

Abstract
Post-translational modifications (PTMs) of High mobility group box 1 (HMGB1) have not been investigated as extensively as those of other HMG proteins but accumulating evidence has shown the remarkable biological significances induced by the post-translational: acetylation, methylation and phosphorylation, oxidation, glycosylation and ADP-ribosylation of the HMGB1 to modulate its interactions with DNA and other proteins. Although HMGB1 is localized in the nucleus in almost all cells at baseline, it can be rapidly mobilized to other sites within the cell, including the cytoplasm and mitochondria, as well as into the extracellular; hence there is an increasing interest by researches into the complex relationship between the PTMs of HMGB1 protein and its diverse biological activities. The PTMs of HMGB1 could also have effects on gene expression following changes in its DNA-binding properties and in extracellular environment displays immunological activity and could serve as a potential target for new therapy. Our reviewed identifies covalent modifications of HMGB1, and highlighted how these PTMs affect the functions of HMGB1 protein in a variety of cellular and extra cellular processes as well as diseases and therapy.
Keywords: HMGB1, post-translational, modification, cancer, autoimmune disease
Introduction
High mobility group box 1 (HMGB1) is the most abundant and well-studied HMG proteins. It senses and coordinates the cellular stress response and plays a critical role inside the cell as a DNA chaperone, chromosome guardian, autophagy sustainer, and protector from apoptotic cell death [1-3]. HMGB1 can also be designated as a damage- (or death-) associated molecular pattern (DAMP) by analogy to a pathogen-associated molecular pattern (PAMP) because of its origin and immunological properties [4]. While PAMPs are exogenous molecules and DAMPs are endogenous molecules, they both responses to physiological and pathological events via interacting with cellular receptors such as the Toll-like receptors (TLRs) as well as induction of similar downstream signaling events. HMGB1 is also the most mobile protein in the nucleus and has the ability translocate into the cytosol within 1-2 seconds [3,5]. Because of the translocational ability of HMGB1, it can be found in the cytosol e.g., mitochondria [6] and lysosome [7]. Furthermore, in the cellular membrane, and extracellular space its nuclear localization signal (NLS) can be modified. Nevertheless, the subcellular location of HMGB1 changes depending on cell type, tissue, and stress signals.
HMGB1 is also a non-histone nuclear protein with multiple functions; it interacts with DNA to modify its structure and regulate transcription inside the cell while outside the cell it serves as an alarmin to activate the innate immune system and promote tissue repair. The translocation of HMGB1 which takes place primarily during cell activation and cell death is vital to it immunological activity and may undergo PTMs similar to those determining the epigenome [8]. Modifications of HMGB1 influence its location in the cell as well as its immunological activity whereas epigenetic modifications usually influence gene expression.
The immunological activity of HMGB1 depends on exposure to cells of the innate immune system hence its translocation or release from cells usually occurs in the settings of activation and cell death. It’s release during cell death makes it play a role as an alarmin. In vitro studies have demonstrated a variety of cell systems with several distinct mechanisms by which HMGB1 is released from cells that importantly may involve PTMs. These modifications can change the location of HMGB1 as well as its interaction with chromatin. It is well noted that over 100 different PTMs like methylation, phosphorylation, acetylation, ribosylation and ubiquitination characterize the epigenome [9,10]. We will discuss in details the PTMs (acetylation, methylation and phosphorylation, oxidation, glycosylation and ADP-ribosylation) of the HMGB1, highlighting how these PTMs affect the functions of the HMGB1 in an array of cellular and extra cellular processes and also look at the role of HMGB1 in disease (e.g. cancer) and therapy.
The redox and oxide status of HMGB1
HMGB1 contains three cysteine residues. Two of these cysteine residues form a disulphide bond, and all three are sensitive to oxidation status in the environment. With recent substandard agreement, the three major isoforms of HMGB1 have been termed ‘disulphide HMGB1’, ‘thiol HMGB1’ and ‘oxidized HMGB1’ [11-14] Figure 1. These isoforms have pleiotropic activities like any other cytokine and the activities depend on the cellular compartment of action, the reciprocal receptor and the specific molecular structure of the isoform. The principal isoform released during necrosis is thiol HMGB1 while the disulphide HMGB1 isoform is the main isoform that accumulates in the extracellular space and serum compartment during acute and chronic inflammation. HMGB1 is a pro-inflammatory cytokine-like molecule that activates macrophages/monocytes and other cells to produce cytokines and additional inflammatory mediators. The oxidized HMGB1 isoform is seen as noninflammatory, although previously unsuspected roles of this molecule are yet to be elucidated. Researchers have established the HMGB1 release from ischemic cells is an early event in response to injury [3,12,15].
Figure 1.
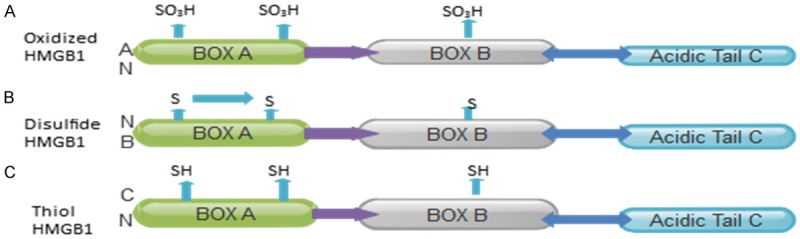
Different HMGB1 redox states: A: The oxidized isoform has all-oxidized cysteine in HMGB1and is seen as noninflammatory; B: The Disulfide isoform has a disulfide bond that induces cytokine cell release; C: The thiol isoform has a reduced cysteine with chemoattractant activity.
