

Abstract
Lung carcinoma has become a more and more serious health problem as platinum-based chemotherapy remains a limited benefit. Accumulating evidences indicate that autophagy plays a significant role in decreased curative effect and chemotherapy failure. Inhibition of autophagy can potentiate anti-proliferation effect and contribute to tumor regression in lung carcinoma. Here, we showed that the expectorant drug ambroxol (Ax) promoted autophagosomes accumulation by blocking late-stage autophagic flux in lung carcinoma cells. Furthermore, Ax treatment caused alkalization of lysosome and impaired lysosomal degradation capacity, which contributed to decreased autophagosomes-lysosomes fusion and interrupted normal cargo degradation. Ax potentiated cell-killing sensitivity of paclitaxel (PTX) and docetaxel (DTX), which had nothing to do with cell uptake but was associated with enhanced autophagy level. Moreover, Ax in combination with PTX exerted a significantly enhanced tumor-shrinking effect and prolonged survival time in subcutaneous and pulmonary metastatic tumor nude mice models. Considering the superiority of lung protection and excellent safety, Ax shows enormous translational potential and preponderance in clinical lung carcinoma therapy. Together, our findings suggested that the novel function of Ax, namely autophagy inhibition, resulted from alkalization and impaired degradation capacity of lysosome. The combination of Ax and PTX showed an enhanced cytotoxicity in vitro and improved satisfactory curative outcome in vivo. Our research provides a promising therapeutic strategy to lung carcinoma, which has clinical transformation potential and practical application value.
Keywords: Ambroxol, autophagy, lysosome, microtubule-stabilizing drug, lung carcinoma, anti-tumor
Introduction
Lung carcinoma is one of the highest morbidity and mortality of diseases worldwide [1]. Platinum-based chemotherapy is the first-line strategy for clinical treatment at present, while accompanying drug resistance and non-selective side effects lead to undesired treatment effect [2,3]. More and more researches have revealed that chemotherapy drugs not only induce tumor cell apoptosis but also activate autophagy [4,5]. Autophagy is a catabolic mechanism that involves degradation of intracellular dysfunctional constituents or extracellular invaders [6]. For cancer therapy, autophagy is considered as a double-edged sword, which is linked to tumor suppression as well as promotion of tumorigenesis and tumor survival [7,8]. The role of autophagy is a dynamic change with tumor progression. Before tumorigenesis, autophagy prevents carcinogenesis and reduces tumor incidence based on controlling benign cell damage, reducing cell endogenous pressure and maintaining cellular microenvironment homeostasis. After tumorigenesis, autophagy supports energy source for hypermetabolic tumor cells as well as removes long-lived proteins and damaged organelles to enhance cancer cell survival [9,10]. Hence, modulating autophagy within tumor may be a valid and promising approach to enhance therapeutic efficacy.
Cancer cells can resist cell death and decrease curative effect by activating autophagy. PTX, a broad-spectrum chemotherapy drug that stabilizes microtubules and promotes hyper-polymerization of tubulin, activates autophagy in a way that weakens phosphorylation of mTOR and its downstream substrates p70s6k also raises Beclin1 and Atg5 at low concentration, causing treatment resistance and chemotherapy failure [11,12]. Cellular response to autophagy modulation depends on tumor type, stage, microenvironment, autophagy inducer or inhibitor and combined tumor treatment [13,14]. In some situations, inhibition of autophagy can overcome drug resistance and enhance the sensitivity of tumor cells to chemotherapy drugs. For nasopharyngeal carcinoma, taxol-induced caspase-dependent apoptosis could be potentiated with autophagy inhibited, leading to partial reversal of the acquired taxol resistance in drug-resistant CNE-1 and HNE-2 cells [15].Therefore, these researches established autophagy as a promising therapeutic target where modulation exhibits new opportunities for cancer treatment.
Chemotherapy drugs in combination with autophagy inhibitors serve as a novel and potent strategy in cancer treatment [16]. As shown in multiple preclinical models, inhibition of autophagy restored chemosensitivity and potentiated curative effect in vitro and in vivo [16,17]. Suppression of autophagy by 3-methyladenine or bafilomycin A1 enhanced sensitivity of gefitinib to MDA-MB-231 and MDA-MB-468 cells, detected from stronger inhibition of cell vitality and colony formation, higher level of G0/G1 arrest and DNA damage, and these improved curative effects were also verified in nude mice vivo [18]. Chloroquine (CQ) blocked autophagic flux and chemosensitized HEC-1A and JEC cells to PTX-mediated cell death [19,20]. These reports suggest that the combination of autophagy inhibitors and chemotherapy drugs can lead to a significant decrease in tumor volume and contribute to tumor regression in various cancers.
Although several drugs can suppress autophagy, most of these drugs lack specificity in lung cancer therapy. As a frequently prescribed drug that can facilitate mucociliary clearance, Ax has been used to cure acute or chronic bronchitis and bronchial asthma with wide therapeutic window and little side effect even used in large dose [21,22]. Ax treatment could activate coordinated lysosomal expression and regulation network by transcription factors EB and modulate lysosomal biochemistry as a molecular chaperone, which improved the performance of patients with Gaucher disease [23,24]. During autophagy process, a part of the cytoplasm is sequestered in autophagosomes that fuse with lysosomes to form autolysosomes, a process termed autophagy flux. Next, the enzymes present in the autolysosome lumen degrade autophagic cargo, thus supplying macromolecules that can be transported into the cytosol [6]. Considering the close connection between autophagy and lysosome, we established hypothesis that Ax might regulate autophagy in lysosome-dependent way. Besides, Ax was also applied to postoperative lung protection for which prevented pulmonary complications of patients with lung cancer after pulmonary lobectomy [25,26]. The secretion of pulmonary surfactant protein could be promoted after Ax treatment, which was considered as a key factor in lung homeostasis and suppressed lung cancer progression [27-29]. Based on the multiple advantages of Ax including lung protection and low toxicity, co-treatment of Ax and PTX shows enormous translational potential and preponderance in clinical lung carcinoma therapy.
In this study, we investigated whether Ax can manipulate autophagy and what the mechanism of Ax modulating autophagy is. We evaluated whether Ax can enhance cytotoxicity sensitivity of A549 cells to PTX and DTX in vitro. Furthermore, we utilized A549 subcutaneous or A549-luciferase pulmonary metastatic tumor nude mice models to assess whether Ax exerted an enhanced tumor-shrinking effect and prolonged survival time of microtubule-stabilizing drug to lung carcinoma in vivo.
Materials and methods
Reagents and antibodies
Ax (427650) and CQ (C6628) were purchased from Boehringer Ingelheim and sigma, respectively. Rapa (KGATGR005) and Earle’s Balanced Salt Solution (KGATGR001) were purchased from KeyGEN BioTECH. PTX was purchased from Jiangsu Yew Pharmaceutical Co., Ltd. Anti-LC3 antibody (12741), Anti-SQSTM1/p62 antibody (8025), Anti-mTOR antibody (2983), Anti-Phospho-mTOR antibody (5536), Anti-p70 S6 kinase antibody (2708), Anti-Phospho-p70 S6 kinase antibody (9234) were purchased from Cell Signaling Technology. Anti-β-actin antibody (AA128) and HRP-conjugated anti-mouse antibody (A0216) were purchased from Beyotime. HRP-conjugated anti-rabbit antibody (BL003A) was purchased from Biosharp.
Cell lines and cell culture
Human lung adenocarcinoma A549 cell lines were purchased from Chinese Academy of Sciences Cell Bank (Shanghai, China) in 2015. A549-luciferase and A549/RFP-GFP-LC3 cells were provided by Dr. Yonghua Yang (School of Pharmacy, Fudan University, Shanghai, China) in 2016. A549, A549-luciferase and A549/RFP-GFP-LC3 cells were all cultured at 37°C with 5% (v/v) CO2 in an RPMI 1640 medium (Gibco, 11875-093) supplemented with 10% fetal bovine serum (Gibco, 10099-141) and 100 units/mL penicillin-streptomycin (Gibco, 15140122).
