

Abstract
Ovarian cancer is the 8th most common cancer in women, and the 5th leading cause of cancer-related deaths among women in the United States. Statins have been shown to have promising anti-tumorigenic activity in many types of cancers. We sought to determine the effects of atorvastatin (ATO) on cell proliferation in ovarian cancer and identify the mechanisms by which ATO inhibits cell growth in this disease. ATO inhibited cell proliferation of both the Hey and SKOV3 ovarian cancer cells in a dose-dependent manner. The anti-proliferative activity of ATO in the ovarian cancer cell lines was associated with induction of apoptosis, autophagy, cellular stress and cell cycle G1 arrest via inhibition of AKT/mTOR and activation of the MAPK pathways. Moreover, ATO inhibited cell adhesion and invasion as well as decreased expression of VEGF and MMP9. c-Myc was downregulated in ovarian cancer cells exposed to ATO. Inhibition of c-Myc by JQ1 synergistically increased the sensitivity of ovarian cancer cells to ATO. This data suggests that ATO may have a therapeutic role in the treatment of ovarian cancer and warrant further exploration in clinical trials.
Keywords: Atorvastatin, ovarian cancer, HMGCR, apoptosis, autophagy, mTOR
Introduction
Ovarian cancer has the highest mortality of all gynecologic cancers, with a dismal 5 year overall survival of only 46% [1]. This makes ovarian cancer the 5th leading cause of cancer deaths in women [1-3]. Despite aggressive surgery and adjuvant treatment, a significant proportion of women with ovarian cancer will ultimately develop disease recurrence and chemo-resistance [4]. Currently there are no chemotherapeutic agents that effectively target chemo-resistant or late stage ovarian cancer [4,5]. The development of new, targeted therapies is paramount to decreasing ovarian cancer related mortality and improving overall survival.
The mevalonate pathway serves as the main pathway for cholesterol production. This pathway also produces numerous non-steroid isoprenoid side products that are critical for functioning in both normal and cancer cells. These isoprenoid derivatives, including cholesterol, dolichol, ubiquinone, isopentenyladenine, geranylgeranyl pyrophosphate and farnesyl pyrophosphate, have been shown to promote tumor cell growth, differentiation, migration, and intracellular trafficking [6,7]. Transformed malignant cells are also highly dependent on the mevalonate pathway for the synthesis of lipid moieties critical for cell proliferation, membrane integrity, cell cycle progression and cell signaling [8]. Upregulated activity of the mevalonate pathway has been shown in a range of different cancers, including ovarian cancer [6,9,10]. Thus, inhibition of the mevalonate pathway using HMG-CoA reductase inhibitors should result in decreased levels of mevalonate and its downstream products, and thus, may have significant inhibitory influences on cancer cell growth [9,11,12].
HMG-CoA reductase (3-hydroxy-3-methylglutharyl-coenzyme A reductase, HMGCR), which is responsible for cholesterol and isoprenoid derivative production, is the rate limiting enzyme in the mevalonate pathway. Inhibition of HMGCR activity results in decreased levels of mevalonate and its downstream products that affects critical cell functions such as membrane integrity, cell signaling, protein synthesis and cell cycle progression [8,13]. The level of HMGCR has been associated with carcinogenesis and tumor progression in several cancers [10,13]. Targeting HMGCR using statins increases tumor specific apoptosis, induces cell cycle arrest and inhibits tumor growth [11,12]. The expression of HMGCR has been found to be an independent predictor of prolonged recurrence-free survival in ovarian cancer [9-12].
Statins are a class of HMGCR inhibitors traditionally used to treat hyperlipidemia and to decrease the risk of cardiovascular disease [8]. In the United States, nearly 30% of adults age 40-75 take a statin [14]. Epidemiologic studies have shown that long term statin use is associated with a decreased risk of developing ovarian cancer [14-16]. Our group has recently shown that simvastatin significantly inhibits cell proliferation and induces apoptosis and cell cycle arrest in endometrial cancer and ovarian cancer in vitro and in vivo, suggesting that simvastatin may serve as a potential agent in the management of gynecologic malignancies [11,12].
Atorvastatin (ATO) is the most commonly used statin approved for cholesterol reduction and has a very favorable toxicity profile [17]. This agent is more effective at lowering LDL and reducing the risk of peripheral artery disease and cardiovascular disease than simvastatin [18]. ATO’s main side effects are GI disturbances, which are usually temporary. As a class of drugs, statins are known for causing myalgias; however, ATO causes appreciably fewer myalgias than simvastatin [18,19]. Previous epidemiological studies indicate that ATO may be associated with a reduced risk of developing cancers, and animal studies have found that ATO effectively inhibits tumor growth in breast, prostate, pancreatic and liver cancer [20-24]. Given that ATO has demonstrated more beneficial effects than simvastatin in the management of lowering lipid levels and reducing side effects, ATO warrants further investigation as a potential therapeutic agent in ovarian cancer. Thus, the objective of this study was to explore the effect of ATO on growth, apoptosis and adhesion/invasion in human ovarian cancer cell lines.
Methods
Cell culture and reagents
Two ovarian cancer cell lines, Hey and SKOV3, were used for all experiments. The Hey cells were grown in RPMI 1640 medium supplemented with 5% fetal bovine serum, 100 units/ml penicillin and 100 ug/ml streptomycin under 5% CO2. The SKOV3 cells were grown in DMEM supplemented with 10% fetal bovine serum, 300 mM l-glutamine, 10,000 U/ml penicillin and 10,000 μg/ml streptomycin under 5% CO2. ATO, RNase and RIPA buffer were purchased from Sigma (St. Louis, MO). Antibodies to phosphorylated-AKT (Ser473), phosphorylated-p44/42 MAPK (Thr202/Tyr204), phosphorylated-S6 (Ser235/236), c-Myc, PERK, Bip, BCL-2, MCL-1, PARP, ATG3, ATG6, cleaved PARP, β-actin, pan-AKT, pan-p44/42 MAPK, pan-S6, VEGF and MMP9 were obtained from Cell Signaling Technology (Beverly, MA). The Annexin V FITC kit was purchased from BioVision (Mountain View, CA). JQ1 was kindly gifted by Dr James E. Bradner (Harvard Medical School, MA). Enhanced chemiluminescence western immunoblotting detection reagents were purchased from Amersham (Arlington Heights, IL). All other chemicals were purchased from Sigma.
Cell proliferation assay
The Hey and SKOV3 cells were plated and grown in 96-well plates at a concentration of 5000 cells/well for 24 hours. Cells were then treated with varying concentrations of ATO for 72 hours. After the addition of MTT dye (5 mg/mL), the 96-well plates were incubated for 1-2 hours at 37°C. 100 uL of DMSO was added to the plates in order to terminate the MTT reaction. The plates were read by measuring absorption at 595 nm. The effect of ATO was calculated as a percentage of control cell growth obtained from phosphate-buffered saline (PBS 1%) treated cells grown in the same 96-well plates. Each experiment was repeated three times to assess for consistency of results.