Function of HMGB1
HMGB1 is a unique, ubiquitous and highly expressed nuclear protein which stabilizes nucleosome formation and facilitates transcription factor binding to DNA. Outside the cell it may function as a potent cytokine with the ability to trigger inflammatory mediators [16]. In damaged or dying cells, passive release of HMGB1 is by cell necrosis and not apoptosis and it’s a chemoattractant for immature dendritic cells (DCs) and promotes their maturation [4]. Studies have shown that HMGB1 lacks a signal sequence. Furthermore, monocytic cells receiving inflammatory signals can acetylate it’s interfering with nuclear localization signals and allowing secretion [17]. Dumitriu et al indicated that DCs can secrete HMGB1, and such secretion promotes proliferation and Th1 polarization of interacting T cells [18]. Additionally, several studies have indicated that HMGB1 can directly or indirectly contribute Th17 expansion [19,20]. When unregulated, HMGB1 can contribute to immune-related pathology. It is also angiogenic and promotes cardiac stem cell growth and differentiation indicating its potential involvement in repairing damaged tissues [21]. It has direct and potent bactericidal activity just like defensins and cathelicidins [8]. Abeyama and colleagues have indicated that vascular thrombin binding protein, thrombomodulin (TM) is responsible for binding and sequestering HMGB1. It has protection effects which partially explains it’s anti-inflammatory effects [22]. Researchers have shown that tissue damage caused by trauma, ischemia, hemorrhage or severe infection leading to sepsis may result in life-threatening out-of-control HMGB1 responses [23-25]. Inhibiting of HMGB1 has been effective in increasing survival in mouse or rat models of sepsis or hemorrhage [26] although 30% of patients do not survive due to organ failure and cardiac arrest even with intensive treatment for severe sepsis. Therefore, therapeutic strategies based on one or more of these inhibitors are attractive, especially considering fact that HMGB1 levels peak later than 24 hours after the initiation of sepsis, potentially allowing time for treatment to occur.
HMGB1 receptor and intracellular signaling
The mechanism by which HMGB1 interacts with target cells is still not well understood. RAGE is a transmembrane protein that is a member of the immunoglobulin (Ig) superfamily and is homologous to a neural cell-adhesion molecule [27]. It is expressed in central nervous system, endothelial cells, smooth muscle cells, and mononuclear phagocytes. It has been found that HMGB1 is a specific and saturable ligand for RAGE. It has higher affinity for RAGE than other known ligands such as advanced glycation end products (AGEs) [28]. Studies have shown that HMGB1-RAGE interaction will also lead to phosphorylation of MAP-kinases p38, p42/p44, and c-jun NH2-terminal kinase, resulting in NF-B activation [29,30]. Furthermore, extracellular proteolytic activity induced by HMGB1 expressed on the leading edge of motile cells has also recently been confirmed in an experimental tumor system [29] (Figure 3A).
Figure 3.
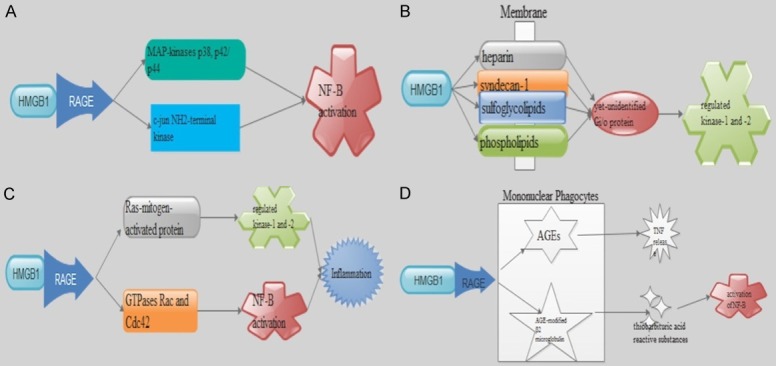
Potential HMGB1 receptor and possible signaling pathways. A: HMGB1-RAGE interaction leads to phosphorylation of MAP-kinases p38, p42/p44, and c-jun NH2-terminal kinase, resulting in NF-B activation. B: HMGB1 binds to many membrane molecules such as heparin, proteoglycans including syndecan-1, sulfoglycolipids, and phospholipid and mediate phosphorylated of extracellular regulated kinase-1 and -2. that involves signaling via an unidentified Gi/o protein. C: HMGB1 through RAGE can activate two different cascades, one involving the involves the Ras-mitogen-activated protein (MAP) kinase pathway and a second that involves a small GTPases Rac and Cdc42 leading to cytoskeletal reorganization and subsequent nuclear factor (NF)-B nuclear translocation-mediating inflammation. D: RAGE is also expressed on mononuclear phagocytes where its interaction with AGEs enhances cellular oxidant stress and generation of thiobarbituric acid reactive substances and activation of NF-B. RAGE signaling has also been shown to stimulates an inflammatory response when AGE-modified β2 microglobulin binds RAGE in mononuclear phagocytes to mediate monocyte chemotaxis and induce TNF release.
Researchers have also indicated that HMGB1 being a “sticky” molecule, binds to many membrane molecules such as heparin, proteoglycans including syndecan-1, sulfoglycolipids, and phospholipids [31,32]. Also, HMGB1-mediated movement of smooth muscle cell involved in the activation of the MAP-kinase pathway. Additionally, nuclear translocation of phosphorylated extracellular regulated kinase-1 and -2. is involved in cell signaling via an unidentified Gi/o protein [30] (Figure 3B). Induction of intracellular signaling by HMGB1 through RAGE can activate two different cascades, one involving the involves the Ras-mitogen-activated protein (MAP) kinase pathway and a second that involves a small GTPases Rac and Cdc42 leading to cytoskeletal reorganization and subsequent nuclear factor (NF)-B nuclear translocation-mediating inflammation [33] (Figure 3C). RAGE is also expressed on mononuclear phagocytes. Also, its interaction with AGEs enhances cellular oxidant stress [34] and generation of thiobarbituric acid reactive substances and activation of NF-B [33] (Figure 3D). RAGE signaling has also been shown to stimulates an inflammatory response when AGE-modified β2 microglobulin binds RAGE in mononuclear phagocytes to mediate monocyte chemotaxis and induce TNF release [34]. However, it has been found that HMGB1-induced differentiation of erythroleukaemia cells is independent on RAGE signaling. This suggest that there may be additional signaling HMGB1 receptors yet to be identified [35]. Meanwhile Tumor growth and metastasis was suppressed when HMGB1 was prevented from interacting with RAGE using RAGE-blocking antibodies or neutralizing anti-HMGB1 antibodies. RAGE has been implicated in the pathogenesis of multiple diseases such as diabetes, atherosclerosis, and Alzheimer’s disease [36,37]. It is also indicated that cell membrane-associated HMGB1 signals neurite outgrowth by interaction with the multiligand receptor RAGE [28]. Potential dangerous signs may occur when local binding of HMGB1 restricts the diffusion of extracellular HMGB1 and inhibit systemic release.