Western blotting
The cells were harvested and lysed in RIPA buffer supplemented with protease inhibitor (Roche, 04693116001). The proteins were separated on SDS-polyacrylamide gel and transferred to PVDF membranes (Millipore, IPVH00010). The membranes were blocked with 5% nonfat dry milk for 1 h and then incubated with primary antibody at 4°C overnight. After washed by TBST, membranes were incubated with corresponding horseradish peroxidase-labeled secondary antibody for 2 h at room temperature. Finally, the bands were detected with an enhanced chemiluminescence kit (Thermo scientific, QL222301).
Immunofluorescence confocal microscopy
The cells were seeded in laser confocal culture dish at a density of 2 × 104 cells per well and treated with drug for indicated time. Then samples were stained by specific stain with Cyto-ID autophagy detection kit (Enzo Life Sciences, ENZ-51031-K200) following the manufacturer’s instructions and further photographed by Laser scanning confocal microscope (Carl Zeiss LSM710, Germany).
Transmission electron microscopy (TEM)
The cells were collected and washed with PBS. Subsequently, cells were fixed with 2.5% glutaraldehyde at 4°C overnight and then post-fixed with 1% osmium tetroxide for 1 h. The cells were dehydrated in graded ethanol and embedded in epoxy resin. The sample was sliced and doubly stained with uranyl acetate and lead citrate, finally, observed under TEM.
Quantitative assay of GFP-RFP-LC3 Dots
The A549 cells transfected with GFP-RFP-LC3 were seeded in laser confocal culture dish. After cells were treated with drug for indicated time, GFP-RFP-LC3 fluorescence was observed using confocal microscope and the average number of dots was quantified by counting 300 cells. The assays were independently carried out by 2 of the authors in a blind manner.
Assessment of lysosomal acidity
The cells were harvested and washed twice with PBS after treated with drug for indicated time. Then, cells were incubated with medium containing 1 μM LysoSensor Green DND-189 (Yeasen, 40767ES50) dye for 30 min. Culture medium containing dye were replaced with fresh ones and cells were observed under fluorescence microscopy (Leica DMI4000B, Germany).
Evaluation of activity of acid phosphatase
The cells were collected and washed twice with PBS after treated with drug for indicated times. Cells were lysed and centrifuged to obtain supernatant. Then the supernatant was examined activity of acid phosphatase according to the manufacturer’s instructions of Fluoride Resistant Acid Phosphatase Assay Kit (Beyotime, P0335).
Cell viability detection
Culture medium was exchanged with fresh one containing 0.5 mg/mL MTT and incubated for 4 h at 37°C. The absorbance was measured at 570 nm with microplate reader (Synergy 2; Bio Tek) after formazan crystal was dissolve with dimethyl sulfoxide.
Cell apoptosis assay
The apoptosis assay was detected with Annexin V-FITC Apoptosis Detection Kit (KeyGEN BioTECH, KGA108). For abbreviation, floating and attached cells were collected and washed with PBS twice. Re-suspended with 195 μL binding buffer, cells were incubated with 5 μL Annexin V-FITC and 10 μL propidium iodide (PI) for 10 min, avoiding exposure to light. Finally, samples were analyzed by flow cytometry (BD Biosciences, FACSAria II, USA) and data were processed with FlowJo software.
Quantitative analysis of cellular uptake
The cells were seeded at a density of 2 × 105 cells per well in 6-well plates. Then, cells were co-treated PTX with Ax for indicated time. The cells were smashed to release cellular PTX after collected and washed. After centrifugation, the contents of PTX in the supernatant were detected by HPLC (Agilent 1260, USA).
In vivo anti-tumor evaluation with A549 subcutaneous tumor nude mice
Male Balb/c nude mice (6 to 8 weeks old), ranging from 18 to 22 g, were obtained from the Experimental Animals Center of Fudan University (Shanghai, China). All animal experiments were approved by Animal Ethics Committee of Fudan University. A549 cells (5 × 106 cells/mouse) were injected subcutaneously to nude mice at the right axilla. Mice were held under specific pathogen-free conditions. Tumor volume was calculated as V = 0.5 × dmax × dmin 2 (dmax and dmin are the longest and the shortest diameters of tumor, respectively). The mice were randomly grouped into six groups (n = 6) and intraperitoneally injected with corresponding therapeutic drug on day 1, 4, 7, 10 when tumor size grew up to 50~100 mm3. Body weight and tumor volume were measured daily. The mice were sacrificed on day 14 and tumors were stripped to be weighed and photographed. The inhibitory rate of tumor growth (IRT%) was calculated as IRT % = (WC - WT)/WC × 100% (WC and WT are the tumor weight of saline group and corresponding treatment group, respectively). Tumor tissues were fixed in formalin for immunohistochemistry (IHC) assay and terminal deoxyribonucleotidyl transferase (TDT)-mediated dUTP-digoxigenin nick end labeling (TUNEL) assay.
Immunohistochemistry assay
The tumors were dehydrated and embedded in paraffin after fixed in 4% paraformaldehyde, then sectioned on glass slide. The non-specific binding sites were blocked after high-temperature antigen retrieval and endogenous peroxidase removal. Slices were incubated with primary antibody and horseradish peroxidase-labeled secondary antibody. Finally, samples were visualized by DAB chromogenic substrate solution.
TUNEL assay
The tumors were prepared into paraffin embedded sections. TUNEL assay was performed by In situ Cell Death Detection Kit, Fluorescein (Roche, 11684795910). Paraffin embedded sections were dewaxed in xylene, rehydrated in graded ethanol series and then incubated with proteinase K for 15 min at 37°C for permeabilization of tissues. After samples were washed with PBS, TUNEL reaction mixture were added to test sections, and then incubated in humidified chamber in dark at 37°C for 60 min. The sections were counterstained with 4’, 6-diamidino-2-phenylindole (DAPI) and observed with fluorescence microscope.
In vivo anti-cancer evaluation with A549-luciferase pulmonary metastatic tumor nude mice
A549 cells stably expressing luciferase (5 × 106 cells/mouse) were injected to nude mice by tail vein to develop lung metastatic tumor nude mice model [30]. A consecutive observation revealed that pulmonary tumors were developed after two weeks of injection. Two weeks post A549-luciferase cells inoculation, mice were randomly divided into six groups (n = 6). The treatment regimen was the same as the experiment of A549 subcutaneous tumor nude mice described previously. Bioluminescence of pulmonary tumor were photographed using In Vivo Imaging System (IVIS) spectrum (PerkinElemer, USA) after luciferase substrate (Sciencelight, luc003) was injected at 0, 5, 10, 15 days to monitor the growth of lung cancer.
Statistical analysis
Results were presented as the mean ± standard deviation of at least three independent experiments unless stated otherwise. Statistical comparisons of data were performed by the Student’s t-test. *P < 0.05, **P < 0.01 and ***P < 0.001 were regarded as statistically significant difference. All analyses were conducted using GraphPad Prism 5 software (GraphPad Software Inc., La Jolla, CA, USA).
Results
Ax induced autophagosomes accumulation
In order to figure out whether Ax could regulate cell autophagy or not, western bolt analysis was routinely performed to measure the level of LC3, the most specific and also the most analyzed autophagy marker. Results showed the level of LC3-II, an endogenous LC3 protein that was selectively incorporated into autophagic membranes, significantly increased in Ax-treated cells, which was both time- and dose-dependent, with the maximal effect observed at 24 h for 300 μM Ax (Figure 1A, 1B). We utilized Cyto-ID, a specific dye that selectively stained autophagic compartments including phagosomes, autophagosomes and autolysosomes in live cells and therefore allowed determination of autophagic flux as accumulation of stained compartments. Confocal microscopy showed that puncta were evenly distributed in control cells, while plenty of bright punctate dots appeared after Ax treated, indicating Ax indeed induced autophagosomes accumulation (Figure 1C). TEM was used to further investigate the morphological changes, and revealed that more autophagosomes and autolysosomes in the cells treated with Ax than in the untreated cells (Figure 1D-F). At the same time, TEM showed the representative ultrastructural morphological features of autophagosomes, which was double-membrane vacuolar structures contained undigested cytoplasmic contents and exhibited increased electron density (Figure 1F). Collectively, these results demonstrated that Ax could promote autophagosomes accumulation.
Figure 1.
