

ABSTRACT
Two Gram-stain-positive, coagulase-negative staphylococcal strains were isolated from abiotic sources comprising stone fragments and sandy soil in James Ross Island, Antarctica. Here, we describe properties of a novel species of the genus Staphylococcus that has a 16S rRNA gene sequence nearly identical to that of Staphylococcus saprophyticus. However, compared to S. saprophyticus and the next closest relatives, the new species demonstrates considerable phylogenetic distance at the whole-genome level, with an average nucleotide identity of <85% and inferred DNA-DNA hybridization of <30%. It forms a separate branch in the S. saprophyticus phylogenetic clade as confirmed by multilocus sequence analysis of six housekeeping genes, rpoB, hsp60, tuf, dnaJ, gap, and sod. Matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) and key biochemical characteristics allowed these bacteria to be distinguished from their nearest phylogenetic neighbors. In contrast to S. saprophyticus subsp. saprophyticus, the novel strains are pyrrolidonyl arylamidase and β-glucuronidase positive and β-galactosidase negative, nitrate is reduced, and acid produced aerobically from d-mannose. Whole-genome sequencing of the 2.69-Mb large chromosome revealed the presence of a number of mobile genetic elements, including the 27-kb pseudo-staphylococcus cassette chromosome mec of strain P5085T (ψSCCmecP5085), harboring the mecC gene, two composite phage-inducible chromosomal islands probably essential to adaptation to extreme environments, and one complete and one defective prophage. Both strains are resistant to penicillin G, ampicillin, ceftazidime, methicillin, cefoxitin, and fosfomycin. We hypothesize that antibiotic resistance might represent an evolutionary advantage against beta-lactam producers, which are common in a polar environment. Based on these results, a novel species of the genus Staphylococcus is described and named Staphylococcus edaphicus sp. nov. The type strain is P5085T (= CCM 8730T = DSM 104441T).
IMPORTANCE The description of Staphylococcus edaphicus sp. nov. enables the comparison of multidrug-resistant staphylococci from human and veterinary sources evolved in the globalized world to their geographically distant relative from the extreme Antarctic environment. Although this new species was not exposed to the pressure of antibiotic treatment in human or veterinary practice, mobile genetic elements carrying antimicrobial resistance genes were found in the genome. The genomic characteristics presented here elucidate the evolutionary relationships in the Staphylococcus genus with a special focus on antimicrobial resistance, pathogenicity, and survival traits. Genes encoded on mobile genetic elements were arranged in unique combinations but retained conserved locations for the integration of mobile genetic elements. These findings point to enormous plasticity of the staphylococcal pangenome, shaped by horizontal gene transfer. Thus, S. edaphicus can act not only as a reservoir of antibiotic resistance in a natural environment but also as a mediator for the spread and evolution of resistance genes.
KEYWORDS: coagulase-negative staphylococci, methicillin resistance, genomics, polyphasic taxonomy, pathogenicity islands, drug resistance evolution, beta-lactams, chromosomal islands, mobile genetic elements, penicillin-binding proteins, phylogenetic analysis
INTRODUCTION
Members of the genus Staphylococcus are widespread in nature and occupy a variety of niches (1). As a result of their ubiquity and adaptability, staphylococci are a major group of bacteria inhabiting the skin, skin glands, and mucous membranes of humans, other mammals, and birds. Most environmental sources also contain small, transient populations of staphylococci, many of which are probably contaminants disseminated by human, other mammal, or bird host carriers (2, 3). Moreover, Staphylococcus succinus subsp. succinus was isolated from plant and soil inclusions in Dominican amber (4). Recently, Staphylococcus argensis was isolated from river sediments (5). A small number of species, such as Staphylococcus xylosus and Staphylococcus sciuri, have occasionally been isolated from soil, beach sand, and natural waters and also from plants (1, 6, 7). They can grow in habitats containing only an inorganic nitrogen source and, thus, might be capable of a free-living existence.
The ubiquity of S. xylosus might be explained by its ability to adapt to different environments. Its capacity to colonize biotic and abiotic surfaces is probably due to its ability to form a biofilm (8) and to genes implicated in ecological fitness (9). It has been considered a nonpathogenic commensal organism (10) and has rarely been reported to be associated with infections (11). On the other hand, Staphylococcus saprophyticus, the closest phylogenetic relative of S. xylosus, is an important opportunistic pathogen, causing human urinary tract infections, wound infections, and septicemia (12). The host range of S. saprophyticus varies from humans to lower mammals and birds (6, 13). It has been isolated from the gastrointestinal tract of both humans and animals, as well as from meat and cheese products, vegetables, and the environment (14).
The aim of this study was to investigate the genomic properties and clarify the taxonomic position of two coagulase-negative staphylococcal isolates belonging to the S. saprophyticus phylogenetic clade that could not be identified to the species level by common diagnostic techniques. The isolates are notable due to their origin from the Antarctic environment and resistance to beta-lactam antibiotics encoded by a novel pseudo-staphylococcus cassette chromosome mec (ψSCCmec) element.
TAXONOMY
Staphylococcus edaphicus (e.da'phi.cus. Gr. n. edaphos, soil; N. L. masc. adj. edaphicus, belonging to soil).
Its cells are Gram stain-positive, spherical or irregular cocci, 880 ± 60 nm in diameter, occurring predominantly in clusters, non-spore forming. Colonies on P agar after 24 h are circular with whole margins, slightly convex, smooth, shiny, whitish, 2 mm in diameter and aerobic. Weak hemolytic activity (production of δ-hemolysin) on blood agar. Good growth at 10°C, 42°C, and in the presence of 11% NaCl; weak growth in the presence of 12% NaCl or at 5°C. No growth at 0 or 45°C. No growth in M9 minimal agar. No growth in thioglycolate medium or in the presence of 14% NaCl on a tryptic soy agar (TSA) plate. Catalase, urease, pyrrolidonyl arylamidase, Voges-Proskauer test (acetoin), nitrate reduction, and hydrolysis of Tween 80 positive. Coagulase, clumping factor, hyaluronidase, thermonuclease, oxidase, arginine dihydrolase, ornithine decarboxylase, and arginine arylamidase negative. Susceptible to polymyxin B (300 IU) and furazolidone (100 μg), but resistant to bacitracin (0.2 IU) and novobiocin (5 μg). Susceptible to lysostaphin (200 mg liter−1) and resistant to lysozyme (400 mg liter−1). Hydrolysis of esculin, DNA, and gelatin negative. Butyrate esterase, caprylate esterase, myristate lipase (weak), β-glucuronidase, acid phosphatase (weak), alkaline phosphatase, and naphthol-AS-BI-phosphohydrolase (7-bromo-3-hydroxy-2-naphthoic-o-anisidide) activities are present, but not leucine arylamidase, valine arylamidase, cystine arylamidase, trypsin, α-chymotrypsin, α-galactosidase, β-galactosidase, α-glucosidase, β-glucosidase, N-acetyl-β-glucosaminidase, α-mannosidase, and α-fucosidase activities. Acid is produced from glycerol, ribose, galactose, d-glucose, fructose, mannose, mannitol, sorbitol (weak), N-acetylglucosamine, salicin, maltose