Cellular sources of HMGB1
Current studies have shown that HMGB1 is normally located in the nucleus and translocates from the nucleus to the cytosol, including mitochondria and lysosome, following various stressors (e.g., cytokine, chemokine, heat, hypoxia, H2O2, and oncogene). Although the function of cytosolic HMGB1 still remains poorly understood, there is evidence that the main function of HMGB1 in cytoplasm is to provide positive regulator of autophagy, which was first reported in 2010 [38]. It is also now clear that HMGB1 is expressed in the nucleus of all vertebrate cells. In contrast, only resting human platelets express cytoplasmic HMGB1, which is exported to the cell surface during platelet activation. On the other hand, activated mononuclear phagocytes and pituicytes have developed the ability to translocate their nuclear HMGB1 to the cytoplasm [39]. Whether there are additional cell lineages that enable resting human platelets to express cytoplasmic HMGB1 that is exported to the cell surface during platelet activation is still not well understood but it is well established that HMGB1 associates with plasminogen and tissue plasminogen activator on cell surfaces and enhances plasminogen generation and proteolysis [40,41]. Understanding of signaling mechanism of cellular HMGB1 focus on the design of sepsis therapeutics that interferes with activation of blood clotting systems but it will be important to delineate the connection between neutralization of HMGB1 and coagulation mechanisms, as these two systems occupy a critical and finally a common pathway to tissue injury and death from sepsis.
Extracellular HMGB1 release
HMGB1 can be passively secreted from the nuclei of necrotic cells, damaged cells or actively secreted from activated macrophages/monocytes or pituicytes which does not involve cell death (Figure 2). These process ware not known until recently [7]. However, HMGB1 does not have a leader sequence and hence not processed via the endoplasmic reticulum/Golgi pathway. This is characterized by a few secreted proteins such as fibroblast growth factor and IL-1β. Pulse-chase labeling with 35S-methionine showed that extracellular HMGB1 was newly synthesized during the first 12 h after TNF stimulation with macrophages from a preformed pool [42]. Further studies have indicated that cultured, activated macrophages will translocate their nuclear HMGB1 to the cytoplasm before extracellular release via lysosomal exocytosis [7].
Figure 2.
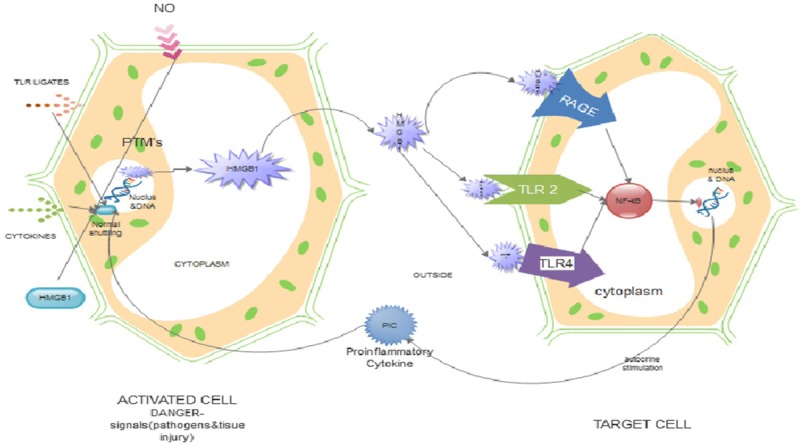
The role of post-translational modification (PTM) in translocation and immune activation by HMGB1. HMGB1 undergoes PTMs (e.g., acetylation, phosphorylation, methylation, etc.) following cell activation induced by external stimuli (necrotic cell or by active secretion from activated macrophages/monocytes). This modification leads to translocation of HMGB1 from the nucleus into the cytoplasm, into secretory endosomes and out of the cell. Extracellular HMGB1 functions as an immune activator by binding TLRs 2 and 4 and RAGE on immune cells like macrophages and neutrophils. Following binding, it leads to activation of gene expression via NF-kB. This explains the pro-inflammatory role HMGB1 during PTMs.
HMGB1 and post translational modifications
PTMs of HMGB1 protein have not been investigated as extensively as those of other HMG proteins but accumulating evidence has shown the remarkable biological significances induced by the post-translational: acetylation, methylation and phosphorylation, oxidation, glycosylation and ADP-ribosylation of the HMGB1 protein to modulate its interactions with DNA and other proteins.
Phosphorylation
Earlier studies indicate that HMGB1 isolated from lamb thymus could be phosphorylated by calcium/phospholipid-dependent protein kinase but not by cAMP-dependent protein kinase. Serine residues were phosphorylate in HMGB1 and a minimum of at least six phosphorylation sites were suggested [15]. Youn and colleagues found that HMGB1 can be phosphorylated in RAW264.7 cells and human monocytes after treatment with TNF-α or okadaic acid, a phosphatase inhibitor, resulting in the transport of HMGB1 to the cytoplasm for eventual secretion [15]. In their study, the phosphorylation sites were not identified but the possible phosphorylation sites were suggested to be Ser-34, Ser-38, Ser-41, Ser-45, Ser-52 and Ser-180, which reside mainly around NLS1 and NLS2 signal regions in the nucleus [15]. There is currently no study about the exact phosphorylation sites as well as the corresponding kinases involved in this process hence further studies will provide a better understanding of phosphorylation-controlled nuclear export of HMGB1.
Acetylation
Sterner et al in 1979 were the first to describe reversible acetylation of HMGB1 by incubating calf thymus homogenates with 3H-labeled acetate. Automated Edman degradation of the intact 3H-labeled HMGB1 revealed that two lysine residues in the N terminal region of the protein, Lys-2 and Lys-11, were acetylated [43]. HMGB1 acetylated/deacetylated by the same enzymes such as those acting on histone H4, indicated the roles of histone acetyltransferases (HATs) and histone deacetylases (HDACs) in the dynamic acetylation of HMGB1 protein [43].
It is also noted that the modification site was Lys-2 when the acetylated HMGB1 protein, isolated from cells grown in the presence of sodium n-butyrate, exhibited significantly enhanced ability to recognize UV light- or cisplatin-damaged DNA and four-way junction [44]. HMGB1 is an in-vitro substrate for CBP, but not for PCAF or Tip60, and the full-length HMGB1 is monoacetylated at Lys-2. Furthermore, the removal of the C terminal acidic tail of HMGB1 resulted in increased acetylation, catalyzed by CBP, at Lys-2 and a novel target site at Lys-81 [45]. HMGB1 is located in the nucleus in most cells and recent studies has demonstrated that HMGB1 could be secreted by monocytes and macrophages [46]. It can also passively leak out of cells during necrosis [5]. HMGB1 lacks a secretory signal peptide and doesn’t traverse the ER-Golgi system hence the secretion of this nuclear protein seems to require a tightly controlled relocation program [17]. It’s also well noted that, in monocytes and macrophages, HMGB1 can be extensively acetylated in such a way that the protein can be relocated from the nucleus to cytoplasm and eventually secreted out of the cell [17]. In resting macrophages, HMB1 can be forced to translocate from the nucleus to the cytosol via hyperacetylation with the aid of different proteolytic enzymes such as trypsin, Glu-C, and Asp-N, alone or in combination. Trypsin, Glu-C, and Asp-N are usually employed for the digestion, and the resulting peptides analyzed by MALDI-MS. Totally 17 lysine residues were suggested to be acetylated, among which Lys-27, Lys-28, Lys-29, Lys-179, Lys-181, Lys-182, Lys-183 and Lys-184 were the major acetylated residues, and all of them were within the nuclear localization signal regions [17].