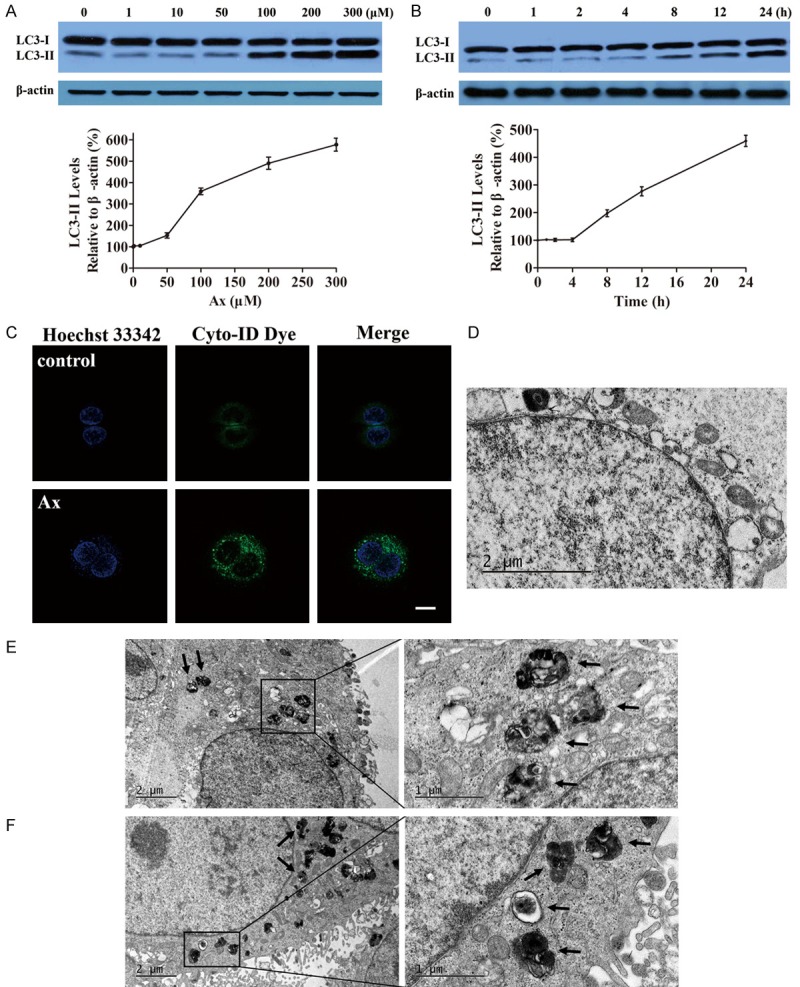
Ax promoted autophagosomes accumulation. (A) A549 cells were treated with various concentrations of Ax for 24 h. Endogenous LC3-II levels were measured by western blotting with anti-LC3 antibodies and quantitative analysis by densitometric analysis relative to β-actin. Mean ± SD, n = 3. (B) A549 cells were treated with 100 μM Ax for various time. Endogenous LC3-II levels were measured by western blotting with anti-LC3 antibodies and quantitative analysis by densitometric analysis relative to β-actin. Mean ± SD, n = 3. (C) A549 cells were seeded into the confocal plates and treated with 100 μM Ax for 24 h. Then, cells were co-stained with Hoechst 33342 and Cyto-ID green dye, and further detected with laser scanning confocal microscope. Scale bar: 20 μm. TEM of cells treated with PBS as control (D) or 100 μM Ax for 24 h (E and F). Arrows indicated autophagosomes and autolysosomes.
Ax treatment blocked autophagic flux and degradation of autophagy substrate
Increased autophagosomes accumulation could be linked to either genuine autophagy induction or blockade of autophagic flux. To distinguish between two possibilities, we performed autophagic flux assay. First, we measured LC3-II turnover in the presence and absence of rapamycin (Rapa), an extensively applied autophagy inducer, which directly inhibited MTORC1 and thus stimulated autophagy. Co-incubated Ax with Rapa led to a further increase in the relative level of LC3-II (Figure 2A), which indicated that normal cargo degradation was interrupted by Ax treatment. Similarly, adding Ax further enhanced starvation-induced accumulation of LC3-II, indicating that autophagic flux was blocked by Ax treatment (Figure 2B). However, adding Ax could not further increased CQ-induced accumulation of LC3-II, as compared to the cells treated with CQ only, suggesting that Ax did not actually induce autophagy (Figure 2C). These results showed that Ax treatment blocked autophagic flux and interrupted normal cargo degradation.
Figure 2.
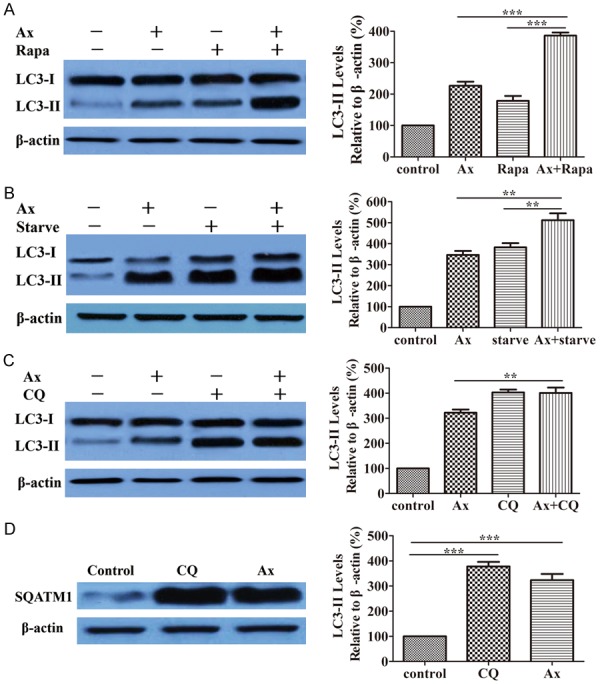
Ax induced autophagosomes accumulation by blocking autophagic flux. A. A549 cells were treated with 100 μM Ax for 24 h in the presence or absence of 200 nM Rapa. Endogenous LC3-II levels were measured by western blotting with anti-LC3 antibodies and quantitative analysis by densitometric analysis relative to β-actin. Mean ± SD, n = 3. ***P < 0.001. B. A549 cells were treated with 100 μM Ax under normal conditions or starvation conditions. Endogenous LC3-II levels were measured by western blotting with anti-LC3 antibodies and quantitative analysis by densitometric analysis relative to β-actin. Mean ± SD, n = 3. **P < 0.01. C. A549 cells were treated with 100 μM Ax for 24 h in the presence or absence of 20 μM CQ. Endogenous LC3-II levels were measured by western blotting with anti-LC3 antibodies and quantitative analysis by densitometric analysis relative to β-actin. Mean ± SD, n = 3. **P < 0.01. D. A549 cells were treated with 100 μM Ax or 20 μM CQ for 24 h. Endogenous SQATM1 levels were measured by western blotting with anti-SQATM1 antibodies and quantitative analysis by densitometric analysis relative to β-actin. Mean ± SD, n = 3. ***P < 0.001.
To further investigate the role of Ax in autophagy, we measured the level of sequestosome 1 (SQSTM1/p62), a protein substrate that is selectively incorporated into the forming autophagosomes and preferentially degraded by autophagy. The increased amount of SQSTM1 was linked to the suppression of autophagic flux. Results revealed that Ax treatment caused an increase in the level of SQSTM1 (Figure 2D), similar to CQ treatment, suggesting that autophagic flux was inhibited and autophagic degradation capacity was possibly impaired. To detect the influence of upstream signaling pathway of autophagy, we selected the level of phospho-mTOR (P-mTOR) and phospho-p70 S6 Kinase (P-p70S6K) as marker for mammalian target of rapamycin (mTOR) activity. Results showed the level of P-mTOR and P-p70S6K kept unchanged after Ax treated while Rapa caused a decrease and altered the activity of both mTOR and p70S6k (Supplementary Figure 1), suggesting Ax induced autophagosomes accumulation in mTOR-independent fashion.