sucrose, trehalose, and β-gentiobiose, but not from erythritol, d-arabinose, l-arabinose, d-xylose, l-xylose, adonitol, β-methyl-d-xyloside, sorbose, rhamnose, dulcitol, inositol, α-methyl-d-mannoside, α-methyl-d-glucoside, amygdaline, arbutine, cellobiose, lactose, melibiose, inulin, melezitose, d-raffinose, glycogen, xylitol, turanose, d-lyxose, d-tagatose, d-fucose, l-fucose, d-arabitol, l-arabitol, gluconate, 2 keto-gluconate, or 5 keto-gluconate. The test result was strain dependent for acid production from starch (CCM 8730T positive). S. edaphicus had the ability to utilize the following carbon sources via respiration as determined by the Biolog GEN III MicroPlate test panel: dextrin, d-maltose, d-trehalose, gentiobiose, sucrose, d-turanose, β-methyl-d-glucoside, d-salicin, N-acetyl-β-d-mannosamine, N-acetyl neuraminic acid, α-d-glucose, mannose, d-fructose, d-galactose, inosine, d-mannitol, d-arabitol, glycerol, glycyl-l-proline, l-alanine, l-arginine, l-aspartic acid, l-glutamic acid, l-histidine, l-serine, pectin, d-gluconic acid, d-glucuronic acid, glucuronamide, l-lactic acid, Tween 40, acetoacetic acid, acetic acid, and formic acid. The negative utilization tests were d-cellobiose, stachyose, d-raffinose, α-d-lactose, d-melibiose, N-acetyl-d-galactosamine, 3-methyl glucose, l-fucose, d-sorbitol, myo-inositol, d-glucose-6-phosphate, d-aspartic acid, d-serine, gelatin, l-pyroglutamic acid, d-galacturonic acid, d-galactonic acid lactone, mucic acid, quinic acid, d-saccharic acid, p-hydroxy phenylacetic acid, d-malic acid, bromo-succinic acid, γ-amino-butyric acid, α-hydroxy-butyric acid, β-hydroxy-d,l-butyric acid, α-keto butyric acid, and propionic acid. The strain-dependent tests were N-acetyl-d-glucosamine, l-rhamnose, d-serine, d-fructose-6-phosphate, rifamycin SV, quanidine HCl, methyl pyruvate, d-lactic acid methyl ester, citric acid, α-keto glutaric acid, and l-malic acid (CCM 8730T positive in all tests), and d-fucose (CCM 8730T negative). Chemical sensitivity assays, performed with the Biolog GEN III MicroPlate test panel, exhibited positive growth at pH 6 and in the presence of 1% NaCl, 4% NaCl, 8% NaCl, 1% sodium lactate, nalidixic acid, lithium chloride, potassium tellurite, aztreonam, and sodium butyrate. Negative growth was exhibited at pH 5 and in the presence of fusidic acid, troleandomycin, minocycline, lincomycin, Niaproof 4, vancomycin, tetrazolium violet, tetrazolium blue, and sodium bromate. The tested strains were susceptible to cephalothin, ciprofloxacin, clindamycin, erythromycin, gentamicin, chloramphenicol, imipenem, kanamycin, neomycin, co-trimoxazole, tetracycline, and vancomycin and resistant to penicillin G, ampicillin, ceftazidime, methicillin, cefoxitin, and fosfomycin.
The type strain CCM 8730T (= P5085T = DSM 104441T) was retrieved in January 2013 from fragments of black porous stone at a hill above Cape Lachman, James Ross Island, Antarctica. It had G+C content of 33.3 mol% calculated from whole genomic sequence. The majority of the characteristics of the type strain are in agreement with the general species description; in addition, the CCM 8730T strain produces acid from starch.
RESULTS AND DISCUSSION
Two isolates were obtained as a by-product in the framework of a project monitoring psychrotolerant bacteria of the phylum Bacteroidetes from abiotic sources in James Ross Island, Antarctica (CzechPolar2 project). Gram staining and phase-contrast and transmission electron microscopy showed that the cells are Gram-positive spherical cocci with the typical appearance of staphylococci, without flagella (Fig. 1). Strain P5085T (= CCM 8730T) was isolated from stone fragments sampled from a hill above Cape Lachman (GPS coordinates 63°46′58″S 57°47′11″W), and strain P5191 (= CCM 8731) was isolated from sandy soil in the Panorama Pass locality (GPS coordinates 63°48′51″S 57°50′45″W). The phenotypic characteristics of the new isolates are summarized in the species description above.
FIG 1.
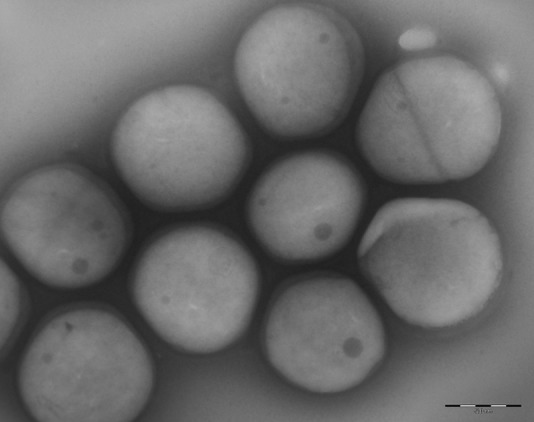
Transmission electron microscopy of type strain Staphylococcus edaphicus CCM 8730T, performed with Morgagni 268D Philips (FEI Company, USA) electron microscope. Negative staining with 2% ammonium molybdate. Bar represents 500 nm (original magnification, ×10,000).
The preliminary identification by sequencing of the 16S rRNA gene assigned both isolates to the Staphylococcus saprophyticus species group defined by Takahashi et al. (15). The whole genomic sequence of the S. saprophyticus type strain (16) even exhibited indistinguishability from that of the strain CCM 8730T 16S rRNA gene in two rrn operons, but there was a 1-residue difference in each of the three other rrn operons (positions 190, 278, and 457). However, the phenotypic results obtained, which are listed in the species description given above, did not allow for the classification of isolates into any known staphylococcal species. The key characteristics differentiating the novel taxon represented by strain CCM 8730T from the phylogenetically closely related species are shown in Table 1.
TABLE 1.
Differentiation of Staphylococcus edaphicus sp. nov. from the phylogenetically closest Staphylococcus spp.
| Test | Result obtained for indicated type strain in this study/from species descriptiona: |
|||||||
|---|---|---|---|---|---|---|---|---|
| S. edaphicus CCM 8730T | S. xylosus CCM 2738T | S. saprophyticus subsp. saprophyticus CCM 883T | S. saprophyticus subsp. bovis CCM 4410T | S. succinus subsp. succinus CCM 7157T | S. succinus subsp. casei CCM 7194T | S. equorum subsp. equorum CCM 3832T | S. equorum subsp. linens CCM 7278T | |
| Nitrate reduction | + | +/D | −/− | +/+ | −/− | +/+ | +/+ | +/+ |
| Pyrrolidonyl arylamidase | + | +/D | −/− | W/+ | −/− | −/− | −/− | −/− |
| Voges-Proskauer test (acetoin) | + | −/D | +/+ | +/D | −/− | −/− | −/− | −/− |
| Esculine hydrolysis | − | −/D | −/− | −/− | +/+ | +/+ | −/D | −/− |
| β-Glucosidase | − | +/+ | −/D | −/D | +/NT | +/NT | +/NT | −/NT |
| β-Glucuronidase | + | +/+ | −/− | −/− | +/+ | +/+ | W/+ | +/+ |
| β-Galactosidase | − | +/+ | +/+ | +/D | +/+ | +/+ | W/D | −/− |
| Acid from mannose | + | +/+ | −/− | −/− | +/D | +/+ | −/+ | +/+ |
Species description data are from Schleifer and Bell (17). +, positive reaction; W, weakly positive reaction; −, negative reaction; D, variable reaction; NT, not tested.
The strains were analyzed by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) and yielded profiles containing signals in the mass range of 2 to 15 kDa. They did not exhibit significant similarity (BioTyper log score of >1.7) to any of the reference entries belonging to Staphylococcus species with valid names included in BioTyper database version 5989 (Bruker Daltonics) and proved S. saprophyticus to be the closest relative (see Fig. S1 in the supplemental material).
Three DNA fingerprinting techniques were used to show the differences between the isolates and similarities to related taxa. Repetitive sequence-based PCR (rep-PCR) fingerprinting using the (GTG)5 primer (Fig. S2) and automated ribotyping with the EcoRI restriction enzyme (Fig. S3) showed high genetic similarity between strains CCM 8730T and CCM 8731, because they gave visually identical fingerprints. At the same time, both DNA fingerprinting techniques also clearly differentiated the strains analyzed from the type strains representing the phylogenetically close Staphylococcus spp. Strains CCM 8730T and CCM 8731 were also undistinguishable by SmaI macrorestriction analysis resolved by pulsed-field gel electrophoresis (Fig. S4).