Methylation
Ito et al in 2007, demonstrated the mono-methylation of lysine in HMGB1 isolated from neutrophils, which regulated its relocalization from the nucleus to cytoplasm, with the methylation site mapped at Lys-42. The methylation led to conformational changes of HMGB1 proteins. They indicated that most methylated HMGB1 resides in the cytoplasm of neutrophil, whereas un-methylated HMGB1 exists in the nucleus. They further stated that the possible mechanism for methylation-controlled distribution was that methylation of Lys-42 altered the conformation of box-A, thereby weakening its ability to bind to DNA. Also, methylated HMGB1 is distributed in the cytoplasm through passive diffusion from the nucleus [47]. Although HMGB1 was found to be methylated only in neutrophils, they suspect that this is not unique because the cytoplasmic release of HMGB1 also exists in other cells, which could also be controlled by its PTMs such as methylation.
Oxidation
It’s now clear that HMGB1 contains three cysteines, Cys23, 45, and 106 with Cys23 and 45 inducing conformational changes in response to oxidative stress, while Cys106 is critical for HMGB1 translocation from nucleus to cytoplasm. Also, reduction in inflammatory activities may be caused by the oxidation of Cys106 within the HMGB1 molecule [48]. Studies have shown that oxidation of Cys106 is necessary to block the stimulatory activity of HMGB1 [48]. Binding of HMGB1 to macrophages toll-like receptor 4 and Cys106 has also been reported [49]. Alterations such as the mutation of Cys106 also prevented the HMGB1-induced activation of cytokine release by cultured macrophages [49]. Injection of rHMGB1 to mice led to an acute inflammatory injuries in the lungs with neutrophil accumulation and development of lung edema as well as increased pulmonary production of inflammatory cytokines [50]. It’s now well documented that oxidation of HMGB1 reduce inflammatory activity both in-vitro and in-vivo. Reactive oxygen species (ROS) is noted to significantly promote HMGB1 translocation and release in activated immune cells or injured cells [51] which means that ROS is a major signal that decreases nuclear HMGB1 DNA binding activation hence promotes cytoplasmic translocation and release. The redox status of HMGB1 in terms of location and release directly influences its extracellular activity, such as immunity and autophagy [52].
Glycosylation
HMGB1 N-glycosylation is a prerequisite critical for nucleocytoplasmic translocation and extracellular secretion. Young and colleagues indicated that HMGB1 can be N-glycosylated at Asn37 and alternatively at Asn134/135 residues. This determines the nucleocytoplasmic transport, extracellular secretion, and protein stability of HMGB1. They noted two N-glycosylations at Asn37 and Asn134, with the consensus motifs of Asn-Xxx-Ser/Thr, and at the non-classical consensus residue Asn135 using recombinant HMGB1 proteins produced in both HEK293T and insect cells [53]. Recently, the sequence requirements of the acceptor substrate for N-glycosylation have become less strict; atypical (non-consensus) Asn-linked glycosylation is possible [54,55]. N-glycans are synthesized and transferred to polypeptides containing a signal peptide via glycosyltransferase to Asn residues within the Asn-Xxx-Ser/Thr sequon in the luminal side of the ER and Golgi apparatus [56]. The interaction of the autophagy-based unconventional secretion of HMGB1 and its glycosylation is needed to further identify and understand the pathophysiologic mechanism of HMGB1-mediated inflammation.
ADP-ribosylation
The addition of one or more ADP-ribose moieties to a protein by ADP-ribosyl transferases is termed ADP-ribosylation. Hassa and colleagues in their study classified ADP-ribosylation reactions into four groups: mono-ADP-ribosylation, poly-ADP-ribosylation, ADP-ribose cyclization, and formation of O-acetyl-ADP-ribose [57]. Hyper ADP-ribosylation of HMGB1 downregulates gene transcription since ADP-ribosylation is generally inversely related to transcription. PARP1-mediated poly-ADP-ribosylation reactions are required for the nuclear export and release of HMGB1 during cell death, especially necrosis and not mono-ADPribosylation alone [58,59]. Hyper poly (ADP-ribosylated) HMGB1 enhances inhibition of efferocytosis by binding to PS and RAGE when released [60] but lack of intracellular HMGB1 leads to excessive PARP1 activation and injury [61] hence cross linkage between HMGB1 and PARP1 in ADP-ribosylation reaction in regulating cell death.
Post translational modifications of HMGB and nuclear export
Bustin and colleagues proposed that there are two DNA-binding motifs on the HMGB1 protein, two nuclear localization signals and two putative nuclear export signals [62], There is enough evidence that the PTMs of HMGB1 also controlled the shuffle of this protein between the nucleus and cytoplasm through PTMs in the HMGB box-domains which regulates HMGB1’s biological function in gene transcription, [7,15,17,47]. Phosphorylation at both NLS sites is important in blocking HMGB1 re-entry to the nucleus and in the accumulation in the cytoplasm [15]. There is indication that the subcellular localizations of HMGB1 depends on its acetylation with deacetylase inhibitors causing the relocalization of HMGB1 from nucleus to the cytoplasm, and mutation of six major acetylation sites to glutamine, which mimics an acetylated lysine thereby inducing the relocalization of HMGB1 to the cytoplasm [17]. Nevertheless, methylation of HMGB1 in neutrophils could weaken its binding to DNA and causing its cytoplasmic relocalization in neutrophil through passive diffusion out of the nucleus [47]. The PTMs of HMGB1 and nuclear export clearly shown in Figure 2. It’s still not clear as to which specific modification is dominant, although all the PTMs plays important but different roles in HMGB1’s localization in cytoplasm under physiological conditions.