Ax impaired autophagosomes-lysosomes fusion and lysosome degradation capacity
To investigate the mechanism that Ax blocked autophagic flux, we used A549 cells transfected with a red fluorescent protein-green fluorescent protein-microtubule associated protein 1 light chain 3 (RFP-GFP-LC3) expression vector to monitor autophagy process. [31,32] In this assay, GFP was sensitive to low pH and tended to quench in the acidic environment of the lysosome while RFP was more resistant to low pH. Therefore, when autophagosomes fused with lysosomes, the yellow puncta of GFP disappeared and red-only puncta of RFP remained. The results showed that autophagy was induced in normal autophagosomes-lysosomes fusion way with Rapa treated. However, treating cells with Ax promoted formation of both GFP and RFP puncta that extensively co-localized with each other and thus appeared yellow (Figure 3A-C). These results suggested that Ax blocked autophagosome maturation and decreased autophagosomes-lysosomes fusion.
Figure 3.
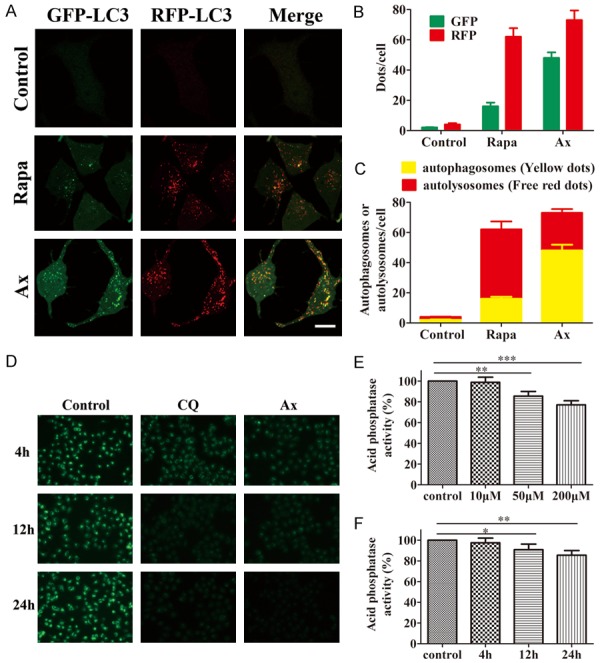
Ax impaired autophagosomes-lysosomes fusion and caused alkalization of lysosome. A. A549 cells transfected with GFP-RFP-LC3 were subjected to 200 nM Rapa or 100 μM Ax for 24 h, and then observed with confocal microscope. Scale bar: 20 μm. B. The statistic number of green puncta and red puncta per cell in each condition were quantified (n = 300). C. The statistic numbers of autolysosomes (GFP- RFP+) and autophagosomes (GFP+ RFP+) per cell in each condition were quantified (n = 300). D. A549 cells were subjected to 20 μM CQ or 100 μM Ax for 4, 12 and 24 h, and then exposed to 1 μM/L LysoSensor Green DND-189 for 30 min. Furthermore, cells were detected with fluorescence microscope. Microscopy 400 × magnification. E. A549 cells were subjected to 10, 50 and 200 μM Ax for 24 h, and then acid phosphate activity was measured. Mean ± SD, n = 3. **P < 0.01, ***P < 0.001. F. A549 cells were subjected to 50 μM Ax for 4, 12 and 24 h and then acid phosphate activity were measured. Mean ± SD, n = 3. *P < 0.05, **P < 0.01.
We analyzed the acidity of lysosome, as lysosomal functions performed demanded an acidic environment. We utilized LysoSensor Green DND-189, an acidotropic dye that selectively accumulated in intracellular acidic organelles and had a fluorescence intensity that is proportional to acidity, to test whether Ax can affect lysosomal pH. Fluorescent microscopic analysis showed that fluorescence intensity decreased in a time-dependent manner, which revealed an increase in lysosomal pH in Ax-treated cells (Figure 3D). Moreover, the activity of acid phosphatase, a lysosome marked enzyme with an optimal pH of 4.5, was greatly lower by Ax treatment (Figure 3E, 3F). On the basis of these results, we concluded that Ax treatment caused alkalization of lysosome and impairment of lysosome degradation capacity, and therefore decreased autophagosomes-lysosomes fusion and blocked autophagic flux.
Ax treatment increased PTX-induced cell death
It is known that inhibition of autophagy increases cytotoxic sensitivity of cancer cells to chemotherapy drugs, and therefore improves treatment effect. PTX, a widely used microtubule-stabilizing chemotherapy drug, is demonstrated to induce autophagy in various cancer cells. Since Ax can block autophagic degradation, combinative therapy of PTX and Ax may promote tumor cells death and enhance curative performance. At the same time, PTX resistance may decrease within tumor cells because of inhibited autophagy.
To investigate whether Ax promoted PTX-induced cell death, we carried out MTT assay. Incubation of cells with PTX apparently decreased survival rates in a time- and dose-dependent manner, while co-treatment of Ax and PTX obviously increased cytotoxicity than PTX alone and caused further reduction in the proliferation rates (Figure 4A). The IC50 values of PTX without and with Ax at different concentrations after 24, 48, 72 h treatment were listed in Table 1, from which we found that IC50 values of PTX without and with 100 μM Ax were 107.00 ± 4.38 and 1.49 ± 0.13 ng/mL after 24 h treatment respectively. This meant that Ax could enhance the in vitro therapeutic effects of PTX by 72-folds for 24 h treatment. The results of another microtubule-stabilizing drug, DTX, showed similar therapy benefits (Table 2). Besides, 0.01 μg/mL PTX treatment caused 27.4% cell apoptosis, while co-treated with 50 μM Ax elicited a 27.3% increase in cell apoptosis assessed by Annexin-V/FITC staining (Supplementary Figure 2). These results strongly demonstrated that Ax strengthened cytotoxicity sensibility of tumor cells to PTX, which contributed to boosted therapeutic effect in vitro.
Figure 4.
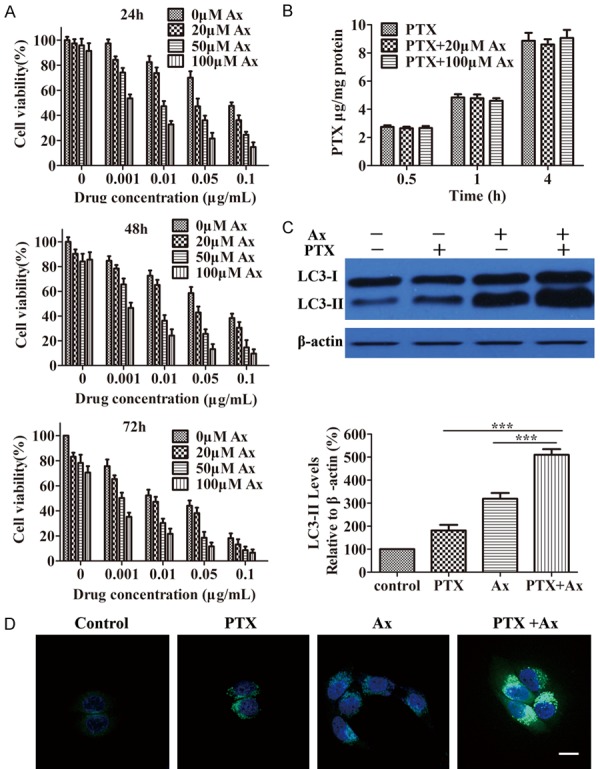
Ax treatment enhanced PTX-induced cell death. A. Viability of A549 cells co-treated 0.001-0.1 μg/mL PTX with 20-100 μM Ax for 24, 48 and 72 h, respectively. B. The cell uptake of 0.01 μg/mL PTX alone and 0.01 μg/mL PTX in combination with 20 or 100 μM Ax for 0.5, 1 and 4 h. C. A549 cells were treated with 50 μM Ax, 0.005 μg/mL PTX and co-treated 0.005 μg/mL PTX with 50 μM Ax for 24 h. Endogenous LC3-II levels were measured by western blotting with anti-LC3 antibodies and quantitative analysis by densitometric analysis relative to β-actin. Mean ± SD, n = 3. ***P < 0.001. D. A549 cells were seeded into the confocal plate and treated with 50 μM Ax, 0.005 μg/mL PTX and co-treated 0.005 μg/mL PTX with 50 μM Ax for 24 h. Then, cells were co-stained with Hoechst 33342 and Cyto-ID green dye. Scale bar: 20 μm.