The fatty acid content of strain CCM 8730T corresponded with those of phylogenetically related species (Table S1). The fatty acid compositions of strains CCM 8730T and CCM 8731 were found to be very similar. The major fatty acids (>10%) were C15:0 anteiso (51.4%), C15:0 iso (13.3%), and C17:0 anteiso (12.9%). C15:0 and C15:0 anteiso were found to be major fatty acids in all members of the S. saprophyticus species group. The amount of C17:0 anteiso differed slightly between members of this group, only reaching the highest value of 10% in strains CCM 8730T and CCM 8731 and both subspecies of S. saprophyticus. The overall content of other fatty acids was found to be comparable in this clade, with the exception of the S. succinus subsp. succinus type strain, where C13:0 iso and C13:0 anteiso were found to be predominant as well (4).
The genome of strain CCM 8730T was shotgun sequenced, and the contigs were compared with S. saprophyticus ATCC 15305T and S. xylosus CCM 2738T whole-genome sequences (Fig. 2). The size of the draft genome of CCM 8730T is 2.69 Mb, comprised of 45 contigs of >500 bp (N50 = 99,210 bp; 309× mean coverage) with an average G+C content of 33.3 mol%. The sequencing and assembly statistics are shown in Table S2. A total of 2,645 genes were predicted. Comparative analysis of the genes with the closest relatives identified 2,280 gene clusters and 328 singletons. The majority of the clusters, up to 2,264, are orthologous and shared with either S. xylosus or S. saprophyticus (Fig. 3). There are 16 unique gene clusters, including 39 open reading frames (ORFs) and 328 singletons specific to the CCM 8730T strain, localized mainly on variable genetic elements, such as the pseudo-Staphylococcus cassette chromosome mec (ψSCCmec), two composite phage-inducible chromosomal islands (PICIs), and one complete and one defective prophage (Fig. 2). High heterogeneity in the gene composition compared to those of related species was detected in the region following the ψSCCmec. Genes for a type II restriction modification system similar to Sau3AI were found, as well as a thiamine biosynthesis operon, two predicted protein-coding genes with an LPXTG motif, and genes for a putative Pls cell antiadhesin and cardiolipin synthase. The pls gene has been associated with virulence (18), and it has been found adjacent to SCCs present in other staphylococci (19). Cardiolipin synthesis can enhance cell survival under acid or salinity stress conditions (20, 21), and mutations in this gene enhance resistance to daptomycin (22).
FIG 2.

Circular display of Staphylococcus edaphicus CCM 8730T genome (GenBank accession number MRZN00000000) compared to the Staphylococcus saprophyticus ATCC 15305T (GenBank accession number NC_007350) and Staphylococcus xylosus CCM 2738T (GenBank accession number MRZO00000000) genomes. The picture shows (from inner to outer circle) GC skew, mol% G+C, unique regions in CCM 8730T genome based on BLASTn analysis, and orthologous regions in S. saprophyticus ATCC 15305T genome and S. xylosus CCM 2738T genome based on BLASTn analysis. Outer circle depicts locations of accessory elements in S. edaphicus CCM 8730T genome.
FIG 3.
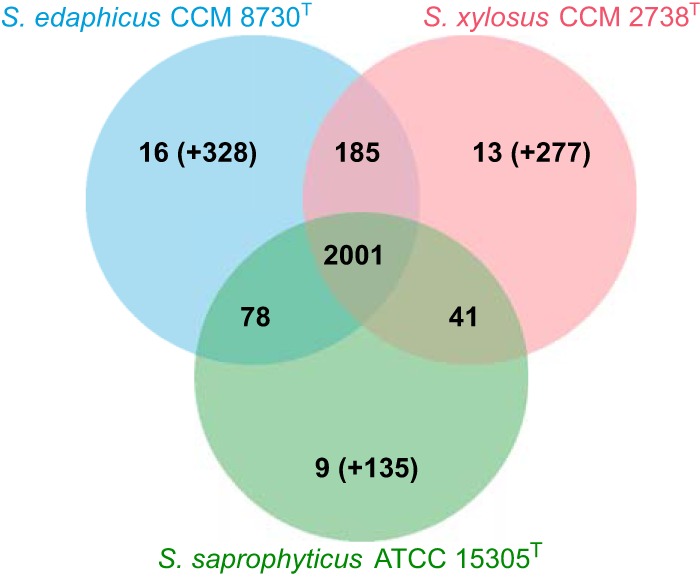
Venn diagram showing orthologous gene clusters for strain Staphylococcus edaphicus CCM 8730T (GenBank accession number MRZN00000000), Staphylococcus saprophyticus ATCC 15305T (GenBank accession number NC_007350), and Staphylococcus xylosus CCM 2738T (GenBank accession number MRZO00000000). The number of singletons specific for each genome is shown in parentheses.
The 27,158-bp ψSCCmec element of strain CCM 8730T (ψSCCmecP5085), bordered by imperfectly matched 27-bp direct repeats, CCGCATCACTTGTGATA(C/T)GCTTC(C/T)CCC, was identified inserted between the rlmH gene, encoding rRNA-methyltransferase, and the gene for putative threonyl-tRNA synthetase. This ψSCCmec contains mec gene complex class E (blaZ-mecC-mecR1-mecI), reflecting the corresponding International Working Group on the Staphylococcal Cassette Chromosome elements (IWG-SCC) designation (23), and it lacks recombinase genes (ccr) or their homologues (Fig. 4). The mecC gene shares 99% nucleotide identity with that of Staphylococcus aureus strain LGA251 (24) and 93% nucleotide identity with the mecC1 gene of S. xylosus strain S04009 (25) and with the mecC2 of S. saprophyticus strain 210 (26). Apart from human and livestock staphylococcal isolates (24), the mecC gene was previously identified in isolates from wildlife, including birds and mammals (27–29), and in waste and river waters (30, 31), but to our knowledge, this is the first time the mecC gene was identified in an isolate from soil.
FIG 4.

Comparison of genetic structure of SCCmec element type XI in Staphylococcus aureus LGA251 (GenBank accession number FR821779), ψSCCmecP5085 in Staphylococcus edaphicus CCM 8730T, and mecC1 region in Staphylococcus xylosus S04009 (GenBank accession number HE993884). Arrows indicate ORFs and their orientation on the genome. ACME, arginine catabolic mobile element.
Besides the mecC gene, very few genes were conserved among ψSCCmecP5085 and related SCCs; these were localized mainly in the mec gene complex (Fig. 4). This implies that SCC elements might serve for foreign DNA integration and exchange, as suggested for other genomic islands. Downstream from the mec gene complex of ψSCCmecP5085, there are 28 predicted ORFs encoding kinase, hydrolase, oxidoreductase, transcriptional regulator, and several hypothetical proteins, whose homologs were identified predominantly in coagulase-negative staphylococcal species. Furthermore, in ψSCCmecP5085, one truncated and other, intact lpl genes coding for tandem lipoprotein-like proteins with amino acid similarities of over 89% to those of S. epidermidis were identified. Lipoprotein-like proteins are usually encoded by νSaα genomic islands (32), and under certain nutrient limitations and environmental conditions, they may be crucial for ion and nutrient transport, allowing growth and survival (33).