HMGB1 measurements and significance in clinical practice
Atkinson et al defined biomarkers as characteristics that are objectively measured and evaluated as indicators of normal biological processes, pathogenic processes, or pharmacologic responses to therapeutic intervention [63]. Many researchers have tried to evaluate the level of HMGB1 in samples (e.g., serum, plasma, cerebrospinal fluid, sputum, urine, fecal, and tissue) as a biomarker of human disease which can be used for detection and diagnosis of disease as well as prediction of response to therapeutic interventions and prognosis of outcome. They found out that circulating HMGB1 levels have been positively or inversely associated with sRAGE levels pointing to a fact that sRAGE not only regulates HMGB1 activity but also eliminates circulating HMGB1 in human disease [64]. Although ELISA and Western blot are the two methods used to detect HMGB1 in serum, plasma, and body fluid, HMGB1 levels in serum or plasma maybe five times higher when analyzed by Western blot as compared to ELISA because serum and plasma components (e.g., immunoglobulins, phospholipids, thrombomodulin, and proteoglycans) can interfere with the detection of HMGB1 detection via ELISA [65].
Barnay-Verdier and colleagues indicated that perchloric acid modified ELISA can detect masked forms of HMGB1 [66] but now several new techniques (e.g., DNA nanostructure-based assay) have shown dominance in detecting HMGB1 concentration in serum or supernatants [67]. Nevertheless, immunohistochemical staining is widely used in the detection of HMGB1 expression and localization in tissues while RT-PCR and q-PCR are widely used to test HMGB1 mRNA expression in tissues. HMGB1 gene polymorphisms are involved systemic inflammatory response syndrome [68,69] and several human disease and notable among this disease are chronic HBV infection [70], trauma [71], allogeneic hematopoietic cell transplantation [72]. Also Serum anti-HMGB1 autoantibody is increased in several autoimmune diseases [73].
The role of HMGB1 as a mediator of disease and target of therapy
Strategies such as antibodies, peptide, RNAi, anti-coagulant agents, endogenous hormones, chemicals including natural product, HMGB1-receptor and signaling pathway inhibition, artificial DNAs, physical methods (e.g., medical hydrogen gas), vagus nerve stimulation, and surgery have been proposed from cell, animal, and human studies to inhibit HMGB1 expression, release, and activity in a direct or indirect manner.
HMGB1 has multifunctional activities in the immune system and can induce a lot of host responses such as cell proliferation, cytokine production and increased expression of cell-surface molecules involved in inflammation. Whereas these activities resemble those of cytokines such as TNF-α and IL-1, there is enough evidence that this protein mediates disease is extensive and derives from two main sources: (1) demonstration that blockade of HMGB1 ameliorates disease in animal models and (2) demonstration of extracellular HMGB1 or cellular translocated HMGB1 in tissue or the blood from either animal models or patients with disease. Studies have now shown that HMGB1 is not only expressed in sepsis but also in wide range of disease such as rheumatoid arthritis, systemic lupus erythematosus, Sjögren’s syndrome and stroke among many others [25,74-79]. As various researches are still on going on HMGB1, the list will undoubtedly grow and it is likely that HMGB1 contributes to the pathogenesis of diseases in which immune-cell activation or cell death occurs.
Systemic lupus erythematosus is one of the diseases where HMGB1 may promote pathogenesis characterized by abnormalities in the extent of apoptotic death as well as impairment in the clearance of apoptotic cells. During the pathological process of the disease, inflammation which is usually a cardinal sign is incites when immune complexes comprised of nuclear macromolecules and anti-nuclear antibodies form and deposit in the tissue and these complexes usually stimulate the production of interferon α/β by plasmacytoid dendritic cells, a response that depends upon TLR9 as well as the Fcg receptor IIα [80,81]. The response above may involve HMGB1, which serves as a component of these complexes and stimulates responses via RAGE and also promote responses to DNA in complexes via its interaction with TLR9 [82].
In diseases where HMGB1 may be pathogenic, mechanisms to block the effects of HMGB1 focus on the antibodies or other agents that bind to it and therefore prevent its interaction with its receptors [23,26,83-85] and both TLR2 and TLR4 can serve as receptors for HMGB1 while RAGE appears to play a major role in the response to this protein [82,86,87]. While antibodies to RAGE have been used widely to treat inflammation in animal models with their efficacy potentially including blocking of HMGB1-receptor interactions, the isolated domain of HMGB1 can block the effect of the intact protein and A-box construct can attenuate disease in animal models of collagen-induced arthritis [88].
Most therapies target HMGB1 after it has left the cell but it important to note that strategies to target the release of HMGB1 is also possible. These mechanisms involve inhibitors of PTMs although their effects maybe broader if the same enzymes modifying HMGB1 also modify histones. Initial studies have demonstrated that gold salts a group of compounds that were initially the standard treatment of rheumatoid arthritis can block the release of HMGB1 from murine macrophages stimulated by LPS in-vitro by gold thiomalate. This effect was specific because gold thiomalate did not affect the release of TNF-α from these cells [89]. Although gold has other immune effects, it is possible that its anti-rheumatic activities is as a result of mechanisms that involved modification or translocation of HMGB1 during activation or a subsequent step in the intracellular trafficking.
Ostberg and colleagues have also demonstrated that Platinum compounds can also block HMGB1 release from macrophages. Therefore, they are very effective in animal models of arthritis [90]. They explained that these compounds can chemically modify DNA and create DNA adducts that avidly bind HMGB1. Although the mechanisms of action of gold and platinum still needs further investigation, these initial studies suggest a potential new target for the therapy of inflammatory and autoimmune disease.
HMGB1 and cancer
Many researchers have implicated the major role HMGB1 players in a number of cancers including colon, breast, lung, prostate, cervical, skin, kidney, stomach, pancreatic, liver, bone, and blood cancers [91-95]. Depending on the context, the study conditions, the location and modification, HMGB1 acts as both a tumor suppressor and an oncogenic factor in tumorigenesis and cancer therapy [96]. Sustainment of proliferative signaling; evasion of growth suppressors; avoidance of immune destruction; enablement of replicative immortality; tumor-promoting inflammation; activation of invasion and metastasis; induction of angiogenesis; genome instability and mutation; resistance to cell death; and deregulation of cellular energetics where the ten fundamental properties that drive tumor development and growth as proposed as Cancer hallmark by Hanahan and Weinberg in their cancer update [2,97].
