk/c* | DMA-PEG400-phosphoric acid (see No 2). | 23 pts w resistant solid tumors; DLT: NP, TCP; PR: 2 pts; SD: 5 pts; Phase II: 52 µg/m2/h, 72 h iv infusion every 3 weeks (21 days). | No 9 [131] |
| 1998 9-AC Phase I | po, d1-5 at 0.2-0.68 mg/m2/d in 2 wk*; | CD formulation (see No 8) | 16 cancer pts; DLT: nausea; OR: 0 pt; conclusion: the CD formulation for iv is not good for po | No 10 [132] |
| 1998 9-AC Phase II | 120 h iv/wk x 3 wks* at 480 µg/m2/d | Not clear, likely used the No 2 recipe | 17 naïve pts w metastatic colorectal cancer; no responses observed; toxicity: granulocytopenia, nausea, vomiting and diarrhea | No 11 [133] |
| 1998 9-AC Phase I | 24 h iv/wk x4 at 0.7-1.9 mg/m2, 5 wk/c* | CD formulation (see No 8) | 16 of 20 pts w 5-Fu resistant colorectal cancer; toxicity: NP and diarrhea; Phase II: 1.65 mg/m2 | No 12 [134] |
| 1998 9-NC Phase I | Po d1-5/wk at 1, 1.5, 2 mg/m2/d in turn for 28, 68 & 159 wks | Not clear (full paper is inaccessible) | 43 pts w resistant metastatic cancer; DLT: anemia, NP, TCP, diarrhea; 5 pts w pancreatic, breast, ovarian & hematologic tumors had response; DS: 14 pts; 1 pt got 18-month TX | No 13 [135] |
| 1998 9-AC Phase II | 72 h iv in 2 wk*; at 59-45.8 µg/m2/h w G-CSF | DMA-PEG400-phosphoric acid (see No 2). | 58 pts w IIIB/IV NSCLC; PR: 5 pts; toxicity: NP, TCP; data not suggested further evaluation w the doses and schedule used. | No 14 [136] |
| 1998 9-AC Phase II | 72 h iv in 3 wk* at 40 µg/m2/h | DMA-PEG400-phosphoric acid (see No 2). | 45 pts w relapsed or refractory lymphomas; PR: 10 pts; G-CSF reduced NP & diarrhea rates, but no help in dose increase; DLT: TCP. | No 15 [137] |
| 1999 9-AC Phase I | po d1-7 or d1-14 at 0.25 to 1.1 mg/m2/d 3 wk/c*; | Gelatin capsules: same as or similar to No 7 | 30 pts w solid tumors; DLT: NP, TCP; PR: 1 pt; (recommended) phase II dose: 0.84 mg/m2/d | No 16 [138] |
| 1999 9-AC Phase I | po at 0.25 to 1.5 mg/m2/d on d1, d6 or on d1, d8; | Gelatin capsules: same as or similar to No 7 | 32 pts w solid tumors; PK focus; linear and dose-independent PK w small intrapatient kinetic variability; lactone form > 10% | No 17 [139] |
| 1999 9-AC Phase I | 0.5 h iv at ≥ 0.4 mg/m(2)/d on d1-5/wk x 3 | CD (see No 8); further dilution w saline if needed | 31 pts w resistant solid tumors; DLT: TCP, NP; PR: 1 pt; recommend phase II: 1.1 mg/m2/d; there is 10% lactone form | No 18 [140] |
| 1999 9-AC Phase II | 72 h iv at 35.4-59 µg/m2/h in 2 wk x 2; | DMA-PEG400-phosphoric acid (see No 2). | 80 pts w solid tumors; NP was the main toxicity; no tumor response; no lactone versus carboxylate information but a total of both were measured. | No 19 [141] |
| 1999 9-NC Phase II | po d1-4/wk* at 1.5 mg/m2/d | Not clear (full paper is inaccessible) | 29 pts w resistant ovarian, tubal or peritoneal cancer; 7% remission and 34% obtained SD. Major toxicity: anemia, NP, TCP and diarrhea. | No 20 [142] |
| 1999 9-AC Phase I | 7 d iv at ≥ 0.2 mg/m2/d in 3-4 wk x ≤ 2*; | CD (see No 8); 100 µg/ml further dilution w sterile water | 39 pts w resistant blood cancer; major toxicity: mucositis and diarrhea; no complete or partial remission was observed. | No 21 [143] |
| 1999 9-NC Phase II | po at 1.5 mg/m2/d d1-5/wk in 8 wk*; | Capsule form (SuperGen provided (see No 7) | 107 pts w advanced pancreatic cancer; 60 pts finished 2 8 wk courses; safe & efficacious; DLT: myelosuppression, interstitial cystitis | No 22 [144] |
| 2000 9-AC Phase II | 72 h iv at 45 µg/m2/h; w G-CSF in 2 wk x ≥ 1*; | Not described in the paper | 18 pts w resistant breast cancer; major toxicity: granulocytopinia and TCP; 2 out of 15 assessable pts showed limited responses. | No 23 [145] |
| 2000 9-AC Phase II | 72 h iv at 35.4 µg/m2/h in 2 wk x ≥ 1*; | Like used the recipe in No 2 | 14 pts w H&N SCC; OR: 0 pt; hematologic toxicity was modest and promptly reversible. | No 24 [146] |
| 2000 9-AC Phase II | 72 h iv or 120 iv at 45.8 or 20 µg/m2/h w or w/o G-CSF, 4 wk/c* | DMA-PEG400-phosphoric acid (see No 2) | 51 naïve pts w metastatic colorectal carcinoma; high toxicity (leukopinia, TCP, NP, diarrhea, hepatotoxicity); 1/40 showed response; 3 pts died from treatment toxicity. | No 25 [147] |
| 2000 9-NC Phase I | Aerosolization d1-5 at 6.7 µg/kg/d in 3 wk x 2-14* | Aerosolized liposomal | 6 pts w tumor metastasis to lung; no side effect higher than grade 2 was observed; plasma 9-NC: 37 to 4.9 ng/ml in 24 h; SD: 2 pts | No 26 [148] |
| 2000 9-NC Phase I | po d1-5 at 1.5 mg/d in 3 wk; Cis 30-60 mg/m2/d x 1 | No formulation information provided in the full paper | 12 pts w unclear cancer type; DLT not reached; 10 pts received ≥ 2 courses; OR: 0 pt; | No 27 [149] |
| 2001 9-AC Phase II | 72 h iv/2 wk x ≥ 2* at 46 µg/m2/h w G-CSF | Formulation is not clear (full paper is inaccessible) | 12 pts w advanced lymphoma; the study was prematurely terminated due to toxicity; 3 pts died due to sepsis after their last 9-AC treatment | No 28 [150] |
| 2001 9-NC Phase II | po d1-5/wk at 1.5-2 mg/m3/d up to 37 wk* | Gelatin capsules (no details in full paper); refer to No 7 | 19 pts w advanced pancreatic cancer; OR: 4/14 pts; subjective responses: 13/14; toxicity terminated treatment in 7 pts | No 29 [151] |
| 2001 9-AC Phase I | 120 h iv/3-4 wk x ≥ 2* at 0.41-0.77 mg/m2/d | Both DMA-PEG400-phosphoric acid and CD, no details (see No 2 & No 8) | 55 pts w solid tumors; OR: 1 pt; minor responses on pts w lung and colon cancer were also observed; DLT: NP, TCP, and diarrhea. | No 30 [152] |
| 2002 9-NC Phase II | po d1-5/wk* at 1.5 mg/m2/d | Formation is not clear (full paper is inaccessible) | 28 pts w metastatic melanoma; SD: 4 pts for 3, 4, 6 and 8 months; diarrhea, moderate hematopoietic toxicity, | No 31 [153] |
| 2002 9-AC Phase I | ip q2d x 6/4 wk x ≥ 1c* at 1.25-13.5 mg/m2 | Formation is not clear (full paper is inaccessible) | 12 pts w peritoneal cancer; DLT: NP; 2 pts had objective evidence of clinical benefit and only one had progressive disease | No 32 [154] |
| 2002 9-NC Phase II | po d1-5/3 wk* at 1.5 mg/m2/d | Formulation was not described in the paper. | 15 eligible pts w advanced glioblastoma multiforme; NP and TCP were common; SD: 5 pts; not support for this disease use | No 33 [155] |
| 2002 9-NC Phase II | po (fast vs. food) d1-5/wk* at 1.5 - 2.0 mg/m2/d | Gelatin capsules w drug-lactose mix inside (see No 7) | 19 pts w naive advanced colorectal cancer; DLT: diarrhea, leucopinia, NP; toxicity well tolerated but no objective response; | No 34 [156] |
| 2002 9-NC Phase I | po d1-5/wk x 2 wk at ≥ 0.75 mg/m2/d; Gem, iv | 9-NC from SuperGen Inc, so likely capsule (see No 7) | 21 pts w advanced malignancies; DLT: NT, TCP; SD: 5/18 evaluable pts; MTD: 9NC 1 mg/m2, Gem 1000 mg/m2 on d1and d8/3 wk | No 35 [157] |
| 2003 9-AC Phase I | 72 h iv/2 wks* at 25-59 µg/m2/h | CD formulation (see No 8) | 20 pts w resistant solid tumors; DLT: granulocytopenia; no antitumor response; (recommended) Phase II dose: 47 µg/m2/h | No 36 [158] |
| 2003 9-NC Phase I | po: d1-5/2 wk; combination w capercitabine | Gelatin capsules w 9-NC and lactose inside. | 21 pts w metastatic solid tumors; DLT: nausea, emesis; SD: 9 pts. | No 37 [159] |
| 2003 9-NC Phase II | po d1-5/wk* at 1.5 mg/m2/d | Gelatin capsules w 9-NC w lactose inside. | 56 pts w GI tumor or STS; well tolerated but inactive in GI; minimal activity in pts w STS. | No 38 [160] |
| 2003 9-AC Phase I | iv followed w po using complex schedules. | CD formulation (see No 8 for details) | 32 pts w advanced solid tumors; DLT: anemia, NP, TCP; SD: 2 pts; lack of activity. | No 39 [161] |
| 2004 9-NC Phase II | po d1-5/wk x 3/c* at 1.5 mg/m2/d | Formulation unclear (full paper is inaccessible). | 20 pts w advanced resistant urothelial tract tumors; acceptable toxicity; PR: 1 pt | No 40 [162] |
| 2004 9-AC Phase II | 72 h iv/2 wk* at 35 µg/m2/h | DMA-PEG400-phosphoric acid (see No 2). | 60 pts w ovarian carcinoma treated; 4 full and 6 partial remissions (none was platinum-resistant); SD: 19 pts; DLT: NP, TCP, anemia | No 41 [163] |
| 2004 9-AC Phase II | 120 h iv/2 wk at 25 µg/m2/h 3 wk/c* | Formulation unclear (full paper is inaccessible). | 15 pts w naïve metastatic gastric cancer; SD: 3 pts lasting 3.4 months; DLT: NP, anemia | No 42 [164] |
| 2004 9-NC Phase I/II | po A: d1-5/wk in 2 wk; B: d1-14 in 4 wk; C: d1-5 in 8 wk | crystalline powder in hard gelatin capsules (see No7 and 34) | Pts w solid tumor: 34 on d1 PK, 11 on d10, d11 PK for A; 9 on d10, d11 PK for B; 4 for phase II on d1; focus PK; big interpatient and intrapatient variation of 9-NC vs. 9-AC | No 43 [165] |
| 2004 9-NC Phase II | po d1-5/wk* at 1.5 mg/m2/d | Formulation unclear (full paper is inaccessible). | 17 pts w resistant metastatic breast cancer; SD: 6 pts; nausea, vomiting, fatigue, diarrhea were common | No 44 [166] |
| 2004 9-NC Phase II | po d1-5/wk x 3* at 1.5 mg/m2/d | Gelatin capsules (see No 7) | 35 pts w SCLC; no objective responses were observed; Toxicity was acceptable (TCP, nausea/vomiting, diarrhea) | No 45 [167] |
| 2004 9-NC Phase I | Aerosolization; 6.7-26.6 µg/kg/d x 5 for 1-6 wk | Liposome using dilauroylphosphatidyl choline (DLPC) | 25 pts w advanced lung cancer; the aerosol route is feasible and safe; 2 pts showed partial remissions; SD: 3 pts | No 46 [168] |
| 2004 9-NC Phase I | po d1-5/wk x 2 in 4 wk or d1-14/4 wk | Unclear in full paper (likely capsule, see No 7 or No34) | 26 pts w advanced solid tumors; DLT: NP, TCP, diarrhea; SD: 3 pts; PR: 1 pt; Phase II: 2.43 and 1.70 mg/m2/d | No 47 [169] |
| 2004 9-NC Phase III | po d1-5/wk x 8 wk* at 1.5 mg/m2/d; | SuperGen involved studies; so likely capsule (see No 7) | Resistant pancreatic cancer: 198 pts (9-NC) vs. 211 pts (best care); no median survival difference but MS and PFS favored to 9-NC pts; Conclusions: The study can achieve tumor growth control with an acceptable risk-benefit ratio for the disease with few treatment options. | No 48 [170] |
| 2005 9-NC Phase III | po d1-5/wk* at 1.5 mg/m2; 5-Fu iv weekly at 600 mg/m2 | Capsules | 224 pts w resistant pancreatic cancer; In the evaluable group, 14 of 35 pts achieved tumor growth control (OR: 4 pts; SD: 10 Pts); no survival improvement evidenced; | No 49 [171] |
| No 50 [121] | ||||
| 2005 9-NC Phase II | po d1-5/wk* at 1.25 mg/m2/d | 9-NC from SuperGen Inc, so likely capsule (see No 7) | 51 pts w advanced chordoma, STS or GIST; OR: 2 pts; major toxicity: anemia, leukopenia, fatigue, nausea, diarrhea | No 51 [172] |
| 2005 9-AC Phase II | 120 h iv/wk in 3 wk* at 25 µg/m2/h | CD formulation (see No 8) | 56 pts w platinum-resistant ovarian cancer; major toxicity: NP, leukopenia, anemia, TCP; CR: 4 pts; PR: 4 pts; SD: 18 pts | No 52 [173] |
| 2005 9-NC Phase I | po d1-5/wk• at 1-1.25 mg/m2/d; 45 Gy/5 wks of radiation | 9-NC from SuperGen Inc, so likely capsule (see No 7) | 8 pts w locally advanced pancreatic cancer; DLT: nausea/vomiting, fatigue, anorexia, leukopenia, dehydration; Conclusion: 1 mg/m2/day can be given w radiation | No 53 [174] |
| 2005 9-NC Phase II | po d1-5/wk* at 1.5, 1.75 and/or 2.0 mg/m2/d | Unclear, likely capsule (see No 7) | 17 pts w IIIB(9), IV(8) naive NSCLC; well tolerated (no NP, TCP); SD: 10 pts; conclusion: inactive at doses used for this type of NSCLC. | No 54 [175] |
| 2005 9-NC Phase II | po d1-5/wk x 8 wk at 1.5 mg/m2/d | Unclear, likely capsule (see No 7) | 58 pts w resistant pancreatic cancer; PR: 3/43 pts; SD: 7/43 pts; common toxicity: gastrointestinal and hematologic toxicity | No 55 [176] |
| 2006 9-AC Phase I | 72 h iv/2 wk* at 46 µg/m2/h | Formulation unclear (full paper is inaccessible). | 14 pts w glioblastoma multiforme (GBM); DLT: lymphopenia, NP; lack activity against GBM, no further trial necessary for 9-AC in GBM | No 56 [177] |
| 2006 9-AC Phase I | 7 d (DMA) iv or 21 d (CD) iv at ≥ 6.2 µg/m2/h | Both DMA & CD formulations used (see No 2 and No 8) | 57 pts w resistant solid tumor; DLT: NP, TCP; OR: 6/57 pts; 9AC/CD has ~2x lactone form of those from 9AC/DMA for the same dose level. | No 57 [178] |
| 2006 9-NC Phase I | po d1-3 (9NC at 0.75-2 mg/m2), then etoposide | Formulation not provided in the paper | 45 pts with advanced cancer; DLT: NP, TCP, nausea, vomiting, diarrhea and fatigue in 6 pts; OR: 2 pts; SD: 13 pts; | No 58 [179] |
| 2006 9-NC Phase I | po w or w/o fast at 1.5 mg/m2; then d1-5/wk | 0.5 mg tablets from SuperGen, Inc. (Dublin, CA, USA) | 16 pts w solid tumors; SD: 2 pts for 8 wks; food recued 9-NC absorption but no 9-AC exposure difference; high inter-patient variability | No 59 [180] |
| 2006 9-NC Phase II | po d1-5/wk* at 1.5 mg/m2/d; | Drug from SuperGen Inc, likely capsule (see No 7) | 16 pts w resistant metastatic breast cancer; SD: 5/13 pts; grade 3/4 toxicity: allergy, pain, diarrhea, TCP(2), myalgia | No 60 [181] |
| 2006 9-NC Phase II | po d1-5/wk* at 1.5 mg/m2/d | Formulation unclear (full paper is inaccessible). | 14 pts w advanced 5Fu-resistant colorectal cancer; well tolerated; DLT: anemia, diarrhea; no response/no seen clinical activity. | No 61 [182] |
| 2008 9-NC Phase I | iv Cis on d1, then po 9NC d1-5/wk x 3 wk | Formulation unclear (full paper is inaccessible) | 51 pts w resistant solid tumors; DLT: TCP, NP; 1 pt w partial remission; SD: 12 pts; Phase II: Cip/9NC, 60 mg/m2/1.25 mg/d or 40 vs. 2.0 | No 62 [183] |
| 2008 9-NC Phase II | po d1-5/wk* at 1.5 mg/m2/d; | Crystal powder in gelatin capsule, likely from Supergene Inc. | 19 pts w resistant metastatic head-&-neck cancer; SD: 3/13 pts & 10 progressed; 3 died shortly after treatment; DLT: anemia NP, TCP | No 63 [184] |
| 2009 9-AC Phase II | 72 h iv/2 wk*; 0.85 mg/m2/d (DMA); 1.1 mg/m2/d (CD) | Both DMA and CD formulations used (see No 2 and No 8) | 37 pts w relapsed lymphoma; OR rate: ~17% similar in both formulations; DLT: NP, anemia, TCP; serum drug level not link to response & toxicity | No 64 [185] |
| 2011 9-NC Phase I | po once at 1.25-1.75 mg/m2; then d1-5/wk at 1.5 mg/m2 | Capsules from Qilu Pharmaceutical Co., Ltd, China | 23 pts w advanced solid tumors; PK focused, no serous toxicity; There was 2-13 fold variabilities in 9-NC and 9-AC exposure among different pts | No 65 [186] |
Belotecan (CKD-602/CKD602/CKD 602, Camtobell) (Figure 1H)
Belotecan is a water-soluble CPT analogue and was found to be a substrate of Pgp/MDR1 and BCRP/ABCG2 [187]. This finding is consistent with the fact that belotecan has never been reported to be orally administered thus far. Recent studies using oral squamous cell carcinoma cell lines indicated that the antiproliferative effects of belotecan is associated with an increase of phospho-cdc2 (Tyr 15), cyclin A2 and cyclin B1 as well as apoptosis in parallel with G2/M arrest [188]. However, it is unclear whether the increased expression of cyclin A2 and cyclin B1 is actually a resistant factor for belotecan, and involved in making belotecan less effective, which could be worthy of further investigation for clarification. Nevertheless, preclinical studies revealed that belotecan had good antitumor activity as a Top1 inhibitor, although in most cases belotecan was only able to delay or transiently regress tumor growth [114,115]. Clinical development of belotecan began sometime before 2000 (Table 2). A large number of belotecan Phase I and Phase II clinical trials were published between 2007 and 2013 (Table 2). The results from these clinical trials obtained mixed results in various cancer types with belotecan monotherapy or in combination with cisplatin (Table 2). Nonetheless, a belotecan Phase III clinical trial was performed using the most promising cancer type of 147 extensive-stage naïve SCLC patients (no past history of chemotherapy or radiotherapy) via randomized open-label for a head-to-head comparison of antitumor efficacy and toxicity between belotecan/cisplatin (BP, 71 patients) and etoposide/cisplatin (EP, 76 patients) in multiple centers [189]. In the BP arm, one patient had a complete response, 41 had a partial response (PR), and 17 had stable disease (SD). In the EP arm, 35 patients had PR and 28 had SD. The response rate (RR) in the BP arm was non-inferior to the EP regimen in patients with ES-SCLC (BP: 59.2%, EP: 46.1%, difference: 13.1%, 90% two-sided confidence interval: -0.3-26.5, meeting the predefined non-inferiority criterion of -15.0%). No significant differences in overall survival (OS) or progression-free survival (PFS) were observed between the treatment arms. Hematologic toxicities, including grade 3/4 anemia and thrombocytopenia (TCP), were significantly more prevalent in the BP arm than the EP arm. The authors concluded that The RR to the BP regimen was non-inferior to the EP regimen in this type of cancer. However, hematologic toxicities were significantly more prevalent in the BP group [189]. In our view, this is a negative result. However, a recent retrospective review of 94 patients with SCLC (with or without prior chemotherapy) who were treated using belotecan monotherapy (n = 59, 188 cycles) or topotecan monotherapy (n = 35, 65 cycles) between September 2003 and December 2011 indicated that TCP occurred during 42% and 61.5% of the belotecan and topotecan cycles, respectively (P = 0.007). Grade 4/5 lung infection (belotecan 3.2% versus topotecan 10.8%, P = 0.003), all-grade headache (belotecan 3.2% versus topotecan 10.8%, P = 0.017), and grade 4/5 increased liver enzymes (belotecan 0.5% versus topotecan 4.6%, P = 0.023). The median time to progressive disease (TTPD), chemotherapy-specific survival (CSS), and OS were 14 months and 11.6 months (P = 0.646), 10 months and 7 months (P = 0.179), and 34.5 months and 21.4 months (P = 0.914) after belotecan and topotecan monotherapy, respectively. These authors concluded that belotecan may be safer than topotecan for monotherapy in SCLC patients, and in terms of efficacy, belotecan could be comparable to topotecan in monotherapy [190]. Nevertheless, based on our review of the relevant information above and in Table 2 for belotecan, additional trials using belotecan will most likely prove to be unproductive.
Table 2.
Clinical trials of belotecan (CKD-602/CKD602, Camtobell)
| Key | Route & dose | Formulation | Cancer type and key clinical trial outcome | Refs |
|---|---|---|---|---|
| 2000 Phase I | 0.5 h iv d1-5 at 0.5-0.9 mg/m2/d; 3 wk/c | Not clear in the publication. | pts w advanced solid cancers; DLT: NP; MTD: 0.7 mg/m2/d; PR in some pts w stomach or ovarian cancer was observed | No 1 [191] |
| 2007 Phase I | 0.5 h iv on d1, d4 at ≥ 0.4 mg/m2/d; Cis 60 mg/m2 on d1; 3 wk/cycle | Not clear in the publication. | 17 pts w SCLC, MTD: 0.5 mg/m2/d; DLT: NP w favor; 13/17 w PR; plasma clearance of belotecan was 5.78 ± 1.32 L/h and terminal half-life was 8.55 ± 2.12 h. warranted for Phase II. | No 2 [192] |
| 2008 Phase II | 0.5 h iv on d1-5 at 0.5 mg/m2/d; 3 wk/cycle | 5% dextrose water infusion | 27 pts w SCLC; 9 PR; 1 CR; most common toxicity: NP; active for SCLC and warranting combination w platinum or other agents. | No 3 [193] |
| 2008 Phase II | 0.5 h iv on d1-5 at 0.5 mg/m2/d; 3 wk/cycle: 94/24 pts | Not clear in the publication. | 24 pts w recurrent ovarian cancer; 4 pts had PR & 5 pts had SD; DLT: NP; against both Cis-sensitive (8) and resistant (1) tumors. | No 4 [194] |
| 2009 Phase I/IIa | 0.5 h iv on d1-5 at ≥ 0.3 mg/m2/d; Cis 60 mg/m2 on d5; 3 wk/cycle: 2-12/pt | 5% dextrose water infusion | 26 pts w recurrent ovarian cancer; MTD: 0.3 mg/m2/d; DLT: NP; grade 3 nausea and anorexia were the most common GI toxicities; against both Cis-sensitive (14) and resistant (4) tumors. | No 5 [195] |
| 2009 Phase I | 1 h iv at 0.1-2.5 mg/m2/d; 3 wk/cycle: 1-8/pt | Pegylated liposomal w 5% dextrose infusion | 45 pts w refractory solid tumors; DLT: mucositis, bone merrow suppression, NP; MTD: 2.1 mg/m2/d; 2 pts w PR; extending exposure | No 6 [196] |
| 2010 Phase II | 0.5 h iv on d1-5 at 0.5 mg/m2/d; 3 wk/cycle: ≤ 6/pt | Not clear in the publication. | 27 pts w relapsing SCLC after irinotecan failure; DLT: NP, TCP; Conclusion: modest activity w manageable toxicities in Asia pts. | No 7 [197] |
| 2010 Phase II | Iv d1-5; at 0.5 (bel), 1.5 (top) mg/m2, 3 wk/cycle | Not clear in the publication. | 45 pts (topotecan) & 35 pts (belotecan) w recurrent ovarian cancer; ORR: topotecan 24% vs. belotecan 45%; no survival differences; | No 8 [198] |
| 2010 Phase II | 0.5 h iv on d1-5 at 0.5 mg/m2/d; 3 wk/cycle: ≥ 3/pt | 5% dextrose water infusion | 62 pts w extensive stage naive SCLC; DLT: NP, TCP; Conclusion: relatively active (ORR: 53%) and well tolerable. | No 9 [199] |
| 2010 Phase II | 0.5 h iv on d1-5 at 0.5 mg/m2/d; 3 wk/cycle: ≥ 3/pt | 5% dextrose water infusion | 63 pts w refractory ovarian cancer; DLT: NP, TCP; active (ORR: 30%, 9 CR); major toxicity: hematopoietic toxicity | No 10 [200] |
| 2010 Phase II | 0.5 h iv on d1-4 at 0.5 mg/m2/d; Cis 60 mg/m2 on d1; 3 wk/cycle: ≤ 6/pt* | Not clear in the publication. | 30 pts w extensive stage naive SCLC; 21 pts (ORR: 70%); PR; DLT: NP, TCP; Conclusion: combination has promising response with a manageable toxicity profile. | No 11 [201] |
| 2010 Phase II | iv on d1-5 at 0.5 or 0.3 mg/m2/d (Cis 50 mg/m2 on d1); 3 wk/cycle: ≤ 6/pt* | Not clear in the publication. | 53 pts w recurrent ovarian cancer; combination better then belotecan alone (16/34: 47.1% vs. 4/19: 21.1%) but belotecan alone has less grade 3 or 4 toxicity than combination. | No 12 [202] |
| 2011 Phase II | 0.5 h iv on d1-5 at 0.3 mg/m2/d; Carbop-latin on d5; 3wk/cycle: > 2/pt* | Not clear in the publication. | 38 pts w recurrent ovarian cancer; CR: 7 pts; PR: 13 pts; SD: 6 pts; progress disease: 9 pts; DLT: NP, TCP, anemia; combination is well-tolerated with activity for the disease. | No 13 [203] |
| 2011 Phase II | 0.5 h iv on d1-5 at 0.5 mg/m2/d; 3 wk/cycle: 1-7/pt | 5% dextrose water infusion | 16 pts w recurrent cervix carcinoma; DLT: NP, anemia; no PR; no CR; Conclusion: belotecan is not active to this disease. | No 14 [204] |
| 2011 Phase II | 0.5 h iv on d1-4 at 0.5 mg/m2/d; 3 wk/cycle: ≥ 3/pt | 5% dextrose water infusion | 25 pts w non-naive SCLC; ORR: 24%; DLT (grade 3/4): NP (88%), TCP (40%); Conclusion: relatively active and well tolerated. | No 15 [205] |
| 2012 Phase II | 0.5 h iv on d1-4 at 0.5 mg/m2/d; Cis 60 mg/m2 on d1; 3 wk/cycle: ≤ 6/pt* | 5% dextrose water infusion | 35 pts w extensive stage naive SCLC; ORR: 71%; DLT (grade 3/4): NP (68%), TCP (28%), anemia (20%); Conclusion: significant efficacy w non-hematologic toxicity improved. | No 16 [206] |
| 2012 Phase I | 0.5 h iv on d1-4 at 0.5 mg/m2/d; etoposid 50 mg/d po d6-10; 3 wk/cycle: ~3/pt* | Sterile water | 9 pts w non-naïve solid tumors; PR: 2 pts; CR: 2 pts; having DLT; conclusion: promising activity for platinum-resistant or heavily pretreated ovarian cancer pts | No 17 [207] |
| 2012 Phase II | 0.5 h iv on d1-4 at 0.5 mg/m2/d; Cis 60 mg/m2 on d1; 3 wk/cycle: ≥ 2/pt* | Not clear in the publication. | 50 pts w relapse/refractory SCLC; ORR: low; DLT (grade 3/4): NP (54%), TCP (38%), anemia (32%); Conclusion: modest activity w an acceptable safety profile. | No 18 [208] |
| 2012 Phase I | 1 h iv at 0.1-2.5 mg/m2/d (PK focus) | Pegylated liposomal w 5% dextrose infusion | 45 pts w solid tumors; pts w liver tumor is 1.5-fold higher to eliminate the drug than pts without liver tumors. | No 19 [209] |
| 2013 Phase II | 0.5 h iv d1-4 at 0.5 mg/m2/d; Cis at 60 mg/m2 on d1; 3 wk/cycle: ≤ 6/pt* | 5% dextrose water infusion | 42 pts w extensive stage naive SCLC; ORR: 62%; DLT (grade ≥ 3): NP (90%), TCP (63%), anemia (34%); Conclusion: combination is effective but toxicity is too high. | No 20 [210] |
| 2013 Phase II | 0.5 h iv on d1-5 at 0.5 mg/m2/d; 3 wk/cycle: ≥ 2/pt | 5% dextrose water infusion | 26 pts w extensive stage naive SCLC; ORR: 35%; DLT (grade 3/4): NP (81%), TCP (15%); Conclusion: modest efficacy w OK toxicity. | No 21 [211] |
| 2016 Phase III | Route: iv; Combination: belotecan/Cis (BP); etoposide/Cis (EP) | 5% dextrose water infusion | 71 pts (BP) & 76 pts (EP) w extensive stage naive SCLC; randomized, open-label, parallel-group studies. Conclusion: No significant difference of BP vs. EP but BP is more toxic. | No 22 [189] |
Exatecan (DX-8951f/DX8951f or DX-8951/DX8951) (Figure 1I)
Unlike belotech being a substrate of both Pgp/MDR1 and BCRP/ABCG2 [187], exatecan is not a substrate of P-gp [114]. However, exatecan induces BCRP/ABCG2 protein, which is associated with reduction of its antitumor activity [212]. Nevertheless, exatecan exhibited activity in multiple cancer cell lines and/or xenografts including human breast, gastric, renal, colon, ovarian, cervical and lung [114,213] as well as in acute myelogenous leukemia (AML) [214] and pancreatic cancer [115]. Interestingly, preclinical toxicological studies revealed that dogs are more sensitive to exatecan than mice [215]. Nevertheless, based on the supportive preclinical studies, exatecan subsequently went into clinical trials (Table 3). Based on the overall information obtained in the Phase I and Phase II clinical trials over time in various types of cancer, high toxicity was always an issue, but it was manageable. As seen from the Phase III clinical trials below, the matter that sends exatecan into the grave is the lack of sufficient antitumor activity. A multicenter randomized open-label phase III clinical trial in 349 patients with advanced pancreatic cancer yielded very disappointing results: Exatecan plus gemcitabine obtains no better patient outcomes than gemcitabine alone, while exatecan plus gemcitabine clearly exhibits more toxic than gemcitabine alone. Patients have locally advanced or metastatic pancreatic adenocarcinoma without prior chemotherapy but may have radiation treatment alone for locally advanced disease; 175 patients were treated with exatecan 2.0 mg/m2 (30 min intravenous infusion) and gemcitabine 1,000 mg/m2 (immediately following exatecan administration) on days 1 and 8, every 3 weeks. Gemcitabine alone for the 174 control patients were dosed at 1,000 mg/m2 up to 7 weeks in the first cycle, then once a week for the first 3 weeks of a 4-week cycle. Tumor assessment was performed every 6 weeks. The primary end point was overall survival. The median survival time was 6.7 months for exatecan plus gemcitabine and 6.2 months for gemcitabine alone (P = 0.52). One complete response (CR, < 1%) and 11 partial responses (PR, 6.3%) were observed in the exatecan plus gemcitabine treatment group, and one CR (< 1%) and eight PR (4.6%) were observed in the gemcitabine-alone group. Grade 3 and 4 toxicities were higher for the two arm versus the gemcitabine alone arm; neutropenia (30% vs. 15%) and thrombocytopenia (15% vs. 4%). From such outcomes, the authors concluded that exatecan plus gemcitabine was not superior to gemcitabine alone with respect to overall survival in the first-line treatment of advanced pancreatic cancer [216].