Noticeably lower G+C content, below 29 mol%, clearly leads to the identification of two genomic islands (Fig. 2). The genomic analysis revealed a high resemblance of both islands to the Staphylococcus aureus pathogenicity islands (SaPIs), the prototypical members of the PICIs (34). The islands of strain CCM 8730T carry genes necessary for the transfer and autonomous replication of the element, namely, integrase, primase, small subunit of the phage terminase complex, and transcriptional regulators, and were thus classified as PICIs. However, the two genomic islands are much longer than SaPIs. The first genomic island, designated SedCIP5085-1 and found in CCM 8730T, is approximately 45 kb long with more than 70 predicted ORFs. The island is integrated into the transfer-messenger RNA (tmRNA) gene within the attachment sequence site TCCCGCCGTCTCCA(T/C)TATAGAGTCTGCAACC(C/-)AT(T/-)GTGGTTGTGGGCTTTTTATTTTTG that corresponds to the attachment site of SaPIm/νSa3 (35). Two additional copies of the att site were found in this island, thus dividing the element into three parts. The first part is 17.9 kb long and resembles canonical SaPI (νSa3) in its gene composition (36). It encodes integrase and contains genes necessary for the transfer, the fosB fosfomycin resistance gene homologue (91% amino acid identity with the fosfomycin resistance protein family of S. saprophyticus), and several ORFs of unknown function. So far only plasmid-borne fosfomycin resistance genes have been detected in Staphylococcus spp., though the possible localization of fosB on other mobile genetic elements has been discussed (37). The second part of SedCIP5085-1, 15.4 kb long, is comprised of phage- and SaPI (νSa2)-related genes, but no integrase has been found. It also carries a remnant of the fosB gene and other genes of unknown function. The last part of SedCIP5085-1 is 11.7 kb with an indistinct left border, because the bracketing sequence of the att site is absent. This part consists of putative genes encoding resistance to arsenic, cadmium, and bleomycin, hypothetical proteins, and a few phage-related genes, such as a transcriptional regulator from the Cro/cI family, which is known to control the switch between the lytic and lysogenic cycle of bacteriophages. The mosaic organization of islands suggests that more than one integration event occurred in the locus.
The second genomic island, designated SedCIP5085-2, is about 30 kb and contains more than 45 predicted ORFs. The attachment site TATATTATTCCCACTCGAT of SedCIP5085-2 is located within the glutamine-hydrolyzing GMP synthase gene, which matches the attachment site for SaPIbov1/νSa2 (35, 38). This genomic island probably enhances the resistance and endurance of the host, because it codes for transporters and transcriptional regulators involved in antibiotic resistance, oxidative stress responses, and the synthesis of virulence factors. It also contains an additional copy of the cspC gene, coding for cold shock protein C, which has been found to be expressed strongly after antibiotic, arsenate, and peroxide induction (39). In addition, many insertion sequences from the IS3 and IS1182 families were found in this genomic island.
A prophage designated ϕSED1 (vB_SedS-P5085-1), located downstream from the tRNALeu gene, was identified. The prophage is 44,424 bp long with an average G+C content of 33.36 mol%, which is comparable to the G+C content of the CCM 8730T strain. The direct repeats of the phage att site ATCCCGACCACCGGTAT flank 67 ORFs encoding essential phage genes, which cluster together into functional modules corresponding to those in staphylococcal Siphoviridae (40). The lysogeny module starts with phage integrase, which has a tyrosine recombinase XerD domain, AP2-like DNA binding domain, and N-terminal S-adenosylmethionine (SAM)-like domain. There are genes in the DNA replication and transcription regulation modules encoding a DNA primase, helicase, HNH endonuclease domain protein, and putative single-stranded binding protein. The DNA packaging and head and tail morphogenesis modules share 78% nucleotide identity with those of an StB20 phage infecting Staphylococcus capitis (41). The holin and amidase from the lysis module exhibit high amino acid identities to S. saprophyticus prophage holins and amidases, 95 to 99% and 77 to 85%, respectively. The last gene downstream from the lysis module encodes a glycosyltransferase family 2 protein. The prophage does not carry any genes encoding tRNA or virulence factors. However, another, incomplete ϕSED2 prophage, 9,241 bp long, bordered by TAGTGTCCTGGGAGG direct repeats, was found in the CCM 8730T genome. This prophage has an average G+C content of 30.73 mol%, lower than that of its host.
Although the strain CCM 8730T comes from a geographically isolated polar environment, it carries genes for a surprisingly high number of antimicrobial resistance factors. Resistance to beta-lactam antibiotics is determined by the class E mec complex, coding for a beta-lactamase and alternative penicillin-binding MecC protein that is more stable and active at lower temperatures than the MecA protein (42). This might represent an evolutionary advantage against beta-lactam producers, which are common in a polar environment. McRae et al. (43) documented that penicillia are major decomposers and represent an important element of the terrestrial nutrient cycle of Antarctica, particularly since a limited range of other microbial taxa can tolerate the harsh Antarctic climate. Penicillia producing beta-lactams, such as Penicillium chrysogenum, have been isolated from sediments of ponds (44), permafrost (45), subglacial ice (46), and oligotrophic and ornithogenic soil in Antarctica (43, 47). Several studies tested for extrolite production and confirmed the antibacterial activity of Penicillium species strains isolated in Antarctica (44, 46, 48). This is consistent with the fact that both localities where the strains were isolated are apical parts of the landscape often visited by skua birds in austral summer, and there is a high probability they are colonized by fungi.
The 16S rRNA analysis had limited discriminatory power for identifying the staphylococcal isolates examined, and therefore, their phylogenetic position was assessed using the concatenated multilocus sequence data of six housekeeping genes, rpoB, hsp60, dnaJ, tuf, gap and sod, that are commonly used in phylogenetic studies of Staphylococcus, as well as the amino acid sequences of their protein products. The neighbor-joining and maximum-likelihood phylogenetic trees for the six housekeeping genes were very similar and confirmed that the isolates under study represented a well-delineated group within the S. saprophyticus phylogenetic clade, clearly separated from known species (Fig. 5).
FIG 5.
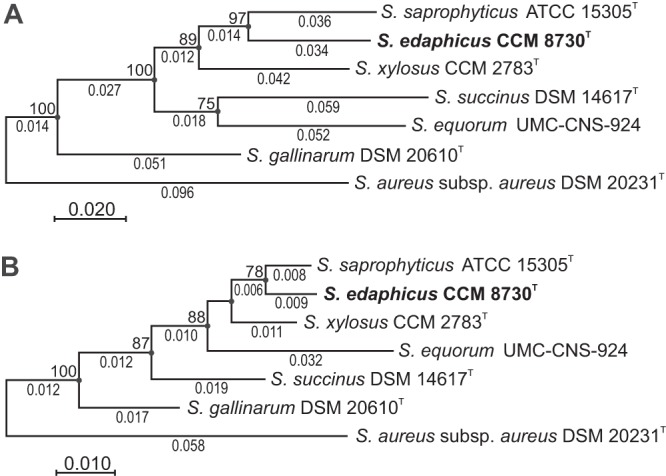
Unrooted maximum-likelihood trees based on concatenated sequences from six multiple loci showing the phylogenetic position of Staphylococcus edaphicus sp. nov. The sections of gene sequences used and their protein products correspond to the following gene coordinates of Staphylococcus aureus subsp. aureus NCTC 8325: 949 to 2748 for rpoB, 16 to 1573 for hsp60, 1 to 1136 for dnaJ, 1 to 1185 for tuf, 4 to 1004 for gap, and 1 to 597 for the sod gene. Bootstrap probability values (percentages of 500 tree replications) greater than 70% are shown at branch points. The evolutionary distances are given as the number of substitutions per site (below the branches). Filled circles indicate that the corresponding nodes are also obtained in the neighbor-joining tree. (A) Evolutionary history inferred from nucleotide sequences by using the maximum-likelihood method based on the Tamura-Nei model. There were a total of 7,175 positions in the final data set. (B) Evolutionary history inferred from amino acid sequences by using the maximum-likelihood method based on the Poisson correction model. There were a total of 2,391 positions in the final data set.
To evaluate the intergenomic distances between the genome sequences of strain CCM 8730T and reference type strains belonging to the phylogenetically closest Staphylococcus spp., the average nucleotide identity (ANI) and digital DNA-DNA hybridization (dDDH) values were determined (Table S3). The calculated ANI and dDDH values are well below the thresholds of 95 to 96% and 70%, respectively, for species delineation (49). This confirms that strain CCM 8730T represents a distinct Staphylococcus species.