Colony formation assay
The Hey and SKOV3 cells growing in log phase were seeded in 6 cm dishes (800 cells/dish) in their standard growth medium. Cells were allowed to adhere for 24 hours, and then were treated with ATO for 24 hours. The cells were cultured at 37°C for 14 days with medium changes every third or fourth day. Cells were stained with 0.5% crystal violet, and colonies were counted under the microscope. Each experiment was repeated three times for consistency of results.
Cell cycle analysis
The effect of ATO on cell cycle progression was assessed using Cellometer (Nexcelom, Lawrence, MA). Cells were plated at a density of 2 × 105 cells/well in 6-well plates overnight and then treated with varying concentrations of ATO for 48 hours. Cells were collected by 0.05% trypsin (Gibco Grand Island, NY), washed with PBS solution, fixed in a 90% methanol solution and then stored at -20°C until cell cycle analysis was performed. On the day of analysis, the cells were washed with PBS and centrifuged, resuspended in 50 ul RNase A solution (250 ug/ml) with 10 mM EDTA, followed by incubation for 30 min at 37°C. After incubation, 50 µl of propidium iodide (PI) staining solution (2 mg/ml PI, 0.1 mg/ml Azide and 0.05% Triton X-100) was added to each tube and incubated for 10 min in the dark. The cells were assessed by Cellometer. The results were analyzed using FCS4 express software (Molecular Devices, Sunnyvale, CA). Each experiment was repeated at least twice for consistency of response.
Annexin V assay
The effect of ATO on cell apoptosis was detected using the Annexin-V FITC kit. Briefly, 2 × 105 cells/well were seeded into 6-well plates overnight and then the cells were cultured in media with varying concentrations of ATO for 24 hours. The cells were collected by 0.25% trypsin without EDTA. After PBS washing, cells were resuspended in 100 ul of Annexin-V and PI dual-stain solution (0.1 ug of Annexin-V FITC and 1 ug of PI) for 15 min in the dark. Apoptotic cells were detected by Cellometer. The results were analyzed by FCS4 express software. Each experiment was repeated at least twice for consistency of response.
Reactive oxygen species (ROS) assay
ROS generation was assessed using the ROS-sensitive fluorescence indicator, DCFH-DA. To determine intracellular ROS scavenging activity, the Hey and SKOV3 cells (1.0 × 104 cells/well) were seeded in black 96-well plates. After 24 hours, the cells were treated with ATO for 24 hours to induce ROS generation. After the cells were incubated with DCFH-DA (20 μM) for 30 minutes, the fluorescence intensity was measured at an excitation wavelength of 485 nm and an emission wavelength of 530 nm using a fluorescence microplate reader. All experiments were performed in duplicate to assess for consistency of response.
Adhesion assay
Each well in a 96-well plate was coated with 100 ul laminin-1 (10 ug/ml) and incubated at 37°C for 1 hour. The fluid was then aspirated, and 200 ul blocking buffer was added to each well for 45-60 min at 37°C. The wells were then washed with PBS, and the plate was allowed to chill on ice. Next, 2.5 × 103 cells were added with PBS and varying concentrations of ATO directly. The plate was then allowed to incubate at 37°C for 2 hours. After this period, the medium was aspirated, and cells were fixed by directly adding 100 ul of 5% glutaraldehyde. The plates were then incubated for 30 min at room temperature. Adherent cells were washed with PBS and stained with 100 ul of 0.1% crystal violet for 30 minutes. The cells were then washed repeatedly with water, and 100 ul of 10% acetic acid was added to each well to solubilize the dye. After 5 minutes of shaking, the absorbance was measured at 570 nm using a micro-plate reader from Tecan (Morrisville, NC). Each experiment was repeated at least twice for consistency of response.
Invasion assay
Cell invasion assays were performed using 96-well HTS transwells (Corning Life Sciences, Durham, NC) coated with 0.5-1 × BME (Trevigen, Gaithersburg, MD). The Hey and SKOV3 cells (50,000 cells/well) were starved for 12 hours and then seeded in the upper chambers of the wells in 50 μl FBS-free medium. The lower chambers were filled with 150 μl standard medium with ATO. The plates were incubated for 16 hours at 37°C to allow invasion into the lower chamber. After washing the upper and lower chambers with PBS, 100 ul Calcein AM solution was added into the lower chambers and incubated at 37°C for 30-60 min. The lower chamber plate was measured by the plate reader for reading fluorescence at EX/EM 485/520 nM. Each experiment was repeated at least twice for consistency of response.
Western immunoblotting
The Hey and SKOV3 cells were plated at 2 × 105 cells/well in 6 well plates in their corresponding medium and were treated for 24 hours with ATO. Cell lysates were prepared in RIPA buffer (1% NP40, 0.5 sodium deoxycholate and 0.1% SDS) plus PhosStop. Equal amounts of protein were separated by gel electrophoresis and transferred onto a PVDF membrane. The membrane was blocked with 5% nonfat dry milk and then incubated with a 1:1000 dilution of primary antibody overnight at 4°C. The membrane was then washed and incubated with a secondary peroxidase conjugated antibody for 1 hour after washing. Antibody binding was detected using an enhanced chemiluminescence detection buffer by Alpha Innotech imaging system (San Leandro, CA). Each experiment was repeated three times to assess for consistency of results.
Statistical analysis
Data are presented as a mean ± the standard error of the mean. Comparisons between groups were determined with the two-sided unpaired student’s t-test using GraphPad software (La Jolla, CA USA). A value of P < 0.05 was considered significant. The combination-index (CI) methods, derived from the median-effect principle of Chou and Talalay, were used to define the pharmacologic interaction between the ATO and JQ1. CI < 1 or CI > 1 indicates synergism or antagonism, respectively.
Results
The effect of ATO on cell proliferation in ovarian cancer cells
The effects of ATO on cell proliferation were examined in the SKOV3 and Hey ovarian cancer cell lines. Both cell lines were exposed to varying doses of ATO (1-250 uM) for 72 hours. MTT assay showed that ATO decreased cell proliferation in a dose-dependent manner in both cell lines after 72 hours of treatment, with IC50 values of 122 uM for the Hey cells and 80 uM for the SKOV3 cells (Figure 1A).
Figure 1.
