

Abstract
The clinical management of pediatric acute lymphoblastic leukemia (ALL) is still changeling and identification of agents that can sensitize standard chemotherapy is needed for its better management. In this work, we demonstrate that tigecycline, a FDA-approved antibiotic, is an attractive candidate for ALL treatment. Tigecycline inhibits growth and induces apoptosis of multiple ALL cell lines. Compared to normal hematopoietic cells, tigecycline is more active against primary lymphocytes and CD34 progenitors from ALL patients through decreasing survival and clonogenic growth. Notably, tigecycline significantly augments the efficacy of chemotherapeutic drugs, such as doxorubicin and vincristine, in ALL cell lines, primary samples and xenograft mouse model. Tigecycline acts on ALL via inhibiting mitochondrial respiration, leading to energy crisis and oxidative stress and damage. We show that the enhanced mitochondrial biogenesis and increased oxygen consumption rate in ALL versus normal hematopoietic cells are important for their different sensitivity to tigecycline. We further show that ATP production and growth rate are largely affected in mitochondrial respiration-deficient ρ0 ALL cells. Our work provides pre-clinical evidence for repurposing tigecycline for ALL treatment and highlights the therapeutic value of targeting mitochondrial metabolism in sensitizing ALL to chemotherapy.
Keywords: Tigecycline, acute lymphoblastic leukemia, mitochondria respiration
Introduction
Acute lymphoblastic leukemia (ALL), characterized by the infiltration of bone marrow, blood, and extra medullary location by malignant cells of the lymphoid lineage is the most common hematological malignance in children [1]. ALL is genetically and clinically heterogeneous, and the mechanisms involved in its progression and resistance are complex [2]. The clinical management of ALL is still challenging and the prognosis is poor, particularly in patients with T cell ALL [3]. Further research is warranted into developing novel therapeutic strategies that can either act independently or in combination with standard therapeutic agents for the treatment of ALL. Over the recent years, there has been developing interest in targeting mitochondrial metabolism in blood cancers. Studies have shown that inhibiting mitochondrial respiration by genetic approach or pharmacological inhibitors are effective in eliminating leukemia stem and bulk cells without affecting normal hematopoietic cells, including acute myeloid leukemia and chronic myeloid leukemia [4,5]. Leukemia-associated changes in mitochondrial biogenesis and increased oxygen consumption in leukemia versus normal hematopoietic cells have been found and demonstrated to be associated with the preferential toxicity of mitochondrial inhibitors in leukemia [4].
Tigecycline is a broad spectrum antibiotic that binds to ribosome and thus suppresses protein synthesis in bacteria [6,7]. It also suppresses mitochondrial protein translation, leading to mitochondrial dysfunction and subsequent cell death in leukaemia and renal cancer with minimal toxicity in normal counterparts [4,8]. Apart from inducing mitochondrial dysfunction, tigecycline has been demonstrated to inhibit Wnt/β-catenin and induce autophagy in cervical and breast cancer [9,10]. The anti-cancer activities of tigecycline has been recently revealed in various studies.
In this work, we investigated the in vitro and in vivo efficacy of tigecycline as drug alone and its combination with approved ALL chemotherapeutic drugs on multiple cell lines, patients samples and xenograft mouse model of human ALL, and analysed the underlying mechanism of tigecycline focusing on mitochondrial functions.
Materials and methods
Cell lines, primary cells and agents
CCRF-CEM (ATCC, US), DND-41 and MOLT (DMSZ, Germany) were cultured in RPMI 1640 medium supplemented with 10% FBS (Invitrogen, US) and 100 U/mL penicillin-streptomycin (Thermo Fisher Scientific, US). CCRF-CEM ρ0 cells were generated by culturing parental cells in the presence of 2 µg/ml ethidium bromide (EtBr), 50 µg/ml uridine and 1 mM sodium pyruvate (Sigma, US) for 8 weeks, and thereafter maintained in above media without EtBr [11]. Normal bone marrow (NBM) CD34 and normal peripheral blood mononuclear cell (PBMC) were purchased form Stem cell Technologies Inc. US. ALL CD34 cells and ALL lymphocytes were isolated from bone marrow or peripheral blood of ALL patients using CD34 MicroBead kit and HLA B/T Cell Isolation Kit (Miltenyi Biotec, Germany), respectively. ALL bone marrow or peripheral blood were obtained from patients seen at the Zhongnan Hospital of Wuhan University. Written informed consent was obtained from all patients under institutional review board-approved protocols. CD34 cells were cultured in StemPro-34 SFM complete medium (Life technologies, US) supplemented with cytokines and growth factors as previously described [12]. Lymphocytes were cultured in LGM-3 Lymphocyte Growth Medium-3 supplemented with 20 ng/ml human IL-2. Tigecycline, vincristine and doxorubicin were purchased from Sigma and dissolved in DMSO.
MTS proliferation and colony-forming cell assays
ALL cell lines (5000/well in 96-well plate) were treated with drug alone or combination for 3 days, followed by measuring proliferation using MTS Cell Proliferation Colorimetri Assay Kit (BioVision, US). Primary CD34 cells (2000/petri dish) together with drugs were plated in HSC complete methylcellulose medium (StemCell Technologies, US), and colonies were manually counted after 12-14 days.
Annexin V labelling and flow cytometry
Cells (100,000/well in 12-well plate) were treated with drug alone or combination for 3 days. Cells were then stained with Annexin V-FITC and 7-AAD (BD Pharmingen, US) and analysed by flow cytometry (Beckman Coulter FC500).