Table 3.
Clinical trials of exatecan (DX-8951f/DX-8951 or DX8951f/DX8951)
| Key | Route & dose | Formulation | Cancer type and key clinical trial outcome | Refs |
|---|---|---|---|---|
| 2000 Phase I | 0.5 h iv at 4-7.1 mg/m2; 3 wk/c: 3/pt | Lyophilized drug in saline | 12 pts w refractory solid tumors; DLT: NP; MTD: 7.1 mg/m2; Phase II dose: 5.33 mg/m2 | No 1 [217] |
| 2000 Phase I | iv w multiple Phase I schedules and doses | Not clear in the publication. | pts w various solid tumors; DLT: NP; Goal: find DLT, MTD, Phase II schedule and dose. | No 2 [218] |
| 2000 Phase I | 0.5 h iv d1-5 at 0.1-0.6 mg/m2/d; 3 wk/c: 3-4/pt | Lyophilized drug w maltose in saline | 36 pts w advanced solid tumors; Phase II dose: ≥ 0.3 mg/m2; DLT: NP, myelosuppression; exhibiting modest antitumor activity. | No 3 [219] |
| 2000 Phase II | 0.5 h iv d1-5 at 0.3 (HP), 0.5 (MP) mg/m2/d | Not clear in the publication. | 14 pts w advanced ovarian, tubal or peritoneal resistant cancers; DLT: NP; SD: 4 pts. HP: heavily pretreated; MP: minimally pretreated | No 4 [220] |
| 2001 Phase I | 0.5 h iv at 3, 5, 6.65 mg/m2; 3 wk/c: ≥ 1/pt | Drug dissolved in saline | 15 pts w advanced solid tumors; DLT: NP; Phase II dose: 5 mg/m2; focus on pharmacokinetics | No 5 [215] |
| 2001 Phase I | 24 h iv at ≥ 0.15 mg/m2; 3 wk/c: ≥ 2/pt | Drug dissolved in saline | 22 pts w advanced solid tumors; SD: 4 pts; DLT: gradulocytopenia; MTD/Phase 2 dose: 2.4 mg/m2 | No 6 [221] |
| 2001 Phase I | 24 h iv at 0.05-1.2 mg/m2; 3/4 wk/c: 3/pt | Drug dissolved in saline | 27 pts w resistant solid tumors; SD: 4 pts; DLT: NP. TCP; Phase 2: 0.8 (MP), 0.53 (HP) mg/m2 | No 7 [222] |
| 2002 Phase I | 0.5 h iv d1-5 at 0.6 -1.35 mg/m2; 3/4 wk/c: ≥ 1/pt | Drug dissolved in saline | 25 pts w advanced leukemia; SD: 4 pts; DLT: 0.9-1.35 mg/m2; PR: 1 pt but no CR; Phase 2 dose: 0.9 mg/m2 | No 8 [223] |
| 2003 Phase I | 0.5 h iv d1 w multi-doses; 4 wk/c: 1-10/pt | Lyophilized drug in saline | 35 pts w advanced solid tumors; PR: 2 pts; SD: 12 pts; DLT: NP (MP), NP & TCP (HP); Phase 2 dose: 2.75 (MP), 2.1 (HP) mg/m2 | No 9 [224] |
| 2003 Phase II | 0.5 h iv d1-5 at 0.5 mg/m2/d; 3 wk/c: ≤ 6/pt | Not clear in the publication. | 39 pts w advanced NSCLC; PR: 2 pts; SD: 20 pts; DLT: NP; Conclusion: limited activity. | No 10 [225] |
| 2003 Phase II | 0.5 h iv d1-5 at 0.5 mg/m2/d; 3 wk/c: 1-16/pt | Drug dissolved in saline | 39 pts w resistant/metastatic breast carcinoma; PR: 3 pts; SD: 8 pts; DLT: NP; moderate activity. | No 11 [226] |
| 2003 Phase I | 21 d iv at 0.15 mg/m2/d | Lyophilized drug w maltose in saline | 31 pts w advanced solid tumors; PR: 2 pts; DLT: N, TCP (unacceptable high) | No 12 [227] |
| 2004 Phase IIa | 0.5 h iv, d1-5 at 0.3 mg/m2/d, 3 wk/c or at 2.1 mg/wk; 3/4 wk/c | Lyophilized drug w maltose in saline | 39 pts w resistant ovarian cancer; poor/modest activity; DLT: NP, myelosupression and emesis. | No 13 [228] |
| 2004 Phase II | 0.5 h iv d1-5 at 0.5 mg/m2/d; 3 wk/c* | Drug dissolved in saline | 15 pts w resistant/metastatic colorectal cancer; SD: 6 pts; DLT: NP; Conclusion: poor activity. | No 14 [229] |
| 2004 Phase II | 0.5 h iv d1-5 at 0.5 mg/m2/d; 3 wk/c: ≥ 2/pt | Lyophilized drug in saline | 16 pts w advanced/resistant ovarian, tubal or peritoneal resistant cancers; SD: 7 pts. DLT: NP, neutropenia; conclusion: poor activity | No 15 [230] |
| 2005 Phase II | 0.5 h iv d1-5 at 0.5 mg/m2/d; 3 wk/c: ≥ 2/pt | Not clear in the publication. | 41 pts w advanced biliary tract cancers; PR: 2 pts; SD: 12 pts. DLT: NP, neutropenia; conclusion: minimal activity w manageable toxicity | No 16 [231] |
| 2005 Phase II | 0.5 h iv d1-5 at 0.5 mg/m2/d; 3 wk/c*: 1-10/pt (median 3) | Not clear in the publication. | 39 pts w metastatic naïve gastric cancer; PR: 2 pts; SD: 18 pts; DLT: NP, neutropenia; conclusion: modest activity; toxicity manageable | No 17 [232] |
| 2006 Phase III | 0.5 h iv (see text for detail) | Not clear in the publication. | 175 pts (exatecan plus Gem) vs. 174 pts (Ge only) w advanced naïve pancreatic cancer; Two drugs no better than Gem alone but more toxic | No 18 [216] |
| 2007 Phase II | 0.5 h iv d1-5 at 0.5 mg/m2/d; 3 wk/c*: median 2 | Not clear in the publication. | 39 pts w advanced soft tissue sarcoma; DLT (grade 3/4): NP (49%), TCP (23%), anemia (15%); modest/non-significant activity | No 19 [233] |
DE-310/DE310
DE-310 is the exatecan/DX-8951f covalently conjugated with a carboxymethyldextran polyalcohol carrier via a peptidyl spacer. Use of a murine Meth A (fibrosarcoma) model demonstrated that DX-8951f at its MTD via a qd × 5 schedule shrank the tumor. In contrast, single treatment (qd × 1) with DE-310 at the MTD or 1/4 MTD shrank the tumor, with no body weight loss at 1/4 MTD [234]. Against 5 human tumor (colon and lung cancer) xenografts in nude mice, DE-310 (qd × 1) was as effective as DX-8951f administered once every 4 days, 4 times [234]. Additionally, DE-310 was also showed to have a better PK profile than exatecan [235]. These results indicated that DE-310 is superior to exatecan in terms of antitumor activity. However, follow-up studies revealed that DE-310 can induce various abnormalities in rat fetuses including menignocele [236]. Furthermore, clinical studies failed to demonstrate a favorable PK or striking antitumor activity (Table 4). Thus, further clinical studies of DE-310 appear to have stopped since 2006 (no more clinical trials were reported).
Table 4.
DE-310/DE310
| Key | Route & dose | Formulation | Cancer type and key clinical trial outcome | Refs |
|---|---|---|---|---|
| 2005 Phase I | 3 h iv at 1-2 mg/m2, 2 wk/c or at 6-9 mg/m2, 6 wk/c (86c/24 pts) | Lyophilized drug w maltose diluted in saline | 27 pts w advanced solid tumors; DLT: NP, TCP, veno-occlusive hepatotoxicity; CR: 1 pt; PR: 1 pt; SD: 14 pts; Phase II: 7.5 mg/m2, 6 wk/c | No 1 [237] |
| 2005 Phase I | 3 h iv at 6 mg/m2; one time for PK studies | Lyophilized drug w maltose diluted in saline | 6 pts w solid tumors; preferential accumulation of DE-310, DX-8951 and G-DX-8951 in human tumor tissues was not observed | No 2 [238] |
Lurtotecan (GI147211/GI-147211, NX211/NX-211, OSI211/OSI-211) (Figure 1J)
Lurtotecan was studied for possible oral administration, but the conclusion of the study was that oral administration of lurtotecan results in a low bioavailability with relatively wide interpatient variation, and the authors advised the intravenous route for further lurtotecan development [239]. Interestingly, a liposomal formulation of lurtotecan (NX211/OSI211) via iv administration was found to be much better than lurtotecan iv administration in terms of antitumor efficacy, PK ad biodistribution in nude mice of human ovary clear cell carcinoma (ES-2) and human ubiquitous KERATIN-forming tumor cell line HeLa subline (KB)-established xenograft tumor models [240] as well as in SCID mice of human leukemia models in later studies [241]. Subsequently, a sensitive fluorescence-based detection of liposomal-formulated lurtotecan (NX211) in human plasma and urine was developed [242]. NX211 is easy to be lysed to 7-methyl-10,11-ethylenedioxy-20 (S)-CPT under normal light and thus, light protection and reconstitution of NX211 immediately before clinical use in a light protection fashion are required [243]. Similar to rubitecan, belotecan and exatecan, while in vitro preclinical studies of lurtotecan showed antitumor activity, clinical trials with lurtotecan or its liposome-formulated version (NX211/OSI211) was unable to demonstrate significantly better antitumor activity in comparison with topotecan and/or irinotecan/SN-38 in various types of cancer (Table 5).
Table 5.
Clinical trials of lurtotecan (GI147211/GI-147211, NX211/NX-211, OSI211/OSI-211)
| Key | Route & dose | Formulation | Cancer type and key clinical trial outcome | Refs |
|---|---|---|---|---|
| 1996 Phase I | 0.5 h iv d1-5 at 0.3-1.5 mg/m2/d; 3 wk/c: ≥ 3/pt | Drug diluted w D5W | 18 pts w refractory solid tumors; DLT: NP, TCP; PR: 1 pt; Phase II: 1.2 mg/m2 on d1-5, 3 wk/c | No 1 [244] |
| 1998 Phase I | 0.5 h iv d1-5 at 0.3-1.75 mg/m2/d; 3 wk/c: ~3/pt | Drug diluted in 5% dextrose in water (D5W) | 24 pts w advanced solid tumors; DLT: NP, TCP; Phase II: 1.0 mg/m2 on d1-5, 3 wk/c; manageable toxicity but need to see distinct efficacy | No 2 [245] |
| 1998 Phase I | 72 h iv at 0.25-2.5 mg/m2/d | Drug diluted in D5W | 44 pts w advanced solid tumors; DLT: NP, TCP; PR: 3 pts; Phase II: 1.75 (MP), 1.2 (HP) mg/m2/d | No 3 [246] |
| 2000 Phase II | 0.5 h iv d1-5 at 1.2 mg/m2/d; 3 wk/c: ≥ 2/pt (267 c/67 pts) | Drug diluted 5% dextrose in water (D5W) | 67 pts w breast/NSCLC/colon tumors; DLT: NP, TCP, anemia; PR: 3/25 (breast), 2/23 (NSCLC), 0/19 (colon); conclusion: modest activity. | No 4 [247] |
| 2000 Phase II | 0.5 h iv d1-5 at 1.2 mg/m2/d; 3 wk/c: ≥ 2/pt - ≤ 4/pt | Drug diluted 5% dextrose in water (D5W) | pts w refractory (28) & chemosensitive (34) SCLC; ORR: 16.6%; PR: observed; DLT: NP (25%), TCP (23%). Conclusion: antitumor efficacy and toxicity is equivalent to topotecan. | No 5 [248] |
| 2002 Phase I | 0.5 h iv at 0.4, 0.8, 1.6, 3.8, 4.3 mg/m2; 3 wk/c: ≥ 2/pt (77 c/29 pts) | Liposomal form w 10 mM NH4Cl 9% sucrose | 29 pts w solid tumors; DLT: NP, TCP; antitumor activity unclear; Phase II dose: 3.8 mg/m3 once every 3 weeks (3 wk/c) | No 6 [249] |
| 2002 Phase II | 0.5 h iv d1-5 at 1.2 mg/m2/d; 3 wk/c: ≥ 2/pt | Drug diluted 5% dextrose in water (D5W) | 173 pts w solid tumors (breast, colon, N/SCLC, ovarian); DLT: myelosuppression; antitumor activity unclear; PK focused. | No 7 [250] |
| 2004 Phase II | 0.5 h iv d1, d8 at 2.4 mg/m2/d; 3 wk/c: ≥ 2/pt | Liposomal form | 46 pts w head & neck squamous cell carcinoma; ORR: 1 pt & SD: 18 pts (18 wk); grade 1/2 anemia in 79%, but minimal antitumor activity | No 8 [251] |
| 2004 Phase I | 0.5 h iv d1-3 at 0.15-2.1 mg/m2/d; 3 wk/c* | Liposomal form | 37 pts w solid tumors; DLT: myelosuppression; MTD: 2.1 (MP), 1.8 (HP) mg/m2/d; PR: 2 pts; | No 9 [252] |
| 2004 Phase I | 0.5 h iv d1-3 at ≥ 1.5 mg/m2/d; 3 wk/c* | Liposomal form | 20 pts w leukemia; DLT: mucositis, diarrhea; MTD: 3.7 mg/m2/d; minimal activity | No 10 [253] |
| 2004 Phase I | iv d1-3 at 0.9 (Cis: 25) mg/m2/d; 3 wk/c | Liposomal form | 14 pts w solid tumors; DLT: NP, TCP; Phase II: 0.7 + 25 (Cis) mg/m2/d; CR: 1 pt; PR: 3 pts | No 11 [254] |
| 2004 Phase II | 0.5 h iv d1, d8 at 2.4 mg/m2/d; 3 wk/c | Liposomal form in D5W | 22 pts w topotecan-resistant ovarian cancer; highly manageable toxicity; no evidence of clinical activity (only 8 pts w SD) | No 12 [255] |
| 2005 Phase II/IIIa | 0.5 h iv d1-3 at 1.8 (arm A) or on d1, d8 (arm B) mg/m2/d; 3 wk/c*: ≥ 2/pt | Liposomal form w 10 mM NH4Cl 9% sucrose | 80 pts w relapse ovarian cancer; hematologic toxicity: arm A (51%) > arm B (22%); ORR: 10% (A: 15.1% vs. B:4.9%) | No 13 [256] |
Gimatecan (ST1481/ST-1481) (Figure 1K)
Gimatecan was initially tested for oral administration with daily schedules in preclinical studies and was found to overcome Pgp/MDR1 resistance [257]. However, cell-based studies indicated that expression of BCRP resulted in 8- to 10-fold resistance to gimatecan [258]. Nevertheless, preclinical studies demonstrated that gimatecan exhibited promising cytotoxicity and antitumor potential in various types of human tumor xenograft models with [257,259] and without [260,261] the use of topotecan as a control drug. Gimatecan induction of less Top1 downregulation than topotecan was reasoned as an additional evidence of gimatecan to be a better Top1 activity inhibitor for its efficacy than topotecan [262]. Gimatecan subsequently went into clinical trials (Table 6). Based on the weak Phase I and Phase II clinical trial result, further development of gimatecan using Phase III clinical trials may have a very high risk.
Table 6.
Gimatecan (ST1481/ST-1481)
| Key | Route & dose | Formulation | Cancer type and key clinical trial outcome | Refs |
|---|---|---|---|---|
| 2007 Phase I | po d1-5 at 0.05-0.48 mg/m2/d at wk1, 2, 3; 4 wk/c: ≥ 2/pt | Formulated in gel caps (gelucire 44/14 as diluent), | 108 pts w solid tumors; DLT: TCP; half-life: 77 h; PR: 6 pts; conclusion: toxicity is schedule-dependent | No 1 [263] |
| 2009 Phase I | po d1/wk at 0.27-3.2 mg/m2/wk at wk1, 2, 3; 4 wk/c: 60 c/33 pts | Formulated in capsules at different doses | 33 pts w advanced solid tumors; DLT: TCP, hyperbilirubinemia, fatigue; ORR: 0; SD: 4 pts; antitumor activity needs further to be defined. | No 2 [264] |
| 2010 Phase I | po d1-5 at 0.05-0.48 mg/m2/d at wk1, 2, 3 | Hard gelatine capsules | 78 pts w solid tumors; half-life: 77 h; PK focused studies; antitumor activity no mentioned. | No 3 [265] |
| 2010 Phase II | po d1-5 at 0.8 mg/m2/d; 4 wk/c: 312 c/69 pts | Hard gelatine capsules | 69 pts w recurrent ovarian, fallopian tube or peritoneal cancer; PR: 17 pts; SD: 22 pts; DLR: NP, TCP | No 4 [266] |
| 2013 Phase II | po d1-5 at 1.22, 1.0 mg/m2/d; 4 wk/c*: ≤ 12/pt | Oral capsules | 29 pts w recurrent glioblastoma; DLR: NP, TCP, leukopenia; 3 pts reached the endpoint of PFS for 6 months; Conclusion: minimal efficacy | No 5 [267] |
Diflomotecan (BN80915/BN-80915) (Figure 1L)
Diflomotecan is a 7-membered lactone ring CPT and was considered one of the most potent Top1 inhibitors described [268]. Various types of apoptosis assay testing with diflomotecan versus SN-38 revealed that diflomotecan induces a more pronounced apoptosis in HL60 cancer cells in comparison with SN-38 [268]. Interestingly, 5 patients with an ABCG2 421C > A heterozygous status had 299% of diflomotecan exposure in plasma in comparison with the 15 patients with wild type allele [269]. Consistently, clinical trial Phase I PK studies indicated that there is a wide inter-patient variability in all doses tested [270,271]. Furthermore, human glioblastoma cell lines with reduced Top1 expression were found to be resistant to diflomotecan [272]. During these studies, 5-Phase I clinical trials were carried out and published between 2003 and 2009 (Table 7). However, up to October 2017, no Phase II clinical trials for diflomotecan were published. One reason for this could be that researchers may have taken the lesson learned from rubitecan (Table 1), belotecan (Table 2), exatecan (Table 3), DE-310 (Table 4), lurtotecan (Table 5), and gimatecan (Table 6), and realized that Phase II clinical trials may be too risky, since the 5-diflomotecan Phase I clinical trials had not obtained the advantages of diflomotecan over rubitecan, belotecan, exatecan, lurtotecan, or gimatecan in terms of either favorable side effect toxicity and/or antitumor activity (Table 7). Interestingly, while diflomotecan has a high possibility of never being moved into Phase II and Phase III clinical trials, a recent report used the data derived from the 5-Phase I clinical trials (Table 7) and made a semi-mechanistic cell-cycle type (proliferative cell population versus stem/latent cell population)-based pharmacokinetic/pharmacodynamic model to study the chemotherapy-induced neutropenic effects of diflomotecan under different dosing schedules [273]. These authors believe that the new model could properly describe the neutropenic effects of diflomotecan after very different dosing scenarios, and can be used to explore the potential impact of dosing schedule dependencies on neutropenia prediction [273]. Of course, the significance of this study to the further development of diflomotecan remains to be seen.
Table 7.
Diflomotecan (BN80915/BN-80915)
| Key | Route & dose | Formulation | Cancer type and key clinical trial outcome | Refs |
|---|---|---|---|---|
| 2003 Phase I | 1/3 h iv d1 once; po d1 -5 at 0.1-0.35 mg/m2/d; 3 wk/c: 57c/24 pts | Drug dissolved in DMA for iv or po routes | 22 pts w solid tumors; DLT: TCP, NP; Phase II/MTD: 0.27 mg/m2/d; SD: 6 pts; PK studies is one major focus | No 1 [274] |
| 2004 Phase I | 1/3 h iv d1 once; po d1 -5 at 0.1-0.35 mg/m2/d; 3 wk/c: 57c/24 pts | Drug dissolved in DMA for iv or po routes | 22 pts w advanced refractory solid tumors; ABCG2 allele polymorphism affect diflomotecan exposure; inter-patient variation could be large | No 2 [269] |
| 2006 Phase I | 1/3 h iv d1 once at 2, 4, 5, 6 mg/m2; 3 wk/c: 75 c/24 pts | Drug dissolved in DMA for iv routes | 24 pts w advanced refractory solid tumors; DLT: hematopoietic toxicity; SD: 7 pts; PD: 17 pts; MTD vs. Phase II: 5 vs. 4 mg/m2 | No 3 [275] |
| 2007 Phase I | 1/3 h iv d1-5 at 0.05-0.15 mg/m2/d; 3 wk/c: 89 c/30 pts | Drug dissolved in DMA for iv routes | 30 pts w advanced solid tumors; SD: 7 pts; PD: 1 pt; SD: 8 pts; MTD vs. Phase II: 0.15 vs. 0.125 mg/m2/d; large inter-patient variation of PK. | No 4 [270] |
| 2009 Phase I | 1/3 h iv d1 once at 2, 4, 7 mg/m2; 3 wk/c: 22 c/13 pts | Not clear in the publication | 13 pts w advanced refractory solid tumors; DLT: NP; only 1 pt w minor response; PK & toxicity prediction is not better than po. | No 5 [271] |
Karenitecin (BNP1350/BNP-1350, BNP1100/BNP-1100) (Figure 1M)
Karenitecin is a 7-silicon-containing lipophilic CPT analogue and was initially engineered via computer modeling as a better Top1 inhibitor due to its potential of better lactone stability and/or insensitivity to Pgp. However, in vivo studies using colon and ovarian cancer cell-established xenografts with or without Pgp expression indicated that BNP1350 has very similar antitumor efficacy to those of irinotecan (CPT-11) via ip. However, we know that irinotecan cannot be orally administered but Karenitecin has similar efficacy either ip or po [276]. Clonogenic analyses revealed that sequential treatment of colon cancer cells first with the thymidylate synthase inhibitor ZD1694 for one cell doubling time followed by karenitecin treatment at clinically achievable concentrations exhibited highly synergistic effects with > 99.9% cell killing. Mechanistically, the pretreatment with ZD1694 increased the amount of DNA-bound Topo I by up to 4-fold and the DNA-damaging capability of karenitecin by up to 15-fold [277]. This finding is consistent with the regulation of head-&-neck A253 carcinoma cell cycle by karenitecin as a Top1 functional inhibitor [278,279]. Nevertheless, in ovarian cancer models (A2780, IGROV-1, OVCAR-3), growth inhibition in all 3 xenografts induced by Karenitecin was ≥ 75%, which was significantly better than that resulting from topotecan (P < 0.05) [280]. Consistent with karenitecin oral available potential [276], using ABCG2/BCRP-overexpressed 2780K32 cells, these authors demonstrated that karenitecin is not a good substrate for BCRP in comparison with topotecan [280]. Nevertheless, further studies of the role of karenitecin in cancer cell cycle regulation revealed that karenitecin induces chk1 phosphorylation at Ser345, which is a karenitecin resistant factor [281]. Later studies demonstrated that 48 h pretreatment of melanoma cells with the histone deacetylase inhibitor valproic acid (VPA) could potentiate Karenitecin-induced DNA strand breaks and apoptosis in melanoma cells and mouse A375 xenografts but Phase I/II clinical trials exhibit minimal anticancer activity although no toxicity issue [282]. The overall clinical trial studies obtained positive results but lacked a robust demonstration of significant superiority to either irinotecan and/or topotecan (Table 8). Interestingly, while further clinical trials appear to have stopped after 2009, a recent study found that karenitecin and flavapridol as cell cycle regulators and radiosensitizers can produce synergistic effects during radiation treatment [283]. However, whether this finding could bring karenitecin back to clinical trials again remains to be seen.