The data from this study demonstrate that although they are highly similar to S. saprophyticus, the two isolates represented by strain CCM 8730T belong to a new taxon that can be distinguished both genotypically and phenotypically from established species of the genus Staphylococcus. We suggest classifying these isolates as a novel species for which the name Staphylococcus edaphicus is proposed, with the strain P5085T (= CCM 8730T = DSM 104441T) as the type strain.
MATERIALS AND METHODS
Sampling and cultivation.
Sampling was carried out by dispersing approximately 1 g of stone fragments or soil in 5 ml of sterile saline solution; 100 μl of the suspension was spread on an R2A agar plate (Oxoid) and cultivated at 15°C for 5 days. Subsequently, individual morphologically diverse colonies were picked up and purified by repeated streaking on R2A medium at 15°C, and the resulting pure cultures were maintained at −70°C until analyzed. Two Gram stain-positive isolates were obtained. Additional cultivation of these isolates for biotyping and genotyping was performed with cells growing on TSA (Oxoid). The type strains of the closely related staphylococcal taxa were obtained from the Czech Collection of Microorganisms (www.sci.muni.cz/ccm) and were simultaneously investigated in all tests.
Phenotypic characterization.
Extensive phenotypic characterization using the commercial kits API 50CH, API ID 32 Staph, and API ZYM (bioMérieux), phenotypic fingerprinting using the Biolog system with the identification test panel GEN III MicroPlate (Biolog), and conventional biochemical, physiological, and growth tests relevant for the genus Staphylococcus were done as described previously (50, 51). All phenotypic data presented were from two replicates using commercial kits and three replicates using conventional tests.
Transmission electron microscopy.
The surface of the plate containing the bacterial culture was washed off and the bacteria resuspended in distilled water. A 200-mesh Formvar-coated grid was placed on a drop of the suspension for 20 min. Bacterial cells located on the grid were negative stained with 2% ammonium molybdate and treated with UV light. A Morgagni 268D Philips (FEI Company) transmission electron microscope was used to visualize bacterial cells.
Antimicrobial susceptibility tests.
The antibiotic resistance pattern was tested by the disc diffusion method on Mueller-Hinton agar (Oxoid). Commercially prepared plates were dried at 35°C for 15 min before use. The direct colony suspension method was used to make a suspension of the organism in saline to the density of a McFarland 0.5 turbidity standard. The antibiotic discs were applied by a disc dispenser (Oxoid) within 15 min of inoculation. Sixteen discs generally used for Gram-positive cocci were applied: ampicillin (10 μg), ceftazidime (10 μg), cefoxitin (30 μg), cephalothin (30 μg), ciprofloxacin (5 μg), clindamycin (2 μg), erythromycin (15 μg), fosfomycin (50 μg), gentamicin (10 μg), chloramphenicol (30 μg), imipenem (10 μg), kanamycin (30 μg), neomycin (10 μg), novobiocin (5 μg), oxacillin (1 μg), penicillin G (1 IU), co-trimoxazole (25 μg), tetracycline (30 μg), and vancomycin (30 μg). The incubation conditions were 16 to 20 h at 36°C in air. The inhibition zone diameters were measured with a caliper. EUCAST/CLSI standards for reading the inhibition zone diameter and interpreting susceptibility to antibiotics were strictly followed (52, 53).
MALDI-TOF MS.
For matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS), cultures were grown on Columbia blood agar (Oxoid) and transferred with a bamboo toothpick onto an MSP 96 polished steel target plate (Bruker Daltonics). The samples were covered with 1 μl of matrix solution, comprised of a saturated solution of α-cyano-4-hydroxy-cinnamic acid (Bruker) in 50% acetonitrile (Sigma-Aldrich) and 2.5% trifluoroacetic acid (VWR). Each strain was spotted in 10 replicates. Samples were processed using a MALDI-TOF mass spectrometer (Microflex LT; Bruker Daltonics). The mass range of spectra analyzed was 2,000 to 20,000 m/z. Each spectrum was obtained after 300 shots in an automatic acquisition mode. Mass spectra were processed using the software Flex Analysis (version 3.4; Bruker Daltonics). The MALDI-TOF MS-based dendrogram was generated using the software BioTyper (version 3.0; Bruker Daltonics). Signals present in at least 70% of analyses for one sample were transformed into main spectrum projections (MSPs). Afterwards, Pearson coefficients based on the MSPs' similarities were calculated and the dendrogram was generated using the unweighted average linkage algorithm.
FAME analysis.
The fatty acid methyl ester (FAME) analysis was performed with cells growing on BBL Trypticase soy agar plates (Becton Dickinson) at 28°C ± 1°C for 24 h. The extraction of fatty acid methyl esters was performed according to the standard protocol of the Sherlock Microbial Identification System (54). Bacterial biomass was harvested from the third sector of growth on agar plates, where the cultures reached the late-exponential stage of growth according to the four-quadrant streak method (54). Cellular fatty acid extracts were analyzed with an Agilent 7890B gas chromatograph using the rapid Sherlock microbial identification system (MIS) (version 6.2B, MIDI database RTSBA 6.21; MIDI Inc.). The quantities of individual fatty acids are given as percentages of all named fatty acids. Strains CCM 8730T and CCM 8731T and the reference strains were tested three times to evaluate the reproducibility of the method.
Genotypic analysis by fingerprinting techniques.
Rep-PCR fingerprinting with the (GTG)5 primer was performed as described previously (55). The automatic ribotyping was performed using a RiboPrinter microbial characterization system (DuPont Qualicon) in accordance with the manufacturer's instructions. Numerical analysis of the fingerprints obtained and dendrogram construction were done using the software BioNumerics 7.6 (Applied Maths). The ribotype patterns were imported into the software BioNumerics using the load samples import script provided by the manufacturer. Pulsed-field gel electrophoresis (PFGE) using SmaI macrorestriction pattern analysis was performed as previously described (56).
Genome sequencing and bioinformatics analysis.
Genomic DNA was extracted using a high pure PCR template preparation kit (Roche) according to the manufacturer's recommendations. The concentration of extracted DNA was estimated with a Qubit 2.0 fluorometer using a Qubit double-stranded DNA (dsDNA) broad-range (BR) assay kit (Invitrogen). The partial 16S rRNA gene was sequenced as described previously (57). Whole-genome shotgun (WGS) sequencing was performed using an Ion Torrent Personal Genome Machine (PGM). The purified genomic DNA was used for preparing a 400-bp sequencing library with an Ion plus fragment library kit (Thermo Fisher Scientific). The sample was loaded onto an Ion 316 chip, version 2, and sequenced using an Ion PGM Hi-Q view sequencing kit (Thermo Fisher Scientific). Quality trimming of the reads was performed with the Ion Torrent Suite software (version 5.0.4) with default settings. The assembly computation and error correction were performed using Assembler SPAdes 3.9 (58) with default parameters for Ion Torrent data and k-mer settings of 21, 33, 55, 77, 99, and 127. Contigs were then reordered according to the S. saprophyticus ATCC 15305T reference genome (GenBank accession number NC_007350) (16) using MauveContigMover (59). Ordering was evaluated using the assembly graph visualized in Bandage (60).
Sequences were manipulated and inspected in the cross-platform bioinformatics software Ugene version 1.23.1 (61). For primary analysis, the genome was annotated using RAST (62). Gene content was further examined by BLASTp (63), ISFinder (64), InterProScan (65), CD-Search (66), and OrthoVenn (67). The complete 16S rRNA gene sequence was extracted from WGS data using RNAmmer version 1.2 (68). Phylogenetic analysis from multilocus sequence data was performed using the software MEGA version 7 (69).
DNA homology studies.
To calculate the ANI value, the OrthoANI algorithm implemented on the EzBioCloud server (http://www.ezbiocloud.net/tools/ani) was used (70). The dDDH values were calculated using the web-based genome-to-genome distance calculator (GGDC) version 2.1 (71), and the recommended formula 2 was taken into account to interpret the results. Whole-genome sequences of related staphylococcal species were obtained from the NCBI database (72–75).
Accession number(s).