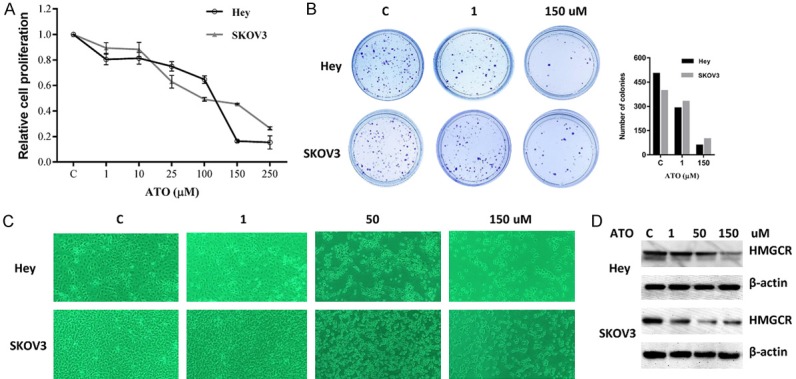
ATO inhibited the proliferation of ovarian cancer cells. The Hey and SKOV3 cells were cultured for 24 hours and then treated with varying concentrations of ATO in 96 well plates for 72 hours. Cell proliferation was assessed by MTT assay (A). The Hey and SKOV3 cells were seeded at low density in 6 cm dishes and treated with ATO for 24 hours. The cells were cultured for 14 days with medium changes every third or fourth day. Colonies were visualized by crystal violet staining (B). Morphologies of the Hey and SKOV3 cells after treatment of ATO for 48 hours (C). The effect of ATO on HMGCR was examined by Western blot analysis. ATO treatment resulted in a dose-dependent decrease in expression of HMGCR protein in both cell lines (D). Each experiment was performed three times.
Given that the colony formation assay is an excellent indicator of long term tumor cell survival, a colony formation assay was performed to investigate the long-term effect of ATO on cell growth in both cell lines. As shown in Figure 1B, the colony-forming ability of Hey and SKOV3 was reduced by 88% and 75%, respectively, after exposure to 150 uM of ATO for 14 days. This data suggest that ATO effectively decreases cell growth in ovarian cancer cells. The effects of ATO on cellular morphology in both cell lines is shown in Figure 1C. Control cells were round or oval shape with large, clear nuclei. Following ATO treatment for 48 hours, the treated cells shrunk and displayed a rounder shape. The size and density of the treated cells were also greatly decreased.
To assess the effect of ATO on HMGCR, we treated Hey and SKOV3 cells with varying doses of ATO for 24 hours. Western blotting results showed a significant decrease in the expression of HMGCR in both cell lines with atorvastatin treatment (Figure 1D).
Atorvastatin induced cell cycle arrest and apoptosis
To assess the underlying mechanism of growth inhibition of the ovarian cancer cells by ATO, the cell cycle profile was analyzed by Cellometer after treating the Hey and SKOV3 cell lines with varying doses of ATO. ATO induced G1 phase arrest and decreased S phase in both cell lines after 24 hours of treatment (Figure 2A). In the Hey cells, G1 arrest increased from 49% in control cells to 68% in cells treated with 150 uM of ATO. In the SKOV3 cells, treatment with ATO increased G1 arrest from 44% in controls to 62% at a dose of 150 μM.
Figure 2.
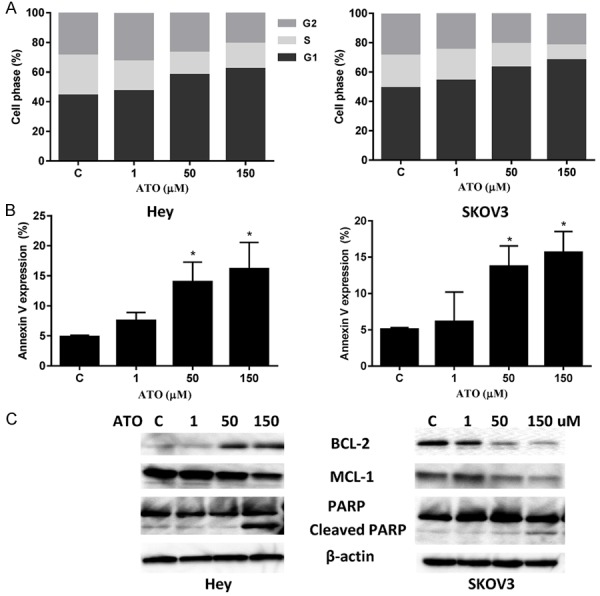
ATO induced cell cycle G1 arrest and apoptosis in ovarian cancer cells. The Hey and SKOV3 cell lines were treated with the indicated doses of ATO (1-150 uM) for 24 hours. Cell cycle analysis was performed by Cellometer. ATO markedly induced cell cycle G1 arrest in both cell lines in a dose dependent manner (A). Apoptosis was detected using Annexin-V FITC assay in both cell lines after 18 hours of treatment. ATO increased Annexin V expression in the Hey and SKOV3 cells (B). Western blotting indicated treatment ATO for 18 hours decreased the expression of MCL-1 and increased Cleaved PARP expression in both cell lines (C). Data are shown as mean + SEM of two experiments (*P < 0.05). Each experiment was performed three times.
We then examined the effects of ATO on cell apoptosis using the Annexin V assay, which detects early apoptotic cells by monitoring fluorescent labeled Annexin V. The Hey and SKOV-3 cells were treated with ATO at varying concentrations (1-150 uM) for 18 hours. ATO increased the percentage of cells in apoptosis in a dose-dependent manner in both the Hey and SKOV3 cell lines, with slightly more apoptosis seen in the Hey cells (Figure 2B).
We also assessed the effects of ATO on the apoptotic proteins, MCL-1, BCL-2 and PARP in both ovarian cancer cell lines. Our western blotting results showed that ATO decreased MCL-1 expression in both cell lines, and increased BCL-2 expression in the Hey cells and reduced BCL-2 expression in the SKOV3 cells. We also found that ATO treatment increased cleaved PARP protein expression at dose of 150 uM in both cell lines (Figure 3C), suggesting that inducing mitochondrial apoptosis may be one mechanism by which ATO inhibits cell proliferation in ovarian cancer cells. These data, combined with the cell cycle results, indicate that ATO inhibits cell proliferation through induction of cell cycle G1 arrest and apoptosis in ovarian cancer cells.
Figure 3.
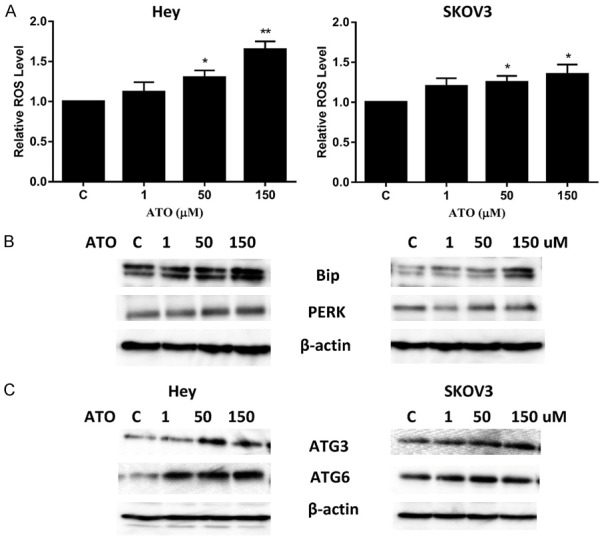
ATO caused cellular stress and autophagy in ovarian cancer cells. The Hey and SKOV3 cell lines were treated with ATO at different concentrations for 24 hours and reactive oxygen species (ROS) levels were determined using DCFH-DA dye on a plate reader (A). PERK and Bip were determined by Western immunoblotting after exposure to ATO for 24 h (B). Autophagy in the Hey and SKOV3 cell lines was analyzed by immunoblotting after treatment with ATO for 24 hours. ATO increased the expression of ATG3 and ATG6 in the cells (C). Each experiment was performed three times. (*P < 0.05, **P < 0.01).