Mito stress and glycolytic stress test assays
Cells (20,000/well in 24-well plate) were treated with drug for 24 h. Oxygen Consumption Rate (OCR) or Extracellular Acidification Rate (ECAR) was measured at 37°C in a CO2-free environment on a Seahorse XF24 extracellular flux analyser (Seahorse Bioscience, US) according to Seahorse Bioscience protocols.
Metabolic assays
Cells (100,000/well in 24-well plate) were treated with drug for 24 h. ATP levels were measured by ATPlite Luminiescent Assay kit (Perkin Elmer, US). ROS levels were measured by flow cytometry of CM-H2DCFDA (Life Technologies, US) staining.
Mitochondrial content and membrane potential
Genomic DNA was isolated from primary cells using the DNeasy Blood and Tissue kit (Qiagen, US). Mitochondrial DNA (mtDNA) copy number was measured by a real-time PCR, and compared relative to nuclear DNA (HGB-1). The sequences of primers for PCR were the same as previously reported [4]. Mitochondrial potential and mass were measured by flow cytometry of 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethyl benzimidazolylcarbocyanine iodide (JC-1, Invitrogen) and Mitotracker Green (Invitrogen, US) staining, respectively.
Measurement of oxidative DNA and protein damage
Cells (100,000/well in 12-well plate) were treated with drug for 24 h. DNA was extracted using the DNEasy Mini Kit (Qiagen, US). Oxidative DNA damage was measured by quantifying 8-hydroxy-2’-deoxyguanosine (8-OHdG) levels using the OxiSelect Oxidative DNA Damage ELISA Kit (Cell Biolabs). Oxidative protein damage was measured by quantifying protein carbonylation levels using the Protein Carbonyl ELISA Kit (Enzo LifeSciences).
ALL xenograft in SCID mouse
Animal experiments were approved by the Institutional Animal Care and Use Committee of Wuhan University. One million CCRF-CEM human ALL cells were injected subcutaneously into the flank of 6 weeks old SCID/NOD mice (Shanghai Experimental Animal Center, Chinese Academy of Sciences). Following development of palpable tumors (~7 days after cell injection), the mice were treated with vehicle (20%/80% DMSO/saline, n=10 per group), intraperitoneal tigecycline, doxorubicin (0.5 mg/kg, three times per week), vincristine (0.5 mg/kg, three times per week) or combination. Tumour size was calculated every two days based on caliper measurements of tumor length and width using the formula: (width)2 × length/2.
Statistical analyses
The data are expressed as mean and standard deviation. Statistical analyses were performed by unpaired Student’s t test. Values were considered statistically significant at P<0.05.
Results
Tigecycline effectively targets multiple pediatric ALL cell lines
To determine the effects of tigecycline in pediatric ALL, we examined the growth and survival after tigecycline treatment on a broader spectrum of cell lines derived from pediatric ALL patients: CCRF-CEM, DND-41 and MOLT-16. There were chosen in our experimental models for malignant T cells because they represent different cellular origin and display heterogeneity, including immunophenotyping and differential pattern [13]. We show that tigecycline significantly inhibits proliferation in ALL cell lines in a dose-dependent manner (Figure 1A). IC50 values of tigecycline in inhibiting growth range from 5 to 10 µM in various ALL cell lines. Tigecycline also induced ALL apoptosis as assessed by flow cytometry of Annexin V/7-AAD (Figure 1B and 1C).
Figure 1.
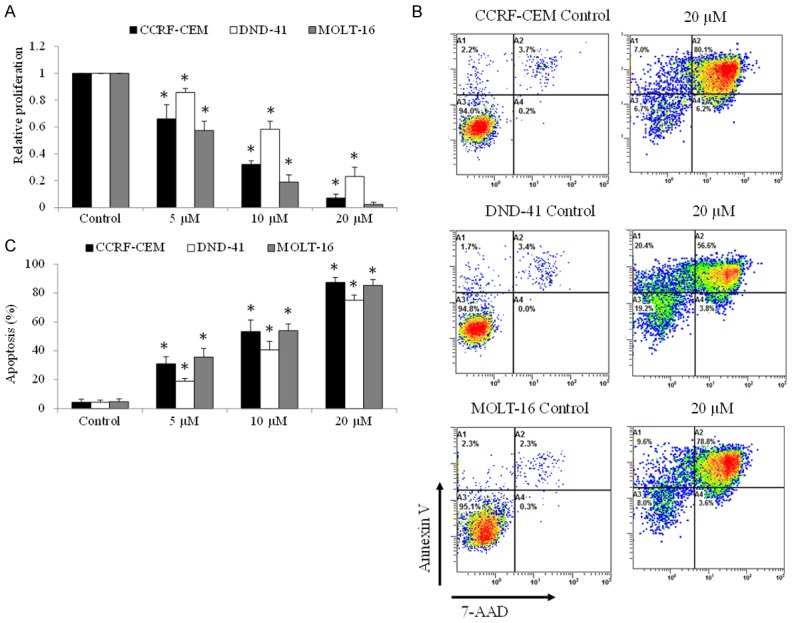
Tigecycline significantly inhibits proliferation and induces apoptosis in pediatric ALL cells. A. Tigecycline at 5, 10 and 20 µM inhibits proliferation of ALL cells as assessed by MTS proliferation assay. B. Representative flow cytometry dot plots showing the percentage of Annexin V and 7-AAD staining in ALL cells exposed to control or 20 µM tigecycline. C. Tigecycline significantly induces apoptosis of ALL cells in a concentration-dependent manner. Three human ALL cell lines were tested: CCRF-CEM, DND-41 and MOLT-16. *P<0.05, compared to control.