Table 1.
IC50 values of PTX in combination with 20, 50 and 100 μM Ax on A549 cells following 24, 48 and 72 h treatment, respectively. Mean ± SD, n = 6
| Incubation Time (h) | IC50 (ng/mL) | |||
|---|---|---|---|---|
|
| ||||
| PTX | PTX + 20 μM Ax | PTX + 50 μM Ax | PTX + 100 μM Ax | |
| 24 | 107.00 ± 4.38 | 41.68 ± 3.35 | 9.67 ± 0.97 | 1.49 ± 0.13 |
| 48 | 61.61 ± 3.59 | 24.92 ± 2.57 | 3.63 ± 0.41 | 0.74 ± 0.11 |
| 72 | 12.77 ± 0.86 | 6.09 ± 0.82 | 1.13 ± 0.15 | 0.22 ± 0.04 |
Table 2.
IC50 values of DTX in combination with 20, 50 and 100 μM Ax on A549 cells following 24, 48 and 72 h treatment, respectively. Mean ± SD, n = 6
| Incubation Time (h) | IC50 (ng/mL) | |||
|---|---|---|---|---|
|
| ||||
| DTX | DTX + 20 μM Ax | DTX + 50 μM Ax | DTX + 100 μM Ax | |
| 24 | 86.24 ± 4.92 | 40.73 ± 3.82 | 8.85 ± 1.03 | 1.36 ± 0.15 |
| 48 | 49.48 ± 3.37 | 20.74 ± 2.38 | 3.34 ± 0.36 | 0.70 ± 0.13 |
| 72 | 10.26 ± 0.62 | 5.11 ± 0.70 | 1.05 ± 0.12 | 0.20 ± 0.03 |
Ax treatment promoted PTX-induced cell death, which may be attributed to enhance cellular uptake. To verify this possibility, we measured cellular uptake of PTX with Ax co-incubated. Interestingly, Ax did not influence cellular uptake of PTX, which remained unchanged (Figure 4B), suggesting Ax promoted PTX-induced cell death had nothing to do with cellular uptake. Furthermore, the level of LC3-II was further increased after co-treating PTX with Ax compared to PTX or Ax treatment alone (Figure 4C). Accordingly, we found considerable autophagosomes induced by PTX treatment, and autophagosomes were further accumulated when PTX was co-incubated with Ax (Figure 4D). These results indicated that Ax potentiated PTX-induced cell death, which might be associated with enhanced autophagy level but had nothing to do with cellular uptake.
Ax treatment enhanced anti-tumor effect of PTX to A549 subcutaneous tumor
In order to evaluate therapeutic benefits of PTX in combination with Ax in vivo, we used A549 subcutaneous tumor nude mice as the models. Different groups of tumor-bearing mice were treated by intraperitoneal injections with i) saline, ii) 100 mg/kg Ax, iii) 3 mg/kg PTX, iv) 3 mg/kg PTX + 20 mg/kg Ax, v) 10 mg/kg PTX, vi) 3 mg/kg PTX + 100 mg/kg Ax every three days for four consecutive cycles. The tumor size of mice was measured every two days for 14 days. Ax alone at the concentration used showed no effects on tumor suppression. iii) and v) showed an effective inhibition of tumor growth with different degrees. At the same time, iv) had a modest tumor-shrinking effect while vi) showed a significant effect on tumor-killing, superior to v) (Figure 5A). All mice were sacrificed, also stripped tumors were photographed and weighed after 14 days of treatment. iii) caused a 33.79% decrease in tumor weight, and iv) showed a 42.23% reduction, which illustrated 8.44% enhancement in anti-tumor efficacy. Notably, vi) Ax led to a 75.48% shrink, representing 33.25% improvement in anti-tumor efficacy compared to iv), which was also superior to v), a 68.66% shrink (Figure 5B, 5C). The body weight of nude mice kept normally rising except the group of v), indicating that v) might have toxicity to some extent while other treatment groups had no accompanied toxicity (Supplementary Figure 3).
Figure 5.
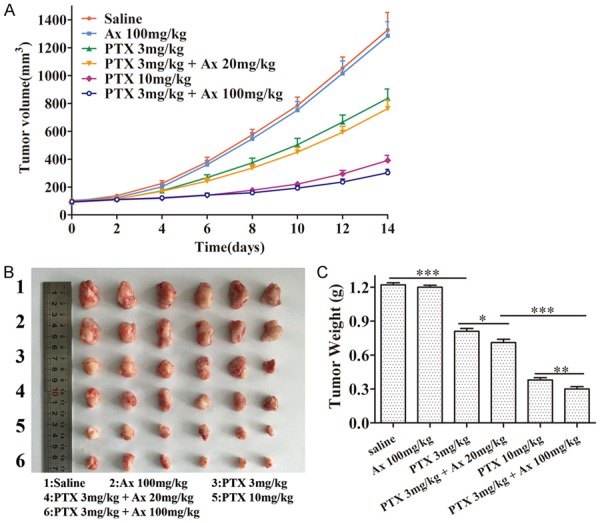
In vivo anti-tumor effects of different treatment groups in A549 subcutaneous nude mice model. A. The changes of tumor volume of subcutaneous tumor-bearing nude mice at the indicated time. Mean ± SD, n = 6. B. Tumor morphology and size at the experimental end point (14th day). C. Tumor weight at the experimental end point (14th day). Mean ± SD, n = 6. *P < 0.05, **P < 0.01, ***P < 0.001.
In accordance with the results of tumor weight, the TUNEL assay revealed that there were the most apoptotic cells in the tumor of mice treated with vi), which were more than those treated with v), while fewer apoptotic cells were observed in the mice given with iii) or iv) (Figure 6A). IHC assay was used to evaluate the distribution of endogenous LC3 proteins for detecting autophagy level within tumor tissues. The level of LC3 within tumor tissues were elevated after Ax or PTX treatment alone. However, a combination of PTX and Ax led to a further increased LC3 level in vivo (Figure 6B). These results suggested that better anti-tumor effect was linked to higher level of LC3 within tumor tissues. Taken together, the above data demonstrated Ax potentiated anti-tumor effect of PTX to A549 subcutaneous tumor with no accompanying toxicity, and higher dosage of Ax exhibited better performance. Detect of tumor tissues by TUNEL and IHC showed Ax further enhanced cell apoptosis and LC3 level.
Figure 6.
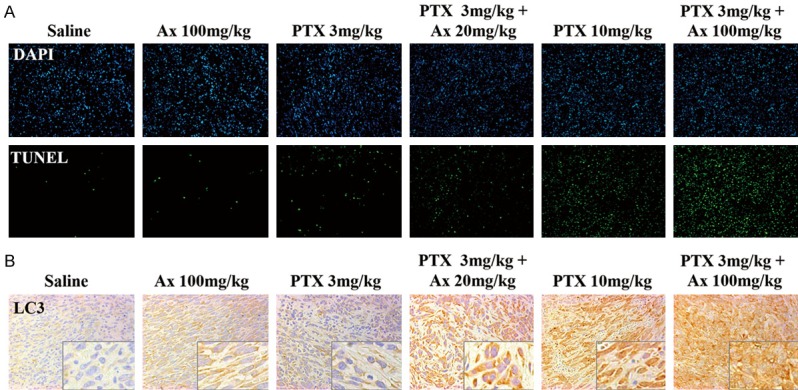
TUNEL and LC3 distribution assay of tumor tissues. A. TUNEL staining (green) of the subcutaneous tumor tissues of each group were performed to show apoptosis cells. Cell nucleus were stained with DAPI (blue). Microscopy 200 × magnification. B. LC3 distribution in the subcutaneous tumor tissues of each group. Microscopy 400 × magnification. The lower-right images were the enlarged ones.