ATO induces cell stress and autophagy
Increased reactive oxygen species (ROS) can cause damage to a variety of cellular components and eventually lead to cell death in cancer cells [25]. The effect of ATO on ROS production in the Hey and SKOV3 cells was evaluated. ATO increased ROS levels in both cell lines in a dose-dependent manner after 24 hours of treatment. The difference in ROS levels between the ATO doses of 50 and 150 uM and control were statistically different in both cell lines (Figure 3A). Additionally, alternations in the expression of the cellular stress proteins, Bip and PERK, was analyzed by western immunoblotting. Treatment with ATO for 24 hours resulted in a significant increase in expression of Bip and PERK in both cell lines. At a dose of 150 μM, ATO increased Bip expression 2.36-fold in the Hey cells and 1.92-fold in the SKOV3 cells. Similarly, 150 uM of ATO increased PERK expression by 1.41-fold in the Hey cells and 1.18-fold in the SKOV3 cells.
Conventional cytotoxic drugs and irradiation have been shown to induce autophagy in cancer cells [26]. The effect of ATO on autophagy in both cell lines was examined. The Hey and SKOV3 cells were treated with different concentrations of ATO for 24 hours. Western immunoblotting results showed that ATO induced ATG3 and ATG6 expression in a dose-dependent manner in both cell lines. At a dose of 50 uM, ATO increased ATG3 expression by 2.1 fold and ATG6 expression by 3.2 fold in the Hey cells. Similarly, ATG3 expression was increased by 1.3 fold and ATG expression by 1.6 fold in the SKOV3 cells. These results suggest that in addition to cell cycle G1 arrest and apoptosis, autophagy may be responsible for inhibition of cell growth induced by ATO in ovarian cancer cells.
The effect of ATO on AKT/mTOR and MAPK pathways
Given that the AKT/mTOR and MAPK pathways have been shown to regulate glucose and lipid metabolism in cancer cells, we investigated whether these pathways were involved in the anti-proliferative effects of ATO in ovarian cancer cells. Western blotting showed that phosphorylation of S6 was repressed by ATO in a dose dependent manner after 24 hours of treatment in both cell lines (Figure 4). ATO increased phosphorylation of AKT in the Hey cells, and decreased phosphorylation of ERK in the SKOV3 cells. Together, these results suggest that ATO targets the mTOR pathway; however, the inhibition of cell proliferation by ATO may involve different pathways in different ovarian cancer cells.
Figure 4.
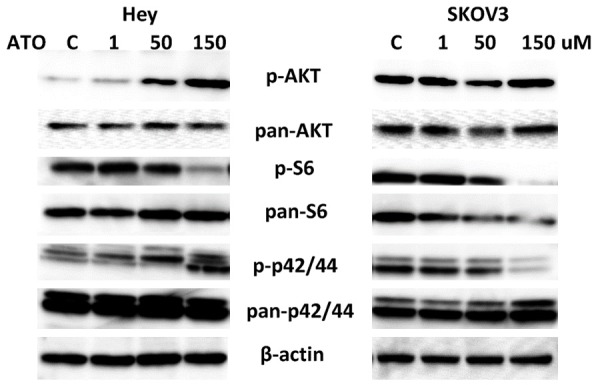
Effect of ATO on MAPK and AKT/mTOR pathways in ovarian cancer cells. The Hey and SKOV3 cells were treated with ATO at different doses for 24 h. Phosphorylated-p42/44, phosphorylated-AKT and phosphorylated-pS6 were assessed by Western blotting. ATO inhibited the activity of AKT/mTOR pathway in both cell lines, increased MAPK activity in the Hey cells and decreased phosphorylation of p42/44 in the SKOV3 cells. Each experiment was performed two times.
ATO inhibited cell adhesion and invasion
In order to determine the effect of ATO on the invasive ability of ovarian cancer cells, a laminin adhesion assay and a transwell invasion system were employed. Incubation of the Hey and SKOV3 cells with ATO (1, 50 and 150 uM) for 2 hours showed significant inhibition of cell adhesion (Figure 5A). ATO significantly blocked ovarian cancer cell invasion after 16 hours of treatment as determined by the transwell invasion assay (Figure 5B). Inhibition of cell invasion was dose-dependent in both cell lines. Since vascular endothelial growth factor (VEGF) and matrix metalloproteinase (MMPs) are important mediators of angiogenesis and invasion in ovarian cancer, we sought to determine the effects of ATO on production of VEGF and MMP9 in both cell lines. ATO significantly reduced the expression of VEGF and MMP9 after 24 hours of treatment (Figure 5C). These results suggest that ATO may function to inhibit adhesion and invasion and decrease angiogenesis, as well as inducing apoptosis, cell cycle arrest and cellular stress in the Hey and SKOV3 cell lines.
Figure 5.
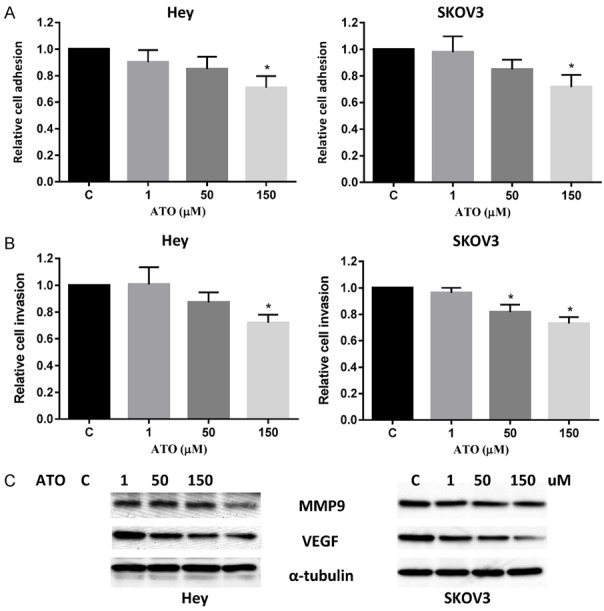
ATO decreased adhesion and invasion in ovarian cancer cells. The Hey and SKOV3 cell lines were cultured for 24 hours and then treated as indicated with ATO in a laminin-coated 96 well plate for 2 hours to assess adhesion (A) or a BME coated 96 transwell plate for 16 hours to assess invasion (B), respectively. The data represents relative inhibition in each cell line. VEGF and MMP9 was measured by Western blotting in cell lysates after a 24 hour exposure to ATO. ATO decreased the expression of VEGF and MMP9 expression in a dose-dependent manner (C). Each experiment was performed three times. (*P < 0.05, **P < 0.01).
c-Myc was involved in inhibition of cell growth by ATO
c-Myc is a central oncogene that acts to control cell proliferation, differentiation, metabolism and apoptosis in ovarian cancer. It has been previously shown that inhibition of HMG-COA by ATO significantly reduces the phosphorylation and activation of c-Myc [27]. We hypothesized that the inhibition of c-Myc by JQ1, a small molecular inhibitor for c-Myc, would increase the sensitivity of ovarian cancer cells to ATO. We conducted MTT assays which demonstrated that the combination of 100 nM of JQ1 with ATO at varying concentrations resulted in synergistic inhibitory effects in both ovarian cancer cell lines after 72 hours of treatment (Figure 6A, CI < 1). The combination of JQ1 and ATO significantly reduced the expression of c-Myc compared to the control (Figure 6B). We also found that combination treatment resulted in greater induction of apoptosis, as seen by increased annexin V expression as well as heightened cell cycle G1 arrest in both cell lines compared with the controls (Figure 6C, 6D). These results support that the inhibition of c-Myc results in increased sensitivity of the ovarian cancer cells to ATO.