Tigecycline targets primary ALL malignant cells more effectively than normal hematopoietic cells
We next examined the effects of tigecycline on primary CD34 and lymphoma cells from 15 pediatric ALL patients with either newly diagnosed (n=10) or treatment-refractory disease (n=5). We found that tigecycline dose-dependently induced apoptosis in primary ALL CD34+ progenitors and lymphoma cells (Figure 2A). In addition, tigecycline significantly inhibited ALL CD34+ progenitors clonogenic growth on methylcellulose matrix (Figure 2B and 2C). To compare the effect of tigecycline on normal hematopoietic cells, the same experiments were performed on normal bone marrow (NBM) CD34+ progenitors and peripheral blood mononuclear cells (PBMC). Of note, both subsets of normal hematopoietic cells were significantly less sensitive to tigecycline (Figure 2).
Figure 2.
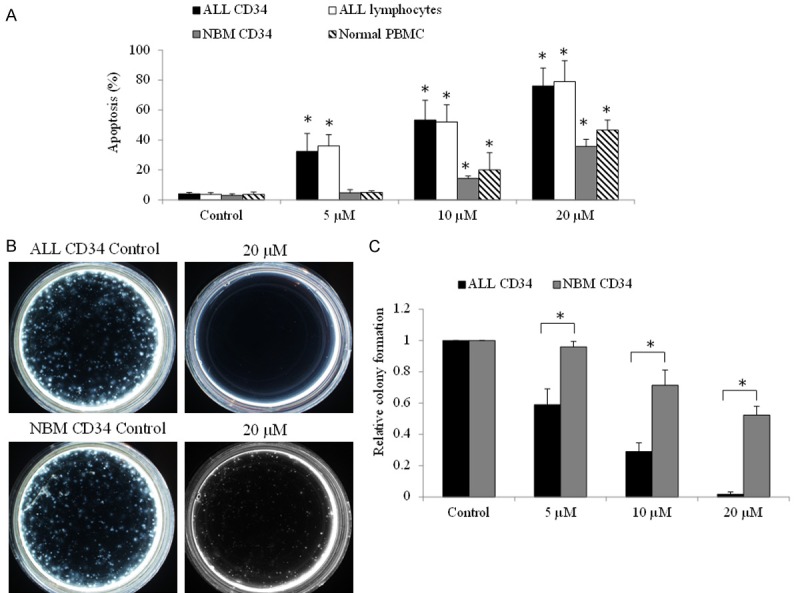
Tigecycline selectively targets pediatric ALL patient cells. Tigecycline induces apoptosis (A) and decrease colony formation (B and C) of pediatric ALL lymphoma and CD34 cells (n=15), and in a significantly greater extent than in normal lymphoma and hematopoietic CD34 cells (n=5). Representative colony formation photos were taken at 2 weeks after seeding cells on methylcellulose. *P<0.05, compared to normal control.
Tigecycline significantly augments the efficacy of standard ALL chemotherapeutic drugs in ALL cell lines and patient samples
To investigate the translational potential of tigecycline in ALL, we tested the combinatory efficacy of tigecycline and commonly used ALL chemotherapeutic drugs: doxorubicin and vincristine. The concentration of each drug we used for combination studies is the concentration that induces sublethal inhibitory effects. We found that the combination of tigecycline with doxorubicin or vincristine is significantly more effective than single drug alone (Figure 3A and 3B). Almost complete inhibition of growth and survival in ALL cell lines were achieved in the combination (Figure 3A and 3B). In addition, the significant augment of chemotherapeutic drugs’ inhibitory effects by tigecycline was observed in primary ALL cell populations (Figure 3C and 3D). Importantly, the combination of tigecycline and doxorubicin or vincristine was not effective to normal hematopoietic cells (Figure 3C and 3D).
Figure 3.
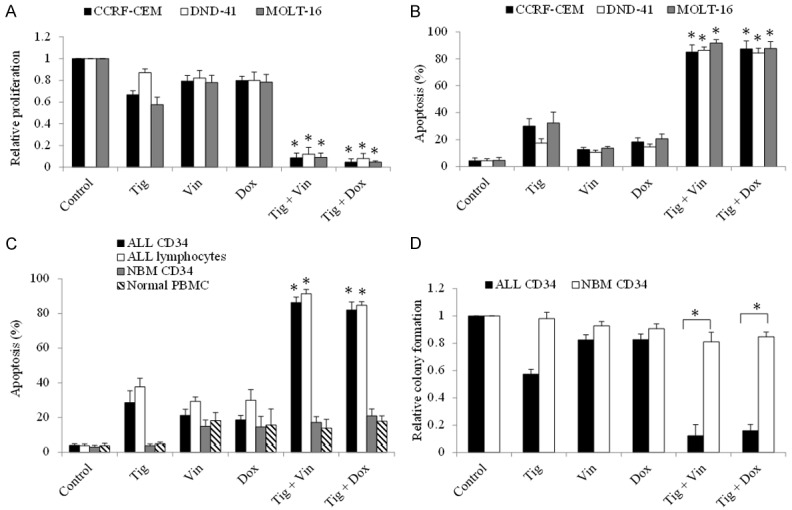
Tigecycline significantly enhances the standard chemotherapeutic drugs in vitro efficacy in ALL cells. Combination of tigecycline (5 µM) with doxorubicin (5 nM) or vincristine (10 nM) inhibits more proliferation (A) and induces more apoptosis (B) in ALL cell lines than doxorubicin or vincristine alone. Combination of tigecycline (5 µM) with doxorubicin (10 nM) or vincristine (20 nM) induces more apoptosis (C) and inhibits more colony formation (D) in ALL CD34 cells or lymphoma cells than doxorubicin or vincristine alone. Combination of tigecycline with doxorubicin or vincristine has minimal effects on normal CD34 or lymphoma cells. Tig, tigecycline; Vin, vincristine; Dox, doxorubicin. *P<0.05, compared to single drug alone.