Table 8.
Karenitecin (BNP1350/BNP-1350)
| Key | Route & dose | Formulation | Cancer type and key clinical trial outcome | Refs |
|---|---|---|---|---|
| 2005 Phase II | (1 h?) iv d1-5 at 1 mg/m2/d; 3 wk/c: ≤ 16 c/pt | Not clear in the publication | 43 pts (most pre-treated) w metastatic melanoma; toxicity (main hemotopoitic) is manageable & reversible; CR: 1 pt; SD: 10 pts; PD: 27 pts. | No 1 [284] |
| 2005 Phase II | 1 h iv d1-5 at 1 mg/m2/d; 3 wk/c: ≤ 6 + 2c/pt | Diluted in D5W | 52 pts w relapsed (28) or refractory (24) NSCLC; PR: 1 pt; SD: 12 pts; major toxicity: NP, TCP. | No 2 [285] |
| 2008 Phase I | 1 h iv d1-5 at ≥1 mg/m2/d; 3 wk/c*: ≥ 2 c/pt | Complex solution | 32 pts w recurrent malignant glioma; DLT: NP, TCP; MTD: 1.5-2 mg/m2/d; little activity shown | No 3 [286] |
| 2008 Phase II | 1 h iv d1-5 at ≥ 1 mg/m2/d; 3wk/c*: ≥ 2 c/pt | Not clear in the publication | 26 pts w recurrent or persistent ovarian cancer; PR: 2 pts; CR: 1 pt; DLT: NP; minimal activity | No 4 [287] |
| 2009 Phase I/II | po VPA d1, d2 at 30-90; 1 h iv d3-7 at 0.8-1 mg/m2/d; 3 wk/c: ≥ 2/pt | Not clear in the publication | 33 pts w stage IV melanoma; SD in one group: 7/15 pts; DLT: somnolence; VPA on d1, d2 at 75 mg/kg/d, followed by karenitecin d1-5 1 mg/kg/d without overlapping toxicities | No 5 [282] |
Silatecans, silatecan (DB-67/DB67) (Figure 1N)
Silatecans are also a class of 7-silyl-modified CPT analogues. The typical one is DB-67 (7-tert-butyldimethylsilyl-10-hydroxy camptothecin). They are all highly lipophilic and have the potential to favor blood-brain barrier transit and more lactone stability in vivo [288,289]. DB-67 was shown to have higher lactone levels in human blood and be considered as an attractive candidate for clinical development [290]. However, similar to SN-38 and topotecan, it was found that DB-67 strongly inhibits Top1 expression and low Top1 level is associated with DB-67 resistance [291]. Nevertheless, it was found that liposomal DB67 is better than free DB67 in terms of inhibition of primary murine CT-26 xenograft tumors but less effective than irinotecan [292]. However, DB67 and liposomal DB67 are more effective than irinotecan in the treatment of liver metastases after resection of the primary tumor [292]. Additional findings include that 1) DB67 is a Top1-targeted radiation sensitizer [293]; and 2) in vivo xenograft testing of DB67 versus 7-membered lactone ring DB67 (DB91) demonstrated that DB91 basically loses antitumor activity [294]. Based on these preclinical research outcomes, clinical studies of DB67 were likely halted or never started, since thus far no clinical studies on DB67 have been reported.
Namitecan (ST1968/ST-1968) (Figure 1O)
Namitecan is a relative new hydrophilic CPT analogue (Figure 1O). Use of a large panel of human cancer cell line-established tumor models including irinotecan-resistant once demonstrated that although less potent than SN-38 in vitro, iv administration of ST1968 caused a marked tumor inhibition (superior to that of irinotecan) in most tested models [295]. Interestingly, yeast spot tests indicated that while both CPT and ST1968 reduced the growth of yeast cells exogenously expressing wild-type human Top1 without affecting cell growth for the yeast cells exogenously expressing the human Top1 G363C and A653P mutants, ST1968 was able to inhibit yeast cells exogenously expressing the human Top1 K720E mutant [295]. This suggested that inhibition of cell growth by ST1968 may rely on Top1 function more than those of CPT. Consistently, ST1968 was shown to be a better Top1 activity inhibitor and also exhibited superior antitumor activity in a panel of human squamous cell carcinoma (SCC) cell line-established xenograft tumors overall in comparison with irinotecan [296]. The studies also found that ST1968 treatment induced a persistent DNA damage response, as documented by phosphorylation of p53, RPA-2 and histone H2AX, which was associated with a marked cellular/tumor drug accumulation [296]. However, results from another study suggest that inhibition of checkpoint kinases by ST1968 may likely be involved in improving the efficacy of ST1968 [297]. Studies using A431 versus topotecan (TPT)-resistant A431/TPT cell pair demonstrated that ST1968 has a comparable accumulation and retention in sensitive (A431) and resistant (A431/TPT) cells, in spite of expression of Pgp in resistant cells, while the uptake and retention of topotecan were dramatically reduced in both tumor cell lines, especially in the resistant one [298]. Consistently, ST1968 exhibited superior antitumor activity in both A431 and A431/TPT-established xenografts in comparison with topotecan [298]. Studies using high Top1-expressing pediatric sarcoma U2OS and RD/TE670 cell line-established xenografts demonstrated that at the optimal and half optimal doses with q4d x 4 schedules, ST1968 showed an efficacy superior to irinotecan/CPT-11, and ST1968 was able to temporarily eliminate U2OS tumor and regress RD/TE670 tumors [299]. Use of pediatric neuroblastoma models obtained similar in vivo results for ST1968 antitumor efficacy [300]. Furthermore, ST1968 in combination with cisplatin or caboplatin [300] in SK-N-AS xenograft models or with cetuximab in A431, A431/TPT, Caski and AiH xenograft models [301] exhibited high synergistic effects to inhibit or eliminate tumors. Based on these preclinical studies, a Phase I clinical trial with various solid tumors was carried out (Table 9). In this study, it is clear that only two patients with PR provide no clue to predict ST1968 antitumor activity. Therefore, Phase II clinical trials for ST1968 remain to be seen for monotherapy or combination treatment for ST1968.
Table 9.
Namitecan (ST1968/ST-1968)
| Key | Route & dose | Formulation | Cancer type and key clinical trial outcome | Refs |
|---|---|---|---|---|
| 2015 Phase I | 2 h iv d1, d8 at 2.5-20 or d1 at 17.5-30 mg (flat); 3 wk/c*: ≥ 1c/pt | Unclear. ST1968 from Sigma-Tau (Rome, Italy) | 34 (schedule 1, S1) + 29 (S2) pts w pretreated solid tumors; DLT: NP; RD: 15 mg (S1) & 23 (S2) mg; PR: 2 pts. | No 1 [302] |
BN80927 (BN 80927, elomotecan) (Figure 1P)
BN80927 was reported to be an inhibitor of both Top1 and Top2, and showed pronounced cytotoxicity against human HT29, SKOV-3, DU145 and MCF7 cancer cell lines [303]. Use of PC3 and Du145 prostate cell line-established xenografts demonstrated that oral administration of BN80927 resulted in more efficacious than topotecan or irinotecan administered via ip in different schedules ((every day for 14 days, twice a day for 14 days, every week for 3 weeks, and 4-days-on/3-days-off for three cycles) [304]. However, clinical studies revealed minimal antitumor activity (Table 10).
Table 10.
BN80927 (BN 80927, elomotecan)
| Key | Route & dose | Formulation | Cancer type and key clinical trial outcome | Refs |
|---|---|---|---|---|
| 2012 Phase I | 0.5 h iv d1 at 1.5-75 mg (flat); 3 wk/c: ≥1 c/pt - ≤ 10 c/pt | Unclear. Drug from Ipsen Pharma | 56 pts w advanced solid tumors; DLT: NP; MTD/RD: 75 mg & 60 mg; pts in the RD cohort got 41.7% SD in duration of 123.6 ± 43.4 d | No 1 [305] |
DRF-1042 (DRF1042) (Figure 1Q)
DRF1042 is an oral active CPT analogue. There are not many preclinical studies on DRF1042 (rather a few meeting abstracts). This compound went into clinical trials in Dr. Reddy’s Laboratories Lid in India as soon as a HPLC-based quantification of the drug in plasma were established [306]. After two Phase I clinical trials in 2004 and 2005, further development of DRF1042 appears to have stopped (Table 11).
Table 11.
DRF-1042 (DRF1042)
| Key | Route & dose | Formulation | Cancer type and key clinical trial outcome | Refs |
|---|---|---|---|---|
| 2004 Phase I | po d1-5 at 1.5-270 mg/m2/d in 2 wk; 3 wk/c: (73 c/25 pts) | Unclear. Trial was done in Dr. Reddy’s Lab | 25 pts w refractory solid tumors; DLT: diarrhea, myelosuppresssion; MTD/Phase II: 120 mg/m2/d vs. 80 mg/m2/d; CR: 2 pts; PR: 2 pts; SD: 4 pts. | No 1 [307] |
| 2005 Phase I | po d1-5 at 81 mg/m2/d in 2 wk; 3 wk/c: (10 c/6 pts) | Capsule. Trial was done in Dr. Reddy’s Lab | 25 pts w refractory solid tumors; DLT: TCP, diarrhea; RD for Phase II: 80 mg/m2/d; SD: 1 pt; capsule correlated with but better than suspension | No 2 [308] |
MAG-CPT (PNU 166148/PNU166148)
MAG-CPT is pro-drug derived from the CPT p20 covalently linked to a water-soluble polymeric carrier. Three Phase I clinical trial results (Table 12) lead to the withdrawal of MAG-CPT from clinical development [309].
Table 12.
MAG-CPT (PNU 166148/PNU166148)
| Key | Route & dose | Formulation | Cancer type and key clinical trial outcome | Refs |
|---|---|---|---|---|
| 2002 Phase I | 0.5 h iv d1-3 at ≥ 17 mg/m2/d; 4 wk/c: 39 c/16 pts | Drug complex reconstituted in saline | 16 pts w malignant solid tumors; PK prolonged; DLT: serious bladder toxicity; no response was found; SD: 1 pt. | No 1 [310] |
| 2003 Phase I | 24 h iv once at 60 mg/m for PK studies | Unclear. | 10 pts w colorectal carcinoma; MAG-CPT was delivered at similar levels to tumor and normal tissue. Not focus antitumor activity. | No 2 [311] |
| 2004 Phase I | 0.5 h iv once at 30-240 mg/m2; 4 wk/c: 47 c/23 pts | Lyophilized drug reconstituted in saline | 16 pts w malignant solid tumors; DLT: diarrhea, myelosuppression NP sepsis; MTD/RD: 200 mg/m2. | No 3 [309] |
BAY 38-3441 (BAY 56-3722)
BAY 38-3441 is a covalently glycoconjugated CPT on CPT p20. As mentioned in the 3 published clinical trials (Table 13), it appears that multiple clinical trials were initiated at approximately the same time under the financial support from Bayer Inc.. The Phase II clinical trials were basically terminated more than 10 years ago without publication. However, the researchers involved in the Phase II studies felt that it was their obligation to share the interrupted phase II study for reporting the fate of this glycoconjugated CPT and presenting the unique situation of a clinical hold during a phase II study [312]. Such reports of negative findings are very helpful and thus important to the research and development field.
Table 13.
BAY 38-3441 (BAY 56-3722)
| Key | Route & dose | Formulation | Cancer type and key clinical trial outcome | Refs |
|---|---|---|---|---|
| 2004 Phase I | 0.5 h iv d1 at 20-600 or d1-3 at 126-416 mg/m2/d; 3 wk/c*: 1-7 c/pt | Lyophilized power dissolved in D5W | 81 pts w advanced solid tumors; DLT: renal toxicity, granulocytopenia, TCP; RD: 320 mg/m2/d (0.5 h iv d1-3); SD: 2 pts (18-21 wk). | No 1 [313] |
| 2005 Phase I | 0.5 h iv d1-5 at 320 mg/m2/d; 3 wk/c*: 1-10 c/pt | Lyophilized power dissolved in D5W | 31 pts w advanced/refractory solid tumors; DLT: diarrhea, granulocytopenia, NP; SD: 9 pt - ~2.7 months (range: 2.3-20.6 months). | No 2 [314] |
| 2012 Phase II | 0.5 h iv d1-3 at 14-295 mg/m2/d; 3 wk/c*: ≥ 1 c/pt | Unclear but likely same as above | 24 pts w irinotecan-resistant advanced colon cancer; 18 pts discontinued due to disease progression; ≥ 1 TX-emergent event in 23 pts | No 3 [312] |
CRLX101 (IT-101)
CRLX101 (IT-101) is a b-cyclodextrin (b-CD)-covalently conjugated CPT on CPT p20 via ester bonds [315,316] and is a self-assembling nanoparticle drug. PK and biodistribution studies revealed that IT-101 iv administration in rats and nude mice bearing human LS174T colon tumors exhibited prolonged plasma half-life and enhanced distribution to tumor tissue compared to CPT alone; and also active CPT is released from the conjugate within the tumor for an extended period of time [316]. Antitumor efficacy of IT-101 was evaluated in nude mice bearing six human cancer cell line-established xenografts (CRC: LS174T and HT29; NSCLC: H1299; SCLC: H69; pancreatic cancer: Panc-1; breast cancer: MDA-MB-231) and a luciferase (luc)-labeled Ewing’s sarcoma (TC71-luc) [317]. Complete tumor regression was reached at the best schedule and dose in all animals bearing H1299 tumors and in the majority of animals with disseminated Ewing’s sarcoma tumors; the studies also found that antitumor activity and toxicity is schedule-dependent [317]. In the xenograft setting condition, IT-101 antitumor activity appeared to be better than irinotecan/CPT-11 [317]. Another study with human lymphoma xenograft models showed that as compared with CPT-11 and SN-38, IT-101 and CPT had higher inhibition of DNA Top1 catalytic activities, and IT-101 significantly prolonged the survival of animals bearing human xenografts when compared with CPT-11 at its MTD in mice [318]. Similarly, in human gastric cancer cell line BGC823-established xenograft model, CRLX101 exhibited antitumor activity better than CPT-11 via iv administration, and the authors also found that CRLX101 significantly decreased the expression of carbonic anhydrase, VEGF, and CD31 proteins in treated tumors indicating an inhibition of hypoxia and angiogenesis [319]. Furthermore, in a human breast cancer mouse model, concurrent administration of CRLX101 (iv) with bevacizumab (ip) impeded bevacizumab-mediated induction of HIF-1a and cancer stem cells (CSCs) in breast tumors, and resulted in greater tumor regression and delayed tumor recurrence in comparison with bevacizumab alone [320]. Tumor reimplantation experiments demonstrated that the combination therapy effectively targets the CSC populations [320]. Similarly, CRLX101 was showed to be as potent as CPT in vitro to radiosensitize CRC cells, and in human CRC xenograft tumor models, addition of CRLX101 to standard chemoradiotherapy significantly increased therapeutic efficacy by inhibiting DNA repair and HIF1a pathway activation in tumor cells [321]. CRLX101 in combination with 5-Fu produced the highest therapeutic efficacy with significant low gastrointestinal toxicity for CRLX101 compared with CPT in combination with radiotherapy [321]. Another comparative study demonstrated that CRLX101 is better in terms of inhibiting HIF1a, suppressing tumor growth, and extending mouse survival compared with topotecan [322]. CRLX101 in combination with bevacizumab obtained significant better results than either alone, and this concept appears to be supported by an ongoing phase I/IIa clinical study of CRLX101 monotherapy that showed measurable tumor reductions in 74% of patients and a 16% RECIST response rate to date [322]. Furthermore, it was found that CRLX101 nanoparticles localize in human tumors and not in adjacent, nonneoplastic tissue after iv administration [323]. In human glioma in vitro and in vivo models, CRLX101 was shown to possess antitumor abilities by inducing cell cycle arrest and apoptosis in glioma cells and inhibiting tumor angiogenesis, and prolonging the lifespan of mice bearing intracranial gliomas from vehicle-treated control for ~31 days to CRLX101-treated mice for ~41 days [324]. In mouse models of orthotopic primary triple-negative breast tumor xenografts, a long-term efficacy evaluation of CRLX101 demonstrated that CRLX101 alone or combined with bevacizumab was highly efficacious, leading to complete tumor regressions, reduced metastasis, and greatly extended survival of mice with metastatic disease [325]. CRLX101 led to improved tumor perfusion and reduced hypoxia by suppressing HIF1a and thus potentially counteracting undesirable effects of elevated tumor hypoxia caused by bevacizumab [325].
During the preclinical studies above, two Phase I/IIa clinical trials were performed and, the outcome was positive, overall, including the case of metastatic RCC (Table 14). Based on the encouraging on-going preclinical and clinical data reviewed above, a comprehensive Phase II studies with two arms across 34 centers in the United States and Korea were launched and the data were published in 2017. Specifically, since in the Phase I/IIa (Ib/II) trial CRLX101 + bevacizumab was well tolerated with encouraging activity in metastatic renal cell carcinoma (mRCC) [326], a randomized phase 2 trial comparing CRLX101 + bevacizumab versus standard of care (SOC) in refractory mRCC were conducted [327]. Patients with mRCC and 2-3 prior lines of therapy were randomized 1:1 to CRLX101 + bevacizumab versus SOC, defined as investigator’s choice of any approved regimen not previously received. The primary endpoint was progression-free survival (PFS) by blinded independent radiological review in patients with clear cell mRCC. Secondary endpoints included overall survival (OS), objective response rate (ORR) and safety. One hundred eleven patients were randomized and received ≥ 1 dose of drug (CRLX101 + bevacizumab, 55; SOC, 56). Within the SOC arm, patients received single-agent bevacizumab (19), axitinib (18), everolimus (7), pazopanib (4), sorafenib (4), sunitinib (2), or temsirolimus (2). In the clear cell population, the median PFS on the CRLX101 + bevacizumab aim was 3.7 months (95% confidence interval [CI]: 2.0-4.3) versus the SOC arm was 3.9 months (95% CI: 2.2-5.4), respectively (stratified Log-rank P = 0.831). The ORR by IRR was 5% with CRLX101 + bevacizumab versus 14% with SOC (Mantel-Haenszel test, P = 0.836) [327]. This appears to be a very disappointing negative result. Nevertheless, consistent with the previous study [326], the CRLX101 + bevacizumab combination was generally well tolerated, and no new safety signal was identified. These authors concluded that despite promising efficacy data on the earlier phase Ib/II (I/IIb) trial of mRCC, this randomized trial did not demonstrate improvement in PFS for the CRLX101 + bevacizumab combination when compared to approved agents in patients with heavily pretreated clear cell mRCC [327].
Table 14.
CRLX101 (IT-101)
| Key | Route & dose | Formulation | Cancer type and key clinical trial outcome | Refs |
|---|---|---|---|---|
| 2013 Phase I/IIa | 1 h iv at 6, 12, 18 mg/m2/wk or at 12, 15, 18 mg/m2/biwk* | Self-assembled nanoparticles | 62 pts w advanced solid tumors; bywkly better tolerated (MTD: 15 mg/m2); DLT: myelosuppression (NP), fatique; SD: 28 pts; | No 1 [328] |
| 2016 Phase I/IIa | 1 h iv at 12, 15 mg/m2, + bevacizumab, 10 mg/kg, biwkly*; | Self-assembled nanoparticles | 22 pts w metastatic renal cell carcinoma (mRCC); DLT not reached; PR: 5 pts; 4 pts obtained > 4 months PFS | No 2 [326] |
| 2017 Phase II | Iv d1, d15 at 15 mg/m2, + bevacizumab, 10 mg/kg, 4 wk/c* | Self-assembled nanoparticles | No improvement in PFS for the CRLX101 + bevacizumab versus the approved agents in pts with heavily pretreated clear cell mRCC | No 3 [327] |
T-0128 (MEN4901)
T-0128 is a pro-drug derived from the CPT analogue T-2513 (delimotecan, Figure 1R) conjugating with carboxymethyl (CM) dextran via a Gly-Gly-Gly linker. It was shown that T-2513 directly interacts with DNA-Top1 complex as CPT, and using rat Walker-256 carcinoma in rat xenograft models, T-0128 was shown to be 10 times as active as T-2513 [329]. Similarly, T-2513 at 80 mg/kg (q7dx3, iv) only delayed human lung tumor cell line LX-1 xenograft growth, while T-0128 at 10 mg/kg (q7dx3, iv) was able to eliminate the tumor in nude mice and also using the CPT-resistant HT29 CRC cell line-established xenograft, T-0128 at 20 mg/kg (q7dx3, iv) was able to significantly regress tumors in nude mice, while T-2513 at 80 mg/kg (q7dx3, iv) and CPT-11 at 100 mg/kg (q7dx3, iv) only slightly delay HT29 tumor growth [329]. PK studies using Walker-256 tumor-bearing rats showed that after iv administration of T-0128, the conjugate continued to circulate at a high concentration for an extended period, resulting in the accumulation of drug in liver, spleen and tumor much higher than in kidney, heart, lung and bone marrow tested; in contrast, T-2513 disappeared rapidly from the body and tumor after iv administration [329]. Another similar study using different human tumor cell line-established xenografts (gastric: H-81; colon: H-110; lung: Mqnu-1, H-74; esophageal: H-204; liver: H-181 and pancreatic: H-48) via iv routes with q7dx4 schedules showed that a marked antitumor activity in each of these tumor models, producing tumor shrinkage in the models of H-204 and H-181 at its MTD of 80 mg/kg via q7d x 4 schedule, and tumor-shrinking or marked growth-inhibitory effects in the models of H-81, H-110, Mqnu-1, H-74, and H-48 carcinomas at its 1/3MTD (q7d x 4) [330]. The third study showed that while the approved dacarbazine drug for metastatic melanoma was ineffective in the Me15392 melanoma xenograft model. T-0128/delimotecan exhibited significant antitumor activity against this xenograft tumor better than or equivalent to CPT-11 [331]. Further studies of the T-2513 release from T-0128 revealed that it is the tumor-associated macrophage playing a major role in update of T-0128 and release of T-2513 [332,333]. More recently, using a GFP-labeled HT29 colon cancer cell for an orthotopic tumor model, it was shown that T-0128 had a high efficacy, better ability than those of irinotecan, to inhibit HT29 cell lymph node metastasis as well as against the primary tumor [334]. One phase I clinical trial was performed in 2008 (Table 15). It is clear that while more clinical trials may be risky, a go or no go decision remains unsolved.
Table 15.
T-0128 (MEN4901, delimotecan)
| Key | Route & dose | Formulation | Cancer type and key clinical trial outcome | Refs |
|---|---|---|---|---|
| 2008 Phase I | 3 h iv once/6wk at 150-5400 mg/m2; 35 c/22 pts in 8 dose levels | Lyophilized powder diluted in saline | 22 pts w refractory solid tumors; DLT: ≥ 2,400 mg/m2; PR: 2 pts; half-life (T1/2) = 109 h; RD: 1,800 mg/m2 once every 6 wk for 3 h iv. | No 1 [335] |
Other CPT analogues, CPT conjugates or CPT nanoparticles
NSC606985 is a CPT analogue (Figure 1S) that was found to induce apoptosis (caspase-3 activation and loss of mitochondrial potential) in acute myeloid leukemia (AML) cell lines NB4 and U937 through rapid activation of protein kinase C d (PKCd), and NSC606985-induced apoptosis can be completely blocked by co-treatment with the PKCd-specific inhibitor rottlerin [336]. Although a number of preclinical studies were followed up [337-341], no clinical studies have yet been reported.
Chimmitecan (Figure 1T) is a 9-substituted lipophilic CPT and is an active metabolite of pro-drug simmitecan (p10 has the same chemical group as irinotecan has on p10). It was found that chimmitecan exhibited more potent cytotoxicity than SN38 and topotecan with comparable effects on Top1, in terms of inhibiting Top1 catalytic activity and trapping and stabilizing covalent Top1-DNA complexes [342]. Nanomolar levels of chimmitecan caused impressive DNA damage, G2/M phase arrest, and apoptosis in human leukemia HL60 cells [342]. In the experimental setting condition using the xenograft tumor models established from HCT-116, MDA-MB-435, BEL-7402, and A549 human cancer cell lines in nude mice via iv administration, chimmitecan showed greater potency than CPT-11 against the BEL-7402 and A549-established tumors [342]. The method used to determine chimmitecan or its prodrug simmitecan in plasma and organ tissues was developed [343] and the PK profile of the drugs in rats, dogs and nude mice were studies [344]. Interestingly, in rats and nude mice bearing human hepatic cancers, most organs had significantly higher concentrations of simmitecan than the corresponding plasma levels. However, in tumor tissues, simmitecan levels were comparable to those of plasma, whereas chimmitecan levels were lower than the simmitecan levels [344]. It is clear that more preclinical studies, especially for antitumor efficacy potential, are needed before making a go decision for clinical trials with chimmitecan and/or simmitecan.
CT-2106 is a poly-L-glutamate-conjugated CPT. This linkage was claimed to stabilize the active lactone form of CPT and enhance aqueous solubility. It was also postulated that the poly-L-glutamate might increase tumor delivery of CPT through enhanced permeability and retention effect in tumor. Therefore, a clinical phase I trial was carried out (Table 16) and found that the PK profile for conjugated CPT did not exhibit significant advantage over unconjugated CPT [345].
Table 16.
CT-2106/CT2106
| Key | Route & dose | Formulation | Cancer type and key clinical trial outcome | Refs |
|---|---|---|---|---|
| 2007 Phase I | 1/6 h iv d1, d8, d15 at 25-35 mg/m2; 4 wk/c*: ≥ 1-12 c/pt | Lyophilized drug reconstituted in sterile water | 26 pts w refractory solid tumors; DLT: TCP, fatigue; MTD: 25 mg/m2 weekly. SD: 3 pts but no CR or PR. | No 1 [345] |
HM910 (HM-910) is a recently published CPT p20-sodium bisulfate-conjugated derivative. HM910 was found to inhibit multiple myeloma (MM) cell growth in vitro at a concentration range of 0.1-10 µM and xenograft tumor growth in nude mouse models at a dose range of 18-35 mg/kg via ip with a schedule of q4d x 2. In the xenograft experimental setting with equivalent body weight change levels to these of topotecan, HM910 exhibited anti-MM tumor activity better than or equivalent to topotecan [346]. Interestingly, similar to but distinct from the CPT analogue NSC606985’s case in AML cancer models [336], HM910 mechanistically reduced the mitochondrial transmembrane potential (DeltaPsim) via an increase in reactive oxygen species (ROS), which induced cytochrome c release and the activation of mitochondrial-dependent apoptotic pathway [336]. HM910 treatment also triggered cell cycle arrest in G1 phase via downregulating the expressions of CDK 4, CDK 6, and cyclin D1. Based on the note provided in the paper [336], HM910 was synthesized by Fangsheng Pharmaceuticals, Inc. and was in Phase I clinical trials in China from 2014. However, the Phase I clinical trial results are currently not available.
ZBH-1205 (ZBH1205) is a CPT analogue with a chemical structure similar to irinotecan and SN-38 (Figure 1U). A recent publication showed that using a panel of human tumor cell lines including the multi-drug resistant cell line SK-OV-3/DPP as well as HEK293, ZBH-1205 exhibited IC50 values ranged from 0.0009 µM to 2.5671 µM, which were consistently lower than IC50 values of CPT-11 or SN38 [347]. The authors also demonstrated that ZBH-1205 was more effective than CPT-11 or SN38 at stabilizing Topo-1-DNA complexes and inducing tumor cell apoptosis [347]. Based on their in vitro studies, these authors claimed that ZBH-1205 is a promising chemotherapeutic agent to be further assessed in large-scale clinical trials. However, in our view more preclinical studies, especially with appropriate human tumor animal models, will be needed before considering whether to initiate Phase I clinical studies for ZBH-1205.