Figure 6.
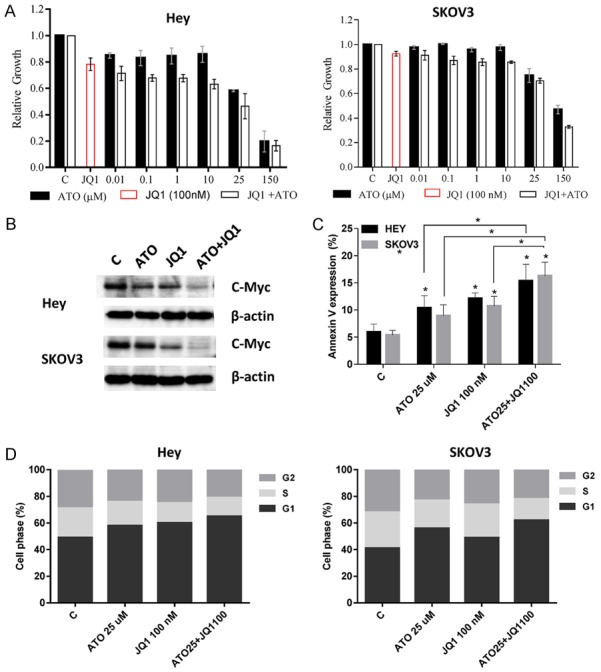
JQ1 sensitized ATO in ovarian cancer cells. The cells were treated with different concentrations of ATO in media with 100 nM JQ1 for 72 h. MTT assays showed that JQ1 increased sensitivity to ATO (A). Western blotting showed that JQ1, ATO and the combination of JQ1 and ATO reduced expression of c-Myc (B). Inhibition of c-Myc by JQ1 significantly increased Annexin V expression (C) and cell cycle G1 arrest (D) induced by ATO in the cells after 24 hours of treatment. Data are shown as mean + SEM of three experiments (*P < 0.05).
Discussion
Statins such as simvastatin, mevastatin, and lovastatin have been shown to decrease cell viability in several ovarian cancer cell lines [11,28,29]. Our recent results found that treatment with simvastatin in an orthotopic mouse model reduced ovarian tumor growth, coincident with decreased Ki-67, HMGCR, phosphorylated-Akt and phosphorylated-p42/44 protein expression [11]. In the present study, our results indicate that ATO, similar to simvastatin, exhibits anti-tumorigenic activity in ovarian cancer cells through induction of apoptosis, cell cycle G1 arrest and autophagy. Furthermore, reduced cell proliferation by ATO was associated with inhibition of downstream targets of the mTOR and MAPK pathways. Intriguingly, inhibition of c-Myc by JQ1 synergized cellular sensitivity to ATO through induction of apoptosis and cell cycle arrest in ovarian cancer cells. Taken together, our data provide insight into the underlying mechanisms of ATO’s anti-proliferative effects in ovarian cancer cells and reveals a potential novel combination strategy in this disease, i.e. inhibition of c-Myc combined with ATO.
Previous investigators have shown that deprivation of mevalonate results in G1 phase cell cycle arrest in the MCF-7 breast cancer cells that is in part mediated by impaired activity of cyclin-dependent kinase 2 (CDK2) and decreased expression of positive regulators of G1 to S phase cell cycle progression [30]. Similarly, targeting HMG-COA by statins also induces malignant cells to undergo apoptosis and cell cycle arrest [31]. Many in vitro studies have shown that statins induce cell cycle G1 or S phase arrest in a time- and dose-dependent manner through affecting cell cycle regulatory proteins and activating apoptosis. Statins accomplish this anti-proliferative effect likely through inhibition of cholesterol synthesis and prenylation of G proteins [31]. Reduction of the proliferative capacity of colon cancer cells by ATO is associated with cell cycle G1 arrest via upregulation of p21(Cip1/Waf1), p27(Kip1) and phospho-JNK, downregulation of phosphosphorylated-AKT, hyperphosphorylation of Rb and activation of the caspase cascade [32,33]. A recent phase II window-of-opportunity trial of ATO in breast cancer confirmed that ATO significantly increases the expression of p27 and decreases cyclin D1 staining in tumor tissues, suggesting that cell cycle regulatory effects may contribute to anti-proliferative activity induced by ATO via cyclin D1 and p27 [34]. In our study, treating ovarian cancer cells with ATO induced cell cycle G1 phase arrest and increased the expression of annexin V and cleaved PARP in a dose-dependent manner. This is consistent with the proposed mechanisms of action of ATO.
Autophagy is a cellular process of self-ingestion that delivers cytoplasmic contents to the lysosome. Autophagy plays an important role in balancing sources of energy and protecting cells in response to metabolic stress [35]. Inhibition of cell proliferation by simvastatin has been associated with an increase in cellular ROS production and induction of autophagy in breast cancer, lung cancer, lymphoma and ovarian cancer [36-38]. We found that ATO exhibits similar effects in induction of autophagy and cellular stress, which has also been reported in lymphoma and glioma cells [37,39]. Additionally, a recent study found that ATO was also shown to elevate activity of autophagy and induce autophagy-associated cell death in PC3 prostate cancer cells [40]. Our findings suggest that ATO functions to inhibit ovarian cancer proliferation, not only by affecting cell cycle and apoptosis, but also by inducing cellular stress and autophagy. The stress induced by ATO led to an increase in cellular ROS production, which was accompanied by increased AGT3 and AGT6 expression and mitochondrial apoptosis, ultimately leading to cell death in the ovarian cancer cell lines. Thus, our results suggest that inhibition of cell proliferation by ATO involves multiple cellular processes, along with its known targeting of HMGCR.
Several studies have suggested that the anti-angiogenic and anti-metastatic activities of statins are achieved by inhibiting proliferation and migration of endothelial cells, inducing cell apoptosis and cellular stress, reducing cell migration and possibly impacting several signaling pathways including the AKT/mTOR, p53, Rho and MAPK pathways, etc. [11,41,42]. It has been reported that treatment with ATO at a 10 uM concentration can significantly decrease migration and invasion of U87 glioblastoma spheroid cells after 48 hours of treatment [43]. Treatment of non-small cell lung carcinoma cells with ATO also significantly inhibited VEGF expression both in vitro and in vivo by affecting ROS production [44]. Additionally, a recent study found that ATO reduced the pro-tumorigenic effects of microglia on glioma migration and invasion by reducing the microglial expression of membrane type 1 metalloproteinase through the p38 MAPK pathway [45]. To determine whether ATO exerted anti-angiogenic and anti-metastatic activities in ovarian cancer cells, we used laminin-1 and transwell assays to access the capacity of adhesion and invasion. As previously seen with simvastatin in ovarian cancer, ATO reduced ovarian cancer cell invasion and migration and inhibited VEGF protein expression in a dose-dependent manner.