Tigecycline acts on ALL dependent of mitochondrial respiration inhibition
The mechanisms of anti-cancer activity of tigecycline seem to be cell type specific, including mitochondrial dysfunction [4,14], inhibition of Wnt/β-catenin [15] and cyclin-dependent kinase inhibitor 1 [16]. Schimmer et al; have shown that compared to normal hematopoietic cells, acute myeloid leukemia cells are more dependent on mitochondrial biogenesis and sensitive to tigecycline [4]. We therefore first analyzed the effects of tigecycline on mitochondrial respiration in ALL cells.
We observed a remarkable reduction on basal level of oxygen consumption rate (OCR, indicative of mitochondrial respiration) as well as maximal OCR (indicative of reserved respiratory capacity) as early as 2 hours tigecycline treatment in ALL cell line, primary lymphocytes and CD34+ progenitors (Figure 4A-D). In contrast, glycolysis rate was increased and then decreased in cell treated with tigecycline (Figure 4E and 4F), suggesting that glycolysis level change is the secondary effect of tigecycline, possibly due to a compensatory response to inhibition of mitochondrial respiration. Consistently, the decreased ATP production and the increased reactive oxygen species (ROS, indicative of oxidative stress) were observed in tigecycline-treated ALL cell line, primary lymphocytes and CD34+ progenitors in the presence of tigecycline (Figure 5A and 5B). The oxidative stress leads to oxidative DNA and protein damage as shown by increased level 8-OHdG (an oxidized DNA by product) and protein carbonyls carbonyls (a modification of proteins resulting from oxidative damage) (Figure 5C and 5D).
Figure 4.
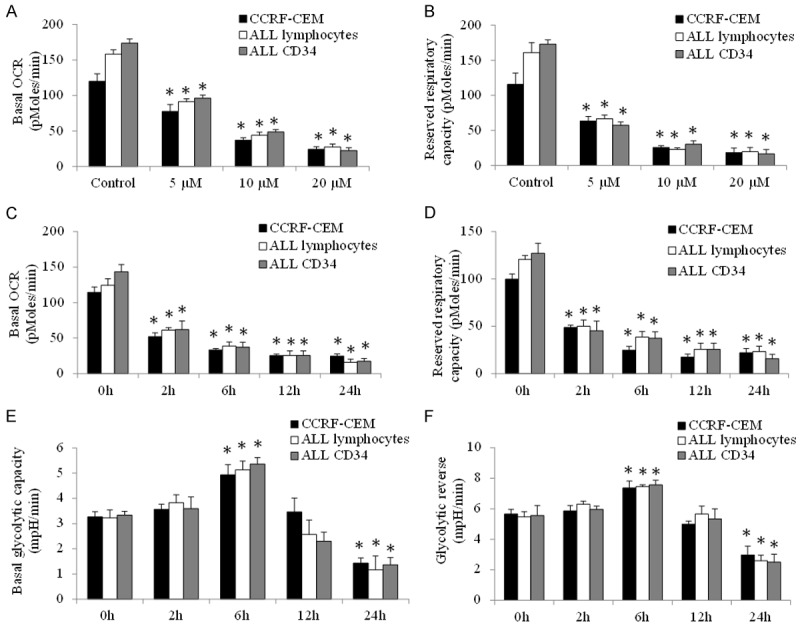
Tigecycline inhibits mitochondrial respiration in ALL cell line and patient samples. Tigecycline significantly decreases basal (A) and maximal (B) OCR in a concentration-dependent manner in CCRF-CEM, ALL lymphoma and CD34 cells. Tigecycline significantly decreases basal (C) and maximal (D) OCR in a time-dependent manner. The effects of tigecycline on basal glycolysis (E) and glycolysis reverse (F) in ALL cells. 20 µM tigecycline was used for time course analysis. *P<0.05, compared to control.
Figure 5.
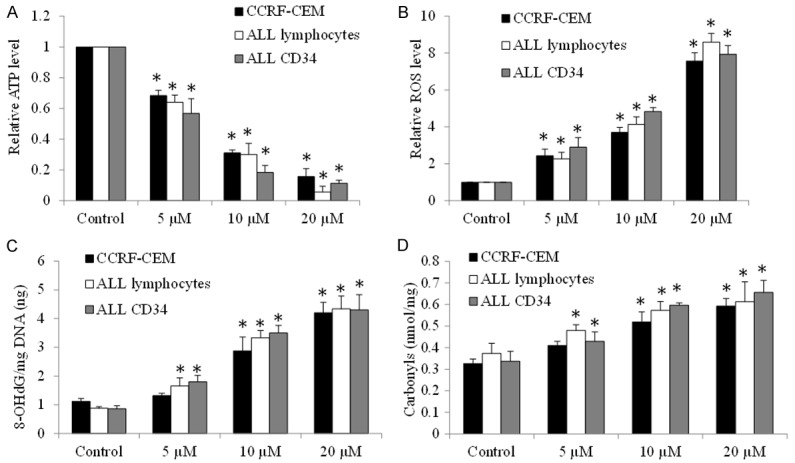
Tigecycline induces energy crisis and oxidative damage in ALL cell line and patient samples. Tigecycline significantly decreases ATP levels (A) and increases intracellular levels of ROS (B), 8-OHdG (C) and carbonylation (D) in CCRF-CEM, ALL lymphoma and CD34 cells. *P<0.05, compared to control.