WCN-21 is a CPT p20 conjugate by introducing a thiocarbamide group to the 20 position of CPT and it appears that WCN-21 nanocrystals increased WCN-21 solubility and efficacy [348], but whether it will be worthy of further development remains to be a question.
A series of CPT derivatives via uracil-1’(N)-acetic acid ester linkage on the p20 of CPT were synthesized and tested for antitumor activity [349]. However, in comparison with other similar studies, their uniqueness and superiority in antitumor activity remain to be explored before thinking any of these CPT analogues to be moved toward clinical trials.
Overall, based on the clinical trial outcomes from CPT p20 conjugates, we feel that more attempts to move a CPT p20 conjugate for treatment of human cancer may likely be a futile effort. Our point is that if a compound itself is not good enough (e.g. possessing significant weakness) to become a drug, it may be risky in terms of obtaining a breakthrough by conjugation.
Additionally, some studies directly made CPT into non-covalent nanoparticles. For example, CPT-TMC is a non-covalent TMC-encapsulated CPT nanoparticles, which was generated by drop-wise addition of CPT/DMSO solution into water-based N-trimethyl chitosan (TMC) solution, and the resulting colloid solution was then ultra-sonicated and dialyzed to obtain the CPT-TMC nanoparticles [350]. The studies demonstrated CPT-TMC is better than free-CPT in terms of stability, anti-melanoma cell proliferation and induction of apoptosis [350].
Novel non-CPT Top1 inhibitors
Genz-644282 is a structurally novel non-CPT Top1 inhibitor (Figure 1V). It was shown that Genz-644282 and its metabolites induce Top1 cleavage at similar, as well as unique genomic positions, compared with CPT [351]. Genz-644282 exhibited partial cross-resistance in cell lines resistant to CPT. In addition, a limited cross-resistance to Genz-644282 was found in the Top1 knockdown HCT116 and MCF7 cell lines, as well as in human adenocarcinoma cells (KB31/KBV1) that overexpress Pgp/MDR1 [351]. Using various human cancer cell line-established xenografts (CRC: HCT116, HT29, HCT15, DLD-1; melanoma: LOX-IMVI; RCC: 786-O; NSCLC: NCI-H460, NCI-H1299), the study demonstrated that Genz-644282 has antitumor activity superior or equivalent to those of the standard drug comparators for the corresponding disease (irinotecan, dacarbazine, docetaxel) [352]. In various pediatric cancer cell line-established xenograft models, using the schedule of q3w x 2 repeated at day 21, Genz-644282 at its MTD (4 mg/kg) exhibited maintained complete responses (MCR) in 6/6 evaluable solid tumor models. At 2 mg/kg Genz-644282 exhibited CR or MCR in 3/3 tumor models that were relatively insensitive to topotecan, but there were no objective responses at 1 mg/kg; further testing at 2 mg/kg showed that Genz-644282 induced objective regressions in 7 of 17 (41%) models [353]. These are encouraging preclinical results and it will be interesting to see whether the encouraging data can be translated into positive clinical results in cancer patients.
A series of 4-substituted anthrax [2, 1-c] [1,2,5] thiadiazole-6,11-dione derivatives were synthesized (e.g. non-CPT1, Figure 1W) and evaluated as novel non-CPT Top1 inhibitors, which showed anti-proliferative activity against various types of cancer cells [354].
Research toward non-CPT Top1-inhibiting compounds is an interesting research area and is still in its early development stage. A weakness of developing such Top1 inhibitors may have the same inherent limitations possessing by the CPTs described above, unless it will be found that some of such non-CPT compounds use novel MOA and act on novel disease-associated key protein targets.
Concluding remarks
Thus far, the field has largely used Top1 inhibition intensity to predict the antitumor potential of a CPT analogue. Now, accumulating evidence supports the possibility that certain CPT analogues can exert significant non-Top1-mediated antitumor activity; in fact, Top1 activity inhibition by such analogues could be involved in the drug side effects, since normal tissue and cell renewal requires Top1 for DNA replication. The fact is that while most (if not all) of the CPT analogues in clinical development that were reviewed in this article exhibited stronger inhibition of Top1 activity than either irinotecan and/or topotecan; yet, extensive clinical trials with these analogues did not show a significant advantage over irinotecan or topotecan in antitumor activity and/or high side-effect toxicities. We predict that if further efforts at finding CPT analogues still focus on stronger inhibition of Top1 function/activity as the primary criterion for preclinical and clinical development of CPT analogues, we may continuously be unable to make a breakthrough in the development of next generation of novel CPT analogues with high efficacy and low toxicities for human disease (e.g. cancer) treatment. Alternatively, we propose to develop CPT derivatives that exhibits low inhibitory effects on Top1 function/activity, while they can target multiple key disease-associated genes and/or gene products. Such molecules could be the key to finding drugs that possess high efficacy and low toxicity for fighting cancer and other human diseases.
Acknowledgements
The authors thank Dr. David Westover (Previous PhD student in Dr. Fengzhi Li’s laboratory and currently a postdoctoral researcher in the Department of Medicine, Vanderbilt University Medical Center, Nashville, Tennessee, USA) for his constructive comments and suggestions for this review article; Dr. Geoffrey White (Interim CEO, Canget BioTekpharma, USA) for critical review of the final manuscript and constructive suggestions; Dr. Yves Pommier (Laboratory of Molecular Pharmacology, Center for Cancer Research, National Cancer Institute, USA) and the lab manager Keli Agama for kindly providing their Du145-derived two cell lines (RC.1 and RC0.1) that have Top1 mutations and resistance to SN-38 and topotecan. We also thank Ms. Amanda Hess for editorial check of this manuscript before submission. This work was supported in part by grants from USA National Cancer Institute (NCI) (R21CA180764, R03CA182552), a subcontract from Canget (NCI. R44CA176937) and grants from Roswell Park Alliance Foundation. The animal model of human tumor in vivo work was performed in Roswell Park Cancer Institute Animal Center which was supported in part by the Institute NCI Core grant (P30CA016056).
Disclosure of conflict of interest
FL118 and FL118 core structure-based analogues are currently under further development in Canget BioTekpharma (www.canget-biotek.com), a Roswell Park Cancer Institute-spinoff company. FL, TJ and XL are initial investors in Canget for development of FL118 and FL118 core structure-relevant anticancer agents.
Abbreviations
- *
until disease progression or treatment termination
- c
cycle(s)/course(s)
- CD
colloidal dispersion
- Cis
cisplatin
- CPT
camptothecin
- CR
complete response
- d
day/days
- d1-5
daily x 5
- D5W
5% dextrose in water
- DLT
dose-limiting toxicity
- DMA
dimethylacetamide
- DMPC
dimyristoylphosphatidyl choline
- DMPG
dimyristoylphosphatidyl glycerol
- Gem
gemcitabine
- GIST
gastrointestinal stromal tumor
- G-CSF
granulocyte colony-stimulating factor
- h
hour/hours
- HP
heavily pretreated
- ip
intraperitoneal
- iv
intravenous injection/infusion
- MP
minimally pretreated
- MS
median survival
- MTD
maximum tolerated dose
- NP
neutropenia
- (N)SCLC
(non-)small cell lung cancer
- OR
overall/objective response
- PD
progressive disease
- PEG
polyethylene glycol
- PFS
progression-free survival
- po
per oral
- PR
partial response
- pt(s)
patient(s)
- RCC
renal cell carcinoma
- RD
recommended dose
- S
schedule
- SD
stable disease
- STS
soft-tissue sarcoma
- TCP
thrombocytopenia
- TX
treatment
- w
with
- w/o
without
- wk
week/weeks
Supporting Information
References
- 1.Wall ME, Wani MC, Cook CE, Palmer KH, McPhail AT, Sim GA Plant Antitumor Agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from camptotheca acuminata 1, 2. Journal of the American Chemical Society. 1966;88:3888–3890. [Google Scholar]
- 2.Wall ME, Wani MC. Camptothecin. Discovery to clinic. Ann N Y Acad Sci. 1996;803:1–12. doi: 10.1111/j.1749-6632.1996.tb26371.x. [DOI] [PubMed] [Google Scholar]
- 3.Schultz AG. Camptothecin. Chem Rev. 1973;73:385–405. doi: 10.1021/cr60284a004. [DOI] [PubMed] [Google Scholar]
- 4.Gottlieb JA, Luce JK. Treatment of malignant melanoma with camptothecin (NSC-100880) Cancer Chemother Rep. 1972;56:103–105. [PubMed] [Google Scholar]
- 5.Muggia FM, Creaven PJ, Hansen HH, Cohen MH, Selawry OS. Phase I clinical trial of weekly and daily treatment with camptothecin (NSC-100880): correlation with preclinical studies. Cancer Chemother Rep. 1972;56:515–521. [PubMed] [Google Scholar]
- 6.Moertel CG, Schutt AJ, Reitemeier RJ, Hahn RG. Phase II study of camptothecin (NSC-100880) in the treatment of advanced gastrointestinal cancer. Cancer Chemother Rep. 1972;56:95–101. [PubMed] [Google Scholar]
- 7.Pettit GR. A view of cancer treatment in the People’s Republic of China. China Q. 1976;68:789–796. [Google Scholar]
- 8.Xu B. In: U.S./China Pharmacology Symposium. Burns JJ, Tsuchiatani PJ, editors. 156. Washington, D.C.: National Academy of Sciences; 1980. [Google Scholar]
- 9.Horwitz SB. Mechanism of action of antimicrobial and antitumor agents. Springer; 1975. Camptothecin; pp. 48–57. [Google Scholar]
- 10.Rozencweig M, Slavik M, Muggia FM, Carter SK. Overview of early and investigational chemotherapeutic agents in solid tumors. Med Pediatr Oncol. 1976;2:417–432. doi: 10.1002/mpo.2950020408. [DOI] [PubMed] [Google Scholar]
- 11.Horwitz SB, Chang CK, Grollman AP. Studies on camptothecin: I. Effects on nucleic acid and protein synthesis. Mol Pharmacol. 1971;7:632–644. [PubMed] [Google Scholar]
- 12.Kessel D. Effects of camptothecin on RNA synthesis in leukemia L1210 cells. Biochim Biophys Acta. 1971;246:225–232. doi: 10.1016/0005-2787(71)90131-6. [DOI] [PubMed] [Google Scholar]
- 13.Abelson HT, Penman S. Selective interruption of high molecular weight RNA synthesis in HeLa cells by camptothecin. Nature. 1972;237:144–146. doi: 10.1038/newbio237144a0. [DOI] [PubMed] [Google Scholar]
- 14.Wu RS, Kumar A, Warner JR. Ribosome formation is blocked by camptothecin, a reversible inhibitor of RNA synthesis. Proc Natl Acad Sci U S A. 1971;68:3009–3014. doi: 10.1073/pnas.68.12.3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horwitz SB, Horwitz MS. Effects of camptothecin on the breakage and repair of DNA during the cell cycle. Cancer Res. 1973;33:2834–2836. [PubMed] [Google Scholar]
- 16.Tsao YP, D’Arpa P, Liu LF. The involvement of active DNA synthesis in camptothecin-induced G2 arrest: altered regulation of p34cdc2/cyclin B. Cancer Res. 1992;52:1823–1829. [PubMed] [Google Scholar]
- 17.Goldwasser F, Shimizu T, Jackman J, Hoki Y, O’Connor PM, Kohn KW, Pommier Y. Correlations between S and G2 arrest and the cytotoxicity of camptothecin in human colon carcinoma cells. Cancer Res. 1996;56:4430–4437. [PubMed] [Google Scholar]
- 18.Froelich-Ammon SJ, Osheroff N. Topoisomerase poisons: harnessing the dark side of enzyme mechanism. J Biol Chem. 1995;270:21429–21432. doi: 10.1074/jbc.270.37.21429. [DOI] [PubMed] [Google Scholar]
- 19.Hsiang YH, Hertzberg R, Hecht S, Liu L. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem. 1985;260:14873–14878. [PubMed] [Google Scholar]
- 20.Morham SG, Kluckman KD, Voulomanos N, Smithies O. Targeted disruption of the mouse topoisomerase I gene by camptothecin selection. Mol Cell Biol. 1996;16:6804–6809. doi: 10.1128/mcb.16.12.6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang CX, Chen AD, Gettel NJ, Hsieh TS. Essential functions of DNA topoisomerase I in Drosophila melanogaster. Dev Biol. 2000;222:27–40. doi: 10.1006/dbio.2000.9704. [DOI] [PubMed] [Google Scholar]
- 22.Koster DA, Croquette V, Dekker C, Shuman S, Dekker NH. Friction and torque govern the relaxation of DNA supercoils by eukaryotic topoisomerase IB. Nature. 2005;434:671–674. doi: 10.1038/nature03395. [DOI] [PubMed] [Google Scholar]
- 23.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 24.Eng WK, Faucette L, Johnson RK, Sternglanz R. Evidence that DNA topoisomerase I is necessary for the cytotoxic effects of camptothecin. Mol Pharmacol. 1988;34:755–760. [PubMed] [Google Scholar]
- 25.Nitiss J, Wang JC. DNA topoisomerase-targeting antitumor drugs can be studied in yeast. Proc Natl Acad Sci U S A. 1988;85:7501–7505. doi: 10.1073/pnas.85.20.7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leteurtre F, Fesen M, Kohlhagen G, Kohn K, Pommier Y. Specific interaction of camptothecin, a topoisomerase I inhibitor, with guanine residues of DNA detected by photoactivation at 365 nm. Biochemistry. 1993;32:8955–8962. doi: 10.1021/bi00085a029. [DOI] [PubMed] [Google Scholar]
- 27.Gongora C, Vezzio-Vie N, Tuduri S, Denis V, Causse A, Auzanneau C, Collod-Beroud G, Coquelle A, Pasero P, Pourquier P. New Topoisomerase I mutations are associated with resistance to camptothecin. Mol Cancer. 2011;10:64. doi: 10.1186/1476-4598-10-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Urasaki Y, Laco GS, Pourquier P, Takebayashi Y, Kohlhagen G, Gioffre C, Zhang H, Chatterjee D, Pantazis P, Pommier Y. Characterization of a novel topoisomerase I mutation from a camptothecin-resistant human prostate cancer cell line. Cancer Res. 2001;61:1964–1969. [PubMed] [Google Scholar]
- 29.Benedetti P, Fiorani P, Capuani L, Wang JC. Camptothecin resistance from a single mutation changing glycine 363 of human DNA topoisomerase I to cysteine. Cancer Res. 1993;53:4343–4348. [PubMed] [Google Scholar]
- 30.Chang JY, Liu JF, Juang SH, Liu TW, Chen LT. Novel mutation of topoisomerase I in rendering cells resistant to camptothecin. Cancer Res. 2002;62:3716–3721. [PubMed] [Google Scholar]
- 31.Arakawa Y, Ozaki K, Okawa Y, Yamada H. Three missense mutations of DNA topoisomerase I in highly camptothecin-resistant colon cancer cell sublines. Oncol Rep. 2013;30:1053–1058. doi: 10.3892/or.2013.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jensen NF, Agama K, Roy A, Smith DH, Pfister TD, Romer MU, Zhang HL, Doroshow JH, Knudsen BR, Stenvang J, Brunner N, Pommier Y. Characterization of DNA topoisomerase I in three SN-38 resistant human colon cancer cell lines reveals a new pair of resistance-associated mutations. J Exp Clin Cancer Res. 2016;35:56. doi: 10.1186/s13046-016-0335-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yanase K, Sugimoto Y, Andoh T, Tsuruo T. Retroviral expression of a mutant (Gly-533) human DNA topoisomerase I cDNA confers a dominant form of camptothecin resistance. Int J Cancer. 1999;81:134–140. doi: 10.1002/(sici)1097-0215(19990331)81:1<134::aid-ijc22>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 34.Wu Y, Wang KY, Li Z, Liu YP, Izumi H, Uramoto H, Nakayama Y, Ito K, Kohno K. Y-box binding protein 1 enhances DNA topoisomerase 1 activity and sensitivity to camptothecin via direct interaction. J Exp Clin Cancer Res. 2014;33:112. doi: 10.1186/s13046-014-0112-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wall ME, Wani MC, Natschke SM, Nicholas AW. Plant antitumor agents. 22. Isolation of 11-hydroxycamptothecin from Camptotheca acuminata Decne: total synthesis and biological activity. J Med Chem. 1986;29:1553–1555. doi: 10.1021/jm00158a044. [DOI] [PubMed] [Google Scholar]
- 36.Wani MC, Nicholas AW, Wall ME. Plant antitumor agents. 23. Synthesis and antileukemic activity of camptothecin analogues. J Med Chem. 1986;29:2358–2363. doi: 10.1021/jm00161a035. [DOI] [PubMed] [Google Scholar]
- 37.Wani MC, Nicholas AW, Manikumar G, Wall ME. Plant antitumor agents. 25. Total synthesis and antileukemic activity of ring a substituted camptothecin analogues. Structure-activity correlations. J Med Chem. 1987;30:1774–1779. doi: 10.1021/jm00393a016. [DOI] [PubMed] [Google Scholar]
- 38.Wani MC, Nicholas AW, Wall ME. Plant antitumor agents. 28. Resolution of a key tricyclic synthon, 5’(RS)-1,5-dioxo-5’-ethyl-5’-hydroxy-2’H,5’H,6’H-6’-oxopyrano[3’, 4’-f] delta 6,8-tetrahydro-indolizine: total synthesis and antitumor activity of 20(S)- and 20(R)-camptothecin. J Med Chem. 1987;30:2317–2319. doi: 10.1021/jm00395a024. [DOI] [PubMed] [Google Scholar]
- 39.Wall ME, Wani MC, Nicholas AW, Manikumar G, Tele C, Moore L, Truesdale A, Leitner P, Besterman JM. Plant antitumor agents. 30. Synthesis and structure activity of novel camptothecin analogs. J Med Chem. 1993;36:2689–2700. doi: 10.1021/jm00070a013. [DOI] [PubMed] [Google Scholar]
- 40.Plattner JJ, Gless RD, Rapoport H. Synthesis of some DE and CDE ring analogs of camptothecin. J Am Chem Soc. 1972;94:8613–8615. doi: 10.1021/ja00779a072. [DOI] [PubMed] [Google Scholar]
- 41.Tang C, Rapoport H. Total synthesis of (+-)-camptothecin. J Am Chem Soc. 1972;94:8615–8616. doi: 10.1021/ja00779a073. [DOI] [PubMed] [Google Scholar]
- 42.Tang CS, Morrow CJ, Rapoport H. Total synthesis of dl-camptothecin. J Am Chem Soc. 1975;97:159–167. doi: 10.1021/ja00834a028. [DOI] [PubMed] [Google Scholar]
- 43.Henegar KE, Ashford DW, Baughman TA, Sih JC, Gu RL. Practical asymmetric synthesis of (S)-4-Ethyl-7,8-dihydro-4-hydroxy-1H-pyrano[3,4-f] indolizine- 3,6,10(4H)-trione, a key intermediate for the synthesis of irinotecan and other camptothecin analogs. J Org Chem. 1997;62:6588–6597. [Google Scholar]
- 44.Tang CJ, Babjak M, Anderson RJ, Greene AE, Kanazawa A. Novel, efficient total synthesis of natural 20(S)-camptothecin. Org Biomol Chem. 2006;4:3757–3759. doi: 10.1039/b611202a. [DOI] [PubMed] [Google Scholar]
- 45.Li QY, Zu YG, Shi RZ, Yao LP. Review camptothecin: current perspectives. Curr Med Chem. 2006;13:2021–2039. doi: 10.2174/092986706777585004. [DOI] [PubMed] [Google Scholar]
- 46.Danishefsky S, Etheredge SJ. Synthesis and biological evaluation of DE-AB-camptothecin. J Org Chem. 1974;39:3430–3432. doi: 10.1021/jo00937a034. [DOI] [PubMed] [Google Scholar]
- 47.Mabb AM, Simon JM, King IF, Lee HM, An LK, Philpot BD, Zylka MJ. Topoisomerase 1 regulates gene expression in neurons through cleavage complex-dependent and-independent mechanisms. PLoS One. 2016;11:e0156439. doi: 10.1371/journal.pone.0156439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li F, Ling X, Harris DL, Liao J, Wang Y, Westover D, Jiang G, Xu B, Boland PM, Jin C. Topoisomerase I (Top1): a major target of FL118 for its antitumor efficacy or mainly involved in its side effects of hematopoietic toxicity? Am J Cancer Res. 2017;7:370–382. [PMC free article] [PubMed] [Google Scholar]
- 49.Morham SG, Kluckman KD, Voulomanos N, Smithies O. Targeted disruption of the mouse topoisomerase I gene by camptothecin selection. Mol Cell Biol. 1996;16:6804–6809. doi: 10.1128/mcb.16.12.6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Egusa Y, Fujiwara Y, Syaharuddin E, Sumiyoshi H, Isobe T, Yamakido M. Mobilization of peripheral blood stem cells in patients with advanced thoracic malignancies after irinotecan (CPT-11) administration. Anticancer Res. 1998;18:481–487. [PubMed] [Google Scholar]
- 51.Cacciari N, Zamagni C, Martoni A. The addition of topotecan to carboplatin and paclitaxel as first-line therapy for advanced ovarian cancer; is it possible only with peripheral blood stem cell support? Eur J Gynaecol Oncol. 2000;21:84–85. [PubMed] [Google Scholar]
- 52.Schilder RJ, Gallo JM, Millenson MM, Bookman MA, Weiner LM, Rogatko A, Rogers B, Padavic-Shallers K, Boente M, Rosenblum N, Adams AL, Ciccotto S, Ozols RF. Phase I trial of multiple cycles of high-dose carboplatin, paclitaxel, and topotecan with peripheral-blood stem-cell support as front-line therapy. J. Clin. Oncol. 2001;19:1183–1194. doi: 10.1200/JCO.2001.19.4.1183. [DOI] [PubMed] [Google Scholar]
- 53.Li F, inventor; USPTO, editor. Compositions and methods for identifying agents that alter expression of survivin (Patent US7569221). USPTO. 7569221. USA: Health Research Inc., Roswell Park Cancer Institute; US. (B2 ed) 2009:1–22.