Ax treatment improved anti-tumor effect of PTX to A549-luciferase pulmonary metastatic tumor
To investigate the treatment effect of PTX in combination with Ax to pulmonary tumor, we utilized A549 cells transfected with luciferase expression vector to visualize development process of lung cancer. A549-luciferase pulmonary metastatic tumor nude mice were used as the models, and the intensity of bioluminescence in lung indicated cancer progression. The treatment groups and dosage regimen were in accordance with the experiment of A549 subcutaneous tumor nude mice described previously. The pulmonary tumor size was photographed every five days for 15 days. The death time of mice was recorded to obtain survival curve and median survival time (MST). ii) alone showed unobvious suppressed effect on tumor development (Figure 7A). The tumor-suppressed effect of iv) was superior to iii), with MST prolonged from 30.0 days to 33.0 days but no statistically significant difference. However, vi) showed stronger tumor-shrinking effects than iv), with MST prolonged from 33.0 days to 44.0 days. Furthermore, vi) had better anti-tumor performance than v), with MST prolonged from 37.5 days to 44.0 days (Figure 7B, 7C). Above all, these results demonstrated Ax enhanced tumor-shrinking effect of PTX to A549-luciferase pulmonary metastatic tumor and prolonged MST, and higher dosage of Ax exhibited better performance.
Figure 7.
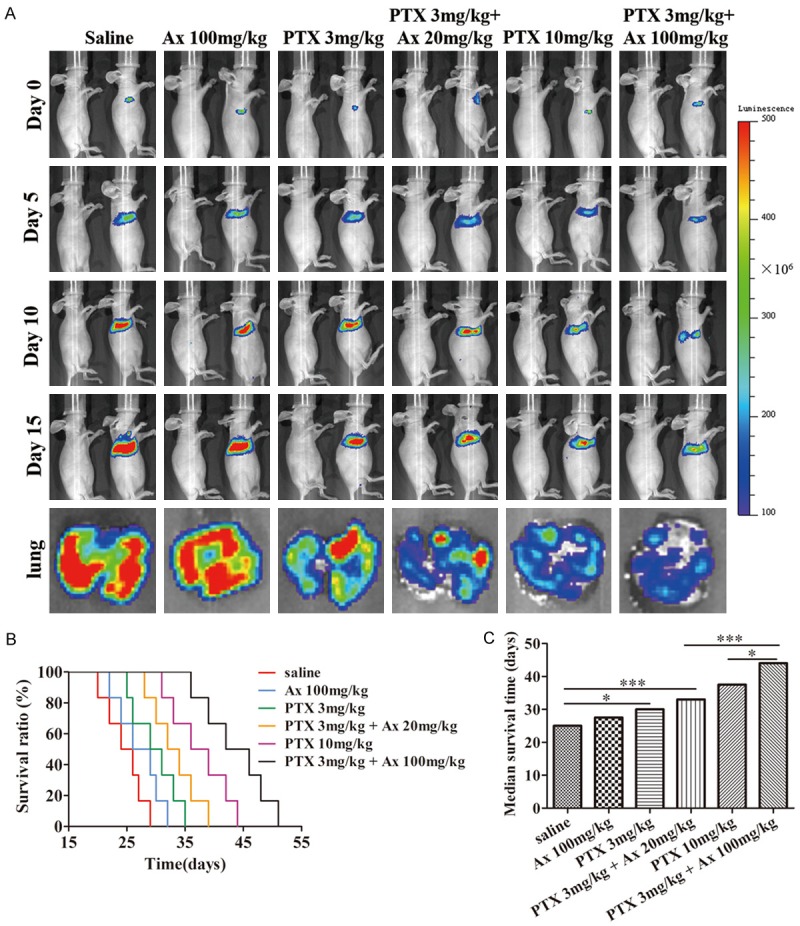
In vivo anti-cancer effects of different treatment groups in A549-luciferase pulmonary metastatic nude mice model. A. The changes of pulmonary tumor size were detected at 0, 5, 10, 15 days by IVIS spectrum. Pulmonary tumor morphology and bioluminescence were photographed at the experimental end point (15th day). B. Survival curves of pulmonary tumor-bearing nude mice at different treatment groups. C. MST of pulmonary tumor-bearing nude mice were processed by GraphPad Prism 5. *P < 0.05, ***P < 0.001.
Discussion
Lung cancer has been a severe public health problem worldwide. Platinum-based chemotherapy provides a survival benefit for patients with advanced lung cancer. However, more than half of the patients fail to survive to one year, as most tumors quickly acquire resistance to chemotherapy drugs [33]. Besides, molecular targeted therapy drugs, like gefitinib and erlotinib, also impose restrictions on patient’s benefits due to development of drug resistance [34,35]. The potential mechanisms of resistance include the ABC transporter family, autophagy induction, cancer stem cell regulation, miRNA regulation, and hypoxia induction etc. [36,37]. Autophagy induction plays a pivotal mechanism among them. In fact, many tumor cells show higher autophagy level than normal cells, and thus are more vulnerable to regulation of autophagy pathways [38]. For pancreatic cancer, enhanced basal levels of autophagy were measured in tumor specimens, and they were shown to enable tumor cell growth by maintaining cellular energy production. Suppression of autophagy in these cells contributed to tumor regression and prolonged survival in pancreatic cancer xenografts and genetic mouse models [10]. In view that autophagy is responsible for drug resistance and reduces treatment effect, co-treatment of chemotherapy drugs, even including drug loading micelles [39], nanoparticles [40], and autophagy inhibitors serve as a promising and potential way to overcome resistance and achieve better therapeutic effects.
Recently, some classical drugs have been explored the novel functions. The most representative drug is aspirin, a non-steroidal anti-inflammatory drug that has been found the new application of tumor chemoprevention and anti-cancer activity, attributed to inhibit the cyclooxygenase 2-prostaglandin axis and nuclear factor ĸB [41,42]. Ax is also a classical drug that is mainly used for expectorant of respiratory diseases. Ax improves the postoperative lung function and optimizes the perioperative airway management for non-small cell lung cancer patients in fast track surgery through reducing the incidence of pulmonary complications and shortening time of hospital stay [43,44]. Using large doses of Ax would lead to better clinical effects than using normal doses in preventing post-operation complications for patients with lung cancer [25]. Compared to other autophagy inhibitors, Ax has excellent safety and has been applied to lung protection for many years, which is appropriate to lung cancer therapy.
In our study, a series of systematic evidences illuminated that Ax indeed promoted autophagosomes accumulation. Ax treatment caused a blocked autophagic flux and interrupted intracellular cargo degradation, which showed that autophagosomes accumulation was derived from decreased autophagosomes turnover but not increased autophagosomes formation. Furthermore, we found that inhibited autophagy by Ax was due to alkalization [45] and impaired degradation capacity of lysosome. Ax treatment exerted no effect on the phosphorylation level of mTOR and its substrate p70 S6k. These results demonstrated for the first time that Ax inhibited autophagy by which impaired lysosomal function and blocked late-stage autophagic flux with mTOR-p70 S6k signaling pathway independent.
Chemotherapy drugs can induce autophagy in either pro-survival or pro-death mechanism [46-48]. PTX induced protective autophagy that hindered eventual death of endometrial carcinoma cell and osteosarcoma cell, and the combination of PTX and autophagy inhibitor achieved a potent and improved treatment outcome [49]. On the other hand, parthenolide induced autophagy in pro-death way which CQ, 3-MA, or LC3-II siRNA could reduce parthenolide-induced apoptosis [50]. For A549 cells, PTX induced autophagy in pro-survival mechanism, and co-treatment with Ax augmented PTX-induced cytotoxicity with time- and dose-dependent. At the same time, co-incubation of PTX and Ax led to a further increased LC3 accumulation, indicating excessive autophagy may be stimulated. Autophagy is a cytoprotective mechanism in most cases while may also contribute to another form of cell death. In some cases, excessive and continuous autophagy lead to cell death for important organelles depleted, activity of lysosomal enzymes reduced and caspase-dependent apoptosis pathway activated, which is called autophagy cell death (ACD) [51,52]. ACD is a non-apoptotic cell death based on autophagy pathways, which plays a crucial role in development and treatment of malignant tumor. Whether enhanced anti-proliferation activity by combining PTX with Ax is associated with ACD needs to be further investigated.