To confirm that tigecycline acts on ALL dependent of mitochondrial respiration inhibition, we generated mitochondrial respiration-deficient CCRF-CML ρ0 cells by prolong exposure of parental cells to ethidium bromide [11] and verified CCRF-CML ρ0 cells by examining the mitochondrial respiration level (Figure 6A and 6B). We further found that CCRF-CML ρ0 cells have less ATP levels with minimal growth (Figure 6C and 6D). Of note, these CCRF-CML ρ0 cells are resistant to apoptosis induction by tigecycline (Figure 6E). Thus, tigecycline inhibits mitochondrial respiration which leads to energy crisis and oxidative damage in ALL cells.
Figure 6.
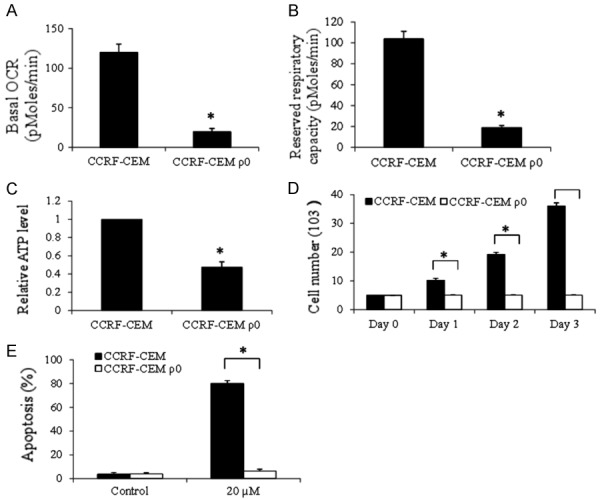
Tigecycline induces ALL cell apoptosis through its suppression of mitochondrial respiration. Basal (A) and maximal (B) OCR analysis confirms of the absence of mitochondrial respiration in CCRF-CEM ρ0 cells. Significant reduction in ATP levels (C) and growth rate (D) in CCRF-CEM ρ0 cells. (E) Tigecycline (20 µM) is ineffective in inducing apoptosis in CCRF-CEM ρ0 cells. *P<0.05, compared to CCRF-CML wildtype.
ALL cells have increased mitochondrial biogenesis than normal counterparts
We further evaluated the baseline of mitochondrial biogenesis in both ALL and normal hematopoietic cells, including mitochondrial membrane potential, mitochondrial mass, respiration and ATP levels. There was no significant difference in resting mitochondrial membrane potential between ALL primary cells and their normal counterparts (Figure 7A). However, compared to normal hematopoietic CD34+ and MNC cells, primary ALL lymphocytes and CD34+ progenitors have significantly increased mitochondrial mass, respiration and ATP levels (Figure 7B-F). Of note, there was no significant difference on glycolysis level (Figure S1), demonstrating the specific changes on mitochondrial biogenesis in ALL cells compared to normal counterparts.
Figure 7.
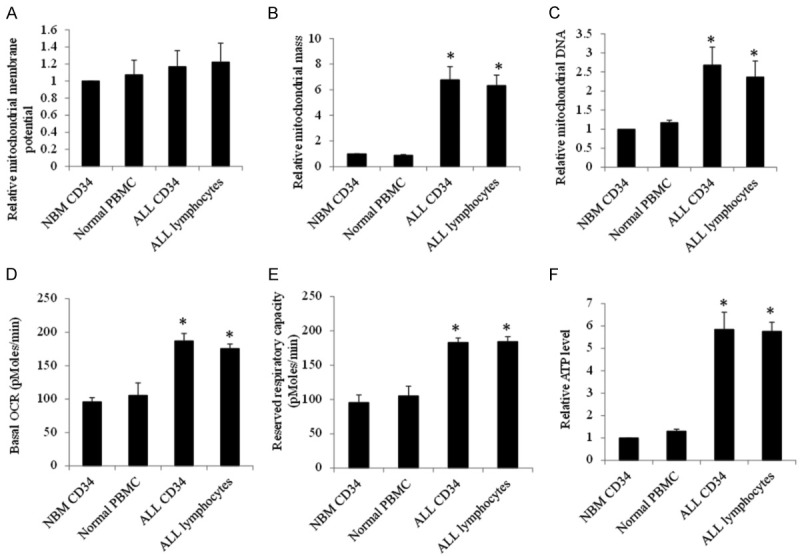
Mitochondrial characteristics of ALL and normal hematopoietic cells. Baseline mitochondrial membrane potential (A), mitochondrial mass (B) and DNA (C), basal (D) and maximal (E), and ATP levels (F) in NBM CD34, normal PBMC, ALL CD34 and ALL lymphocytes. n=5 for ALL and n=3 for normal. *P<0.05, compared to NBM CD34.