- 54.Ling X, Cao S, Cheng Q, Keefe JT, Rustum YM, Li F. A novel small molecule FL118 that selectively inhibits survivin, Mcl-1, XIAP and cIAP2 in a p53-independent manner, shows superior antitumor activity. PLoS One. 2012;7:e45571. doi: 10.1371/journal.pone.0045571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, Altieri DC. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:580–584. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 56.Li F, Ackermann EJ, Bennett CF, Rothermel AL, Plescia J, Tognin S, Villa A, Marchisio PC, Altieri DC. Pleiotropic cell-division defects and apoptosis induced by interference with survivin function. Nat Cell Biol. 1999;1:461–466. doi: 10.1038/70242. [DOI] [PubMed] [Google Scholar]
- 57.Rodel F, Hoffmann J, Distel L, Herrmann M, Noisternig T, Papadopoulos T, Sauer R, Rodel C. Survivin as a radioresistance factor, and prognostic and therapeutic target for radiotherapy in rectal cancer. Cancer Res. 2005;65:4881–4887. doi: 10.1158/0008-5472.CAN-04-3028. [DOI] [PubMed] [Google Scholar]
- 58.Wu J, Apontes P, Song L, Liang P, Yang L, Li F. Molecular mechanism of upregulation of survivin transcription by the AT-rich DNA-binding ligand, Hoechst33342: evidence for survivin involvement in drug resistance. Nucleic Acids Res. 2007;35:2390–2402. doi: 10.1093/nar/gkm149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu J, Ling X, Pan D, Apontes P, Song L, Liang P, Altieri DC, Beerman T, Li F. Molecular mechanism of inhibition of survivin transcription by the GC-rich sequence selective DNAbinding antitumor agent, hedamycin: evidence of survivin downregulation associated with drug sensitivity. J Biol Chem. 2005;280:9745–9751. doi: 10.1074/jbc.M409350200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lu J, Tan M, Huang WC, Li P, Guo H, Tseng LM, Su XH, Yang WT, Treekitkarnmongkol W, Andreeff M, Symmans F, Yu D. Mitotic deregulation by survivin in ErbB2-overexpressing breast cancer cells contributes to Taxol resistance. Clin Cancer Res. 2009;15:1326–1334. doi: 10.1158/1078-0432.CCR-08-0954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang M, Latham DE, Delaney MA, Chakravarti A. Survivin mediates resistance to antiandrogen therapy in prostate cancer. Oncogene. 2005;24:2474–2482. doi: 10.1038/sj.onc.1208490. [DOI] [PubMed] [Google Scholar]
- 62.Park E, Gang EJ, Hsieh YT, Schaefer P, Chae S, Klemm L, Huantes S, Loh M, Conway EM, Kang ES, Koo HH, Hofmann WK, Heisterkamp N, Pelus L, Keerthivasan G, Crispino J, Kahn M, Muschen M, Kim YM. Targeting survivin overcomes drug resistance in acute lymphoblastic leukemia. Blood. 2011;118:2191–2199. doi: 10.1182/blood-2011-04-351239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ye Q, Cai W, Zheng Y, Evers BM, She QB. ERK and AKT signaling cooperate to translationally regulate survivin expression for metastatic progression of colorectal cancer. Oncogene. 2014;33:1828–1839. doi: 10.1038/onc.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang T, Otevrel T, Gao Z, Ehrlich SM, Fields JZ, Boman BM. Evidence that APC regulates survivin expression: a possible mechanism contributing to the stem cell origin of colon cancer. Cancer Res. 2001;61:8664–8667. [PubMed] [Google Scholar]
- 65.Li F, Cheng Q, Ling X, Stablewski A, Tang L, Foster BA, Johnson CS, Rustum YM, Porter CW. Generation of a novel transgenic mouse model for bioluminescent monitoring of survivin gene activity in vivo at various pathophysiological processes. Survivin expression overlaps with stem cell markers. Am J Pathol. 2010;176:1629–1638. doi: 10.2353/ajpath.2010.090414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Y, Chen HX, Zhou SY, Wang SX, Zheng K, Xu DD, Liu YT, Wang XY, Wang X, Yan HZ, Zhang L, Liu QY, Chen WQ, Wang YF. Sp1 and c-Myc modulate drug resistance of leukemia stem cells by regulating survivin expression through the ERK-MSK MAPK signaling pathway. Mol Cancer. 2015;14:56. doi: 10.1186/s12943-015-0326-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee MR, Ji SY, Mia-Jan K, Cho MY. Chemoresistance of CD133(+) colon cancer may be related with increased survivin expression. Biochem Biophys Res Commun. 2015;463:229–234. doi: 10.1016/j.bbrc.2015.05.031. [DOI] [PubMed] [Google Scholar]
- 68.Martini E, Schneider E, Neufert C, Neurath MF, Becker C. Survivin is a guardian of the intestinal stem cell niche and its expression is regulated by TGF-beta. Cell Cycle. 2016;15:2875–2881. doi: 10.1080/15384101.2016.1231286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Siddharth S, Das S, Nayak A, Kundu CN. SURVIVIN as a marker for quiescent-breast cancer stem cells-An intermediate, adherent, pre-requisite phase of breast cancer metastasis. Clin Exp Metastasis. 2016;33:661–675. doi: 10.1007/s10585-016-9809-7. [DOI] [PubMed] [Google Scholar]
- 70.Kanwar JR, Mahidhara G, Roy K, Sasidharan S, Krishnakumar S, Prasad N, Sehgal R, Kanwar RK. Fe-bLf nanoformulation targets survivin to kill colon cancer stem cells and maintains absorption of iron, calcium and zinc. Nanomedicine (Lond) 2015;10:35–55. doi: 10.2217/nnm.14.132. [DOI] [PubMed] [Google Scholar]
- 71.Zhang Y, Zhou SY, Yan HZ, Xu DD, Chen HX, Wang XY, Wang X, Liu YT, Zhang L, Wang S, Zhou PJ, Fu WY, Ruan BB, Ma DL, Wang Y, Liu QY, Ren Z, Liu Z, Zhang R, Wang YF. miR-203 inhibits proliferation and self-renewal of leukemia stem cells by targeting survivin and Bmi-1. Sci Rep. 2016;6:19995. doi: 10.1038/srep19995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Glinsky GV. Genomic models of metastatic cancer: functional analysis of death-from-cancer signature genes reveals aneuploid, anoikis-resistant, metastasis-enabling phenotype with altered cell cycle control and activated Polycomb Group (PcG) protein chromatin silencing pathway. Cell Cycle. 2006;5:1208–1216. doi: 10.4161/cc.5.11.2796. [DOI] [PubMed] [Google Scholar]
- 73.Li F. Survivin study: what is the next wave? J Cell Physiol. 2003;197:8–29. doi: 10.1002/jcp.10327. [DOI] [PubMed] [Google Scholar]
- 74.Li F, Brattain MG. Role of the survivin gene in pathophysiology. Am J Pathol. 2006;169:1–11. doi: 10.2353/ajpath.2006.060121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Altieri DC. Survivin in apoptosis control and cell cycle regulation in cancer. Prog Cell Cycle Res. 2003;5:447–452. [PubMed] [Google Scholar]
- 76.Wheatley SP, McNeish IA. Survivin: a protein with dual roles in mitosis and apoptosis. Int Rev Cytol. 2005;247:35–88. doi: 10.1016/S0074-7696(05)47002-3. [DOI] [PubMed] [Google Scholar]
- 77.Novel Formulations of waterinsoluble chemical compounds and methods of using a formulation of compound FL118 for cancer therapy (PCT/US11/58558). USPTO. USA: Roswell Park Cancer Institute; 2011. [Google Scholar]
- 78.Ling X, Liu XJ, Zhong K, Smith N, Prey J, Li F. FL118, a novel camptothecin analogue, overcomes irinotecan and topotecan resistance in human tumor xenograft models. Am J Transl Res. 2015;7:1765–1781. [PMC free article] [PubMed] [Google Scholar]
- 79.Sugimoto Y, Tsukahara S, Oh-hara T, Isoe T, Tsuruo T. Decreased expression of DNA topoisomerase I in camptothecin-resistant tumor cell lines as determined by a monoclonal antibody. Cancer Res. 1990;50:6925–6930. [PubMed] [Google Scholar]
- 80.Kanzawa F, Sugimoto Y, Minato K, Kasahara K, Bungo M, Nakagawa K, Fujiwara Y, Liu LF, Saijo N. Establishment of a camptothecin analogue (CPT-11)-resistant cell line of human non-small cell lung cancer: characterization and mechanism of resistance. Cancer Res. 1990;50:5919–5924. [PubMed] [Google Scholar]
- 81.Woessner RD, Eng WK, Hofmann GA, Rieman DJ, McCabe FL, Hertzberg RP, Mattern MR, Tan KB, Johnson RK. Camptothecin hyper-resistant P388 cells: drug-dependent reduction in topoisomerase I content. Oncol Res. 1992;4:481–488. [PubMed] [Google Scholar]
- 82.Ando K, Shah AK, Sachdev V, Kleinstiver BP, Taylor-Parker J, Welch MM, Hu Y, Salgia R, White FM, Parvin JD, Ozonoff A, Rameh LE, Joung JK, Bharti AK. Camptothecin resistance is determined by the regulation of topoisomerase I degradation mediated by ubiquitin proteasome pathway. Oncotarget. 2017;8:43733–43751. doi: 10.18632/oncotarget.16376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Desai SD, Li TK, Rodriguez-Bauman A, Rubin EH, Liu LF. Ubiquitin/26S proteasome-mediated degradation of topoisomerase I as a resistance mechanism to camptothecin in tumor cells. Cancer Res. 2001;61:5926–5932. [PubMed] [Google Scholar]
- 84.Desai SD, Liu LF, Vazquez-Abad D, D’Arpa P. Ubiquitin-dependent destruction of topoisomerase I is stimulated by the antitumor drug camptothecin. J Biol Chem. 1997;272:24159–24164. doi: 10.1074/jbc.272.39.24159. [DOI] [PubMed] [Google Scholar]
- 85.Sugimoto Y, Tsukahara S, Oh-hara T, Liu LF, Tsuruo T. Elevated expression of DNA topoisomerase II in camptothecin-resistant human tumor cell lines. Cancer Res. 1990;50:7962–7965. [PubMed] [Google Scholar]
- 86.Kapoor R, Slade DL, Fujimori A, Pommier Y, Harker WG. Altered topoisomerase I expression in two subclones of human CEM leukemia selected for resistance to camptothecin. Oncol Res. 1995;7:83–95. [PubMed] [Google Scholar]
- 87.Liao Z, Robey RW, Guirouilh-Barbat J, To KK, Polgar O, Bates SE, Pommier Y. Reduced expression of DNA topoisomerase I in SF295 human glioblastoma cells selected for resistance to homocamptothecin and diflomotecan. Mol Pharmacol. 2008;73:490–497. doi: 10.1124/mol.107.041178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kotoh S, Naito S, Yokomizo A, Kumazawa J, Asakuno K, Kohno K, Kuwano M. Increased expression of DNA topoisomerase I gene and collateral sensitivity to camptothecin in human cisplatin-resistant bladder cancer cells. Cancer Res. 1994;54:3248–3252. [PubMed] [Google Scholar]
- 89.Sakai A, Kasahara K, Ohmori T, Kimura H, Sone T, Fujimura M, Nakao S. MET increases the sensitivity of gefitinib-resistant cells to SN-38, an active metabolite of irinotecan, by up-regulating the topoisomerase I activity. J Thorac Oncol. 2012;7:1337–1344. doi: 10.1097/JTO.0b013e31825cca4c. [DOI] [PubMed] [Google Scholar]
- 90.Smith PJ, Makinson TA, Watson JV. Enhanced sensitivity to camptothecin in ataxiatelangiectasia cells and its relationship with the expression of DNA topoisomerase I. Int J Radiat Biol. 1989;55:217–231. doi: 10.1080/09553008914550271. [DOI] [PubMed] [Google Scholar]
- 91.Urasaki Y, Laco GS, Pourquier P, Takebayashi Y, Kohlhagen G, Gioffre C, Zhang H, Chatterjee D, Pantazis P, Pommier Y. Characterization of a novel topoisomerase I mutation from a camptothecin-resistant human prostate cancer cell line. Cancer Res. 2001;61:1964–1969. [PubMed] [Google Scholar]
- 92.Li F. Anticancer drug FL118 is more than a survivin inhibitor: where is the Achilles’ heel of cancer? Am J Cancer Res. 2014;4:304–311. [PMC free article] [PubMed] [Google Scholar]
- 93.Zhao J, Ling X, Cao S, Liu X, Wan S, Jiang T, Li F. Antitumor activity of FL118, a survivin, Mcl-1, XIAP, cIAP2 selective inhibitor, is highly dependent on its primary structure and steric configuration. Mol Pharm. 2014;11:457–467. doi: 10.1021/mp4004282. [DOI] [PubMed] [Google Scholar]
- 94.Ling X, Xu C, Fan C, Zhong K, Li F, Wang X. FL118 induces p53-dependent senescence in colorectal cancer cells by promoting degradation of MdmX. Cancer Res. 2014;74:7487–7497. doi: 10.1158/0008-5472.CAN-14-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Troelstra C, van Gool A, de Wit J, Vermeulen W, Bootsma D, Hoeijmakers JH. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne’s syndrome and preferential repair of active genes. Cell. 1992;71:939–953. doi: 10.1016/0092-8674(92)90390-x. [DOI] [PubMed] [Google Scholar]
- 96.Orren DK, Dianov GL, Bohr VA. The human CSB (ERCC6) gene corrects the transcriptioncoupled repair defect in the CHO cell mutant UV61. Nucleic Acids Res. 1996;24:3317–3322. doi: 10.1093/nar/24.17.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhao Z, Zhang G, Li W. Elevated expression of ERCC6 confers resistance to 5-Fluorouracil and is associated with poor patient survival in colorectal cancer. DNA Cell Biol. 2017;36:781–786. doi: 10.1089/dna.2017.3768. [DOI] [PubMed] [Google Scholar]
- 98.Houghton PJ, Germain GS, Harwood FC, Schuetz JD, Stewart CF, Buchdunger E, Traxler P. Imatinib mesylate is a potent inhibitor of the ABCG2 (BCRP) transporter and reverses resistance to topotecan and SN-38 in vitro. Cancer Res. 2004;64:2333–2337. doi: 10.1158/0008-5472.can-03-3344. [DOI] [PubMed] [Google Scholar]
- 99.Su Y, Hu P, Lee SH, Sinko PJ. Using novobiocin as a specific inhibitor of breast cancer resistant protein to assess the role of transporter in the absorption and disposition of topotecan. J Pharm Pharm Sci. 2007;10:519–536. doi: 10.18433/j3qp4w. [DOI] [PubMed] [Google Scholar]
- 100.Su Y, Lee SH, Sinko PJ. Inhibition of efflux transporter ABCG2/BCRP does not restore mitoxantrone sensitivity in irinotecan-selected human leukemia CPT-K5 cells: evidence for multifactorial multidrug resistance. Eur J Pharm Sci. 2006;29:102–110. doi: 10.1016/j.ejps.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 101.Yoshikawa M, Ikegami Y, Sano K, Yoshida H, Mitomo H, Sawada S, Ishikawa T. Transport of SN-38 by the wild type of human ABC transporter ABCG2 and its inhibition by quercetin, a natural flavonoid. J Exp Ther Oncol. 2004;4:25–35. [PubMed] [Google Scholar]
- 102.Shishido Y, Ueno S, Yamazaki R, Nagaoka M, Matsuzaki T. ABCG2 inhibitor YHO-13351 sensitizes cancer stem/initiating-like side population cells to irinotecan. Anticancer Res. 2013;33:1379–1386. [PubMed] [Google Scholar]
- 103.Maliepaard M, van Gastelen MA, de Jong LA, Pluim D, van Waardenburg RC, Ruevekamp-Helmers MC, Floot BG, Schellens JH. Overexpression of the BCRP/MXR/ABCP gene in a topotecan-selected ovarian tumor cell line. Cancer Res. 1999;59:4559–4563. [PubMed] [Google Scholar]
- 104.Kruijtzer CM, Beijnen JH, Rosing H, ten Bokkel Huinink WW, Schot M, Jewell RC, Paul EM, Schellens JH. Increased oral bioavailability of topotecan in combination with the breast cancer resistance protein and P-glycoprotein inhibitor GF120918. J. Clin. Oncol. 2002;20:2943–2950. doi: 10.1200/JCO.2002.12.116. [DOI] [PubMed] [Google Scholar]
- 105.de Vries NA, Zhao J, Kroon E, Buckle T, Beijnen JH, van Tellingen O. P-glycoprotein and breast cancer resistance protein: two dominant transporters working together in limiting the brain penetration of topotecan. Clin Cancer Res. 2007;13:6440–6449. doi: 10.1158/1078-0432.CCR-07-1335. [DOI] [PubMed] [Google Scholar]
- 106.Takeba Y, Sekine S, Kumai T, Matsumoto N, Nakaya S, Tsuzuki Y, Yanagida Y, Nakano H, Asakura T, Ohtsubo T, Kobayashi S. Irinotecan-induced apoptosis is inhibited by increased P-glycoprotein expression and decreased p53 in human hepatocellular carcinoma cells. Biol Pharm Bull. 2007;30:1400–1406. doi: 10.1248/bpb.30.1400. [DOI] [PubMed] [Google Scholar]
- 107.Tagen M, Zhuang Y, Zhang F, Harstead KE, Shen J, Schaiquevich P, Fraga CH, Panetta JC, Waters CM, Stewart CF. P-glycoprotein, but not multidrug resistance protein 4, plays a role in the systemic clearance of irinotecan and SN-38 in mice. Drug Metab Lett. 2010;4:195–201. doi: 10.2174/187231210792928251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Filipski E, Berland E, Ozturk N, Guettier C, van der Horst GT, Levi F, Okyar A. Optimization of irinotecan chronotherapy with P-glycoprotein inhibition. Toxicol Appl Pharmacol. 2014;274:471–479. doi: 10.1016/j.taap.2013.12.018. [DOI] [PubMed] [Google Scholar]
- 109.Hendricks CB, Rowinsky EK, Grochow LB, Donehower RC, Kaufmann SH. Effect of Pglycoprotein expression on the accumulation and cytotoxicity of topotecan (SK&F 104864), a new camptothecin analogue. Cancer Res. 1992;52:2268–2278. [PubMed] [Google Scholar]
- 110.Westover D, Ling X, Lam H, Welch J, Jin C, Gongora C, Del Rio M, Wani M, Li F. FL118, a novel camptothecin derivative, is insensitive to ABCG2 expression and shows improved efficacy in comparison with irinotecan in colon and lung cancer models with ABCG2-induced resistance. Mol Cancer. 2015;14:92. doi: 10.1186/s12943-015-0362-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Westover D, Li F. New trends for overcoming ABCG2/BCRP-mediated resistance to cancer therapies. J Exp Clin Cancer Res. 2015;34:159. doi: 10.1186/s13046-015-0275-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jaxel C, Kohn KW, Wani MC, Wall ME, Pommier Y. Structure-activity study of the actions of camptothecin derivatives on mammalian topoisomerase I: evidence for a specific receptor site and a relation to antitumor activity. Cancer Res. 1989;49:1465–1469. [PubMed] [Google Scholar]
- 113.Bennett RP, Stewart RA, Hogan PA, Ptak RG, Mankowski MK, Hartman TL, Buckheit RW Jr, Snyder BA, Salter JD, Morales GA, Smith HC. An analog of camptothecin inactive against Topoisomerase I is broadly neutralizing of HIV-1 through inhibition of Vif-dependent APOBEC3G degradation. Antiviral Res. 2016;136:51–59. doi: 10.1016/j.antiviral.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Legarza K, Yang LX. Novel camptothecin derivatives. In Vivo. 2005;19:283–292. [PubMed] [Google Scholar]
- 115.Venditto VJ, Simanek EE. Cancer therapies utilizing the camptothecins: a review of the in vivo literature. Mol Pharm. 2010;7:307–349. doi: 10.1021/mp900243b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liu YQ, Li WQ, Morris-Natschke SL, Qian K, Yang L, Zhu GX, Wu XB, Chen AL, Zhang SY, Nan X, Lee KH. Perspectives on biologically active camptothecin derivatives. Med Res Rev. 2015;35:753–789. doi: 10.1002/med.21342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hinz HR, Harris NJ, Natelson EA, Giovanella BC. Pharmacokinetics of the in vivo and in vitro conversion of 9-nitro-20(S)-camptothecin to 9-amino-20(S)-camptothecin in humans, dogs, and mice. Cancer Res. 1994;54:3096–3100. [PubMed] [Google Scholar]
- 118.Rajendra R, Gounder MK, Saleem A, Schellens JH, Ross DD, Bates SE, Sinko P, Rubin EH. Differential effects of the breast cancer resistance protein on the cellular accumulation and cytotoxicity of 9-aminocamptothecin and 9-nitrocamptothecin. Cancer Res. 2003;63:3228–3233. [PubMed] [Google Scholar]
- 119.Chen J, Ping Q, Guo J, Chu X, Song M. Pharmacokinetics of lactone, carboxylate and total 9-nitrocamptothecin with different doses and administration routes in rats. Biopharm Drug Dispos. 2006;27:53–59. doi: 10.1002/bdd.480. [DOI] [PubMed] [Google Scholar]
- 120.Kemp KR, Liehr JG, Giovanella B. Combined radiation and 9-nitrocamptothecin (rubitecan) in the treatment of locally advanced pancreatic cancer. Ann N Y Acad Sci. 2000;922:320–323. doi: 10.1111/j.1749-6632.2000.tb07054.x. [DOI] [PubMed] [Google Scholar]
- 121.Pharmaceuticals A. Rubitecan Astex website. 2004-2005. Available from: https://astx.com/?s=Rubitecan.
- 122.Clark JW. Rubitecan. Expert Opin Investig Drugs. 2006;15:71–79. doi: 10.1517/13543784.15.1.71. [DOI] [PubMed] [Google Scholar]
- 123.Rubin E, Wood V, Bharti A, Trites D, Lynch C, Hurwitz S, Bartel S, Levy S, Rosowsky A, Toppmeyer D, et al. A phase I and pharmacokinetic study of a new camptothecin derivative, 9-aminocamptothecin. Clin Cancer Res. 1995;1:269–276. [PubMed] [Google Scholar]
- 124.Dahut W, Harold N, Takimoto C, Allegra C, Chen A, Hamilton JM, Arbuck S, Sorensen M, Grollman F, Nakashima H, Lieberman R, Liang M, Corse W, Grem J. Phase I and pharmacologic study of 9-aminocamptothecin given by 72-hour infusion in adult cancer patients. J. Clin. Oncol. 1996;14:1236–1244. doi: 10.1200/JCO.1996.14.4.1236. [DOI] [PubMed] [Google Scholar]
- 125.Natelson EA, Giovanella BC, Verschraegen CF, Fehir KM, De Ipolyi PD, Harris N, Stehlin JS. Phase I clinical and pharmacological studies of 20-(S)-camptothecin and 20-(S)-9-nitrocamptothecin as anticancer agents. Ann N Y Acad Sci. 1996;803:224–230. doi: 10.1111/j.1749-6632.1996.tb26392.x. [DOI] [PubMed] [Google Scholar]
- 126.Takimoto CH, Dahut W, Marino MT, Nakashima H, Liang MD, Harold N, Lieberman R, Arbuck SG, Band RA, Chen AP, Hamilton JM, Cantilena LR, Allegra CJ, Grem JL. Pharmacodynamics and pharmacokinetics of a 72-hour infusion of 9-aminocamptothecin in adult cancer patients. J. Clin. Oncol. 1997;15:1492–1501. doi: 10.1200/JCO.1997.15.4.1492. [DOI] [PubMed] [Google Scholar]
- 127.Pazdur R, Diaz-Canton E, Ballard WP, Bradof JE, Graham S, Arbuck SG, Abbruzzese JL, Winn R. Phase II trial of 9-aminocamptothecin administered as a 72-hour continuous infusion in metastatic colorectal carcinoma. J. Clin. Oncol. 1997;15:2905–2909. doi: 10.1200/JCO.1997.15.8.2905. [DOI] [PubMed] [Google Scholar]
- 128.Saltz LB, Kemeny NE, Tong W, Harrison J, Berkery R, Kelsen DP. 9-Aminocamptothecin by 72-hour continuous intravenous infusion is inactive in the treatment of patients with 5-fluorouracil-refractory colorectal carcinoma. Cancer. 1997;80:1727–1732. doi: 10.1002/(sici)1097-0142(19971101)80:9<1727::aid-cncr5>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 129.Sparreboom A, de Jonge MJ, Punt CJ, Nooter K, Loos WJ, Porro MG, Verweij J. Pharmacokinetics and bioavailability of oral 9-aminocamptothecin capsules in adult patients with solid tumors. Clin Cancer Res. 1998;4:1915–1919. [PubMed] [Google Scholar]
- 130.Eder JP Jr, Supko JG, Lynch T, Bryant M, Vosburgh E, Shulman LN, Xu G, Kufe DW. Phase I trial of the colloidal dispersion formulation of 9-amino-20(S)-camptothecin administered as a 72-hour continuous intravenous infusion. Clin Cancer Res. 1998;4:317–324. [PubMed] [Google Scholar]
- 131.Langevin AM, Casto DT, Thomas PJ, Weitman SD, Kretschmar C, Grier H, Pratt C, Dubowy R, Bernstein M, Blaney S, Vietti T. Phase I trial of 9-aminocamptothecin in children with refractory solid tumors: a Pediatric Oncology Group study. J. Clin. Oncol. 1998;16:2494–2499. doi: 10.1200/JCO.1998.16.7.2494. [DOI] [PubMed] [Google Scholar]
- 132.Mani S, Iyer L, Janisch L, Wang X, Fleming GF, Schilsky RL, Ratain MJ. Phase I clinical and pharmacokinetic study of oral 9-aminocamptothecin (NSC-603071) Cancer Chemother Pharmacol. 1998;42:84–87. doi: 10.1007/s002800050789. [DOI] [PubMed] [Google Scholar]
- 133.Pazdur R, Medgyesy DC, Winn RJ, Dakhil SR, Moore DF Jr, Scalzo A, Hoff PM, Arbuck SG, Abbruzzese JL. Phase II trial of 9-aminocamptothecin (NSC 603071) administered as a 120-hr continuous infusion weekly for three weeks in metastatic colorectal carcinoma. Invest New Drugs. 1998;16:341–346. doi: 10.1023/a:1006248700232. [DOI] [PubMed] [Google Scholar]
- 134.Siu LL, Oza AM, Eisenhauer EA, Firby PS, Thiessen JJ, Michael M, Wainman N, Manzo J, Feld R, Goldberg RA, Moore MJ. Phase I and pharmacologic study of 9-aminocamptothecin colloidal dispersion formulation given as a 24-hour continuous infusion weekly times four every 5 weeks. J. Clin. Oncol. 1998;16:1122–1130. doi: 10.1200/JCO.1998.16.3.1122. [DOI] [PubMed] [Google Scholar]
- 135.Verschraegen CF, Natelson EA, Giovanella BC, Kavanagh JJ, Kudelka AP, Freedman RS, Edwards CL, Ende K, Stehlin JS. A phase I clinical and pharmacological study of oral 9-nitrocamptothecin, a novel water-insoluble topoisomerase I inhibitor. Anticancer Drugs. 1998;9:36–44. doi: 10.1097/00001813-199801000-00004. [DOI] [PubMed] [Google Scholar]
- 136.Vokes EE, Ansari RH, Masters GA, Hoffman PC, Klepsch A, Ratain MJ, Sciortino DF, Lad TE, Krauss S, Fishkin PA, Golomb HM. A phase II study of 9-aminocamptothecin in advanced non-small-cell lung cancer. Ann Oncol. 1998;9:1085–1090. doi: 10.1023/a:1008432729754. [DOI] [PubMed] [Google Scholar]
- 137.Wilson WH, Little R, Pearson D, Jaffe ES, Steinberg SM, Cheson BD, Humphrey R, Kohler DR, Elwood P. Phase II and dose-escalation with or without granulocyte colony-stimulating factor study of 9-aminocamptothecin in relapsed and refractory lymphomas. J. Clin. Oncol. 1998;16:2345–2351. doi: 10.1200/JCO.1998.16.7.2345. [DOI] [PubMed] [Google Scholar]
- 138.de Jonge MJ, Punt CJ, Gelderblom AH, Loos WJ, van Beurden V, Planting AS, van der Burg ME, van Maanen LW, Dallaire BK, Verweij J, Wagener DJ, Sparreboom A. Phase I and pharmacologic study of oral (PEG-1000) 9-aminocamptothecin in adult patients with solid tumors. J. Clin. Oncol. 1999;17:2219–2226. doi: 10.1200/JCO.1999.17.7.2219. [DOI] [PubMed] [Google Scholar]