A549 subcutaneous or A549-luciferase pulmonary metastatic tumor nude mice were utilized to evaluate anticancer activity in vivo. Both in two tumor models, low dose Ax showed a modest increased anti-tumor effect when combined with PTX, while high dose one exerted a more significant curative effect, indicting the dose of Ax was a vital role in therapeutic schedules. Smaller volume of tumor tissues exhibited higher level of LC3, which indicated the improvement of curative outcome was associated with the enhancement of autophagy level. Actually, unsatisfied toxicity and side effect impose restrictions on clinical application of PTX. In our study, 10 mg/kg PTX exerted a valid anti-tumor effect with a certain toxicity resulting in reduced weight, while 3 mg/kg PTX + 100 mg/kg Ax achieved stronger anti-tumor effect and longer MST with unobserved toxicity. Therefore, adding Ax to standard lung cancer treatment regimen can enhance therapy outcome and reduce side effect.
Our present data highlighted that Ax was a novel autophagy inhibitor that caused alkalization of lysosome lumen and blocked late-stage autophagic flux. Based on the multiple advantages including lung protection and low toxicity, Ax shows enormous potential and preponderance in clinical lung cancer treatment. The combination of PTX and Ax exerted an enhanced anti-proliferation activity in vitro and achieved a potent and improved curative outcome in A549 subcutaneous or pulmonary metastatic tumor nude mice models. Our research provides a promising therapeutic strategy to lung cancer, which has clinical transformation potential and practical application value.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81773201, 81573006).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 2.Chen Y, Zhang W, Huang Y, Gao F, Fang X. Dual-functional c(RGDyK)-decorated Pluronic micelles designed for antiangiogenesis and the treatment of drug-resistant tumor. Int J Nanomedicine. 2015;10:4863–4881. doi: 10.2147/IJN.S86827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen L, Sha X, Jiang X, Chen Y, Ren Q, Fang X. Pluronic P105/F127 mixed micelles for the delivery of docetaxel against Taxol-resistant non-small cell lung cancer: optimization and in vitro, in vivo evaluation. Int J Nanomedicine. 2013;8:73–84. doi: 10.2147/IJN.S38221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao X, Fang Y, Yang Y, Qin Y, Wu P, Wang T, Lai H, Meng L, Wang D, Zheng Z, Lu X, Zhang H, Gao Q, Zhou J, Ma D. Elaiophylin, a novel autophagy inhibitor, exerts antitumor activity as a single agent in ovarian cancer cells. Autophagy. 2015;11:1849–1863. doi: 10.1080/15548627.2015.1017185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang M, Su L, Xiao Z, Liu X. Methyl jasmonate induces apoptosis and pro-apoptotic autophagy via the ROS pathway in human nonsmall cell lung cancer. Am J Cancer Res. 2016;6:187–199. [PMC free article] [PubMed] [Google Scholar]
- 6.Peynshaert K, Manshian BB, Joris F, Braeckmans K, De Smedt SC, Demeester J, Soenen SJ. Exploiting intrinsic nanoparticle toxicity: the pros and cons of nanoparticle-induced autophagy in biomedical research. Chem Rev. 2014;114:7581–7609. doi: 10.1021/cr400372p. [DOI] [PubMed] [Google Scholar]
- 7.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10:1533–1541. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65–75. doi: 10.1016/j.cell.2014.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kubisch J, Turei D, Foldvari-Nagy L, Dunai ZA, Zsakai L, Varga M, Vellai T, Csermely P, Korcsmaros T. Complex regulation of autophagy in cancer-integrated approaches to discover the networks that hold a double-edged sword. Semin Cancer Biol. 2013;23:252–261. doi: 10.1016/j.semcancer.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 10.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell’antonio G, Mautner J, Tonon G, Haigis M, Shirihai OS, Doglioni C, Bardeesy N, Kimmelman AC. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–729. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Q, Si S, Schoen S, Chen J, Jin XB, Wu G. Suppression of autophagy enhances preferential toxicity of paclitaxel to folliculin-deficient renal cancer cells. J Exp Clin Cancer Res. 2013;32:99. doi: 10.1186/1756-9966-32-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang SF, Wang XY, Fu ZQ, Peng QH, Zhang JY, Ye F, Fu YF, Zhou CY, Lu WG, Cheng XD, Xie X. TXNDC17 promotes paclitaxel resistance via inducing autophagy in ovarian cancer. Autophagy. 2015;11:225–238. doi: 10.1080/15548627.2014.998931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Apel A, Zentgraf H, Buchler MW, Herr I. Autophagy-A double-edged sword in oncology. Int J Cancer. 2009;125:991–995. doi: 10.1002/ijc.24500. [DOI] [PubMed] [Google Scholar]
- 14.Zhao X, He Y, Chen H. Autophagic tumor stroma: mechanisms and roles in tumor growth and progression. Int J Cancer. 2013;132:1–8. doi: 10.1002/ijc.27664. [DOI] [PubMed] [Google Scholar]
- 15.Song Y, Li W, Peng X, Xie J, Li H, Tan G. Inhibition of autophagy results in a reversal of taxol resistance in nasopharyngeal carcinoma by enhancing taxol-induced caspase-dependent apoptosis. Am J Transl Res. 2017;9:1934–1942. [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Z, Zhao J, Xue J, Zhao XR, Liu PS. Autophagy inhibition promotes epithelial-mesenchymal transition through ROS/HO-1 pathway in ovarian cancer cells. Am J Cancer Res. 2016;6:2162–2177. [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Z, Zhu S, Zhang G, Liu S. Inhibition of autophagy enhances the anticancer activity of bortezomib in B-cell acute lymphoblastic leukemia cells. Am J Cancer Res. 2015;5:639–650. [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Z, He K, Ma Q, Yu Q, Liu C, Ndege I, Wang X, Yu Z. Autophagy inhibitor facilitates gefitinib sensitivity in vitro and in vivo by activating mitochondrial apoptosis in triple negative breast cancer. PLoS One. 2017;12:e0177694. doi: 10.1371/journal.pone.0177694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu S, Li X. Autophagy inhibition enhances sensitivity of endometrial carcinoma cells to paclitaxel. Int J Oncol. 2015;46:2399–2408. doi: 10.3892/ijo.2015.2937. [DOI] [PubMed] [Google Scholar]
- 20.Gao M, Xu Y, Qiu L. Enhanced combination therapy effect on paclitaxel-resistant carcinoma by chloroquine co-delivery via liposomes. Int J Nanomedicine. 2015;10:6615–6632. doi: 10.2147/IJN.S91463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baranwal AK, Murthy AS, Singhi SC. Highdose oral ambroxol for early treatment of pulmonary acute respiratory distress syndrome: an exploratory, randomized, controlled pilot Trial. J Trop Pediatr. 2015;61:339–350. doi: 10.1093/tropej/fmv033. [DOI] [PubMed] [Google Scholar]
- 22.Li Q, Yao GQ, Zhu X. High-dose ambroxol reduces pulmonary complications in patients with acute cervical spinal cord injury after surgery. Neurocrit Care. 2012;16:267–272. doi: 10.1007/s12028-011-9642-4. [DOI] [PubMed] [Google Scholar]
- 23.McNeill A, Magalhaes J, Shen C, Chau KY, Hughes D, Mehta A, Foltynie T, Cooper JM, Abramov AY, Gegg M, Schapira AH. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain. 2014;137:1481–1495. doi: 10.1093/brain/awu020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanchez-Martinez A, Beavan M, Gegg ME, Chau KY, Whitworth AJ, Schapira AH. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci Rep. 2016;6:31380. doi: 10.1038/srep31380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, Wang L, Wang H, Zhang H. Perioperative lung protection provided by high-dose ambroxol in patients with lung cancer. Cell Biochem Biophys. 2015;73:281–284. doi: 10.1007/s12013-015-0590-z. [DOI] [PubMed] [Google Scholar]