Recent studies have implicated several signaling pathways as mediators of statin-dependent anti-proliferative and pro-apoptotic effects [37,46]. Simvastatin has been found to activate AMP-activated protein kinase (AMPK), downregulate AKT and inhibit mTOR activity in glioma cells [46]. Fluvastatin induced apoptosis by increasing the activation of caspase-3 and by enhancing Bim expression through inhibition of the Ras/ERK and Ras/mTOR pathways in human head and neck squamous cell carcinoma cell lines [47]. ATO effectively suppressed anchorage-independent growth of ovarian cancer cells in soft agar along with activation of Jun N-terminal kinases (JNK) [48]. In this study, we examined the effect of ATO on the phosphorylation of AKT, ERK and S6, which are frequently overexpressed in many forms of ovarian cancer. Our findings reveal a significant decrease in S6 phosphorylation in both cell lines with an increase in AKT and ERK in the Hey cells and reduction of ERK phosphorylation in the SKOV3 cells. The diverse effects of ATO on these signaling pathways suggests that ATO inhibits cell proliferation via multiple signaling pathways in various ovarian cancer cells.
The activity of HMGCR regulates c-Myc phosphorylation via Rac. C-Myc phosphorylation is a critical mechanism by which the inhibition of HMG-CoA reductase by ATO mediates its anti-neoplastic effects [27]. Inhibition of HMG-CoA reductase by ATO inhibits c-Myc phosphorylation and activation, resulting in blocked c-Myc-induced initiation and growth of liver cancer and lymphomagenesis in vivo [27,49]. Hence, we speculated that by inhibiting c-Myc activity using JQ1, ATO might enhance its anti-proliferative effect in ovarian cancer cells. After treating the ovarian cancer cells with different concentrations of ATO in combination with JQ1, we found that ATO reduced c-Myc expression after 24 hours of treatment, yielding the lowest overall level of c-Myc with combination treatment. Importantly, inhibition of c-Myc activity by JQ1 in combination with ATO exhibited a synergistic inhibitory effect through increasing of apoptosis and cell cycle G1 phase arrest in the Hey and SKOV3 cell lines (CI < 1). Together, these results suggest that inhibition of c-MYC activity appears to be a potential mechanism by which ATO inhibits cell viability in ovarian cancer cells.
Currently, statins are being studied clinically for use in the prevention and treatment of cancer and as an adjuvant treatment in combination with chemotherapeutic agents [11]. ATO’s side effects are well tolerated in the long-term management of dyslipidemia, and multiple clinical trials have shown an acceptable safety profile at even high doses of ATO [39,50,51]. ATO at 100 ppm (close to a clinically acceptable intermediate dose range, 40 mg/d of atorvastatin) significantly blocks pancreatic carcinogenesis and increases survival in Pankras/p53 mice [52]. Combinations of atorvastatin (0.02% in diet) and celecoxib had a stronger inhibitory effect on the formation and growth of androgen-independent LNCaP tumors than either drug alone in SCID mice [53]. Breast cancer patients who were given atorvastatin two weeks prior to surgery showed anti-proliferative effects, with a decrease in Ki-67 staining in those tumors that were HMGCR positive [54]. These findings indicate that ATO is a potential novel and well-tolerated chemotherapeutic agent for use in the prevention and treatment of cancers including ovarian cancer and warrants further investigation in clinical trials for ovarian cancer.
Acknowledgements
This work was generously supported by NIH/NCI 1K23CA143154-01A1 and the Steelman fund.
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Ataseven B, Chiva LM, Harter P, Gonzalez-Martin A, du Bois A. FIGO stage IV epithelial ovarian, fallopian tube and peritoneal cancer revisited. Gynecol Oncol. 2016;142:597–607. doi: 10.1016/j.ygyno.2016.06.013. [DOI] [PubMed] [Google Scholar]
- 3.Narod S. Can advanced-stage ovarian cancer be cured? Nat Rev Clin Oncol. 2016;13:255–261. doi: 10.1038/nrclinonc.2015.224. [DOI] [PubMed] [Google Scholar]
- 4.Sehouli J, Grabowski JP. Surgery for recurrent ovarian cancer: options and limits. Best Pract Res Clin Obstet Gynaecol. 2017;41:88–95. doi: 10.1016/j.bpobgyn.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 5.Oronsky B, Ray CM, Spira AI, Trepel JB, Carter CA, Cottrill HM. A brief review of the management of platinum-resistant-platinum-refractory ovarian cancer. Med Oncol. 2017;34:103. doi: 10.1007/s12032-017-0960-z. [DOI] [PubMed] [Google Scholar]
- 6.Mullen PJ, Yu R, Longo J, Archer MC, Penn LZ. The interplay between cell signalling and the mevalonate pathway in cancer. Nat Rev Cancer. 2016;16:718–731. doi: 10.1038/nrc.2016.76. [DOI] [PubMed] [Google Scholar]
- 7.Dimitroulakos J, Lorimer IA, Goss G. Strategies to enhance epidermal growth factor inhibition: targeting the mevalonate pathway. Clin Cancer Res. 2006;12:4426s–4431s. doi: 10.1158/1078-0432.CCR-06-0089. [DOI] [PubMed] [Google Scholar]
- 8.Pisanti S, Picardi P, Ciaglia E, D’Alessandro A, Bifulco M. Novel prospects of statins as therapeutic agents in cancer. Pharmacol Res. 2014;88:84–98. doi: 10.1016/j.phrs.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 9.Greenaway JB, Virtanen C, Osz K, Revay T, Hardy D, Shepherd T, DiMattia G, Petrik J. Ovarian tumour growth is characterized by mevalonate pathway gene signature in an orthotopic, syngeneic model of epithelial ovarian cancer. Oncotarget. 2016;7:47343–47365. doi: 10.18632/oncotarget.10121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brennan DJ, Brandstedt J, Rexhepaj E, Foley M, Ponten F, Uhlen M, Gallagher WM, O’Connor DP, O’Herlihy C, Jirstrom K. Tumour-specific HMG-CoAR is an independent predictor of recurrence free survival in epithelial ovarian cancer. BMC Cancer. 2010;10:125. doi: 10.1186/1471-2407-10-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stine JE, Guo H, Sheng X, Han X, Schointuch MN, Gilliam TP, Gehrig PA, Zhou C, Bae-Jump VL. The HMG-CoA reductase inhibitor, simvastatin, exhibits anti-metastatic and antitumorigenic effects in ovarian cancer. Oncotarget. 2016;7:946–960. doi: 10.18632/oncotarget.5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schointuch MN, Gilliam TP, Stine JE, Han X, Zhou C, Gehrig PA, Kim K, Bae-Jump VL. Simvastatin, an HMG-CoA reductase inhibitor, exhibits anti-metastatic and anti-tumorigenic effects in endometrial cancer. Gynecol Oncol. 2014;134:346–355. doi: 10.1016/j.ygyno.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Altwairgi AK. Statins are potential anticancerous agents (review) Oncol Rep. 2015;33:1019–1039. doi: 10.3892/or.2015.3741. [DOI] [PubMed] [Google Scholar]