- 139.de Jonge MJ, Verweij J, Loos WJ, Dallaire BK, Sparreboom A. Clinical pharmacokinetics of encapsulated oral 9-aminocamptothecin in plasma and saliva. Clin Pharmacol Ther. 1999;65:491–499. doi: 10.1016/S0009-9236(99)70068-8. [DOI] [PubMed] [Google Scholar]
- 140.Herben VM, van Gijn R, Schellens JH, Schot M, Lieverst J, Hillebrand MJ, Schoemaker NE, Porro MG, Beijnen JH, ten Bokkel Huinink WW. Phase I and pharmacokinetic study of a daily times 5 short intravenous infusion schedule of 9-aminocamptothecin in a colloidal dispersion formulation in patients with advanced solid tumors. J. Clin. Oncol. 1999;17:1906–1914. doi: 10.1200/JCO.1999.17.6.1906. [DOI] [PubMed] [Google Scholar]
- 141.Minami H, Lad TE, Nicholas MK, Vokes EE, Ratain MJ. Pharmacokinetics and pharmacodynamics of 9-aminocamptothecin infused over 72 hours in phase II studies. Clin Cancer Res. 1999;5:1325–1330. [PubMed] [Google Scholar]
- 142.Verschraegen CF, Gupta E, Loyer E, Kavanagh JJ, Kudelka AP, Freedman RS, Edwards CL, Harris N, Steger M, Steltz V, Giovanella BC, Stehlin JS. A phase II clinical and pharmacological study of oral 9-nitrocamptothecin in patients with refractory epithelial ovarian, tubal or peritoneal cancer. Anticancer Drugs. 1999;10:375–383. doi: 10.1097/00001813-199904000-00005. [DOI] [PubMed] [Google Scholar]
- 143.Vey N, Kantarjian H, Tran H, Beran M, O’Brien S, Bivins C, Giles F, Cortes J, Cheson B, Arbuck S, Estey E. Phase I and pharmacologic study of 9-aminocamptothecin colloidal dispersion formulation in patients with refractory or relapsed acute leukemia. Ann Oncol. 1999;10:577–583. doi: 10.1023/a:1026406920321. [DOI] [PubMed] [Google Scholar]
- 144.Stehlin JS, Giovanella BC, Natelson EA, De Ipolyi PD, Coil D, Davis B, Wolk D, Wallace P, Trojacek A. A study of 9-nitrocamptothecin (RFS-2000) in patients with advanced pancreatic cancer. Int J Oncol. 1999;14:821–831. doi: 10.3892/ijo.14.5.821. [DOI] [PubMed] [Google Scholar]
- 145.Kraut EH, Balcerzak SP, Young D, O’Rourke MA, Petrus JJ, Kuebler JP, Mayernik DG. A phase II study of 9-aminocamptothecin in patients with refractory breast cancer. Cancer Invest. 2000;18:28–31. doi: 10.3109/07357900009023059. [DOI] [PubMed] [Google Scholar]
- 146.Lad T, Rosen F, Sciortino D, Brockstein B, Keubler JP, Arietta R, Vokes E. Phase II trial of aminocamptothecin (9-AC/DMA) in patients with advanced squamous cell head and neck cancer. Invest New Drugs. 2000;18:261–263. doi: 10.1023/a:1006481924287. [DOI] [PubMed] [Google Scholar]
- 147.Pitot HC, Knost JA, Mahoney MR, Kugler J, Krook JE, Hatfield AK, Sargent DJ, Goldberg RM. A North Central Cancer Treatment Group Phase II trial of 9-aminocamptothecin in previously untreated patients with measurable metastatic colorectal carcinoma. Cancer. 2000;89:1699–1705. [PubMed] [Google Scholar]
- 148.Verschraegen CF, Gilbert BE, Huaringa AJ, Newman R, Harris N, Leyva FJ, Keus L, Campbell K, Nelson-Taylor T, Knight V. Feasibility, phase I, and pharmacological study of aerosolized liposomal 9-nitro-20(S)-camptothecin in patients with advanced malignancies in the lungs. Ann N Y Acad Sci. 2000;922:352–354. doi: 10.1111/j.1749-6632.2000.tb07063.x. [DOI] [PubMed] [Google Scholar]
- 149.Verschraegen CF, Vincent M, Abbruzzese JL, Siegler D, Kavanagh JJ, Loyer E, Kudelka AP, Rubin E. Phase I study of 9-nitro-20(S)-camptothecin in combination with cisplatin for patients with advanced malignancies. Ann N Y Acad Sci. 2000;922:345–348. doi: 10.1111/j.1749-6632.2000.tb07061.x. [DOI] [PubMed] [Google Scholar]
- 150.Argiris A, Heald P, Kuzel T, Foss FM, DiStasio S, Cooper DL, Arbuck S, Murren JR. Phase II trial of 9-aminocamptothecin as a 72-h infusion in cutaneous T-cell lymphoma. Invest New Drugs. 2001;19:321–326. doi: 10.1023/a:1010613912335. [DOI] [PubMed] [Google Scholar]
- 151.Konstadoulakis MM, Antonakis PT, Tsibloulis BG, Stathopoulos GP, Manouras AP, Mylonaki DB, Golematis BX. A phase II study of 9-nitrocamptothecin in patients with advanced pancreatic adenocarcinoma. Cancer Chemother Pharmacol. 2001;48:417–420. doi: 10.1007/s002800100360. [DOI] [PubMed] [Google Scholar]
- 152.Thomas RR, Dahut W, Harold N, Grem JL, Monahan BP, Liang M, Band RA, Cottrell J, Llorens V, Smith JA, Corse W, Arbuck SG, Wright J, Chen AP, Shapiro JD, Hamilton JM, Allegra CJ, Takimoto CH. A phase I and pharmacologic study of 9-aminocamptothecin administered as a 120-h infusion weekly to adult cancer patients. Cancer Chemother Pharmacol. 2001;48:215–222. doi: 10.1007/s002800100329. [DOI] [PubMed] [Google Scholar]
- 153.Ellerhorst JA, Bedikian AY, Smith TM, Papadopoulos NE, Plager C, Eton O. Phase II trial of 9-nitrocamptothecin (RFS 2000) for patients with metastatic cutaneous or uveal melanoma. Anticancer Drugs. 2002;13:169–172. doi: 10.1097/00001813-200202000-00009. [DOI] [PubMed] [Google Scholar]
- 154.Muggia FM, Liebes L, Hazarika M, Wadler S, Hamilton A, Hornreich G, Sorich J, Chiang C, Newman E, Potmesil M, Hochster H. Phase I and pharmacologic study of i. p. 9-aminocamptothecin given as six fractions over 14 days. Anticancer Drugs. 2002;13:819–825. doi: 10.1097/00001813-200209000-00006. [DOI] [PubMed] [Google Scholar]
- 155.Raymond E, Campone M, Stupp R, Menten J, Chollet P, Lesimple T, Fety-Deporte R, Lacombe D, Paoletti X, Fumoleau P. Multicentre phase II and pharmacokinetic study of RFS2000 (9-nitro-camptothecin) administered orally 5 days a week in patients with glioblastoma multiforme. Eur J Cancer. 2002;38:1348–1350. doi: 10.1016/s0959-8049(02)00070-9. [DOI] [PubMed] [Google Scholar]
- 156.Schoffski P, Herr A, Vermorken JB, Van den Brande J, Beijnen JH, Rosing H, Volk J, Ganser A, Adank S, Botma HJ, Wanders J. Clinical phase II study and pharmacological evaluation of rubitecan in non-pretreated patients with metastatic colorectal cancer-significant effect of food intake on the bioavailability of the oral camptothecin analogue. Eur J Cancer. 2002;38:807–813. doi: 10.1016/s0959-8049(02)00022-9. [DOI] [PubMed] [Google Scholar]
- 157.Fracasso PM, Rader JS, Govindan R, Herzog TJ, Arquette MA, Denes A, Mutch DG, Picus J, Tan BR, Fears CL, Goodner SA, Sun SL. Phase I study of rubitecan and gemcitabine in patients with advanced malignancies. Ann Oncol. 2002;13:1819–1825. doi: 10.1093/annonc/mdf342. [DOI] [PubMed] [Google Scholar]
- 158.Leguizamo J, Quinn M, Takimoto CH, Liang MD, Ismail AS, Pang J, Dahut W, Grem JL. A phase I study of 9-aminocamptothecin as a colloidal dispersion formulation given as a fortnightly 72-h infusion. Cancer Chemother Pharmacol. 2003;52:333–338. doi: 10.1007/s00280-003-0657-1. [DOI] [PubMed] [Google Scholar]
- 159.Michaelson MD, Ryan DP, Fuchs CS, Supko JG, Garcia-Carbonero R, Paul Eder J, Clark JW. A Phase I study of 9-nitrocamptothecin given concurrently with capecitabine in patients with refractory, metastatic solid tumors. Cancer. 2003;97:148–154. doi: 10.1002/cncr.11038. [DOI] [PubMed] [Google Scholar]
- 160.Patel SR, Beach J, Papadopoulos N, Burgess MA, Trent J, Jenkins J, Benjamin RS. Results of a 2-arm Phase II study of 9-nitrocamptothecin in patients with advanced soft-tissue sarcomas. Cancer. 2003;97:2848–2852. doi: 10.1002/cncr.11385. [DOI] [PubMed] [Google Scholar]
- 161.Xiong HQ, Tran HT, Madden TL, Newman RA, Abbruzzese JL. Phase I and pharmacological study of oral 9-aminocamptothecin colloidal dispersion (NSC 603071) in patients with advanced solid tumors. Clin Cancer Res. 2003;9:2066–2071. [PubMed] [Google Scholar]
- 162.de Jonge MJ, Droz JP, Paz-Ares L, van Oosterom AT, de Wit R, Chollet P, Baron B, Lacombe D, Mettinger K, Fumoleau P EORTC-New Drug Development Group/New Drug Development Program. Phase II study on 9-nitrocamptothecin (RFS 2000) in patients with advanced or metastatic urothelial tract tumors. Invest New Drugs. 2004;22:329–333. doi: 10.1023/B:DRUG.0000026260.24275.02. [DOI] [PubMed] [Google Scholar]
- 163.Hochster H, Plimack ER, Runowicz CD, Speyer J, Wallach RC, Sorich J, Mandeli J, Wadler S, Wright J, Muggia FM. Biweekly 72-hour 9-aminocamptothecin infusion as second-line therapy for ovarian carcinoma: phase II study of the New York Gynecologic Oncology Group and the Eastern Cooperative Oncology Group. J. Clin. Oncol. 2004;22:120–126. doi: 10.1200/JCO.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 164.Kindler HL, Avadhani A, Wade-Oliver K, Karrison T, Mani S, Vokes EE. 9-Aminocamptothecin (9-AC) given as a 120-hour continuous infusion in patients with advanced adenocarcinomas of the stomach and gastroesophageal junction: a phase II trial of the University of Chicago phase II consortium. Invest New Drugs. 2004;22:323–327. doi: 10.1023/B:DRUG.0000026259.28395.c2. [DOI] [PubMed] [Google Scholar]
- 165.Jung LL, Ramanathan RK, Egorin MJ, Jin R, Belani CP, Potter DM, Strychor S, Trump DL, Walko C, Fakih M, Zamboni WC. Pharmacokinetic studies of 9-nitrocamptothecin on intermittent and continuous schedules of administration in patients with solid tumors. Cancer Chemother Pharmacol. 2004;54:487–496. doi: 10.1007/s00280-004-0835-9. [DOI] [PubMed] [Google Scholar]
- 166.Miller KD, Soule SE, Haney LG, Guiney P, Murry DJ, Lenaz L, Sun SL, Sledge GW Jr. A phase II study of 9-nitro-camptothecin in patients with previously treated metastatic breast cancer. Invest New Drugs. 2004;22:69–73. doi: 10.1023/b:drug.0000006176.84915.71. [DOI] [PubMed] [Google Scholar]
- 167.Punt CJ, de Jonge MJ, Monfardini S, Daugaard G, Fiedler W, Baron B, Lacombe D, Fumoleau P EORTC New Drug Development Group. RFS2000 (9-nitrocamptothecin) in advanced small cell lung cancer, a phase II study of the EORTC New Drug Development Group. Eur J Cancer. 2004;40:1332–1334. doi: 10.1016/j.ejca.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 168.Verschraegen CF, Gilbert BE, Loyer E, Huaringa A, Walsh G, Newman RA, Knight V. Clinical evaluation of the delivery and safety of aerosolized liposomal 9-nitro-20(s)-camptothecin in patients with advanced pulmonary malignancies. Clin Cancer Res. 2004;10:2319–2326. doi: 10.1158/1078-0432.ccr-0929-3. [DOI] [PubMed] [Google Scholar]
- 169.Zamboni WC, Jung LL, Egorin MJ, Potter DM, Friedland DM, Belani CP, Agarwala SS, Wong MM, Fakih M, Trump DL, Jin R, Strychor S, Vozniak M, Troetschel M, Ramanathan RK. Phase I and pharmacologic study of intermittently administered 9-nitrocamptothecin in patients with advanced solid tumors. Clin Cancer Res. 2004;10:5058–5064. doi: 10.1158/1078-0432.CCR-03-0288. [DOI] [PubMed] [Google Scholar]
- 170.Jacobs AD, Burris HA, Rivkin S, Ritch PS, Eisenberg PD, Mettinger KL. A randomized phase III study of rubitecan (ORA) vs. best choice (BC) in 409 patients with refractory pancreatic cancer report from a North-American multi-center study. J. Clin. Oncol. 2004;22:4013–4013. [Google Scholar]
- 171.Papish SW, Ramanathan RK, Pincus J, Hirmand M, Burris HA. Patients rescued by crossover to rubitecan in phase III study of rubitecan capsules versus 5-FU in pancreatic cancer. American Society of Clinical Oncology (ASCO) 2005 Abstract: 4165. [Google Scholar]
- 172.Chugh R, Dunn R, Zalupski MM, Biermann JS, Sondak VK, Mace JR, Leu KM, Chandler WF, Baker LH. Phase II study of 9-nitro-camptothecin in patients with advanced chordoma or soft tissue sarcoma. J. Clin. Oncol. 2005;23:3597–3604. doi: 10.1200/JCO.2005.02.170. [DOI] [PubMed] [Google Scholar]
- 173.Miller DS, Blessing JA, Waggoner S, Schilder J, Sorosky J, Bloss J, Schilder R. Phase II evaluation of 9-aminocamptothecin (9-AC, NSC #603071) in platinum-resistant ovarian and primary peritoneal carcinoma: a Gynecologic Oncology Group Study. Gynecol Oncol. 2005;96:67–71. doi: 10.1016/j.ygyno.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 174.Tedesco KL, Berlin J, Rothenberg M, Choy H, Wyman K, Scott Pearson A, Daniel Beauchamp R, Merchant N, Lockhart AC, Shyr Y, Caillouette C, Chakravarthy B. A phase I study of concurrent 9-nitro-20(s)-camptothecin (9NC/Orathecin) and radiation therapy in the treatment of locally advanced adenocarcinoma of the pancreas. Radiother Oncol. 2005;76:54–58. doi: 10.1016/j.radonc.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 175.Baka S, Ranson M, Lorigan P, Danson S, Linton K, Hoogendam I, Mettinger K, Thatcher N. A phase II trial with RFS2000 (rubitecan) in patients with advanced non-small cell lung cancer. Eur J Cancer. 2005;41:1547–1550. doi: 10.1016/j.ejca.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 176.Burris HA 3rd, Rivkin S, Reynolds R, Harris J, Wax A, Gerstein H, Mettinger KL, Staddon A. Phase II trial of oral rubitecan in previously treated pancreatic cancer patients. Oncologist. 2005;10:183–190. doi: 10.1634/theoncologist.10-3-183. [DOI] [PubMed] [Google Scholar]
- 177.Farray D, Ahluwalia MS, Snyder J, Barnett GH, Cohen BH, Suh JH, Peereboom DM. Pre-irradiation 9-amino [20s] camptothecin (9-AC) in patients with newly diagnosed glioblastoma multiforme. Invest New Drugs. 2006;24:177–180. doi: 10.1007/s10637-005-2464-5. [DOI] [PubMed] [Google Scholar]
- 178.Sewak S, Sorich J, O’Leary J. Phase I trial of continuous infusion 9-aminocamptothecin in patients with advanced solid tumors: 21-day infusion is an active well-tolerated regimen. Anticancer Drugs. 2006;17:571–579. doi: 10.1097/00001813-200606000-00012. [DOI] [PubMed] [Google Scholar]
- 179.Simon GR, Lush RM, Gump J, Tetteh L, Williams C, Cantor A, Antonia S, Garrett C, Rocha-Lima C, Fishman M, Sullivan DM, Munster PN. Sequential oral 9-nitrocamptothecin and etoposide: a pharmacodynamic- and pharmacokinetic-based phase I trial. Mol Cancer Ther. 2006;5:2130–2137. doi: 10.1158/1535-7163.MCT-06-0034. [DOI] [PubMed] [Google Scholar]
- 180.Zamboni WC, Goel S, Iqbal T, Parise RA, Strychor S, Repinski TV, Egorin MJ, Mani S. Clinical and pharmacokinetic study evaluating the effect of food on the disposition of 9-nitrocamptothecin and its 9-aminocamptothecin metabolite in patients with solid tumors. Cancer Chemother Pharmacol. 2006;57:631–639. doi: 10.1007/s00280-005-0084-6. [DOI] [PubMed] [Google Scholar]
- 181.Chedid S, Rivera E, Frye DK, Ibrahim N, Esteva F, Valero V, Hortobagyi G, Mettinger KL, Cristofanilli M. Minimal clinical benefit of single agent Orathecin (Rubitecan) in heavily pretreated metastatic breast cancer. Cancer Chemother Pharmacol. 2006;57:540–544. doi: 10.1007/s00280-005-0064-x. [DOI] [PubMed] [Google Scholar]
- 182.Patel H, Stoller R, Auber M, Potter D, Cai C, Zamboni W, Kiefer G, Matin K, Schmotzer A, Ramanathan RK. Phase II study of rubitecan, an oral camptothecin in patients with advanced colorectal cancer who have failed previous 5-fluorouracil based chemotherapy. Invest New Drugs. 2006;24:359–363. doi: 10.1007/s10637-006-6451-2. [DOI] [PubMed] [Google Scholar]
- 183.Lee SJ, Gounder M, Rubin EH, Li JM, Gu Z, Thalasila A, Loyer E, Kudelka AP, Verschraegen CF. Optimal modeling for phase I design of a two drug combination-results of a phase I study of cisplatin with 9-nitrocamptothecin. Invest New Drugs. 2008;26:541–551. doi: 10.1007/s10637-008-9147-y. [DOI] [PubMed] [Google Scholar]
- 184.Caponigro F, Carteni G, Droz JP, Milano A, Davis WB, Pollard P. Phase II study of rubitecan in recurrent or metastatic head and neck cancer. Cancer Chemother Pharmacol. 2008;62:209–214. doi: 10.1007/s00280-007-0592-7. [DOI] [PubMed] [Google Scholar]
- 185.Bartlett NL, Johnson JL, Wagner-Johnston N, Ratain MJ, Peterson BA Cancer and Leukemia Group B. Phase II study of 9-aminocamptothecin in previously treated lymphomas: results of cancer and Leukemia Group B 9551. Cancer Chemother Pharmacol. 2009;63:793–798. doi: 10.1007/s00280-008-0803-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Yan Z, Zhu Z, Li K, Chen P, Wang L, Huang C, Xue J, Liu M. A phase I pharmacokinetics study of 9-nitrocamptothecin in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2011;67:955–961. doi: 10.1007/s00280-010-1546-z. [DOI] [PubMed] [Google Scholar]
- 187.Li H, Jin HE, Kim W, Han YH, Kim DD, Chung SJ, Shim CK. Involvement of P-glycoprotein, multidrug resistance protein 2 and breast cancer resistance protein in the transport of belotecan and topotecan in Caco-2 and MDCKII cells. Pharm Res. 2008;25:2601–2612. doi: 10.1007/s11095-008-9678-0. [DOI] [PubMed] [Google Scholar]
- 188.Kim YK, Koo NY, Yun PY. Anticancer effects of CKD-602 (Camtobell(R)) via G2/M phase arrest in oral squamous cell carcinoma cell lines. Oncol Lett. 2015;9:136–142. doi: 10.3892/ol.2014.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Oh IJ, Kim KS, Park CK, Kim YC, Lee KH, Jeong JH, Kim SY, Lee JE, Shin KC, Jang TW, Lee HK, Lee KY, Lee SY. Belotecan/cisplatin versus etoposide/cisplatin in previously untreated patients with extensive-stage small cell lung carcinoma: a multi-center randomized phase III trial. BMC Cancer. 2016;16:690. doi: 10.1186/s12885-016-2741-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Park YH, Chung CU, Park BM, Park MR, Park DI, Moon JY, Park HS, Kim JH, Jung SS, Kim JO, Kim SY, Lee JE. Lesser toxicities of belotecan in patients with small cell lung cancer: a retrospective single-center study of camptothecin analogs. Can Respir J. 2016;2016:3576201. doi: 10.1155/2016/3576201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lee JH, Lee JM, Lim KH, Kim JK, Ahn SK, Bang YJ, Hong CI. Preclinical and phase I clinical studies with Ckd-602, a novel camptothecin derivative. Ann N Y Acad Sci. 2000;922:324–325. doi: 10.1111/j.1749-6632.2000.tb07055.x. [DOI] [PubMed] [Google Scholar]
- 192.Lee DH, Kim SW, Bae KS, Hong JS, Suh C, Kang YK, Lee JS. A phase I and pharmacologic study of belotecan in combination with cisplatin in patients with previously untreated extensive-stage disease small cell lung cancer. Clin Cancer Res. 2007;13:6182–6186. doi: 10.1158/1078-0432.CCR-07-0534. [DOI] [PubMed] [Google Scholar]
- 193.Lee DH, Kim SW, Suh C, Lee JS, Lee JH, Lee SJ, Ryoo BY, Park K, Kim JS, Heo DS, Kim NK. Belotecan, new camptothecin analogue, is active in patients with small-cell lung cancer: results of a multicenter early phase II study. Ann Oncol. 2008;19:123–127. doi: 10.1093/annonc/mdm437. [DOI] [PubMed] [Google Scholar]
- 194.Lee HP, Seo SS, Ryu SY, Kim JH, Bang YJ, Park SY, Nam JH, Kang SB, Lee KH, Song YS. Phase II evaluation of CKD-602, a camptothecin analog, administered on a 5-day schedule to patients with platinum-sensitive or -resistant ovarian cancer. Gynecol Oncol. 2008;109:359–363. doi: 10.1016/j.ygyno.2007.11.023. [DOI] [PubMed] [Google Scholar]
- 195.Kim HS, Kang SB, Seo SS, Han SS, Kim JW, Park NH, Kang SB, Lee HP, Song YS. Phase I/IIa study of combination chemotherapy with CKD-602 and cisplatin in patients with recurrent epithelial ovarian cancer. Ann N Y Acad Sci. 2009;1171:627–634. doi: 10.1111/j.1749-6632.2009.04885.x. [DOI] [PubMed] [Google Scholar]
- 196.Zamboni WC, Ramalingam S, Friedland DM, Edwards RP, Stoller RG, Strychor S, Maruca L, Zamboni BA, Belani CP, Ramanathan RK. Phase I and pharmacokinetic study of pegylated liposomal CKD-602 in patients with advanced malignancies. Clin Cancer Res. 2009;15:1466–1472. doi: 10.1158/1078-0432.CCR-08-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Jeong J, Cho BC, Sohn JH, Choi HJ, Kim SH, Lee YJ, Jung MK, Shin SJ, Park MS, Kim SK, Chang J, Kim JH. Belotecan for relapsing smallcell lung cancer patients initially treated with an irinotecan-containing chemotherapy: a phase II trial. Lung Cancer. 2010;70:77–81. doi: 10.1016/j.lungcan.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 198.Kim HS, Park NH, Kang S, Seo SS, Chung HH, Kim JW, Song YS, Kang SB. Comparison of the efficacy between topotecan- and belotecan-, a new camptothecin analog, based chemotherapies for recurrent epithelial ovarian cancer: a single institutional experience. J Obstet Gynaecol Res. 2010;36:86–93. doi: 10.1111/j.1447-0756.2009.01101.x. [DOI] [PubMed] [Google Scholar]
- 199.Kim SJ, Kim JS, Kim SC, Kim YK, Kim YK, Kang JY, Yoon HK, Song JS, Lee SH, Moon HS, Kim JW, Kim KH, Kim CH, Shim BY, Kim HK. A multicenter phase II study of belotecan, new camptothecin analogue, in patients with previously untreated extensive stage disease small cell lung cancer. Lung Cancer. 2010;68:446–449. doi: 10.1016/j.lungcan.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 200.Kim YM, Lee SW, Kim DY, Kim JH, Nam JH, Kim YT. The efficacy and toxicity of belotecan (CKD-602), a camptothericin analogue topoisomerase I inhibitor, in patients with recurrent or refractory epithelial ovarian cancer. J Chemother. 2010;22:197–200. doi: 10.1179/joc.2010.22.3.197. [DOI] [PubMed] [Google Scholar]
- 201.Lee DH, Kim SW, Suh C, Lee JS, Ahn JS, Ahn MJ, Park K, Na II, Lee JC, Ryoo BY, Yang SH. Multicenter phase 2 study of belotecan, a new camptothecin analog, and cisplatin for chemotherapy-naive patients with extensive-disease small cell lung cancer. Cancer. 2010;116:132–136. doi: 10.1002/cncr.24719. [DOI] [PubMed] [Google Scholar]
- 202.Nam EJ, Kim JW, Kim JH, Kim S, Kim SW, Jang SY, Lee DW, Jung YW, Kim YT. Efficacy and toxicity of belotecan with and without cisplatin in patients with recurrent ovarian cancer. Am J Clin Oncol. 2010;33:233–237. doi: 10.1097/COC.0b013e3181a650bc. [DOI] [PubMed] [Google Scholar]
- 203.Choi CH, Lee YY, Song TJ, Park HS, Kim MK, Kim TJ, Lee JW, Lee JH, Bae DS, Kim BG. Phase II study of belotecan, a camptothecin analogue, in combination with carboplatin for the treatment of recurrent ovarian cancer. Cancer. 2011;117:2104–2111. doi: 10.1002/cncr.25710. [DOI] [PubMed] [Google Scholar]
- 204.Hwang JH, Lim MC, Seo SS, Park SY, Kang S. Phase II study of belotecan (CKD 602) as a single agent in patients with recurrent or progressive carcinoma of uterine cervix. Jpn J Clin Oncol. 2011;41:624–629. doi: 10.1093/jjco/hyr017. [DOI] [PubMed] [Google Scholar]
- 205.Rhee CK, Lee SH, Kim JS, Kim SJ, Kim SC, Kim YK, Kang HH, Yoon HK, Song JS, Moon HS, Kim JW, Kim CH, Shim BY, Kim HK, Sun DS, Kim KH. A multicenter phase II study of belotecan, a new camptothecin analogue, as a secondline therapy in patients with small cell lung cancer. Lung Cancer. 2011;72:64–67. doi: 10.1016/j.lungcan.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 206.Hong J, Jung M, Kim YJ, Sym SJ, Kyung SY, Park J, Lee SP, Park JW, Cho EK, Jeong SH, Shin DB, Lee JH. Phase II study of combined belotecan and cisplatin as first-line chemotherapy in patients with extensive disease of small cell lung cancer. Cancer Chemother Pharmacol. 2012;69:215–220. doi: 10.1007/s00280-011-1689-6. [DOI] [PubMed] [Google Scholar]
- 207.Hwang JH, Yoo HJ, Lim MC, Seo SS, Park SY, Kang S. Phase I clinical trial of alternating belotecan and oral etoposide in patients with platinum-resistant or heavily treated ovarian cancer. Anticancer Drugs. 2012;23:321–325. doi: 10.1097/CAD.0b013e32834ea5d0. [DOI] [PubMed] [Google Scholar]
- 208.Kim GM, Kim YS, Ae Kang Y, Jeong JH, Kim SM, Hong YK, Sung JH, Lim ST, Kim JH, Kim SK, Cho BC. Efficacy and toxicity of belotecan for relapsed or refractory small cell lung cancer patients. J Thorac Oncol. 2012;7:731–736. doi: 10.1097/JTO.0b013e31824b23cb. [DOI] [PubMed] [Google Scholar]
- 209.Wu H, Ramanathan RK, Zamboni BA, Strychor S, Ramalingam S, Edwards RP, Friedland DM, Stoller RG, Belani CP, Maruca LJ, Bang YJ, Zamboni WC. Population pharmacokinetics of pegylated liposomal CKD-602 (S-CKD602) in patients with advanced malignancies. J Clin Pharmacol. 2012;52:180–194. doi: 10.1177/0091270010394851. [DOI] [PubMed] [Google Scholar]
- 210.Lim S, Cho BC, Jung JY, Kim GM, Kim SH, Kim HR, Kim HS, Lim SM, Park JS, Lee JH, Kim D, Kim EY, Park MS, Kim YS, Kim SK, Chang J, Kim JH. Phase II study of camtobell inj. (belotecan) in combination with cisplatin in patients with previously untreated, extensive stage small cell lung cancer. Lung Cancer. 2013;80:313–318. doi: 10.1016/j.lungcan.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 211.Yeo CD, Lee SH, Kim JS, Kim SJ, Kim SC, Kim YK, Kang HH, Yoon HK, Song JS, Moon HS, Kim JW, Kim KH, Shim BY, Kim CH. A multicenter phase II study of belotecan, a new camptothecin analogue, in elderly patients with previously untreated, extensive-stage small cell lung cancer. Cancer Chemother Pharmacol. 2013;72:809–814. doi: 10.1007/s00280-013-2256-0. [DOI] [PubMed] [Google Scholar]