The GenBank accession number for the 16S rRNA genes of isolate CCM 8730T is KY315825. The data from WGS of strains Staphylococcus edaphicus CCM 8730T and Staphylococcus xylosus CCM 2738T were recorded in the GenBank WGS project under the accession numbers MRZN00000000 and MRZO00000000.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for the scientific infrastructure of the J.G. Mendel Czech Antarctic Station as part of the Czech Polar Research Infrastructure (CzechPolar2) and its crew for their assistance, supported by the Ministry of Education, Youth and Sports of the Czech Republic (grant number LM2015078), as well as the Czech Antarctic Foundation for their support. Access to computing and storage facilities owned by parties and projects contributing to the National Grid Infrastructure MetaCentrum provided under the program Projects of Large Research, Development, and Innovations Infrastructures CESNET (grant number LM2015042) is greatly appreciated. Daniel Krsek (NRL for the Diagnostic Electron Microscopy of Infectious Agents, National Institute of Public Health, Prague, Czech Republic) is gratefully acknowledged for transmission electron microscopy and Bernhard Schink (University of Constance) for name correction.
This work was supported by the Ministry of Health of the Czech Republic (grant number NT16-29916A). S.K. is a beneficiary of Brno Ph.D. Talent financial aid.
I.S. isolated and preliminarily identified the strains. I.S., J.K., and P.P. performed phenotypic characterization. Whole-genome sequencing was performed by V.V., and V.K. and A.I. analyzed the data. Phylogenetic analyses were performed by I.M. and V.V. P.S. performed genotyping by fingerprinting techniques and analyzed the data. S.K. performed FAME analysis. P.P. and L.K. performed MALDI-TOF identification and analyzed the data. R.P. and I.S. designed the study, R.P., I.S., A.I., V.V., I.M., and J.D. wrote the manuscript. All the authors approved the final manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01746-17.
REFERENCES
- 1.Götz F, Bannerman T, Schleifer K-H. 2006. The genera Staphylococcus and Macrococcus, p 5–75. In Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (ed), The Prokaryotes: Bacteria, Firmicutes, Cyanobacteria, vol 4 Springer US, New York, NY. doi: 10.1007/0-387-30744-3_1. [DOI] [Google Scholar]
- 2.Emmett M, Kloos WE. 1975. Amino acid requirements of staphylococci isolated from human skin. Can J Microbiol 21:729–733. doi: 10.1139/m75-107. [DOI] [PubMed] [Google Scholar]
- 3.Emmett M, Kloos WE. 1979. The nature of arginine auxotrophy in cutaneous populations of staphylococci. J Gen Microbiol 110:305–314. doi: 10.1099/00221287-110-2-305. [DOI] [PubMed] [Google Scholar]
- 4.Lambert LH, Cox T, Mitchell K, Rossello-Mora RA, Del Cueto C, Dodge DE, Orkand P, Cano RJ. 1998. Staphylococcus succinus sp. nov., isolated from Dominican amber. Int J Syst Bacteriol 48:511–518. doi: 10.1099/00207713-48-2-511. [DOI] [PubMed] [Google Scholar]
- 5.Hess S, Gallert C. 2015. Staphylococcus argensis sp nov., a hovel staphylococcal species isolated from an aquatic environment. Int J Syst Evol Microbiol 65:2661–2665. doi: 10.1099/ijs.0.000319. [DOI] [PubMed] [Google Scholar]
- 6.Kloos WE. 1980. Natural populations of the genus Staphylococcus. Annu Rev Microbiol 34:559–592. doi: 10.1146/annurev.mi.34.100180.003015. [DOI] [PubMed] [Google Scholar]
- 7.Kloos WE, Schleifer KH, Smith RF. 1976. Characterization of Staphylococcus sciuri sp. nov. and its subspecies. Int J Syst Bacteriol 26:22–37. doi: 10.1099/00207713-26-1-22. [DOI] [Google Scholar]
- 8.Planchon S, Gaillard-Martinie B, Dordet-Frisoni E, Bellon-Fontaine MN, Leroy S, Labadie J, Hebraud M, Talon R. 2006. Formation of biofilm by Staphylococcus xylosus. Int J Food Microbiol 109:88–96. doi: 10.1016/j.ijfoodmicro.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 9.Dordet-Frisoni E, Dorchies G, De Araujo C, Talon R, Leroy S. 2007. Genomic diversity in Staphylococcus xylosus. Appl Environ Microbiol 73:7199–7209. doi: 10.1128/AEM.01629-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Irlinger F. 2008. Safety assessment of dairy microorganisms: coagulase-negative staphylococci. Int J Food Microbiol 126:302–310. doi: 10.1016/j.ijfoodmicro.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 11.Giordano N, Corallo C, Miracco C, Papakostas P, Montella A, Figura N, Nuti R. 2016. Erythema nodosum associated with Staphylococcus xylosus septicemia. J Microbiol Immunol Infect 49:134–137. doi: 10.1016/j.jmii.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 12.Becker K, von Eiff C. 2011. Staphylococcus, Micrococcus, and other catalase-positive cocci, p 308–330. In Versalovic J, Carroll KC, Funke G, Jorgensen JH, Landry ML, Warnock DW (ed), Manual of clinical microbiology, 10th ed ASM Press, Washington, DC. doi: 10.1128/9781555816728.ch19. [DOI] [Google Scholar]
- 13.Schleifer KH, Kloos WE. 1975. Isolation and characterization of staphylococci from human skin. 1. Amended descriptions of Staphylococcus epidermidis and Staphylococcus saprophyticus and descriptions of 3 new species—Staphylococcus cohnii, Staphylococcus haemolyticus, and Staphylococcus xylosus. Int J Syst Bacteriol 25:50–61. doi: 10.1099/00207713-25-1-50. [DOI] [Google Scholar]
- 14.Widerström M, Wiström J, Sjöstedt A, Monsen T. 2012. Coagulase-negative staphylococci: update on the molecular epidemiology and clinical presentation, with a focus on Staphylococcus epidermidis and Staphylococcus saprophyticus. Eur J Clin Microbiol Infect Dis 31:7–20. doi: 10.1007/s10096-011-1270-6. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi T, Satoh I, Kikuchi N. 1999. Phylogenetic relationships of 38 taxa of the genus Staphylococcus based on 16S rRNA gene sequence analysis. Int J Syst Bacteriol 49:725–728. doi: 10.1099/00207713-49-2-725. [DOI] [PubMed] [Google Scholar]
- 16.Kuroda M, Yamashita A, Hirakawa H, Kumano M, Morikawa K, Higashide M, Maruyama A, Inose Y, Matoba K, Toh H, Kuhara S, Hattori M, Ohta T. 2005. Whole genome sequence of Staphylococcus saprophyticus reveals the pathogenesis of uncomplicated urinary tract infection. Proc Natl Acad Sci U S A 102:13272–13277. doi: 10.1073/pnas.0502950102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schleifer K-H, Bell JA. 2009. Genus I. Staphylococcus Rosenbach 1884, 18AL (Nom. Cons. Opin 17 Jud. Comm. 1958, 153.), p 392–421. In De Vos P, Garrity GM, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer K-H, Whitman WB (ed), Bergey's manual of systematic bacteriology, vol 3 The Firmicutes Springer-Verlag, New York, NY. doi: 10.1007/978-0-387-68489-5. [DOI] [Google Scholar]
- 18.Foster TJ, Geoghegan JA, Ganesh VK, Hook M. 2014. Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nat Rev Microbiol 12:49–62. doi: 10.1038/nrmicro3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson LK, Coombs GW, Christiansen K, Grubb WB, O'Brien FG. 2016. Characterization of a novel staphylococcal cassette chromosome composite island from community-associated MRSA isolated in aged care facilities in Western Australia. J Antimicrob Chemother 71:3372–3375. doi: 10.1093/jac/dkw317. [DOI] [PubMed] [Google Scholar]
- 20.Ohniwa RL, Kitabayashi K, Morikawa K. 2013. Alternative cardiolipin synthase Cls1 compensates for stalled Cls2 function in Staphylococcus aureus under conditions of acute acid stress. FEMS Microbiol Lett 338:141–146. doi: 10.1111/1574-6968.12037. [DOI] [PubMed] [Google Scholar]