Tigecycline display anti-ALL activity in xenograft mouse model
To assess the in vivo efficacy of tigecycline in ALL, we generated xenograft mouse model of human ALL by subcutaneously transplanting CCRF-CEM cells into SCID mice. After development of palpable tumor (~7 days after cell transplant), we started treatment including tigecycline at various doses, doxorubicin alone, vincristine alone, combination of tigecycline with doxorubicin and combination of tigecycline with vincristine. The dose of each drug did not alter the body weight, appearance or behaviour of the mice (Figure S2). However, tigecycline inhibited ALL tumor growth in a dose-dependent manner (Figure 8A). Compared to doxorubicin or vincristine alone, the combination is significantly more effective in inhibiting ALL tumor growth (Figure 8B).
Figure 8.
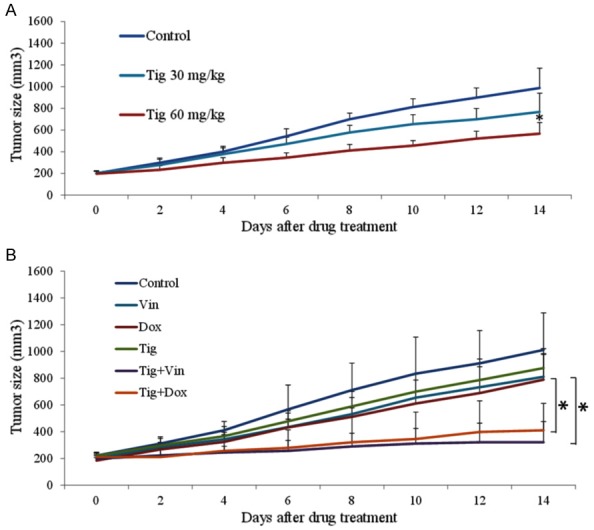
Tigecycline has in vivo activity in human xenograft ALL in mice. A. Tigecycline as drug alone inhibits ALL tumor growth in vivo in a concentration-dependent manner. B. Tigecycline (30 mg/kg, once per day) significantly enhances the in vivo efficacy of vincristine (0.5 mg/kg, three time per week) or doxorubicin (0.5 mg/kg, three time per week) in ALL tumor. *P<0.05, compared to control or single drug alone.
Discussion
Although there have been recent advances in the treatment of ALL, early relapse is common in children and is invariably associated with poor prognosis. We are in need of more effective and selective treatment strategies. In particular, identifying drugable targets that sensitize efficacy of chemotherapeutic drugs at sublethal concentrations is a promising strategy for ALL treatment. In this work, we evaluated tigecycline as a potential agent for overcoming chemotherapy resistance in ALL cells. Tigecycline is an attractive candidate as it is already available for clinical use in the treatment of board spectrum of bacterial infection [17]. It has also been shown to have toxicity in both acute myeloid leukemia and chronic myeloid leukemia stem cells and bulk cells [4,18]. The effect of tigecycline on ALL cells had not yet been investigated. We report that tigecycline is active against human ALL cells in both in vitro and in vivo preclinical models, and synergizes with standard chemotherapeutic drugs, without affecting normal hematologic cells.
We evaluated the efficacy of tigecycline in ALL cell lines as well as patient samples. The cell lines, CCRF-CEM, DND-41 and MOLT-16, we selected to demonstrate the biological effects of tigecycline represent ALL heterogeneity, including cellular origin, immunophenotyping and genetic profiling [13]. In our study, all tested ALL cell lines are sensitive to tigecycline treatment with IC50~5-10 µM (Figure 1), demonstrating the potency of tigecycline in a wide range of ALL cells. Patient-derived primary cultures (PDC) has successfully predicted tumor progression in patient with head and neck squamous cell carcinomas and could guide clinical practice [19]. We further show that tigecycline is also active against ALL PDC, such as CD34 cells and lymphocytes (Figure 2). Importantly, tigecycline remarkably sensitizes ALL cell lines and patient samples to multiple standard chemotherapeutic drugs (Figures 2 and 3). The morbidity suffered by patients treated with current chemotherapy regimens remains a challenge. Notably, the combination of both drugs at sublethal concentration achieving almost complete inhibition in ALL cells suggests that tigecycline is a promising candidate to overcome chemo-resistance in ALL cells meanwhile to minimize non-specific toxicity of chemotherapeutic drugs. Beside haematological malignancies, the anti-cancer activities of tigecycline have been shown in various solid cancers, including liver cancer and renal cell carcinoma [8,14]. Our study together with previous reports demonstrate that tigecycline is a novel type of anti-cancer drug.
A significant finding in this study, in agreement with previous studies [4,8], is that tigecycline is significantly less toxic and its combination with chemotherapeutic drugs at sublethal concentration has no inhibitory effects to normal hematopoietic CD34 and mononuclear cells, as shown by apoptosis and clonogenecity assays (Figures 2 and 3). The selective toxicity is also observed in the xenograft mouse model of human ALL. Tigecycline at dose that effectively inhibits ALL tumor growth and acts synergistically with chemotherapeutic drugs is not toxic to mice (Figures 8 and S2). The exact reason for its selective toxicity in tumour versus normal counterparts is not fully clear. We speculate that tigecycline targets mitochondrial respiration which tumor cells are more dependent on than normal cells. Although the underlying mechanisms of tigecycline in anti-cancer activities vary in different types of cancers [4,14-16]. We clearly show that tigecycline acts on ALL cells via inhibiting mitochondrial respiration, which in turn, induces energy crisis, oxidative stress and damage on not only ALL cell lines but also primary patient samples (Figures 4, 5 and 6). This is consistent with the majority of studies on the mechanisms of tigecycline’s action in cancer [8,14,20]. Like other mitochondrial respiration inhibitors (eg, phenformin) [21], we also observed the increased glycolysis at early time point of tigecycline treatment, possibly due to a compensatory response to mitochondrial respiration inhibition.