- 26.Wang S, Huang D, Ma Q, Chen X. Does ambroxol confer a protective effect on the lungs in patients undergoing cardiac surgery or having lung resection? Interact Cardiovasc Thorac Surg. 2014;18:830–834. doi: 10.1093/icvts/ivu061. [DOI] [PubMed] [Google Scholar]
- 27.Nathan N, Taytard J, Duquesnoy P, Thouvenin G, Corvol H, Amselem S, Clement A. Surfactant protein A: a key player in lung homeostasis. Int J Biochem Cell Biol. 2016;81:151–155. doi: 10.1016/j.biocel.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 28.Mitsuhashi A, Goto H, Kuramoto T, Tabata S, Yukishige S, Abe S, Hanibuchi M, Kakiuchi S, Saijo A, Aono Y, Uehara H, Yano S, Ledford JG, Sone S, Nishioka Y. Surfactant protein A suppresses lung cancer progression by regulating the polarization of tumor-associated macrophages. Am J Pathol. 2013;182:1843–1853. doi: 10.1016/j.ajpath.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hasegawa Y, Takahashi M, Ariki S, Asakawa D, Tajiri M, Wada Y, Yamaguchi Y, Nishitani C, Takamiya R, Saito A, Uehara Y, Hashimoto J, Kurimura Y, Takahashi H, Kuroki Y. Surfactant protein D suppresses lung cancer progression by downregulation of epidermal growth factor signaling. Oncogene. 2015;34:838–845. doi: 10.1038/onc.2014.20. [DOI] [PubMed] [Google Scholar]
- 30.Patel AR, Chougule MB, I T, Patlolla R, Wang G, Singh M. Efficacy of aerosolized celecoxib encapsulated nanostructured lipid carrier in nonsmall cell lung cancer in combination with docetaxel. Pharm Res. 2013;30:1435–1446. doi: 10.1007/s11095-013-0984-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu M, Jiang Y, Feng Q, Ouyang Y, Gan J. DRAM1 protects neuroblastoma cells from oxygen-glucose deprivation/reperfusion-induced injury via autophagy. Int J Mol Sci. 2014;15:19253–19264. doi: 10.3390/ijms151019253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan Q, Joshua AM, Saggar JK, Yu M, Wang M, Kanga N, Zhang JY, Chen X, Wouters BG, Tannock IF. Effect of pantoprazole to enhance activity of docetaxel against human tumour xenografts by inhibiting autophagy. Br J Cancer. 2015;112:832–840. doi: 10.1038/bjc.2015.17. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Han BH, Jin B, Chu TQ, Niu YJ, Dong Y, Xu JL, Gu AQ, Zhong H, Wang HM, Zhang XY, Shi CL, Zhang YW, Zhang W, Lou YQ, Zhu L, Pei J. Combination of chemotherapy and gefitinib as first-line treatment for patients with advanced lung adenocarcinoma and sensitive EGFR mutations: a randomized controlled trial. Int J Cancer. 2017;141:1249–1256. doi: 10.1002/ijc.30806. [DOI] [PubMed] [Google Scholar]
- 34.Sechler M, Cizmic AD, Avasarala S, Van Scoyk M, Brzezinski C, Kelley N, Bikkavilli RK, Winn RA. Non-small-cell lung cancer: molecular targeted therapy and personalized medicine-drug resistance, mechanisms, and strategies. Pharmgenomics Pers Med. 2013;6:25–36. doi: 10.2147/PGPM.S26058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spaans JN, Goss GD. Drug resistance to molecular targeted therapy and its consequences for treatment decisions in non-smallcell lung cancer. Front Oncol. 2014;4:190. doi: 10.3389/fonc.2014.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu Q, Yang Z, Nie Y, Shi Y, Fan D. Multidrug resistance in cancer chemotherapeutics: mechanisms and lab approaches. Cancer Lett. 2014;347:159–166. doi: 10.1016/j.canlet.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 37.Panzarini E, Dini L. Nanomaterial-induced autophagy: a new reversal MDR tool in cancer therapy? Mol Pharm. 2014;11:2527–2538. doi: 10.1021/mp500066v. [DOI] [PubMed] [Google Scholar]
- 38.Wu YN, Yang LX, Shi XY, Li IC, Biazik JM, Ratinac KR, Chen DH, Thordarson P, Shieh DB, Braet F. The selective growth inhibition of oral cancer by iron core-gold shell nanoparticles through mitochondria-mediated autophagy. Biomaterials. 2011;32:4565–4573. doi: 10.1016/j.biomaterials.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 39.Zhang X, Zeng X, Liang X, Yang Y, Li X, Chen H, Huang L, Mei L, Feng SS. The chemotherapeutic potential of PEG-b-PLGA copolymer micelles that combine chloroquine as autophagy inhibitor and docetaxel as an anti-cancer drug. Biomaterials. 2014;35:9144–9154. doi: 10.1016/j.biomaterials.2014.07.028. [DOI] [PubMed] [Google Scholar]
- 40.Lin J, Huang Z, Wu H, Zhou W, Jin P, Wei P, Zhang Y, Zheng F, Zhang J, Xu J, Hu Y, Wang Y, Li Y, Gu N, Wen L. Inhibition of autophagy enhances the anticancer activity of silver nanoparticles. Autophagy. 2014;10:2006–2020. doi: 10.4161/auto.36293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu L, Shi L, Zeng J, Wen Z. Aspirin as a potential modality for the chemoprevention of breast cancer: a dose-response meta-analysis of cohort studies from 857,831 participants. Oncotarget. 2017;8:40389–40401. doi: 10.18632/oncotarget.16315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zong M, Fan DD, Lin S, Song YP, Wang ZY, Ma XL, Qiu WH, Bai YH, Li L, Li S. Anti-cancer activity and potential mechanism of a novel aspirin derivative. Eur J Pharmacol. 2016;791:137–146. doi: 10.1016/j.ejphar.2016.07.050. [DOI] [PubMed] [Google Scholar]
- 43.Wang JY, Hong X, Chen GH, Li QC, Liu ZM. Mucosolvan serves to optimize perioperative airway management for NSCLC patients in fast track surgery: a randomized placebo controlled study. Eur Rev Med Pharmacol Sci. 2015;19:2875–2881. [PubMed] [Google Scholar]
- 44.Gao Y, Cheng Y, Dong S, He Z, Zhou W, Liang L, Zhang C. [Effect of perioperative treatment with ambroxol on lung cancer patients after video-assisted thoracic surgery lobectomy] . Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2014;39:849–854. doi: 10.3969/j.issn.1672-7347.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 45.Fois G, Hobi N, Felder E, Ziegler A, Miklavc P, Walther P, Radermacher P, Haller T, Dietl P. A new role for an old drug: ambroxol triggers lysosomal exocytosis via pH-dependent Ca(2) (+) release from acidic Ca(2)(+) stores. Cell Calcium. 2015;58:628–637. doi: 10.1016/j.ceca.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 46.Swart C, Du Toit A, Loos B. Autophagy and the invisible line between life and death. Eur J Cell Biol. 2016;95:598–610. doi: 10.1016/j.ejcb.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 47.Lin S, Yang L, Shi H, Du W, Qi Y, Qiu C, Liang X, Shi W, Liu J. Endoplasmic reticulum-targeting photosensitizer Hypericin confers chemosensitization towards oxaliplatin through inducing pro-death autophagy. Int J Biochem Cell Biol. 2017;87:54–68. doi: 10.1016/j.biocel.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 48.O’Donovan TR, O’Sullivan GC, McKenna SL. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy. 2011;7:509–524. doi: 10.4161/auto.7.6.15066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo Y, Huang C, Li G, Chen T, Li J, Huang Z. Paxilitaxel induces apoptosis accompanied by protective autophagy in osteosarcoma cells through hypoxia-inducible factor-1alpha pathway. Mol Med Rep. 2015;12:3681–3687. doi: 10.3892/mmr.2015.3860. [DOI] [PubMed] [Google Scholar]
- 50.Liu W, Wang X, Sun J, Yang Y, Li W, Song J. Parthenolide suppresses pancreatic cell growth by autophagy-mediated apoptosis. Onco Targets Ther. 2017;10:453–461. doi: 10.2147/OTT.S117250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 2015;22:367–376. doi: 10.1038/cdd.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhai B, Hu F, Jiang X, Xu J, Zhao D, Liu B, Pan S, Dong X, Tan G, Wei Z, Qiao H, Jiang H, Sun X. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol Cancer Ther. 2014;13:1589–1598. doi: 10.1158/1535-7163.MCT-13-1043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.