- 21.Tsai M, Ohniwa RL, Kato Y, Takeshita SL, Ohta T, Saito S, Hayashi H, Morikawa K. 2011. Staphylococcus aureus requires cardiolipin for survival under conditions of high salinity. BMC Microbiol 11:13. doi: 10.1186/1471-2180-11-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peleg AY, Miyakis S, Ward DV, Earl AM, Rubio A, Cameron DR, Pillai S, Moellering RC Jr, Eliopoulos GM. 2012. Whole genome characterization of the mechanisms of daptomycin resistance in clinical and laboratory derived isolates of Staphylococcus aureus. PLoS One 7:e28316. doi: 10.1371/journal.pone.0028316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.International Working Group on the Classification of Staphylococcal Cassette Chromosome E. 2009. Classification of staphylococcal cassette chromosome mec (SCCmec): guidelines for reporting novel SCCmec elements. Antimicrob Agents Chemother 53:4961–4967. doi: 10.1128/AAC.00579-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.García-Álvarez L, Holden MT, Lindsay H, Webb CR, Brown DF, Curran MD, Walpole E, Brooks K, Pickard DJ, Teale C, Parkhill J, Bentley SD, Edwards GF, Girvan EK, Kearns AM, Pichon B, Hill RL, Larsen AR, Skov RL, Peacock SJ, Maskell DJ, Holmes MA. 2011. Meticillin-resistant Staphylococcus aureus with a novel mecA homologue in human and bovine populations in the UK and Denmark: a descriptive study. Lancet Infect Dis 11:595–603. doi: 10.1016/S1473-3099(11)70126-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrison EM, Paterson GK, Holden MT, Morgan FJ, Larsen AR, Petersen A, Leroy S, De Vliegher S, Perreten V, Fox LK, Lam TJ, Sampimon OC, Zadoks RN, Peacock SJ, Parkhill J, Holmes MA. 2013. A Staphylococcus xylosus isolate with a new mecC allotype. Antimicrob Agents Chemother 57:1524–1528. doi: 10.1128/AAC.01882-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malyszko I, Schwarz S, Hauschild T. 2014. Detection of a new mecC allotype, mecC2, in methicillin-resistant Staphylococcus saprophyticus. J Antimicrob Chemother 69:2003–2005. doi: 10.1093/jac/dku043. [DOI] [PubMed] [Google Scholar]
- 27.Paterson GK, Larsen AR, Robb A, Edwards GE, Pennycott TW, Foster G, Mot D, Hermans K, Baert K, Peacock SJ, Parkhill J, Zadoks RN, Holmes MA. 2012. The newly described mecA homologue, mecALGA251, is present in methicillin-resistant Staphylococcus aureus isolates from a diverse range of host species. J Antimicrob Chemother 67:2809–2813. doi: 10.1093/jac/dks329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monecke S, Gavier-Widen D, Mattsson R, Rangstrup-Christensen L, Lazaris A, Coleman DC, Shore AC, Ehricht R. 2013. Detection of mecC-positive Staphylococcus aureus (CC130-MRSA-XI) in diseased European hedgehogs (Erinaceus europaeus) in Sweden. PLoS One 8:e66166. doi: 10.1371/journal.pone.0066166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loncaric I, Kubber-Heiss A, Posautz A, Stalder GL, Hoffmann D, Rosengarten R, Walzer C. 2013. Characterization of methicillin-resistant Staphylococcus spp. carrying the mecC gene, isolated from wildlife. J Antimicrob Chemother 68:2222–2225. doi: 10.1093/jac/dkt186. [DOI] [PubMed] [Google Scholar]
- 30.Porrero MC, Mentaberre G, Sanchez S, Fernandez-Llario P, Casas-Diaz E, Mateos A, Vidal D, Lavin S, Fernandez-Garayzabal JF, Dominguez L. 2014. Carriage of Staphylococcus aureus by free-living wild animals in Spain. Appl Environ Microbiol 80:4865–4870. doi: 10.1128/AEM.00647-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Porrero MC, Valverde A, Fernandez-Llario P, Diez-Guerrier A, Mateos A, Lavin S, Canton R, Fernandez-Garayzabal JF, Dominguez L. 2014. Staphylococcus aureus carrying mecC gene in animals and urban wastewater, Spain. Emerg Infect Dis 20:899–901. doi: 10.3201/eid2005.130426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Babu MM, Priya ML, Selvan AT, Madera M, Gough J, Aravind L, Sankaran K. 2006. A database of bacterial lipoproteins (DOLOP) with functional assignments to predicted lipoproteins. J Bacteriol 188:2761–2773. doi: 10.1128/JB.188.8.2761-2773.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shahmirzadi SV, Nguyen MT, Götz F. 2016. Evaluation of Staphylococcus aureus lipoproteins: role in nutritional acquisition and pathogenicity. Front Microbiol 7:1404. doi: 10.3389/fmicb.2016.01404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Penadés JR, Christie GE. 2015. The phage-inducible chromosomal islands: a family of highly evolved molecular parasites. Annu Rev Virol 2:181–201. doi: 10.1146/annurev-virology-031413-085446. [DOI] [PubMed] [Google Scholar]
- 35.Baba T, Takeuchi F, Kuroda M, Yuzawa H, Aoki K, Oguchi A, Nagai Y, Iwama N, Asano K, Naimi T, Kuroda H, Cui L, Yamamoto K, Hiramatsu K. 2002. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet 359:1819–1827. doi: 10.1016/S0140-6736(02)08713-5. [DOI] [PubMed] [Google Scholar]
- 36.Novick RP, Christie GE, Penadés JR. 2010. The phage-related chromosomal islands of Gram-positive bacteria. Nat Rev Microbiol 8:541–551. doi: 10.1038/nrmicro2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fu Z, Liu Y, Chen C, Guo Y, Ma Y, Yang Y, Hu F, Xu X, Wang M. 2016. Characterization of fosfomycin resistance gene, fosB, in methicillin-resistant Staphylococcus aureus isolates. PLoS One 11:e0154829. doi: 10.1371/journal.pone.0154829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fitzgerald JR, Monday SR, Foster TJ, Bohach GA, Hartigan PJ, Meaney WJ, Smyth CJ. 2001. Characterization of a putative pathogenicity island from bovine Staphylococcus aureus encoding multiple superantigens. J Bacteriol 183:63–70. doi: 10.1128/JB.183.1.63-70.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chanda PK, Mondal R, Sau K, Sau S.. 2009. Antibiotics, arsenate and H2O2 induce the promoter of Staphylococcus aureus cspC gene more strongly than cold. J Basic Microbiol 49:205–211. doi: 10.1002/jobm.200800065. [DOI] [PubMed] [Google Scholar]
- 40.Kahánková J, Pantůček R, Goerke C, Růžičková V, Holochová P, Doškař J. 2010. Multilocus PCR typing strategy for differentiation of Staphylococcus aureus siphoviruses reflecting their modular genome structure. Environ Microbiol 12:2527–2538. doi: 10.1111/j.1462-2920.2010.02226.x. [DOI] [PubMed] [Google Scholar]
- 41.Deghorain M, Bobay LM, Smeesters PR, Bousbata S, Vermeersch M, Perez-Morga D, Dreze PA, Rocha EP, Touchon M, Van Melderen L. 2012. Characterization of novel phages isolated in coagulase-negative staphylococci reveals evolutionary relationships with Staphylococcus aureus phages. J Bacteriol 194:5829–5839. doi: 10.1128/JB.01085-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim C, Milheirico C, Gardete S, Holmes MA, Holden MT, de Lencastre H, Tomasz A. 2012. Properties of a novel PBP2A protein homolog from Staphylococcus aureus strain LGA251 and its contribution to the beta-lactam-resistant phenotype. J Biol Chem 287:36854–36863. doi: 10.1074/jbc.M112.395962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McRae CF, Hocking AD, Seppelt RD. 1999. Penicillium species from terrestrial habitats in the Windmill Islands, East Antarctica, including a new species, Penicillium antarcticum. Polar Biol 21:97–111. doi: 10.1007/s003000050340. [DOI] [Google Scholar]