Targeting metabolic pathways for cancer therapy has attracted attention even since Warburg’s seminal discovery of aerobic glycolysis. Although the Warburg hypothesis proposes that malignant cells reply on glycolysis and are less dependent on oxidative phosphorylation for survival, more recent studies indicate that some tumors are highly dependent on oxidative phosphorylation for survival [22-24]. Notably, various studies have shown that tumor cells have increased mitochondrial biogenesis and basal level of oxygen consumption than normal cells [4,25,26]. In line with these findings, we demonstrate that CD34 and lymphocytes from ALL patients have significantly increased mitochondrial mass and oxygen consumption rate than normal CD34 and mononuclear cells from healthy donors whereas (Figure 7). Consistently, ALL mitochondrial respiration-deficient p0 cells display significant less levels of ATP production and growth (Figure 6C and 6D). Our results together with previous work demonstrate that inhibiting mitochondrial metabolism is effective and selective in targeting various types of cancer.
In conclusion, our work provides the pre-clinical evidence on the selective anti-ALL activity of tigecycline and supports advancing this drug into clinical trial for ALL. Our work also demonstrate that mitochondria-regulated biochemical pathways is a selective target in ALL and warrants further therapeutic exploitation.
Acknowledgements
This work was supported by research grants provided by Hubei Province Development Planning & Research Institute (220100455) and Nature Science Foundation of Hubei Province (2011CDB526).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.PDQ Cancer Information Summaries. Bethesda (MD): 2002. Childhood acute lymphoblastic leukemia treatment (PDQ(R)): Health professional version. [Google Scholar]
- 2.Gutierrez-Camino A, Martin-Guerrero I, Garcia-Orad A. Genetic susceptibility in childhood acute lymphoblastic leukemia. Med Oncol. 2017;34:179. doi: 10.1007/s12032-017-1038-7. [DOI] [PubMed] [Google Scholar]
- 3.Li MJ, Liu HC, Yen HJ, Jaing TH, Lin DT, Yang CP, Lin KH, Hung IJ, Jou ST, Lu MY, Hsiao CC, Peng CT, Chang TT, Wang SC, Lin MT, Chen JS, Chang TK, Hung GY, Wu KH, Yang YL, Chang HH, Chen SH, Yeh TC, Cheng CN, Lin PC, Chiou SS, Sheen JM, Cheng SN, Chen SH, Chang YH, Ho WL, Chao YH, Chen RL, Chen BW, Wang JL, Hsieh YL, Liao YM, Yang SH, Chang WH, Chao YY, Liang DC. Treatment for childhood acute lymphoblastic leukemia in Taiwan: Taiwan Pediatric Oncology Group ALL-2002 study emphasizing optimal reinduction therapy and central nervous system preventive therapy without cranial radiation. Pediatr Blood Cancer. 2017;64:234–241. doi: 10.1002/pbc.26142. [DOI] [PubMed] [Google Scholar]
- 4.Skrtic M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, Hurren R, Jitkova Y, Gronda M, Maclean N, Lai CK, Eberhard Y, Bartoszko J, Spagnuolo P, Rutledge AC, Datti A, Ketela T, Moffat J, Robinson BH, Cameron JH, Wrana J, Eaves CJ, Minden MD, Wang JC, Dick JE, Humphries K, Nislow C, Giaever G, Schimmer AD. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2011;20:674–688. doi: 10.1016/j.ccr.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiang W, Cheong JK, Ang SH, Teo B, Xu P, Asari K, Sun WT, Than H, Bunte RM, Virshup DM, Chuah C. Pyrvinium selectively targets blast phase-chronic myeloid leukemia through inhibition of mitochondrial respiration. Oncotarget. 2015;6:33769–33780. doi: 10.18632/oncotarget.5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaewpoowat Q, Ostrosky-Zeichner L. Tigecycline : a critical safety review. Expert Opin Drug Saf. 2015;14:335–342. doi: 10.1517/14740338.2015.997206. [DOI] [PubMed] [Google Scholar]
- 7.Lamb R, Ozsvari B, Lisanti CL, Tanowitz HB, Howell A, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget. 2015;6:4569–4584. doi: 10.18632/oncotarget.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang B, Ao J, Yu D, Rao T, Ruan Y, Yao X. Inhibition of mitochondrial translation effectively sensitizes renal cell carcinoma to chemotherapy. Biochem Biophys Res Commun. 2017;490:767–773. doi: 10.1016/j.bbrc.2017.06.115. [DOI] [PubMed] [Google Scholar]
- 9.Li H, Jiao S, Li X, Banu H, Hamal S, Wang X. Therapeutic effects of antibiotic drug tigecycline against cervical squamous cell carcinoma by inhibiting Wnt/beta-catenin signaling. Biochem Biophys Res Commun. 2015;467:14–20. doi: 10.1016/j.bbrc.2015.09.140. [DOI] [PubMed] [Google Scholar]