- 14.Habis M, Wroblewski K, Bradaric M, Ismail N, Yamada SD, Litchfield L, Lengyel E, Romero IL. Statin therapy is associated with improved survival in patients with non-serouspapillary epithelial ovarian cancer: a retrospective cohort analysis. PLoS One. 2014;9:e104521. doi: 10.1371/journal.pone.0104521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elmore RG, Ioffe Y, Scoles DR, Karlan BY, Li AJ. Impact of statin therapy on survival in epithelial ovarian cancer. Gynecol Oncol. 2008;111:102–105. doi: 10.1016/j.ygyno.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 16.Liu Y, Qin A, Li T, Qin X, Li S. Effect of statin on risk of gynecologic cancers: a meta-analysis of observational studies and randomized controlled trials. Gynecol Oncol. 2014;133:647–655. doi: 10.1016/j.ygyno.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 17.Chen X, Liu Y, Wu J, Huang H, Du Z, Zhang K, Zhou D, Hung K, Goodin S, Zheng X. Mechanistic study of inhibitory effects of atorvastatin and docetaxel in combination on prostate cancer. Cancer Genomics Proteomics. 2016;13:151–160. [PMC free article] [PubMed] [Google Scholar]
- 18.Stoekenbroek RM, Boekholdt SM, Fayyad R, Laskey R, Tikkanen MJ, Pedersen TR, Hovingh GK Incremental Decrease in End Points Through Aggressive Lipid Lowering Study Group. High-dose atorvastatin is superior to moderate-dose simvastatin in preventing peripheral arterial disease. Heart. 2015;101:356–362. doi: 10.1136/heartjnl-2014-306906. [DOI] [PubMed] [Google Scholar]
- 19.Bakker-Arkema RG, Best J, Fayyad R, Heinonen TM, Marais AD, Nawrocki JW, Black DM. A brief review paper of the efficacy and safety of atorvastatin in early clinical trials. Atherosclerosis. 1997;131:17–23. doi: 10.1016/s0021-9150(97)06066-8. [DOI] [PubMed] [Google Scholar]
- 20.Mohammed A, Qian L, Janakiram NB, Lightfoot S, Steele VE, Rao CV. Atorvastatin delays progression of pancreatic lesions to carcinoma by regulating PI3/AKT signaling in p48Cre/+ LSL-KrasG12D/+ mice. Int J Cancer. 2012;131:1951–1962. doi: 10.1002/ijc.27456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khosropanah I, Falahatkar S, Farhat B, Heidari Bateni Z, Enshaei A, Allahkhah AA, Khosropanah D. Assessment of atorvastatin effectiveness on serum PSA level in hypercholesterolemic males. Acta Med Iran. 2011;49:789–794. [PubMed] [Google Scholar]
- 22.Braeuning A, Bucher P, Hofmann U, Buchmann A, Schwarz M. Chemically induced mouse liver tumors are resistant to treatment with atorvastatin. BMC Cancer. 2014;14:766. doi: 10.1186/1471-2407-14-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghalali A, Wiklund F, Zheng H, Stenius U, Hogberg J. Atorvastatin prevents ATP-driven invasiveness via P2X7 and EHBP1 signaling in PTEN-expressing prostate cancer cells. Carcinogenesis. 2014;35:1547–1555. doi: 10.1093/carcin/bgu019. [DOI] [PubMed] [Google Scholar]
- 24.Brown M, Hart C, Tawadros T, Ramani V, Sangar V, Lau M, Clarke N. The differential effects of statins on the metastatic behaviour of prostate cancer. Br J Cancer. 2012;106:1689–1696. doi: 10.1038/bjc.2012.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Panieri E, Santoro MM. ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death Dis. 2016;7:e2253. doi: 10.1038/cddis.2016.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10:1533–1541. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao Z, Fan-Minogue H, Bellovin DI, Yevtodiyenko A, Arzeno J, Yang Q, Gambhir SS, Felsher DW. MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG-CoA reductase. Cancer Res. 2011;71:2286–2297. doi: 10.1158/0008-5472.CAN-10-3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gauthaman K, Richards M, Wong J, Bongso A. Comparative evaluation of the effects of statins on human stem and cancer cells in vitro. Reprod Biomed Online. 2007;15:566–581. doi: 10.1016/s1472-6483(10)60390-2. [DOI] [PubMed] [Google Scholar]
- 29.Martirosyan A, Clendening JW, Goard CA, Penn LZ. Lovastatin induces apoptosis of ovarian cancer cells and synergizes with doxorubicin: potential therapeutic relevance. BMC Cancer. 2010;10:103. doi: 10.1186/1471-2407-10-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duncan RE, Lau D, El-Sohemy A, Archer MC. Geraniol and beta-ionone inhibit proliferation, cell cycle progression, and cyclin-dependent kinase 2 activity in MCF-7 breast cancer cells independent of effects on HMG-CoA reductase activity. Biochem Pharmacol. 2004;68:1739–1747. doi: 10.1016/j.bcp.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 31.Matusewicz L, Meissner J, Toporkiewicz M, Sikorski AF. The effect of statins on cancer cells--review. Tumour Biol. 2015;36:4889–4904. doi: 10.1007/s13277-015-3551-7. [DOI] [PubMed] [Google Scholar]
- 32.Xiao H, Zhang Q, Lin Y, Reddy BS, Yang CS. Combination of atorvastatin and celecoxib synergistically induces cell cycle arrest and apoptosis in colon cancer cells. Int J Cancer. 2008;122:2115–2124. doi: 10.1002/ijc.23315. [DOI] [PubMed] [Google Scholar]
- 33.Yang Z, Xiao H, Jin H, Koo PT, Tsang DJ, Yang CS. Synergistic actions of atorvastatin with gamma-tocotrienol and celecoxib against human colon cancer HT29 and HCT116 cells. Int J Cancer. 2010;126:852–863. doi: 10.1002/ijc.24766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feldt M, Bjarnadottir O, Kimbung S, Jirstrom K, Bendahl PO, Veerla S, Grabau D, Hedenfalk I, Borgquist S. Statin-induced anti-proliferative effects via cyclin D1 and p27 in a window-of-opportunity breast cancer trial. J Transl Med. 2015;13:133. doi: 10.1186/s12967-015-0486-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanchez CA, Rodriguez E, Varela E, Zapata E, Paez A, Masso FA, Montano LF, Loopez-Marure R. Statin-induced inhibition of MCF-7 breast cancer cell proliferation is related to cell cycle arrest and apoptotic and necrotic cell death mediated by an enhanced oxidative stress. Cancer Invest. 2008;26:698–707. doi: 10.1080/07357900701874658. [DOI] [PubMed] [Google Scholar]