- 44.Montemartini Corte AM, Liotta M, Venturi CB, Calegari L. 2000. Antibacterial activity of Penicillium spp. strains isolated in extreme environments. Polar Biol 23:294–297. doi: 10.1007/s003000050447. [DOI] [Google Scholar]
- 45.Zucconi L, Selbmann L, Buzzini P, Turchetti B, Guglielmin M, Frisvad JC, Onofri S. 2012. Searching for eukaryotic life preserved in Antarctic permafrost. Polar Biol 35:749–757. doi: 10.1007/s00300-011-1119-6. [DOI] [Google Scholar]
- 46.Sonjak S, Frisvad JC, Gunde-Cimerman N. 2006. Penicillium mycobiota in arctic subglacial ice. Microb Ecol 52:207–216. doi: 10.1007/s00248-006-9086-0. [DOI] [PubMed] [Google Scholar]
- 47.Godinho VM, Goncalves VN, Santiago IF, Figueredo HM, Vitoreli GA, Schaefer CE, Barbosa EC, Oliveira JG, Alves TM, Zani CL, Junior PA, Murta SM, Romanha AJ, Kroon EG, Cantrell CL, Wedge DE, Duke SO, Ali A, Rosa CA, Rosa LH. 2015. Diversity and bioprospection of fungal community present in oligotrophic soil of continental Antarctica. Extremophiles 19:585–596. doi: 10.1007/s00792-015-0741-6. [DOI] [PubMed] [Google Scholar]
- 48.Brunati M, Rojas JL, Sponga F, Ciciliato I, Losi D, Gottlich E, de Hoog S, Genilloud O, Marinelli F. 2009. Diversity and pharmaceutical screening of fungi from benthic mats of Antarctic lakes. Mar Genomics 2:43–50. doi: 10.1016/j.margen.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 49.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. 2007. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 50.Pantůček R, Švec P, Dajcs JJ, Machová I, Černohlávková J, Šedo O, Gelbíčová T, Mašlaňová I, Doškař J, Zdráhal Z, Růžičková V, Sedláček I. 2013. Staphylococcus petrasii sp. nov. including S. petrasii subsp. petrasii subsp. nov. and S. petrasii subsp. croceilyticus subsp. nov., isolated from human clinical specimens and human ear infections. Syst Appl Microbiol 36:90–95. doi: 10.1016/j.syapm.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 51.Mannerová S, Pantůček R, Doškař J, Švec P, Snauwaert C, Vancanneyt M, Swings J, Sedláček I. 2003. Macrococcus brunensis sp. nov., Macrococcus hajekii sp. nov. and Macrococcus lamae sp. nov., from the skin of llamas. Int J Syst Evol Microbiol 53:1647–1654. doi: 10.1099/ijs.0.02683-0. [DOI] [PubMed] [Google Scholar]
- 52.The European Committee on Antimicrobial Susceptibility Testing. 2017. Breakpoint tables for interpretation of MICs and zone diameters, version 7.1. http://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v_7.1_Breakpoint_Tables.pdf.
- 53.CLSI. 2015. Performance standards for antimicrobial disk susceptibility tests; approved standard, 12th ed CLSI document M02-A12. Clinical and Laboratory Standards Institute, Wayne, PA: https://clsi.org/standards/products/microbiology/documents/m02/. [Google Scholar]
- 54.Sasser M. 1990. Identification of bacteria by gas chromatography of cellular fatty acids. MIDI technical note 101. Microbial ID, Inc., Newark, DE. [Google Scholar]
- 55.Švec P, Pantůček R, Petráš P, Sedláček I, Nováková D. 2010. Identification of Staphylococcus spp. using (GTG)5-PCR fingerprinting. Syst Appl Microbiol 33:451–456. doi: 10.1016/j.syapm.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 56.Pantůček R, Götz F, Doškař J, Rosypal S. 1996. Genomic variability of Staphylococcus aureus and the other coagulase-positive Staphylococcus species estimated by macrorestriction analysis using pulsed-field gel electrophoresis. Int J Syst Bacteriol 46:216–222. doi: 10.1099/00207713-46-1-216. [DOI] [PubMed] [Google Scholar]
- 57.Pantůček R, Sedláček I, Petráš P, Koukalová D, Švec P, Štětina V, Vancanneyt M, Chrastinová L, Vokurková J, Růžicková V, Doškař J, Swings J, Hájek V. 2005. Staphylococcus simiae sp. nov., isolated from South American squirrel monkeys. Int J Syst Evol Microbiol 55:1953–1958. doi: 10.1099/ijs.0.63590-0. [DOI] [PubMed] [Google Scholar]
- 58.Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A, Prjibelski AD, Pyshkin A, Sirotkin A, Sirotkin Y, Stepanauskas R, Clingenpeel SR, Woyke T, Mclean JS, Lasken R, Tesler G, Alekseyev MA, Pevzner PA. 2013. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol 20:714–737. doi: 10.1089/cmb.2013.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rissman AI, Mau B, Biehl BS, Darling AE, Glasner JD, Perna NT. 2009. Reordering contigs of draft genomes using the Mauve aligner. Bioinformatics 25:2071–2073. doi: 10.1093/bioinformatics/btp356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wick RR, Schultz MB, Zobel J, Holt KE. 2015. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31:3350–3352. doi: 10.1093/bioinformatics/btv383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Okonechnikov K, Golosova O, Fursov M, UGENE Team. 2012. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28:1166–1167. doi: 10.1093/bioinformatics/bts091. [DOI] [PubMed] [Google Scholar]
- 62.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 64.Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. 2006. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res 34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, Pesseat S, Quinn AF, Sangrador-Vegas A, Scheremetjew M, Yong SY, Lopez R, Hunter S. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Marchler GH, Song JS, Thanki N, Wang Z, Yamashita RA, Zhang D, Zheng C, Bryant SH. 2015. CDD: NCBI's conserved domain database. Nucleic Acids Res 43:D222–D226. doi: 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang Y, Coleman-Derr D, Chen G, Gu YQ. 2015. OrthoVenn: a web server for genome wide comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res 43:W78–W84. doi: 10.1093/nar/gkv487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. 2007. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee I, Kim YO, Park SC, Chun J. 2015. OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Microbiol doi: 10.1099/ijsem.0.000760. [DOI] [PubMed] [Google Scholar]
- 71.Meier-Kolthoff JP, Auch AF, Klenk HP, Goker M. 2013. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Calcutt MJ, Foecking MF, Hsieh HY, Perry J, Stewart GC, Middleton JR. 2013. Genome sequence analysis of Staphylococcus equorum bovine mastitis isolate UMC-CNS-924. Genome Announc 1:e00840-13. doi: 10.1128/genomeA.00840-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou H, Yao Z, Shi H, Wang B, Li D, Hou J, Ma S. 2017. Draft genome sequence of Staphylococcus succinus subsp. succinus type strain DSM 14617, isolated from plant and soil inclusions within 25- to 35-million-year-old Dominican amber. Genome Announc 5:e01521-16. doi: 10.1128/genomeA.01521-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shi D, Fang D, Hu X, Li A, Lv L, Guo J, Chen Y, Wu W, Guo F, Li L. 2015. Draft genome sequence of Staphylococcus gallinarum DSM 20610T, originally isolated from the skin of a chicken. Genome Announc 3:e00580-15. doi: 10.1128/genomeA.00580-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shiroma A, Terabayashi Y, Nakano K, Shimoji M, Tamotsu H, Ashimine N, Ohki S, Shinzato M, Teruya K, Satou K, Hirano T. 2015. First complete genome sequences of Staphylococcus aureus subsp. aureus Rosenbach 1884 (DSM 20231T), determined by PacBio single-molecule real-time technology. Genome Announc 3:e00800-15. doi: 10.1128/genomeA.00800-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.