Oncogenic roles in tumorigenesis
Studies have shown that the tumor microenvironment is usually made up of tumor cells, nontumor cells and several immune cells and HMGB1 is released together with autocrine from the tumor cells and the surrounding cells under hypoxia or other environmental stimuli [98-101]. It’s well noted that extracellular HMGB1 mediates communication between cells in the tumor microenvironment by several receptors (e.g., RAGE and TLR4). These receptors contributes to tumor growth and spreads by several mechanisms including sustenance of the inflammatory microenvironment [102-104], fulfillment of metabolic requirements [96,105], promotion of invasion and metastasis [106] inhibition of antitumor immunity [107], and promotion of angiogenesis [100,108], hence inhibition of HMGB1 release and activity can block tumor growth and development.
Tumor suppressor roles in tumorigenesis
Jiao and colleagues have indicated that HMGB1 binds to tumor suppressor RB, which leads to RB-dependent G1 arrest and apoptosis induction and prevents tumorigenesis in breast cancer cells in vitro and in vivo [109] which support early finding that intracellular HMGB1 maybe a tumor suppressor. Studies have shown that nuclear HMGB1 is an important architectural factor with DNA chaperone activity therefore loss of HMGB1 leads to genome instability with telomere shortening, which is major driving force in tumorigenesis [110] and that deficiencies of autophagy gene (e.g., Beclin-1, ATG5, UVRAG, Bif-1) increase tumorigenesis due to genome instability, inflammation, and organelle injury [111]. HMGB1 deficiency leads to autophagy dysfunction and may cause genome instability and inflammation which promotes tumorigenesis since HMGB1 is a positive regulator of autophagy [38,105]. The translational potential of the above findings is still not clear hence further research is needed to come with concrete explanations.
Sensitivity to anticancer therapy
Cancer cell death can be immunogenic or nonimmunogenic depending on the type of anticancer therapy [2,112,113]. HMGB1 when released by dead or dying cells can mediate immunogenic cell death and subsequent anti-tumor immunity and tumor clearance by binding to TLR4 [13,114-116]. It is now clear that TLR2, but not TLR4 in DCs, mediates the T-cell-dependent antitumor immune response that induces brain tumor regression [13,117] which suggest that HMGB1 release contributes to anticancer immunity. On the other hand, HMGB1 released during cell death may mediate immunogenic tolerance if it binds to TIM-3 or undergoes a redox transformation to oxidized form [118]. Luo et al demonstrated that remnant cancer cells has the ability to regrow and metastasize in a RAGE-dependent way when HMGB1 is released during chemotherapy [119] therefore the inhibition of HMGB1-RAGE pathway improves the effectiveness of chemotherapy [120]. It’s now clear that the activity of HMGB1 in anticancer immune response depends on many factors including receptor, death type, and redox state.
Resistance to anticancer therapy
Studies have shown that intracellular HMGB1 is generally negative regular for the effectiveness of anticancer therapy while extracellular HMGB1 play a significant role in anticancer therapy. Many chemotherapy agents including platinating agents have proven by researchers to increase HMGB1 expression [2,121] hence HMGB1 is becoming a recognized therapeutic target for chemotherapy resistance. It’s well known that RNAi down regulates HMGB1 expression and increased the anticancer activity of cytotoxic agents, while gene transfection up regulates HMGB1 expression and increase drug resistance [122,123]. It’s clear that increased HMGB1 expression in cancer cells facilitates chemotherapy resistance partly through inhibition of apoptosis and promotion of autophagy, which determine cell fate in anticancer therapy [120,122-125] but differs in the regulation of chemotherapeutic agent toxicity in cancer cells and normal cells [126]. The actual processes by which these differences occur need further investigation.
Conclusion
Our review points out clearly that HMGB1 protein is essential in chromatin dynamics and influences various DNA processes in the context of chromatin, which include PTMs. Up or down regulation of HMGB1 levels modifies the cellular phenotype and lead to developmental abnormalities and diseases. Therefore, characterization of these chemical modifications of HMGB1 protein will provide significant insights into the mechanism of action of this protein which may eventually lead to improved detection, therapy and prognosis of human diseases. It’s now also clear that HMGB1 can be inhibited by administration of specific and nonspecific binding proteins, including antibodies, HMGB1 A box, sRAGE, thrombomodulin and haptoglobin and can be a potential for target in sterile inflammation and injury. The development of future therapeutic agents targeting intracellular HMGB1 biology will require further understanding in the following directions: (i) the role of HMGB1 in autophagy, (ii) sensing of intracellular foreign nucleic acids and (iii) nucleosome formation. While we have outlined the role of PTMs of HMGB1 and several disease conditions such as inflammations, autoimmune diseases and cancer further studies is still needed to gain more insight into these modifications of HMGB1 and specific diseases entities.
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. 81370084, 81502663), the Social Development Foundation of Jiangsu Province (Grant no. BE2015668); Six talent peaks project in Jiangsu Province (2013-WSN-002, 2015-WSN-005); maternal and child project in Jiangsu Province (F201511).
Disclosure of conflict of interest
None.