- 10.Tang CL, Yang LQ, Jiang XL, Xu C, Wang M, Wang QR, Zhou ZS, Xiang ZH, Cui HJ. Antibiotic drug tigecycline inhibited cell proliferation and induced autophagy in gastric cancer cells. Biochem Biophys Res Commun. 2014;446:105–112. doi: 10.1016/j.bbrc.2014.02.043. [DOI] [PubMed] [Google Scholar]
- 11.Hashiguchi K, Zhang-Akiyama QM. Establishment of human cell lines lacking mitochondrial DNA. Methods Mol Biol. 2009;554:383–391. doi: 10.1007/978-1-59745-521-3_23. [DOI] [PubMed] [Google Scholar]
- 12.Chu S, Holtz M, Gupta M, Bhatia R. BCR/ABL kinase inhibition by imatinib mesylate enhances MAP kinase activity in chronic myelogenous leukemia CD34+ cells. Blood. 2004;103:3167–3174. doi: 10.1182/blood-2003-04-1271. [DOI] [PubMed] [Google Scholar]
- 13.Burger R, Hansen-Hagge TE, Drexler HG, Gramatzki M. Heterogeneity of T-acute lymphoblastic leukemia (T-ALL) cell lines: suggestion for classification by immunophenotype and Tcell receptor studies. Leuk Res. 1999;23:19–27. doi: 10.1016/s0145-2126(98)00133-7. [DOI] [PubMed] [Google Scholar]
- 14.Tan J, Song M, Zhou M, Hu Y. Antibiotic tigecycline enhances cisplatin activity against human hepatocellular carcinoma through inducing mitochondrial dysfunction and oxidative damage. Biochem Biophys Res Commun. 2017;483:17–23. doi: 10.1016/j.bbrc.2017.01.021. [DOI] [PubMed] [Google Scholar]
- 15.Li H, Jiao S, Li X, Banu H, Hamal S, Wang X. Therapeutic effects of antibiotic drug tigecycline against cervical squamous cell carcinoma by inhibiting Wnt/beta-catenin signaling. Biochem Biophys Res Commun. 2015;467:14–20. doi: 10.1016/j.bbrc.2015.09.140. [DOI] [PubMed] [Google Scholar]
- 16.Hu H, Dong Z, Tan P, Zhang Y, Liu L, Yang L, Liu Y, Cui H. Antibiotic drug tigecycline inhibits melanoma progression and metastasis in a p21CIP1/Waf1-dependent manner. Oncotarget. 2016;7:3171–3185. doi: 10.18632/oncotarget.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rose WE, Rybak MJ. Tigecycline: first of a new class of antimicrobial agents. Pharmacotherapy. 2006;26:1099–1110. doi: 10.1592/phco.26.8.1099. [DOI] [PubMed] [Google Scholar]
- 18.Lu Z, Xu N, He B, Pan C, Lan Y, Zhou H, Liu X. Inhibition of autophagy enhances the selective anti-cancer activity of tigecycline to overcome drug resistance in the treatment of chronic myeloid leukemia. J Exp Clin Cancer Res. 2017;36:43. doi: 10.1186/s13046-017-0512-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chia S, Low JL, Zhang X, Kwang XL, Chong FT, Sharma A, Bertrand D, Toh SY, Leong HS, Thangavelu MT, Hwang JS, Lim KH, Skanthakumar T, Tan HK, Su Y, Hui Choo S, Hentze H, Tan IB, Lezhava A, Tan P, Tan DS, Periyasamy G, Koh JL, Gopalakrishna Iyer N, DasGupta R. Phenotype-driven precision oncology as a guide for clinical decisions one patient at a time. Nat Commun. 2017;8:435. doi: 10.1038/s41467-017-00451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jaras M, Ebert BL. Power cut: inhibiting mitochondrial translation to target leukemia. Cancer Cell. 2011;20:555–556. doi: 10.1016/j.ccr.2011.10.028. [DOI] [PubMed] [Google Scholar]
- 21.Miskimins WK, Ahn HJ, Kim JY, Ryu S, Jung YS, Choi JY. Synergistic anti-cancer effect of phenformin and oxamate. PLoS One. 2014;9:e85576. doi: 10.1371/journal.pone.0085576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson JM, Lai SY, Cotzia P, Cognetti D, Luginbuhl A, Pribitkin EA, Zhan T, Mollaee M, Domingo-Vidal M, Chen Y, Campling B, Bar-Ad V, Birbe R, Tuluc M, Martinez Outschoorn U, Curry J. Mitochondrial metabolism as a treatment target in anaplastic thyroid cancer. Semin Oncol. 2015;42:915–922. doi: 10.1053/j.seminoncol.2015.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang X, de Milito A, Olofsson MH, Gullbo J, D’Arcy P, Linder S. Targeting mitochondrial function to treat quiescent tumor cells in solid tumors. Int J Mol Sci. 2015;16:27313–27326. doi: 10.3390/ijms161126020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;11:9–15. doi: 10.1038/nchembio.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao M, Zhang L, Zhou Y, Rajoria P, Wang C. Pyrvinium selectively induces apoptosis of lymphoma cells through impairing mitochondrial functions and JAK2/STAT5. Biochem Biophys Res Commun. 2016;469:716–722. doi: 10.1016/j.bbrc.2015.12.059. [DOI] [PubMed] [Google Scholar]
- 26.Yu M, Li R, Zhang J. Repositioning of antibiotic levofloxacin as a mitochondrial biogenesis inhibitor to target breast cancer. Biochem Biophys Res Commun. 2016;471:639–45. doi: 10.1016/j.bbrc.2016.02.072. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.