- 212.van Hattum AH, Hoogsteen IJ, Schluper HM, Maliepaard M, Scheffer GL, Scheper RJ, Kohlhagen G, Pommier Y, Pinedo HM, Boven E. Induction of breast cancer resistance protein by the camptothecin derivative DX-8951f is associated with minor reduction of antitumour activity. Br J Cancer. 2002;87:665–672. doi: 10.1038/sj.bjc.6600508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Kumazawa E, Jimbo T, Ochi Y, Tohgo A. Potent and broad antitumor effects of DX-8951f, a water-soluble camptothecin derivative, against various human tumors xenografted in nude mice. Cancer Chemother Pharmacol. 1998;42:210–220. doi: 10.1007/s002800050807. [DOI] [PubMed] [Google Scholar]
- 214.Vey N, Giles FJ, Kantarjian H, Smith TL, Beran M, Jeha S. The topoisomerase I inhibitor DX-8951f is active in a severe combined immunodeficient mouse model of human acute myelogenous leukemia. Clin Cancer Res. 2000;6:731–736. [PubMed] [Google Scholar]
- 215.Minami H, Fujii H, Igarashi T, Itoh K, Tamanoi K, Oguma T, Sasaki Y. Phase I and pharmacological study of a new camptothecin derivative, exatecan mesylate (DX-8951f), infused over 30 minutes every three weeks. Clin Cancer Res. 2001;7:3056–3064. [PubMed] [Google Scholar]
- 216.Abou-Alfa GK, Letourneau R, Harker G, Modiano M, Hurwitz H, Tchekmedyian NS, Feit K, Ackerman J, De Jager RL, Eckhardt SG, O’Reilly EM. Randomized phase III study of exatecan and gemcitabine compared with gemcitabine alone in untreated advanced pancreatic cancer. J. Clin. Oncol. 2006;24:4441–4447. doi: 10.1200/JCO.2006.07.0201. [DOI] [PubMed] [Google Scholar]
- 217.Boige V, Raymond E, Faivre S, Gatineau M, Meely K, Mekhaldi S, Pautier P, Ducreux M, Rixe O, Armand JP. Phase I and pharmacokinetic study of the camptothecin analog DX-8951f administered as a 30-minute infusion every 3 weeks in patients with advanced cancer. J. Clin. Oncol. 2000;18:3986–3992. doi: 10.1200/JCO.2000.18.23.3986. [DOI] [PubMed] [Google Scholar]
- 218.De Jager R, Cheverton P, Tamanoi K, Coyle J, Ducharme M, Sakamoto N, Satomi M, Suzuki M DX-8931f Investigators. DX-8951f: summary of phase I clinical trials. Ann N Y Acad Sci. 2000;922:260–273. doi: 10.1111/j.1749-6632.2000.tb07044.x. [DOI] [PubMed] [Google Scholar]
- 219.Rowinsky EK, Johnson TR, Geyer CE Jr, Hammond LA, Eckhardt SG, Drengler R, Smetzer L, Coyle J, Rizzo J, Schwartz G, Tolcher A, Von Hoff DD, De Jager RL. DX-8951f, a hexacyclic camptothecin analog, on a daily-times-five schedule: a phase I and pharmacokinetic study in patients with advanced solid malignancies. J. Clin. Oncol. 2000;18:3151–3163. doi: 10.1200/JCO.2000.18.17.3151. [DOI] [PubMed] [Google Scholar]
- 220.Verschraegen CF, Levenback C, Vincent M, Wolf J, Bevers M, Loyer E, Kudelka AP, Kavanagh JJ. Phase II study of intravenous DX-8951f in patients with advanced ovarian, tubal, or peritoneal cancer refractory to platinum, taxane, and topotecan. Ann N Y Acad Sci. 2000;922:349–351. doi: 10.1111/j.1749-6632.2000.tb07062.x. [DOI] [PubMed] [Google Scholar]
- 221.Royce ME, Hoff PM, Dumas P, Lassere Y, Lee JJ, Coyle J, Ducharme MP, De Jager R, Pazdur R. Phase I and pharmacokinetic study of exatecan mesylate (DX-8951f): a novel camptothecin analog. J. Clin. Oncol. 2001;19:1493–1500. doi: 10.1200/JCO.2001.19.5.1493. [DOI] [PubMed] [Google Scholar]
- 222.Sharma S, Kemeny N, Schwartz GK, Kelsen D, O’Reilly E, Ilson D, Coyle J, De Jager RL, Ducharme MP, Kleban S, Hollywood E, Saltz LB. Phase I study of topoisomerase I inhibitor exatecan mesylate (DX-8951f) given as weekly 24-hour infusions three of every four weeks. Clin Cancer Res. 2001;7:3963–3970. [PubMed] [Google Scholar]
- 223.Giles FJ, Cortes JE, Thomas DA, Garcia-Manero G, Faderl S, Jeha S, De Jager RL, Kantarjian HM. Phase I and pharmacokinetic study of DX-8951f (exatecan mesylate), a hexacyclic camptothecin, on a daily-times-five schedule in patients with advanced leukemia. Clin Cancer Res. 2002;8:2134–2141. [PubMed] [Google Scholar]
- 224.Braybrooke JP, Boven E, Bates NP, Ruijter R, Dobbs N, Cheverton PD, Pinedo HM, Talbot DC. Phase I and pharmacokinetic study of the topoisomerase I inhibitor, exatecan mesylate (DX-8951f), using a weekly 30-minute intravenous infusion, in patients with advanced solid malignancies. Ann Oncol. 2003;14:913–921. doi: 10.1093/annonc/mdg243. [DOI] [PubMed] [Google Scholar]
- 225.Braybrooke JP, Ranson M, Manegold C, Mattson K, Thatcher N, Cheverton P, Sekiguchi M, Suzuki M, Oyama R, Talbot DC. Phase II study of exatecan mesylate (DX-8951f) as first line therapy for advanced non-small cell lung cancer. Lung Cancer. 2003;41:215–219. doi: 10.1016/s0169-5002(03)00190-9. [DOI] [PubMed] [Google Scholar]
- 226.Esteva FJ, Rivera E, Cristofanilli M, Valero V, Royce M, Duggal A, Colucci P, DeJager R, Hortobagyi GN. A Phase II study of intravenous exatecan mesylate (DX-8951f) administered daily for 5 days every 3 weeks to patients with metastatic breast carcinoma. Cancer. 2003;98:900–907. doi: 10.1002/cncr.11557. [DOI] [PubMed] [Google Scholar]
- 227.Garrison MA, Hammond LA, Geyer CE Jr, Schwartz G, Tolcher AW, Smetzer L, Figueroa JA, Ducharme M, Coyle J, Takimoto CH, De Jager RL, Rowinsky EK. A Phase I and pharmocokinetic study of exatecan mesylate administered as a protracted 21-day infusion in patients with advanced solid malignancies. Clin Cancer Res. 2003;9:2527–2537. [PubMed] [Google Scholar]
- 228.Clamp A, Adams M, Atkinson R, Boven E, Calvert AH, Cervantes A, Ganesan T, Lotz J, Vasey P, Cheverton P, Jayson GC. A phase IIA study of the topoisomerase I inhibitor, exatecan mesylate (DX-8951f), administered at two different dose schedules in patients with platinum- and taxane-resistant/refractory ovarian cancer. Gynecol Oncol. 2004;95:114–119. doi: 10.1016/j.ygyno.2004.06.047. [DOI] [PubMed] [Google Scholar]
- 229.Royce ME, Rowinsky EK, Hoff PM, Coyle J, DeJager R, Pazdur R, Saltz LB. A phase II study of intravenous exatecan mesylate (DX-8951f) administered daily for five days every three weeks to patients with metastatic adenocarcinoma of the colon or rectum. Invest New Drugs. 2004;22:53–61. doi: 10.1023/b:drug.0000006174.87869.6b. [DOI] [PubMed] [Google Scholar]
- 230.Verschraegen CF, Kudelka AP, Hu W, Vincent M, Kavanagh JJ, Loyer E, Bastien L, Duggal A, De Jager R. A phase II study of intravenous exatecan mesylate (DX-8951f) administered daily for 5 days every 3 weeks to patients with advanced ovarian, tubal or peritoneal cancer resistant to platinum, taxane and topotecan. Cancer Chemother Pharmacol. 2004;53:1–7. doi: 10.1007/s00280-003-0696-7. [DOI] [PubMed] [Google Scholar]
- 231.Abou-Alfa GK, Rowinsky EK, Patt YZ, Schwartz GK, Kelsen DP, Sharma S, Siegel E, Becerra CR, Eckhardt SG, Feit K, De Jager R, O’Reilly EM. A Phase II study of intravenous exatecan administered daily for 5 days, every 3 weeks to patients with biliary tract cancers. Am J Clin Oncol. 2005;28:334–339. doi: 10.1097/01.coc.0000158829.63542.2c. [DOI] [PubMed] [Google Scholar]
- 232.Ajani JA, Takimoto C, Becerra CR, Silva A, Baez L, Cohn A, Major P, Kamida M, Feit K, De Jager R. A phase II clinical and pharmacokinetic study of intravenous exatecan mesylate (DX-8951f) in patients with untreated metastatic gastric cancer. Invest New Drugs. 2005;23:479–484. doi: 10.1007/s10637-005-2907-z. [DOI] [PubMed] [Google Scholar]
- 233.Reichardt P, Nielsen OS, Bauer S, Hartmann JT, Schöffski P, Christensen TB, Pink D, Daugaard S, Marreaud S, Van Glabbeke M, Blay JY EORTC Soft Tissue and Bone Sarcoma Group. Exatecan in pretreated adult patients with advanced soft tissue sarcoma: results of a phase II--study of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2007;43:1017–1022. doi: 10.1016/j.ejca.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 234.Kumazawa E, Ochi Y. DE-310, a novel macromolecular carrier system for the camptothecin analog DX-8951f: potent antitumor activities in various murine tumor models. Cancer Sci. 2004;95:168–175. doi: 10.1111/j.1349-7006.2004.tb03199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Masubuchi N. Pharmacokinetics of DE-310, a novel macromolecular carrier system for the camptothecin analog DX-8951f, in tumor-bearing mice. Pharmazie. 2004;59:374–377. [PubMed] [Google Scholar]
- 236.Kato M, Matsuhashi K, Shimomura K, Shimada M, Hagiwara M, Fujikawa K, Furuhama K. Examination of meningocele induced by the antitumor agent DE-310 in rat fetuses. Reprod Toxicol. 2005;20:495–502. doi: 10.1016/j.reprotox.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 237.Soepenberg O, de Jonge MJ, Sparreboom A, de Bruin P, Eskens FA, de Heus G, Wanders J, Cheverton P, Ducharme MP, Verweij J. Phase I and pharmacokinetic study of DE-310 in patients with advanced solid tumors. Clin Cancer Res. 2005;11:703–711. [PubMed] [Google Scholar]
- 238.Wente MN, Kleeff J, Buchler MW, Wanders J, Cheverton P, Langman S, Friess H. DE-310, a macromolecular prodrug of the topoisomerase-I-inhibitor exatecan (DX-8951), in patients with operable solid tumors. Invest New Drugs. 2005;23:339–347. doi: 10.1007/s10637-005-1442-2. [DOI] [PubMed] [Google Scholar]
- 239.Gerrits CJ, Schellens JH, Creemers GJ, Wissel P, Planting AS, Pritchard JF, DePee S, de Boer-Dennert M, Harteveld M, Verweij J. The bioavailability of oral GI147211 (GG211), a new topoisomerase I inhibitor. Br J Cancer. 1997;76:946–951. doi: 10.1038/bjc.1997.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Emerson DL, Bendele R, Brown E, Chiang S, Desjardins JP, Dihel LC, Gill SC, Hamilton M, LeRay JD, Moon-McDermott L, Moynihan K, Richardson FC, Tomkinson B, Luzzio MJ, Baccanari D. Antitumor efficacy, pharmacokinetics, and biodistribution of NX 211: a lowclearance liposomal formulation of lurtotecan. Clin Cancer Res. 2000;6:2903–2912. [PubMed] [Google Scholar]
- 241.Tomkinson B, Bendele R, Giles FJ, Brown E, Gray A, Hart K, LeRay JD, Meyer D, Pelanne M, Emerson DL. OSI-211, a novel liposomal topoisomerase I inhibitor, is active in SCID mouse models of human AML and ALL. Leuk Res. 2003;27:1039–1050. doi: 10.1016/s0145-2126(03)00092-4. [DOI] [PubMed] [Google Scholar]
- 242.Loos WJ, Kehrer D, Brouwer E, Verweij J, de Bruijn P, Hamilton M, Gill S, Nooter K, Stoter G, Sparreboom A. Liposomal lurtotecan (NX211): determination of total drug levels in human plasma and urine by reversed-phase highperformance liquid chromatography. J Chromatogr B Biomed Sci Appl. 2000;738:155–163. doi: 10.1016/s0378-4347(99)00513-7. [DOI] [PubMed] [Google Scholar]
- 243.Loos WJ, Verweij J, Kehrer DF, de Bruijn P, de Groot FM, Hamilton M, Nooter K, Stoter G, Sparreboom A. Structural identification and biological activity of 7-methyl-10,11-ethylenedioxy-20(S)-camptothecin, a photodegradant of lurtotecan. Clin Cancer Res. 2002;8:856–862. [PubMed] [Google Scholar]
- 244.Gerrits CJ, Creemers GJ, Schellens JH, Wissel P, Planting AS, Kunka R, Selinger K, de Boer-Dennert M, Marijnen Y, Harteveld M, Verweij J. Phase I and pharmacological study of the new topoisomerase I inhibitor GI147211, using a daily x 5 intravenous administration. Br J Cancer. 1996;73:744–750. doi: 10.1038/bjc.1996.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Eckhardt SG, Baker SD, Eckardt JR, Burke TG, Warner DL, Kuhn JG, Rodriguez G, Fields S, Thurman A, Smith L, Rothenberg ML, White L, Wissel P, Kunka R, DePee S, Littlefield D, Burris HA, Von Hoff DD, Rowinsky EK. Phase I and pharmacokinetic study of GI147211, a watersoluble camptothecin analogue, administered for five consecutive days every three weeks. Clin Cancer Res. 1998;4:595–604. [PubMed] [Google Scholar]
- 246.Paz-Ares L, Kunka R, DeMaria D, Cassidy J, Alden M, Beranek P, Kaye S, Littlefield D, Reilly D, Depee S, Wissel P, Twelves C, O’Dwyer P. A phase I clinical and pharmacokinetic study of the new topoisomerase inhibitor GI147211 given as a 72-h continuous infusion. Br J Cancer. 1998;78:1329–1336. doi: 10.1038/bjc.1998.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Gamucci T, Paridaens R, Heinrich B, Schellens JH, Pavlidis N, Verweij J, Sessa C, Kaye S, Roelvink M, Wanders J, Hanauske A. Activity and toxicity of GI147211 in breast, colorectal and non-small-cell lung cancer patients: an EORTC-ECSG phase II clinical study. Ann Oncol. 2000;11:793–797. doi: 10.1023/a:1008373031714. [DOI] [PubMed] [Google Scholar]
- 248.Sessa C, Wanders J, Roelvink M, Dombernowsky P, Nielsen D, Morant R, Drings P, Wissel P, Hanauske AR. Second-line treatment of small-cell lung cancer with the camptothecin-derivative GI147211: a study of the EORTC Early Clinical Studies Group (ECSG) Ann Oncol. 2000;11:207–210. doi: 10.1023/a:1008372404504. [DOI] [PubMed] [Google Scholar]
- 249.Kehrer DF, Bos AM, Verweij J, Groen HJ, Loos WJ, Sparreboom A, de Jonge MJ, Hamilton M, Cameron T, de Vries EG. Phase I and pharmacologic study of liposomal lurtotecan, NX 211: urinary excretion predicts hematologic toxicity. J. Clin. Oncol. 2002;20:1222–1231. doi: 10.1200/JCO.2002.20.5.1222. [DOI] [PubMed] [Google Scholar]
- 250.Schellens JH, Heinrich B, Lehnert M, Gore ME, Kaye SB, Dombernowsky P, Paridaens R, van Oosterom AT, Verweij J, Loos WJ, Calvert H, Pavlidis N, Cortes-Funes H, Wanders J, Roelvink M, Sessa C, Selinger K, Wissel PS, Gamucci T, Hanauske AR. Population pharmacokinetic and dynamic analysis of the topoisomerase I inhibitor lurtotecan in phase II studies. Invest New Drugs. 2002;20:83–93. doi: 10.1023/a:1014454821885. [DOI] [PubMed] [Google Scholar]
- 251.Duffaud F, Borner M, Chollet P, Vermorken JB, Bloch J, Degardin M, Rolland F, Dittrich C, Baron B, Lacombe D, Fumoleau P EORTC-New Drug Development Group/New Drug Development Program. Phase II study of OSI-211 (liposomal lurtotecan) in patients with metastatic or loco-regional recurrent squamous cell carcinoma of the head and neck. An EORTC New Drug Development Group study. Eur J Cancer. 2004;40:2748–2752. doi: 10.1016/j.ejca.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 252.Gelmon K, Hirte H, Fisher B, Walsh W, Ptaszynski M, Hamilton M, Onetto N, Eisenhauer E. A phase 1 study of OSI-211 given as an intravenous infusion days 1, 2, and 3 every three weeks in patients with solid cancers. Invest New Drugs. 2004;22:263–275. doi: 10.1023/B:DRUG.0000026252.86842.e2. [DOI] [PubMed] [Google Scholar]
- 253.Giles FJ, Tallman MS, Garcia-Manero G, Cortes JE, Thomas DA, Wierda WG, Verstovsek S, Hamilton M, Barrett E, Albitar M, Kantarjian HM. Phase I and pharmacokinetic study of a low-clearance, unilamellar liposomal formulation of lurtotecan, a topoisomerase 1 inhibitor, in patients with advanced leukemia. Cancer. 2004;100:1449–1458. doi: 10.1002/cncr.20132. [DOI] [PubMed] [Google Scholar]
- 254.MacKenzie MJ, Hirte HW, Siu LL, Gelmon K, Ptaszynski M, Fisher B, Eisenhauer E. A phase I study of OSI-211 and cisplatin as intravenous infusions given on days 1, 2 and 3 every 3 weeks in patients with solid cancers. Ann Oncol. 2004;15:665–670. doi: 10.1093/annonc/mdh133. [DOI] [PubMed] [Google Scholar]
- 255.Seiden MV, Muggia F, Astrow A, Matulonis U, Campos S, Roche M, Sivret J, Rusk J, Barrett E. A phase II study of liposomal lurtotecan (OSI-211) in patients with topotecan resistant ovarian cancer. Gynecol Oncol. 2004;93:229–232. doi: 10.1016/j.ygyno.2003.12.037. [DOI] [PubMed] [Google Scholar]
- 256.Dark GG, Calvert AH, Grimshaw R, Poole C, Swenerton K, Kaye S, Coleman R, Jayson G, Le T, Ellard S, Trudeau M, Vasey P, Hamilton M, Cameron T, Barrett E, Walsh W, McIntosh L, Eisenhauer EA. Randomized trial of two intravenous schedules of the topoisomerase I inhibitor liposomal lurtotecan in women with relapsed epithelial ovarian cancer: a trial of the national cancer institute of Canada clinical trials group. J. Clin. Oncol. 2005;23:1859–1866. doi: 10.1200/JCO.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 257.De Cesare M, Pratesi G, Perego P, Carenini N, Tinelli S, Merlini L, Penco S, Pisano C, Bucci F, Vesci L, Pace S, Capocasa F, Carminati P, Zunino F. Potent antitumor activity and improved pharmacological profile of ST1481, a novel 7-substituted camptothecin. Cancer Res. 2001;61:7189–7195. [PubMed] [Google Scholar]
- 258.Marchetti S, Oostendorp RL, Pluim D, van Eijndhoven M, van Tellingen O, Schinkel AH, Versace R, Beijnen JH, Mazzanti R, Schellens JH. In vitro transport of gimatecan (7-t-butoxyiminomethylcamptothecin) by breast cancer resistance protein, P-glycoprotein, and multidrug resistance protein 2. Mol Cancer Ther. 2007;6:3307–3313. doi: 10.1158/1535-7163.MCT-07-0461. [DOI] [PubMed] [Google Scholar]
- 259.Pratesi G, De Cesare M, Carenini N, Perego P, Righetti SC, Cucco C, Merlini L, Pisano C, Penco S, Carminati P, Vesci L, Zunino F. Pattern of antitumor activity of a novel camptothecin, ST1481, in a large panel of human tumor xenografts. Clin Cancer Res. 2002;8:3904–3909. [PubMed] [Google Scholar]
- 260.De Cesare M, Pratesi G, Veneroni S, Bergottini R, Zunino F. Efficacy of the novel camptothecin gimatecan against orthotopic and metastatic human tumor xenograft models. Clin Cancer Res. 2004;10:7357–7364. doi: 10.1158/1078-0432.CCR-04-0962. [DOI] [PubMed] [Google Scholar]
- 261.Zhao Y, Lau LF, Dai X, Li B. In vitro and in vivo anticancer activity of gimatecan against hepatocellular carcinoma. Asian Pac J Cancer Prev. 2016;17:4853–4856. doi: 10.22034/APJCP.2016.17.11.4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Ulivi P, Zoli W, Fabbri F, Brigliadori G, Ricotti L, Tesei A, Rosetti M, De Cesare M, Beretta GL, Corna E, Supino R, Zunino F. Cellular basis of antiproliferative and antitumor activity of the novel camptothecin derivative, gimatecan, in bladder carcinoma models. Neoplasia. 2005;7:152–161. doi: 10.1593/neo.04397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Sessa C, Cresta S, Cerny T, Baselga J, Rota Caremoli E, Malossi A, Hess D, Trigo J, Zucchetti M, D’Incalci M, Zaniboni A, Capri G, Gatti B, Carminati P, Zanna C, Marsoni S, Gianni L. Concerted escalation of dose and dosing duration in a phase I study of the oral camptothecin gimatecan (ST1481) in patients with advanced solid tumors. Ann Oncol. 2007;18:561–568. doi: 10.1093/annonc/mdl418. [DOI] [PubMed] [Google Scholar]
- 264.Zhu AX, Ready N, Clark JW, Safran H, Amato A, Salem N, Pace S, He X, Zvereva N, Lynch TJ, Ryan DP, Supko JG. Phase I and pharmacokinetic study of gimatecan given orally once a week for 3 of 4 weeks in patients with advanced solid tumors. Clin Cancer Res. 2009;15:374–381. doi: 10.1158/1078-0432.CCR-08-1024. [DOI] [PubMed] [Google Scholar]
- 265.Frapolli R, Zucchetti M, Sessa C, Marsoni S, Vigano L, Locatelli A, Rulli E, Compagnoni A, Bello E, Pisano C, Carminati P, D’Incalci M. Clinical pharmacokinetics of the new oral camptothecin gimatecan: the inter-patient variability is related to alpha1-acid glycoprotein plasma levels. Eur J Cancer. 2010;46:505–516. doi: 10.1016/j.ejca.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 266.Pecorelli S, Ray-Coquard I, Tredan O, Colombo N, Parma G, Tisi G, Katsaros D, Lhomme C, Lissoni AA, Vermorken JB, du Bois A, Poveda A, Frigerio L, Barbieri P, Carminati P, Brienza S, Guastalla JP. Phase II of oral gimatecan in patients with recurrent epithelial ovarian, fallopian tube or peritoneal cancer, previously treated with platinum and taxanes. Ann Oncol. 2010;21:759–765. doi: 10.1093/annonc/mdp514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Hu J, Wen PY, Abrey LE, Fadul CE, Drappatz J, Salem N, Supko JG, Hochberg F. A phase II trial of oral gimatecan for recurrent glioblastoma. J Neurooncol. 2013;111:347–353. doi: 10.1007/s11060-012-1023-0. [DOI] [PubMed] [Google Scholar]
- 268.Lansiaux A, Facompre M, Wattez N, Hildebrand MP, Bal C, Demarquay D, Lavergne O, Bigg DC, Bailly C. Apoptosis induced by the homocamptothecin anticancer drug BN80915 in HL-60 cells. Mol Pharmacol. 2001;60:450–461. [PubMed] [Google Scholar]
- 269.Sparreboom A, Gelderblom H, Marsh S, Ahluwalia R, Obach R, Principe P, Twelves C, Verweij J, McLeod HL. Diflomotecan pharmacokinetics in relation to ABCG2 421C > A genotype. Clin Pharmacol Ther. 2004;76:38–44. doi: 10.1016/j.clpt.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 270.Scott L, Soepenberg O, Verweij J, de Jonge MJ, Th Planting AS, McGovern D, Principe P, Obach R, Twelves C. A multicentre phase I and pharmacokinetic study of BN80915 (diflomotecan) administered daily as a 20-min intravenous infusion for 5 days every 3 weeks to patients with advanced solid tumours. Ann Oncol. 2007;18:569–575. doi: 10.1093/annonc/mdl439. [DOI] [PubMed] [Google Scholar]
- 271.Graham JS, Falk S, Samuel LM, Cendros JM, Evans TR. A multi-centre dose-escalation and pharmacokinetic study of diflomotecan in patients with advanced malignancy. Cancer Chemother Pharmacol. 2009;63:945–952. doi: 10.1007/s00280-008-0795-6. [DOI] [PubMed] [Google Scholar]
- 272.Liao Z, Robey RW, Guirouilh-Barbat J, To KK, Polgar O, Bates SE, Pommier Y. Reduced expression of DNA topoisomerase I in SF295 human glioblastoma cells selected for resistance to homocamptothecin and diflomotecan. Mol Pharmacol. 2008;73:490–497. doi: 10.1124/mol.107.041178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Mangas-Sanjuan V, Buil-Bruna N, Garrido MJ, Soto E, Troconiz IF. Semimechanistic cellcycle type-based pharmacokinetic/pharmacodynamic model of chemotherapy-induced neutropenic effects of diflomotecan under different dosing schedules. J Pharmacol Exp Ther. 2015;354:55–64. doi: 10.1124/jpet.115.223776. [DOI] [PubMed] [Google Scholar]
- 274.Gelderblom H, Salazar R, Verweij J, Pentheroudakis G, de Jonge MJ, Devlin M, van Hooije C, Seguy F, Obach R, Prunonosa J, Principe P, Twelves C. Phase I pharmacological and bioavailability study of oral diflomotecan (BN80915), a novel E-ring-modified camptothecin analogue in adults with solid tumors. Clin Cancer Res. 2003;9:4101–4107. [PubMed] [Google Scholar]
- 275.Troconiz IF, Garrido MJ, Segura C, Cendros JM, Principe P, Peraire C, Obach R. Phase I dose-finding study and a pharmacokinetic/pharmacodynamic analysis of the neutropenic response of intravenous diflomotecan in patients with advanced malignant tumours. Cancer Chemother Pharmacol. 2006;57:727–735. doi: 10.1007/s00280-005-0112-6. [DOI] [PubMed] [Google Scholar]
- 276.Van Hattum AH, Pinedo HM, Schluper HM, Hausheer FH, Boven E. New highly lipophilic camptothecin BNP1350 is an effective drug in experimental human cancer. Int J Cancer. 2000;88:260–266. doi: 10.1002/1097-0215(20001015)88:2<260::aid-ijc18>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 277.Matsui S, Endo W, Wrzosek C, Haridas K, Seetharamulu P, Hausheer FH, Rustum YM. Characterisation of a synergistic interaction between a thymidylate synthase inhibitor, ZD1694, and a novel lipophilic topoisomerase I inhibitor karenitecin, BNP1100: mechanisms and clinical implications. Eur J Cancer. 1999;35:984–993. doi: 10.1016/s0959-8049(99)00018-0. [DOI] [PubMed] [Google Scholar]
- 278.Yin MB, Guo B, Vanhoefer U, Azrak RG, Minderman H, Frank C, Wrzosek C, Slocum HK, Rustum YM. Characterization of protein kinase chk1 essential for the cell cycle checkpoint after exposure of human head and neck carcinoma A253 cells to a novel topoisomerase I inhibitor BNP1350. Mol Pharmacol. 2000;57:453–459. doi: 10.1124/mol.57.3.453. [DOI] [PubMed] [Google Scholar]
- 279.Yin M, Hapke G, Guo B, Azrak RG, Frank C, Rustum YM. The Chk1-Cdc25C regulation is involved in sensitizing A253 cells to a novel topoisomerase I inhibitor BNP1350 by bax gene transfer. Oncogene. 2001;20:5249–5257. doi: 10.1038/sj.onc.1204686. [DOI] [PubMed] [Google Scholar]
- 280.Van Hattum AH, Schluper HM, Hausheer FH, Pinedo HM, Boven E. Novel camptothecin derivative BNP1350 in experimental human ovarian cancer: determination of efficacy and possible mechanisms of resistance. Int J Cancer. 2002;100:22–29. doi: 10.1002/ijc.10434. [DOI] [PubMed] [Google Scholar]
- 281.Yin MB, Hapke G, Wu J, Azrak RG, Frank C, Wrzosek C, Rustum YM. Chk1 signaling pathways that mediated G(2)M checkpoint in relation to the cellular resistance to the novel topoisomerase I poison BNP1350. Biochem Biophys Res Commun. 2002;295:435–444. doi: 10.1016/s0006-291x(02)00683-6. [DOI] [PubMed] [Google Scholar]
- 282.Daud AI, Dawson J, DeConti RC, Bicaku E, Marchion D, Bastien S, Hausheer FA 3rd, Lush R, Neuger A, Sullivan DM, Munster PN. Potentiation of a topoisomerase I inhibitor, karenitecin, by the histone deacetylase inhibitor valproic acid in melanoma: translational and phase I/II clinical trial. Clin Cancer Res. 2009;15:2479–2487. doi: 10.1158/1078-0432.CCR-08-1931. [DOI] [PubMed] [Google Scholar]