References
- 1.Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, Huang J, Yu Y, Fan Xg, Yan Z, Sun X, Wang H, Wang Q, Tsung A, Billiar TR, Zeh HJ 3rd, Lotze MT, Tang D. HMGB1 in health and disease. Mol Aspects Med. 2014;40:1–116. doi: 10.1016/j.mam.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richard SA, Xiang LH, Yun JX, Zhou SS, Jiang YY, Wang J, Su ZL, Xu HX. Carcinogenic and therapeutic role of high-mobility group Box 1 in cancer: is it a cancer facilitator, a cancer inhibitor or both? World Cancer Research Journal. 2017;4:e919. [Google Scholar]
- 3.Richard SA, Sackey M, Su Z, Xu H. Pivotal neuroinflammatory and therapeutic role of high mobility group box 1 in ischemic stroke. Biosci Rep. 2017:37. doi: 10.1042/BSR20171104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 5.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 6.Stumbo AC, Cortez E, Rodrigues CA, Maria das Graças M, Porto LC, Barbosa HS, Carvalho L. Mitochondrial localization of non-histone protein HMGB1 during human endothelial cell-Toxoplasma gondii infection. Cell Biol Int. 2008;32:235–238. doi: 10.1016/j.cellbi.2007.08.031. [DOI] [PubMed] [Google Scholar]
- 7.Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, Rubartelli A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;3:995–1001. doi: 10.1093/embo-reports/kvf198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ullala AJ, Pisetskyb DS. Post-translational modification of HMGB1 and its role in immune activation. The Epigenetics of Autoimmune Diseases. 2009:165–178. [Google Scholar]
- 9.Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 10.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Antoine DJ, Harris HE, Andersson U, Tracey KJ, Bianchi ME. A systematic nomenclature for the redox states of high mobility group box (HMGB) proteins. Mol Med. 2014;20:135–137. doi: 10.2119/molmed.2014.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richard SA, Min W, Su Z, Xu H. High mobility group box 1 and traumatic brain injury. Journal of Behavioral and Brain Science. 2017;7:50. [Google Scholar]
- 13.Seidu RA, Wu M, Su Z, Xu H. Paradoxical role of high mobility group box 1 in glioma: a suppressor or a promoter? Oncol Rev. 2017;11:325. doi: 10.4081/oncol.2017.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richard AS, Min W, Su Z, Xu HX. Epochal neuroinflammatory role of high mobility group box 1 in central nervous system diseases. AIMS Molecular Science. 2017;4:185–218. [Google Scholar]
- 15.Youn JH, Shin JS. Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J Immunol. 2006;177:7889–7897. doi: 10.4049/jimmunol.177.11.7889. [DOI] [PubMed] [Google Scholar]
- 16.Oppenheim JJ, Yang D. Alarmins: chemotactic activators of immune responses. Curr Opin Immunol. 2005;17:359–365. doi: 10.1016/j.coi.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 17.Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dumitriu IE, Baruah P, Valentinis B, Voll RE, Herrmann M, Nawroth PP, Arnold B, Bianchi ME, Manfredi AA, Rovere-Querini P. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J Immunol. 2005;174:7506–7515. doi: 10.4049/jimmunol.174.12.7506. [DOI] [PubMed] [Google Scholar]
- 19.Su Z, Sun C, Zhou C, Liu Y, Zhu H, Sandoghchian S, Zheng D, Peng T, Zhang Y, Jiao Z, Wang S, Xu H. HMGB1 blockade attenuates experimental autoimmune myocarditis and suppresses Th17-cell expansion. Eur J Immunol. 2011;41:3586–3595. doi: 10.1002/eji.201141879. [DOI] [PubMed] [Google Scholar]
- 20.Su Z, Zhang P, Yu Y, Lu H, Liu Y, Ni P, Su X, Wang D, Wang J, Shen H, Xu W, Xu H. HMGB1 facilitated macrophage reprogramming towards a proinflammatory M1-like phenotype in experimental autoimmune myocarditis development. Sci Rep. 2016;6:21884. doi: 10.1038/srep21884. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Mitola S, Belleri M, Urbinati C, Coltrini D, Sparatore B, Pedrazzi M, Melloni E, Presta M. Cutting edge: extracellular high mobility group box-1 protein is a proangiogenic cytokine. J Immunol. 2006;176:12–15. doi: 10.4049/jimmunol.176.1.12. [DOI] [PubMed] [Google Scholar]
- 22.Abeyama K, Stern DM, Ito Y, Kawahara KI, Yoshimoto Y, Tanaka M, Uchimura T, Ida N, Yamazaki Y, Yamada S, Yamamoto Y, Yamamoto H, Iino S, Taniguchi N, Maruyama I. The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J Clin Invest. 2005;115:1267–1274. doi: 10.1172/JCI22782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qin S, Wang H, Yuan R, Li H, Ochani M, Ochani K, Rosas-Ballina M, Czura CJ, Huston JM, Miller E, Lin X, Sherry B, Kumar A, Larosa G, Newman W, Tracey KJ, Yang H. Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp Med. 2006;203:1637–1642. doi: 10.1084/jem.20052203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ueno H, Matsuda T, Hashimoto S, Amaya F, Kitamura Y, Tanaka M, Kobayashi A, Maruyama I, Yamada S, Hasegawa N, Soejima J, Koh H, Ishizaka A. Contributions of high mobility group box protein in experimental and clinical acute lung injury. Am J Respir Crit Care Med. 2004;170:1310–1316. doi: 10.1164/rccm.200402-188OC. [DOI] [PubMed] [Google Scholar]
- 25.Goldstein RS, Gallowitsch-Puerta M, Yang L, Rosas-Ballina M, Huston JM, Czura CJ, Lee DC, Ward MF, Bruchfeld AN, Wang H, Lesser ML, Church AL, Litroff AH, Sama AE, Tracey KJ. Elevated high-mobility group box 1 levels in patients with cerebral and myocardial ischemia. Shock. 2006;25:571–574. doi: 10.1097/01.shk.0000209540.99176.72. [DOI] [PubMed] [Google Scholar]
- 26.Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, Susarla SM, Ulloa L, Wang H, DiRaimo R, Czura CJ, Wang H, Roth J, Warren HS, Fink MP, Fenton MJ, Andersson U, Tracey KJ. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neeper M, Schmidt A, Brett J, Yan S, Wang F, Pan Y, Elliston K, Stern D, Shaw A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem. 1992;267:14998–15004. [PubMed] [Google Scholar]
- 28.Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, Nagashima M, Lundh ER, Vijay S, Nitecki D, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995;270:25752–25761. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- 29.Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, Tanji N, Lu Y, Lalla E, Fu C, Hofmann MA, Kislinger T, Ingram M, Lu A, Tanaka H, Hori O, Ogawa S, Stern DM, Schmidt AM. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature. 2000;405:354–360. doi: 10.1038/35012626. [DOI] [PubMed] [Google Scholar]
- 30.Degryse B, Bonaldi T, Scaffidi P, Müller S, Resnati M, Sanvito F, Arrigoni G, Bianchi ME. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J Cell Biol. 2001;152:1197–1206. doi: 10.1083/jcb.152.6.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bianchi ME. Interaction of a protein from rat liver nuclei with cruciform DNA. EMBO J. 1988;7:843–9. doi: 10.1002/j.1460-2075.1988.tb02883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salmivirta M, Rauvala H, Elenius K, Jalkanen M. Neurite growth-promoting protein (amphoterin, p30) binds syndecan. Exp Cell Res. 1992;200:444–451. doi: 10.1016/0014-4827(92)90194-d. [DOI] [PubMed] [Google Scholar]