- 37.Qi XF, Zheng L, Lee KJ, Kim DH, Kim CS, Cai DQ, Wu Z, Qin JW, Yu YH, Kim SK. HMG-CoA reductase inhibitors induce apoptosis of lymphoma cells by promoting ROS generation and regulating Akt, Erk and p38 signals via suppression of mevalonate pathway. Cell Death Dis. 2013;4:e518. doi: 10.1038/cddis.2013.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hwang KE, Kim YS, Jung JW, Kwon SJ, Park DS, Cha BK, Oh SH, Yoon KH, Jeong ET, Kim HR. Inhibition of autophagy potentiates pemetrexed and simvastatin-induced apoptotic cell death in malignant mesothelioma and nonsmall cell lung cancer cells. Oncotarget. 2015;6:29482–29496. doi: 10.18632/oncotarget.5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oliveira KA, Dal-Cim T, Lopes FG, Ludka FK, Nedel CB, Tasca CI. Atorvastatin promotes cytotoxicity and reduces migration and proliferation of human A172 glioma cells. Mol Neurobiol. 2017 doi: 10.1007/s12035-017-0423-8. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 40.He Z, Yuan J, Qi P, Zhang L, Wang Z. Atorvastatin induces autophagic cell death in prostate cancer cells in vitro. Mol Med Rep. 2015;11:4403–4408. doi: 10.3892/mmr.2015.3334. [DOI] [PubMed] [Google Scholar]
- 41.Lee SJ, Lee I, Lee J, Park C, Kang WK. Statins, 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors, potentiate the anti-angiogenic effects of bevacizumab by suppressing angiopoietin2, BiP, and Hsp90alpha in human colorectal cancer. Br J Cancer. 2014;111:497–505. doi: 10.1038/bjc.2014.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fang Z, Tang Y, Fang J, Zhou Z, Xing Z, Guo Z, Guo X, Wang W, Jiao W, Xu Z, Liu Z. Simvastatin inhibits renal cancer cell growth and metastasis via AKT/mTOR, ERK and JAK2/STAT3 pathway. PLoS One. 2013;8:e62823. doi: 10.1371/journal.pone.0062823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bayat N, Ebrahimi-Barough S, Norouzi-Javidan A, Saberi H, Tajerian R, Ardakan MM, Shirian S, Ai A, Ai J. Apoptotic effect of atorvastatin in glioblastoma spheroids tumor cultured in fibrin gel. Biomed Pharmacother. 2016;84:1959–1966. doi: 10.1016/j.biopha.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 44.Chen J, Liu B, Yuan J, Yang J, Zhang J, An Y, Tie L, Pan Y, Li X. Atorvastatin reduces vascular endothelial growth factor (VEGF) expression in human non-small cell lung carcinomas (NSCLCs) via inhibition of reactive oxygen species (ROS) production. Mol Oncol. 2012;6:62–72. doi: 10.1016/j.molonc.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yongjun Y, Shuyun H, Lei C, Xiangrong C, Zhilin Y, Yiquan K. Atorvastatin suppresses glioma invasion and migration by reducing microglial MT1-MMP expression. J Neuroimmunol. 2013;260:1–8. doi: 10.1016/j.jneuroim.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 46.Misirkic M, Janjetovic K, Vucicevic L, Tovilovic G, Ristic B, Vilimanovich U, Harhaji-Trajkovic L, Sumarac-Dumanovic M, Micic D, Bumbasirevic V, Trajkovic V. Inhibition of AMPK-dependent autophagy enhances in vitro antiglioma effect of simvastatin. Pharmacol Res. 2012;65:111–119. doi: 10.1016/j.phrs.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 47.Tsubaki M, Fujiwara D, Takeda T, Kino T, Tomonari Y, Itoh T, Imano M, Satou T, Sakaguchi K, Nishida S. The sensitivity of head and neck carcinoma cells to statins is related to the expression of their Ras expression status, and statin-induced apoptosis is mediated via suppression of the Ras/ERK and Ras/mTOR pathways. Clin Exp Pharmacol Physiol. 2017;44:222–234. doi: 10.1111/1440-1681.12690. [DOI] [PubMed] [Google Scholar]
- 48.Liu H, Liang SL, Kumar S, Weyman CM, Liu W, Zhou A. Statins induce apoptosis in ovarian cancer cells through activation of JNK and enhancement of Bim expression. Cancer Chemother Pharmacol. 2009;63:997–1005. doi: 10.1007/s00280-008-0830-7. [DOI] [PubMed] [Google Scholar]
- 49.Shachaf CM, Perez OD, Youssef S, Fan AC, Elchuri S, Goldstein MJ, Shirer AE, Sharpe O, Chen J, Mitchell DJ, Chang M, Nolan GP, Steinman L, Felsher DW. Inhibition of HMGcoA reductase by atorvastatin prevents and reverses MYC-induced lymphomagenesis. Blood. 2007;110:2674–2684. doi: 10.1182/blood-2006-09-048033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koren MJ, Hunninghake DB ALLIANCE Investigators. Clinical outcomes in managed-care patients with coronary heart disease treated aggressively in lipid-lowering disease management clinics: the alliance study. J Am Coll Cardiol. 2004;44:1772–1779. doi: 10.1016/j.jacc.2004.07.053. [DOI] [PubMed] [Google Scholar]
- 51.Bernini F, Poli A, Paoletti R. Safety of HMGCoA reductase inhibitors: focus on atorvastatin. Cardiovasc Drugs Ther. 2001;15:211–218. doi: 10.1023/a:1011908004965. [DOI] [PubMed] [Google Scholar]
- 52.Liao J, Chung YT, Yang AL, Zhang M, Li H, Zhang W, Yan L, Yang GY. Atorvastatin inhibits pancreatic carcinogenesis and increases survival in LSL-KrasG12D-LSL-Trp53R172H-Pdx1-Cre mice. Mol Carcinog. 2013;52:739–750. doi: 10.1002/mc.21916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zheng X, Cui XX, Gao Z, Zhao Y, Shi Y, Huang MT, Liu Y, Wagner GC, Lin Y, Shih WJ, Rao CV, Yang CS, Conney AH. Inhibitory effect of dietary atorvastatin and celecoxib together with voluntary running wheel exercise on the progression of androgen-dependent LNCaP prostate tumors to androgen independence. Exp Ther Med. 2011;2:221–228. doi: 10.3892/etm.2011.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bjarnadottir O, Romero Q, Bendahl PO, Jirstrom K, Ryden L, Loman N, Uhlen M, Johannesson H, Rose C, Grabau D, Borgquist S. Targeting HMG-CoA reductase with statins in a window-of-opportunity breast cancer trial. Breast Cancer Res Treat. 2013;138:499–508. doi: 10.1007/s10549-013-2473-6. [DOI] [PubMed] [Google Scholar]