- 283.Rajesh D, Robins HI, Howard SP. Karenitecin (bnp1350) and flavopridol as radiosensitizers in malignant glioma. J Neurol Neuromedicine. 2016;1:1–10. doi: 10.29245/2572.942x/2016/6.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Daud A, Valkov N, Centeno B, Derderian J, Sullivan P, Munster P, Urbas P, Deconti RC, Berghorn E, Liu Z, Hausheer F, Sullivan D. Phase II trial of karenitecin in patients with malignant melanoma: clinical and translational study. Clin Cancer Res. 2005;11:3009–3016. doi: 10.1158/1078-0432.CCR-04-1722. [DOI] [PubMed] [Google Scholar]
- 285.Miller AA, Herndon JE 2nd, Gu L, Green MR Cancer and Leukemia Group B. Phase II trial of karenitecin in patients with relapsed or refractory non-small cell lung cancer (CALGB 30004) Lung Cancer. 2005;48:399–407. doi: 10.1016/j.lungcan.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 286.Grossman SA, Carson KA, Phuphanich S, Batchelor T, Peereboom D, Nabors LB, Lesser G, Hausheer F, Supko JG New Approaches to Brain Tumor Therapy CNS Consortium. Phase I and pharmacokinetic study of karenitecin in patients with recurrent malignant gliomas. Neuro Oncol. 2008;10:608–616. doi: 10.1215/15228517-2008-030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Kavanagh JJ, Sill MW, Ramirez PT, Warshal D, Pearl ML, Morgan MA. Phase II multicenter open-label study of karenitecin in previously treated epithelial ovarian and primary peritoneal cancer: a Gynecologic Oncology Group Study. Int J Gynecol Cancer. 2008;18:460–464. doi: 10.1111/j.1525-1438.2007.01053.x. [DOI] [PubMed] [Google Scholar]
- 288.Pollack IF, Erff M, Bom D, Burke TG, Strode JT, Curran DP. Potent topoisomerase I inhibition by novel silatecans eliminates glioma proliferation in vitro and in vivo. Cancer Res. 1999;59:4898–4905. [PubMed] [Google Scholar]
- 289.Bom D, Curran DP, Kruszewski S, Zimmer SG, Thompson Strode J, Kohlhagen G, Du W, Chavan AJ, Fraley KA, Bingcang AL, Latus LJ, Pommier Y, Burke TG. The novel silatecan 7-tert-butyldimethylsilyl-10-hydroxycamptothecin displays high lipophilicity, improved human blood stability, and potent anticancer activity. J Med Chem. 2000;43:3970–3980. doi: 10.1021/jm000144o. [DOI] [PubMed] [Google Scholar]
- 290.Bom D, Curran DP, Zhang J, Zimmer SG, Bevins R, Kruszewski S, Howe JN, Bingcang A, Latus LJ, Burke TG. The highly lipophilic DNA topoisomerase I inhibitor DB-67 displays elevated lactone levels in human blood and potent anticancer activity. J Control Release. 2001;74:325–333. doi: 10.1016/s0168-3659(01)00343-1. [DOI] [PubMed] [Google Scholar]
- 291.Bence AK, Mattingly CA, Burke TG, Adams VR. The effect of DB-67, a lipophilic camptothecin derivative, on topoisomerase I levels in non-small-cell lung cancer cells. Cancer Chemother Pharmacol. 2004;54:354–360. doi: 10.1007/s00280-004-0804-3. [DOI] [PubMed] [Google Scholar]
- 292.Lopez-Barcons LA, Zhang J, Siriwitayawan G, Burke TG, Perez-Soler R. The novel highly lipophilic topoisomerase I inhibitor DB67 is effective in the treatment of liver metastases of murine CT-26 colon carcinoma. Neoplasia. 2004;6:457–467. doi: 10.1593/neo.04139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Chen AY, Shih SJ, Garriques LN, Rothenberg ML, Hsiao M, Curran DP. Silatecan DB-67 is a novel DNA topoisomerase I-targeted radiation sensitizer. Mol Cancer Ther. 2005;4:317–324. [PubMed] [Google Scholar]
- 294.Yeh TK, Li CM, Chen CP, Chuu JJ, Huang CL, Wang HS, Shen CC, Lee TY, Chang CY, Chang CM, Chao YS, Lin CT, Chang JY, Chen CT. Antitumor activities and pharmacokinetics of silatecans DB-67 and DB-91. Pharmacol Res. 2010;61:108–115. doi: 10.1016/j.phrs.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 295.Pisano C, De Cesare M, Beretta GL, Zuco V, Pratesi G, Penco S, Vesci L, Fodera R, Ferrara FF, Guglielmi MB, Carminati P, Dallavalle S, Morini G, Merlini L, Orlandi A, Zunino F. Preclinical profile of antitumor activity of a novel hydrophilic camptothecin, ST1968. Mol Cancer Ther. 2008;7:2051–2059. doi: 10.1158/1535-7163.MCT-08-0266. [DOI] [PubMed] [Google Scholar]
- 296.Pisano C, Zuco V, De Cesare M, Benedetti V, Vesci L, Fodera R, Bucci F, Aulicino C, Penco S, Carminati P, Zunino F. Intracellular accumulation and DNA damage persistence as determinants of human squamous cell carcinoma hypersensitivity to the novel camptothecin ST1968. Eur J Cancer. 2008;44:1332–1340. doi: 10.1016/j.ejca.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 297.Zuco V, Benedetti V, Zunino F. ATM- and ATR-mediated response to DNA damage induced by a novel camptothecin, ST1968. Cancer Lett. 2010;292:186–196. doi: 10.1016/j.canlet.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 298.Zuco V, Supino R, Favini E, Tortoreto M, Cincinelli R, Croce AC, Bucci F, Pisano C, Zunino F. Efficacy of ST1968 (namitecan) on a topotecan-resistant squamous cell carcinoma. Biochem Pharmacol. 2010;79:535–541. doi: 10.1016/j.bcp.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 299.Cassinelli G, Zuco V, Petrangolini G, De Cesare M, Tortoreto M, Lanzi C, Cominetti D, Zaffaroni N, Orlandi A, Passeri D, Meco D, Di Francesco AM, Riccardi R, Bucci F, Pisano C, Zunino F. The curative efficacy of namitecan (ST1968) in preclinical models of pediatric sarcoma is associated with antiangiogenic effects. Biochem Pharmacol. 2012;84:163–171. doi: 10.1016/j.bcp.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 300.Meco D, Di Francesco AM, Cusano G, Bucci F, Pierri F, Patriarca V, Torella AR, Pisano C, Riccardi R. Preclinical evaluation of the novel 7-substituted camptothecin Namitecan (ST1968) in paediatric tumour models. Cancer Chemother Pharmacol. 2012;70:811–822. doi: 10.1007/s00280-012-1973-0. [DOI] [PubMed] [Google Scholar]
- 301.De Cesare M, Lauricella C, Veronese SM, Cominetti D, Pisano C, Zunino F, Zaffaroni N, Zuco V. Synergistic antitumor activity of cetuximab and namitecan in human squamous cell carcinoma models relies on cooperative inhibition of EGFR expression and depends on high EGFR gene copy number. Clin Cancer Res. 2014;20:995–1006. doi: 10.1158/1078-0432.CCR-13-1684. [DOI] [PubMed] [Google Scholar]
- 302.Joerger M, Hess D, Delmonte A, Gallerani E, Barbieri P, Pace S, Sessa C. Phase-I dose finding and pharmacokinetic study of the novel hydrophilic camptothecin ST-1968 (namitecan) in patients with solid tumors. Invest New Drugs. 2015;33:472–479. doi: 10.1007/s10637-015-0219-5. [DOI] [PubMed] [Google Scholar]
- 303.Lavergne O, Harnett J, Rolland A, Lanco C, Lesueur-Ginot L, Demarquay D, Huchet M, Coulomb H, Bigg DC. BN 80927: a novel homocamptothecin with inhibitory activities on both topoisomerase I and topoisomerase II. Bioorg Med Chem Lett. 1999;9:2599–2602. doi: 10.1016/s0960-894x(99)00428-x. [DOI] [PubMed] [Google Scholar]
- 304.Demarquay D, Huchet M, Coulomb H, Lesueur-Ginot L, Lavergne O, Camara J, Kasprzyk PG, Prevost G, Bigg DC. BN80927: a novel homocamptothecin that inhibits proliferation of human tumor cells in vitro and in vivo. Cancer Res. 2004;64:4942–4949. doi: 10.1158/0008-5472.CAN-03-3872. [DOI] [PubMed] [Google Scholar]
- 305.Troconiz IF, Cendros JM, Soto E, Prunonosa J, Perez-Mayoral A, Peraire C, Principe P, Delavault P, Cvitkovic F, Lesimple T, Obach R. Population pharmacokinetic/pharmacodynamic modeling of drug-induced adverse effects of a novel homocamptothecin analog, elomotecan (BN80927), in a Phase I dose finding study in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2012;70:239–250. doi: 10.1007/s00280-012-1906-y. [DOI] [PubMed] [Google Scholar]
- 306.Upreti VV, Mamidi RN, Katneni K, Srinivas NR. Quantitative determination of DRF-1042 in human plasma by HPLC: validation and application in clinical pharmacokinetics. Biomed Chromatogr. 2003;17:385–390. doi: 10.1002/bmc.253. [DOI] [PubMed] [Google Scholar]
- 307.Chatterjee A, Digumarti R, Mamidi RN, Katneni K, Upreti VV, Surath A, Srinivas ML, Uppalapati S, Jiwatani S, Subramaniam S, Srinivas NR. Safety, tolerability, pharmacokinetics, and pharmacodynamics of an orally active novel camptothecin analog, DRF-1042, in refractory cancer patients in a phase I dose escalation study. J Clin Pharmacol. 2004;44:723–736. doi: 10.1177/0091270004265647. [DOI] [PubMed] [Google Scholar]
- 308.Chatterjee A, Digumarti R, Katneni K, Upreti VV, Mamidi RN, Mullangi R, Surath A, Srinivas ML, Uppalapati S, Jiwatani S, Srinivas NR. Safety, tolerability, and pharmacokinetics of a capsule formulation of DRF-1042, a novel camptothecin analog, in refractory cancer patients in a bridging phase I study. J Clin Pharmacol. 2005;45:453–460. doi: 10.1177/0091270004270225. [DOI] [PubMed] [Google Scholar]
- 309.Bissett D, Cassidy J, de Bono JS, Muirhead F, Main M, Robson L, Fraier D, Magne ML, Pellizzoni C, Porro MG, Spinelli R, Speed W, Twelves C. Phase I and pharmacokinetic (PK) study of MAG-CPT (PNU 166148): a polymeric derivative of camptothecin (CPT) Br J Cancer. 2004;91:50–55. doi: 10.1038/sj.bjc.6601922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Schoemaker NE, van Kesteren C, Rosing H, Jansen S, Swart M, Lieverst J, Fraier D, Breda M, Pellizzoni C, Spinelli R, Grazia Porro M, Beijnen JH, Schellens JH, ten Bokkel Huinink WW. A phase I and pharmacokinetic study of MAG-CPT, a water-soluble polymer conjugate of camptothecin. Br J Cancer. 2002;87:608–614. doi: 10.1038/sj.bjc.6600516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Sarapa N, Britto MR, Speed W, Jannuzzo M, Breda M, James CA, Porro M, Rocchetti M, Wanders A, Mahteme H, Nygren P. Assessment of normal and tumor tissue uptake of MAG-CPT, a polymer-bound prodrug of camptothecin, in patients undergoing elective surgery for colorectal carcinoma. Cancer Chemother Pharmacol. 2003;52:424–430. doi: 10.1007/s00280-003-0685-x. [DOI] [PubMed] [Google Scholar]
- 312.Slingerland M, Gelderblom H. The fate of camptothecin glycoconjugate: report of a clinical hold during a phase II study of BAY 56-3722 (formerly BAY 38-3441), in patients with recurrent or metastatic colorectal cancer resistant/refractory to irinotecan. Invest New Drugs. 2012;30:1208–1210. doi: 10.1007/s10637-011-9679-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Mross K, Richly H, Schleucher N, Korfee S, Tewes M, Scheulen ME, Seeber S, Beinert T, Schweigert M, Sauer U, Unger C, Behringer D, Brendel E, Haase CG, Voliotis D, Strumberg D. A phase I clinical and pharmacokinetic study of the camptothecin glycoconjugate, BAY 38-3441, as a daily infusion in patients with advanced solid tumors. Ann Oncol. 2004;15:1284–1294. doi: 10.1093/annonc/mdh313. [DOI] [PubMed] [Google Scholar]
- 314.Chen EX, Batist G, Siu LL, Bangash N, Maclean M, McIntosh L, Miller WH Jr, Oza AM, Lathia C, Petrenciuc O, Seymour L. Phase I and pharmacokinetic study of Bay 38-3441, a camptothecin glycoconjugate, administered as a 30-minute infusion daily for five days every 3 weeks in patients with advanced solid malignancies. Invest New Drugs. 2005;23:455–465. doi: 10.1007/s10637-005-2905-1. [DOI] [PubMed] [Google Scholar]
- 315.Cheng J, Khin KT, Jensen GS, Liu A, Davis ME. Synthesis of linear, beta-cyclodextrinbased polymers and their camptothecin conjugates. Bioconjug Chem. 2003;14:1007–1017. doi: 10.1021/bc0340924. [DOI] [PubMed] [Google Scholar]
- 316.Schluep T, Cheng J, Khin KT, Davis ME. Pharmacokinetics and biodistribution of the camptothecin-polymer conjugate IT-101 in rats and tumor-bearing mice. Cancer Chemother Pharmacol. 2006;57:654–662. doi: 10.1007/s00280-005-0091-7. [DOI] [PubMed] [Google Scholar]
- 317.Schluep T, Hwang J, Cheng J, Heidel JD, Bartlett DW, Hollister B, Davis ME. Preclinical efficacy of the camptothecin-polymer conjugate IT-101 in multiple cancer models. Clin Cancer Res. 2006;12:1606–1614. doi: 10.1158/1078-0432.CCR-05-1566. [DOI] [PubMed] [Google Scholar]
- 318.Numbenjapon T, Wang J, Colcher D, Schluep T, Davis ME, Duringer J, Kretzner L, Yen Y, Forman SJ, Raubitschek A. Preclinical results of camptothecin-polymer conjugate (IT-101) in multiple human lymphoma xenograft models. Clin Cancer Res. 2009;15:4365–4373. doi: 10.1158/1078-0432.CCR-08-2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Gaur S, Chen L, Yen T, Wang Y, Zhou B, Davis M, Yen Y. Preclinical study of the cyclodextrinpolymer conjugate of camptothecin CRLX101 for the treatment of gastric cancer. Nanomedicine. 2012;8:721–730. doi: 10.1016/j.nano.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 320.Conley SJ, Baker TL, Burnett JP, Theisen RL, Lazarus D, Peters CG, Clouthier SG, Eliasof S, Wicha MS. CRLX101, an investigational camptothecin-containing nanoparticle-drug conjugate, targets cancer stem cells and impedes resistance to antiangiogenic therapy in mouse models of breast cancer. Breast Cancer Res Treat. 2015;150:559–567. doi: 10.1007/s10549-015-3349-8. [DOI] [PubMed] [Google Scholar]
- 321.Tian X, Nguyen M, Foote HP, Caster JM, Roche KC, Peters CG, Wu P, Jayaraman L, Garmey EG, Tepper JE, Eliasof S, Wang AZ. CRLX101, a nanoparticle-drug conjugate containing camptothecin, improves rectal cancer chemoradiotherapy by inhibiting DNA repair and HIF1alpha. Cancer Res. 2017;77:112–122. doi: 10.1158/0008-5472.CAN-15-2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Pham E, Birrer MJ, Eliasof S, Garmey EG, Lazarus D, Lee CR, Man S, Matulonis UA, Peters CG, Xu P, Krasner C, Kerbel RS. Translational impact of nanoparticle-drug conjugate CRLX101 with or without bevacizumab in advanced ovarian cancer. Clin Cancer Res. 2015;21:808–818. doi: 10.1158/1078-0432.CCR-14-2810. [DOI] [PubMed] [Google Scholar]
- 323.Clark AJ, Wiley DT, Zuckerman JE, Webster P, Chao J, Lin J, Yen Y, Davis ME. CRLX101 nanoparticles localize in human tumors and not in adjacent, nonneoplastic tissue after intravenous dosing. Proc Natl Acad Sci U S A. 2016;113:3850–3854. doi: 10.1073/pnas.1603018113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Lin CJ, Lin YL, Luh F, Yen Y, Chen RM. Preclinical effects of CRLX101, an investigational camptothecin-containing nanoparticle drug conjugate, on treating glioblastoma multiforme via apoptosis and antiangiogenesis. Oncotarget. 2016;7:42408–42421. doi: 10.18632/oncotarget.9878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Pham E, Yin M, Peters CG, Lee CR, Brown D, Xu P, Man S, Jayaraman L, Rohde E, Chow A, Lazarus D, Eliasof S, Foster FS, Kerbel RS. Preclinical efficacy of bevacizumab with CRLX101, an investigational nanoparticle-drug conjugate, in treatment of metastatic triple-negative breast cancer. Cancer Res. 2016;76:4493–4503. doi: 10.1158/0008-5472.CAN-15-3435. [DOI] [PubMed] [Google Scholar]
- 326.Keefe SM, Hoffman-Censits J, Cohen RB, Mamtani R, Heitjan D, Eliasof S, Nixon A, Turnbull B, Garmey EG, Gunnarsson O, Waliki M, Ciconte J, Jayaraman L, Senderowicz A, Tellez AB, Hennessy M, Piscitelli A, Vaughn D, Smith A, Haas NB. Efficacy of the nanoparticledrug conjugate CRLX101 in combination with bevacizumab in metastatic renal cell carcinoma: results of an investigator-initiated phase IIIa clinical trial. Ann Oncol. 2016;27:1579–1585. doi: 10.1093/annonc/mdw188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Voss MH, Hussain A, Vogelzang N, Lee JL, Keam B, Rha SY, Vaishampayan U, Harris WB, Richey S, Randall JM, Shaffer D, Cohn A, Crowell T, Li J, Senderowicz A, Stone E, Figlin R, Motzer RJ, Haas NB, Hutson T. A randomized phase 2 trial of CRLX101 in combination with bevacizumab versus standard of care in patients with advanced renal cell carcinoma. Ann Oncol. 2017;28:2754–2760. doi: 10.1093/annonc/mdx493. [DOI] [PubMed] [Google Scholar]
- 328.Weiss GJ, Chao J, Neidhart JD, Ramanathan RK, Bassett D, Neidhart JA, Choi CH, Chow W, Chung V, Forman SJ, Garmey E, Hwang J, Kalinoski DL, Koczywas M, Longmate J, Melton RJ, Morgan R, Oliver J, Peterkin JJ, Ryan JL, Schluep T, Synold TW, Twardowski P, Davis ME, Yen Y. First-in-human phase 1/2a trial of CRLX101, a cyclodextrin-containing polymercamptothecin nanopharmaceutical in patients with advanced solid tumor malignancies. Invest New Drugs. 2013;31:986–1000. doi: 10.1007/s10637-012-9921-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Okuno S, Harada M, Yano T, Yano S, Kiuchi S, Tsuda N, Sakamura Y, Imai J, Kawaguchi T, Tsujihara K. Complete regression of xenografted human carcinomas by camptothecin analogue-carboxymethyl dextran conjugate (T-0128) Cancer Res. 2000;60:2988–2995. [PubMed] [Google Scholar]
- 330.Fujita F, Koike M, Fujita M, Sakamoto Y, Okuno S, Kawaguchi T, Yano S, Yano T, Kiuchi S, Fujiwara T, Kudoh S, Kakushima M. MEN4901/T-0128, a new camptothecin derivative-carboxymethyldextran conjugate, has potent antitumor activities in a panel of human tumor xenografts in nude mice. Clin Cancer Res. 2005;11:1650–1657. doi: 10.1158/1078-0432.CCR-04-1756. [DOI] [PubMed] [Google Scholar]
- 331.Bigioni M, Parlani M, Bressan A, Bellarosa D, Rivoltini L, Animati F, Crea A, Bugianesi R, Maggi CA, Manzini S, Binaschi M. Antitumor activity of delimotecan against human metastatic melanoma: pharmacokinetics and molecular determinants. Int J Cancer. 2009;125:2456–2464. doi: 10.1002/ijc.24661. [DOI] [PubMed] [Google Scholar]
- 332.Harada M, Imai J, Okuno S, Suzuki T. Macrophage-mediated activation of camptothecin analogue T-2513-carboxymethyl dextran conjugate (T-0128): possible cellular mechanism for antitumor activity. J Control Release. 2000;69:389–397. doi: 10.1016/s0168-3659(00)00320-5. [DOI] [PubMed] [Google Scholar]
- 333.Binaschi M, Parlani M, Bellarosa D, Bigioni M, Salvatore C, Palma C, Crea A, Maggi CA, Manzini S, Goso C. Human and murine macrophages mediate activation of MEN 4901/T-0128: a new promising camptothecin analogue-polysaccharide conjugate. Anticancer Drugs. 2006;17:1119–1126. doi: 10.1097/01.cad.0000236307.20339.b4. [DOI] [PubMed] [Google Scholar]
- 334.Ma H, Li X, Yang Z, Okuno S, Kawaguchi T, Yagi S, Bouvet M, Hoffman RM. High antimetastatic efficacy of MEN4901/T-0128, a novel camptothecin carboxymethyldextran conjugate. J Surg Res. 2011;171:684–690. doi: 10.1016/j.jss.2010.05.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Veltkamp SA, Witteveen EO, Capriati A, Crea A, Animati F, Voogel-Fuchs M, van den Heuvel IJ, Beijnen JH, Voest EE, Schellens JH. Clinical and pharmacologic study of the novel prodrug delimotecan (MEN 4901/T-0128) in patients with solid tumors. Clin Cancer Res. 2008;14:7535–7544. doi: 10.1158/1078-0432.CCR-08-0438. [DOI] [PubMed] [Google Scholar]
- 336.Song MG, Gao SM, Du KM, Xu M, Yu Y, Zhou YH, Wang Q, Chen Z, Zhu YS, Chen GQ. Nanomolar concentration of NSC606985, a camptothecin analog, induces leukemic-cell apoptosis through protein kinase Cdelta-dependent mechanisms. Blood. 2005;105:3714–3721. doi: 10.1182/blood-2004-10-4011. [DOI] [PubMed] [Google Scholar]
- 337.Liu W, Zhu YS, Guo M, Yu Y, Chen GQ. Therapeutic efficacy of NSC606985, a novel camptothecin analog, in a mouse model of acute promyelocytic leukemia. Leuk Res. 2007;31:1565–1574. doi: 10.1016/j.leukres.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 338.Yu Y, Wang LS, Shen SM, Xia L, Zhang L, Zhu YS, Chen GQ. Subcellular proteome analysis of camptothecin analogue NSC606985-treated acute myeloid leukemic cells. J Proteome Res. 2007;6:3808–3818. doi: 10.1021/pr0700100. [DOI] [PubMed] [Google Scholar]
- 339.Tan C, Cai LQ, Wu W, Qiao Y, Imperato-McGinley J, Chen GQ, Zhu YS. NSC606985, a novel camptothecin analog, induces apoptosis and growth arrest in prostate tumor cells. Cancer Chemother Pharmacol. 2009;63:303–312. doi: 10.1007/s00280-008-0740-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Zhang N, Zhang H, Xia L, Zheng Y, Yu Y, Zhu Y, Chen G, Di W. NSC606985 induces apoptosis, exerts synergistic effects with cisplatin, and inhibits hypoxia-stabilized HIF-1alpha protein in human ovarian cancer cells. Cancer Lett. 2009;278:139–144. doi: 10.1016/j.canlet.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 341.Wang L, Fu P, Zhao Y, Wang G, Yu R, Wang X, Tang Z, Imperato-McGinley J, Zhu YS. Dissociation of NSC606985 induces atypical ERstress and cell death in prostate cancer cells. Int J Oncol. 2016;49:529–538. doi: 10.3892/ijo.2016.3555. [DOI] [PubMed] [Google Scholar]
- 342.Huang M, Gao H, Chen Y, Zhu H, Cai Y, Zhang X, Miao Z, Jiang H, Zhang J, Shen H, Lin L, Lu W, Ding J. Chimmitecan, a novel 9-substituted camptothecin, with improved anticancer pharmacologic profiles in vitro and in vivo. Clin Cancer Res. 2007;13:1298–1307. doi: 10.1158/1078-0432.CCR-06-1277. [DOI] [PubMed] [Google Scholar]
- 343.Hu Z, Sun Y, Du F, Niu W, Xu F, Huang Y, Li C. Accurate determination of the anticancer prodrug simmitecan and its active metabolite chimmitecan in various plasma samples based on immediate deactivation of blood carboxylesterases. J Chromatogr A. 2011;1218:6646–6653. doi: 10.1016/j.chroma.2011.07.042. [DOI] [PubMed] [Google Scholar]
- 344.Hu ZY, Li XX, Du FF, Yang JL, Niu W, Xu F, Wang FQ, Li C, Sun Y. Pharmacokinetic evaluation of the anticancer prodrug simmitecan in different experimental animals. Acta Pharmacol Sin. 2013;34:1437–1448. doi: 10.1038/aps.2013.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Homsi J, Simon GR, Garrett CR, Springett G, De Conti R, Chiappori AA, Munster PN, Burton MK, Stromatt S, Allievi C, Angiuli P, Eisenfeld A, Sullivan DM, Daud AI. Phase I trial of poly-Lglutamate camptothecin (CT-2106) administered weekly in patients with advanced solid malignancies. Clin Cancer Res. 2007;13:5855–5861. doi: 10.1158/1078-0432.CCR-06-2821. [DOI] [PubMed] [Google Scholar]
- 346.Li J, Ouyang Y, Zhang X, Zhou W, Wang F, Huang Z, Wang X, Chen Y, Zhang H, Fu L. Effect of HM910, a novel camptothecin derivative, on the inhibition of multiple myeloma cell growth in vitro and in vivo. Am J Cancer Res. 2015;5:1000–1016. [PMC free article] [PubMed] [Google Scholar]
- 347.Wu D, Shi W, Zhao J, Wei Z, Chen Z, Zhao D, Lan S, Tai J, Zhong B, Yu H. Assessment of the chemotherapeutic potential of a new camptothecin derivative, ZBH-1205. Arch Biochem Biophys. 2016;604:74–85. doi: 10.1016/j.abb.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 348.You J, Chen Y, Mohamed Alsayeh ZM, Shen X, Li C, Zhao P, Chen F, Liu Y, Xu C. Nanocrystals of a new camptothecin derivative WCN-21 enhance its solubility and efficacy. Oncotarget. 2017;8:29808–29822. doi: 10.18632/oncotarget.16159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Li DZ, Zhang QZ, Wang CY, Zhang YL, Li XY, Huang JT, Liu HY, Fu ZD, Song HX, Lin JP, Ji TF, Pan XD. Synthesis and antitumor activity of novel substituted uracil-1’(N)-acetic acid ester derivatives of 20(S)-camptothecins. Eur J Med Chem. 2017;125:1235–1246. doi: 10.1016/j.ejmech.2016.11.013. [DOI] [PubMed] [Google Scholar]
- 350.Liu XP, Zhou ST, Li XY, Chen XC, Zhao X, Qian ZY, Zhou LN, Li ZY, Wang YM, Zhong Q, Yi T, He X, Wei YQ. Anti-tumor activity of N-trimethyl chitosan-encapsulated camptothecin in a mouse melanoma model. J Exp Clin Cancer Res. 2010;29:76. doi: 10.1186/1756-9966-29-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Sooryakumar D, Dexheimer TS, Teicher BA, Pommier Y. Molecular and cellular pharmacology of the novel noncamptothecin topoisomerase I inhibitor Genz-644282. Mol Cancer Ther. 2011;10:1490–1499. doi: 10.1158/1535-7163.MCT-10-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Kurtzberg LS, Roth S, Krumbholz R, Crawford J, Bormann C, Dunham S, Yao M, Rouleau C, Bagley RG, Yu XJ, Wang F, Schmid SM, Lavoie EJ, Teicher BA. Genz-644282, a novel noncamptothecin topoisomerase I inhibitor for cancer treatment. Clin Cancer Res. 2011;17:2777–2787. doi: 10.1158/1078-0432.CCR-10-0542. [DOI] [PubMed] [Google Scholar]
- 353.Houghton PJ, Lock R, Carol H, Morton CL, Gorlick R, Anders Kolb E, Keir ST, Reynolds CP, Kang MH, Maris JM, Billups CA, Zhang MX, Madden SL, Teicher BA, Smith MA. Testing of the topoisomerase 1 inhibitor Genz-644282 by the pediatric preclinical testing program. Pediatr Blood Cancer. 2012;58:200–209. doi: 10.1002/pbc.23016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Dong G, Fang Y, Liu Y, Liu N, Wu S, Zhang W, Sheng C. Design, synthesis and evaluation of 4-substituted anthra [2, 1-c] [1, 2, 5] thiadiazole-6, 11-dione derivatives as novel non-camptothecin topoisomerase I inhibitors. Bioorg Med Chem Lett. 2017;27:1929–1933. doi: 10.1016/j.bmcl.2017.03.039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.