- 33.Huttunen HJ, Fages C, Rauvala H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-κB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J Biol Chem. 1999;274:19919–19924. doi: 10.1074/jbc.274.28.19919. [DOI] [PubMed] [Google Scholar]
- 34.Miyata T, Hori O, Zhang J, Yan S, Ferran L, Iida Y, Schmidt AM. The receptor for advanced glycation end products (RAGE) is a central mediator of the interaction of AGE-beta2microglobulin with human mononuclear phagocytes via an oxidant-sensitive pathway. Implications for the pathogenesis of dialysis-related amyloidosis. J Clin Invest. 1996;98:1088–94. doi: 10.1172/JCI118889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sparatore B, Pedrazzi M, Passalacqua M, Gaggero D, Patrone M, Pontremoli S, Melloni E. Stimulation of erythroleukaemia cell differentiation by extracellular high-mobility group-box protein 1 is independent of the receptor for advanced glycation end-products. Biochem J. 2002;363:529–535. doi: 10.1042/0264-6021:3630529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmidt AM, Du Yan S, Yan SF, Stern DM. The biology of the receptor for advanced glycation end products and its ligands. Biochim Biophys Acta. 2000;1498:99–111. doi: 10.1016/s0167-4889(00)00087-2. [DOI] [PubMed] [Google Scholar]
- 37.Richard SA, Zheng S, Su Z, Gao J, Xu H. The pivotal neuroinflammatory, therapeutic and neuroprotective role of alpha-mangostin. Journal of Neurology Research. 2017;7:67–79. [Google Scholar]
- 38.Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ 3rd, Lotze MT. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881–892. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rouhiainen A, Imai S, Rauvala H, Parkkinen J. Occurrence of amphoterin (HMG1) as an endogenous protein of human platelets that is exported to the cell surface upon platelet activation. Thromb Haemost. 2000;84:1087–1094. [PubMed] [Google Scholar]
- 40.Parkkinen J, Rauvala H. Interactions of plasminogen and tissue plasminogen activator (t-PA) with amphoterin. Enhancement of t-PAcatalyzed plasminogen activation by amphoterin. J Biol Chem. 1991;266:16730–16735. [PubMed] [Google Scholar]
- 41.Parkkinen J, Raulo E, Merenmies J, Nolo R, Kajander EO, Baumann M, Rauvala H. Amphoterin, the 30-kDa protein in a family of HMG1-type polypeptides. Enhanced expression in transformed cells, leading edge localization, and interactions with plasminogen activation. J Biol Chem. 1993;268:19726–19738. [PubMed] [Google Scholar]
- 42.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 43.Boffa LC, Sterner R, Vidali G, Allfrey VG. Post-synthetic modifications of nuclear proteins high mobility group proteins are methylated. Biochem Biophys Res Commun. 1979;89:1322–1327. doi: 10.1016/0006-291x(79)92153-3. [DOI] [PubMed] [Google Scholar]
- 44.Ugrinova I, Pasheva EA, Armengaud J, Pashev IG. In vivo acetylation of HMG1 protein enhances its binding affinity to distorted DNA structures. Biochemistry. 2001;40:14655–14660. doi: 10.1021/bi0113364. [DOI] [PubMed] [Google Scholar]
- 45.Pasheva E, Sarov M, Bidjekov K, Ugrinova I, Sarg B, Lindner H, Pashev IG. In vitro acetylation of HMGB-1 and-2 proteins by CBP: the role of the acidic tail. Biochemistry. 2004;43:2935–2940. doi: 10.1021/bi035615y. [DOI] [PubMed] [Google Scholar]
- 46.Müller S, Scaffidi P, Degryse B, Bonaldi T, Ronfani L, Agresti A, Beltrame M, Bianchi ME. The double life of HMGB1 chromatin protein: architectural factor and extracellular signal. EMBO J. 2001;20:4337–4340. doi: 10.1093/emboj/20.16.4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ito I, Fukazawa J, Yoshida M. Post-translational methylation of high mobility group box 1 (HMGB1) causes its cytoplasmic localization in neutrophils. J Biol Chem. 2007;282:16336–16344. doi: 10.1074/jbc.M608467200. [DOI] [PubMed] [Google Scholar]
- 48.Hoppe G, Talcott KE, Bhattacharya SK, Crabb JW, Sears JE. Molecular basis for the redox control of nuclear transport of the structural chromatin protein Hmgb1. Exp Cell Res. 2006;312:3526–3538. doi: 10.1016/j.yexcr.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 49.Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, Lu B, Chavan S, Rosas-Ballina M, Al-Abed Y, Akira S, Bierhaus A, Erlandsson-Harris H, Andersson U, Tracey KJ. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A. 2010;107:11942–11947. doi: 10.1073/pnas.1003893107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ. Cutting edge: HMG-1 as a mediator of acute lung inflammation. J Immunol. 2000;165:2950–2954. doi: 10.4049/jimmunol.165.6.2950. [DOI] [PubMed] [Google Scholar]
- 51.Tang D, Kang R, Zeh HJ 3rd, Lotze MT. High-mobility group box 1, oxidative stress, and disease. Antioxid Redox Signal. 2011;14:1315–1335. doi: 10.1089/ars.2010.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, Liu J, Antonelli A, Preti A, Raeli L, Shams SS, Yang H, Varani L, Andersson U, Tracey KJ, Bachi A, Uguccioni M, Bianchi ME. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. 2012;209:1519–1528. doi: 10.1084/jem.20120189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim YH, Kwak MS, Park JB, Lee SA, Choi JE, Cho HS, Shin JS. N-linked glycosylation plays a crucial role in the secretion of HMGB1. J Cell Sci. 2016;129:29–38. doi: 10.1242/jcs.176412. [DOI] [PubMed] [Google Scholar]
- 54.Valliere-Douglass JF, Eakin CM, Wallace A, Ketchem RR, Wang W, Treuheit MJ, Balland A. Glutamine-linked and non-consensus asparagine-linked oligosaccharides present in human recombinant antibodies define novel protein glycosylation motifs. J Biol Chem. 2010;285:16012–16022. doi: 10.1074/jbc.M109.096412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schwarz F, Aebi M. Mechanisms and principles of N-linked protein glycosylation. Curr Opin Struct Biol. 2011;21:576–582. doi: 10.1016/j.sbi.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 56.Hebert DN, Lamriben L, Powers ET, Kelly JW. The intrinsic and extrinsic effects of N-linked glycans on glycoproteostasis. Nat Chem Biol. 2014;10:902–910. doi: 10.1038/nchembio.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hassa PO, Haenni SS, Elser M, Hottiger MO. Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev. 2006;70:789–829. doi: 10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ditsworth D, Zong WX, Thompson CB. Activation of poly (ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. J Biol Chem. 2007;282:17845–17854. doi: 10.1074/jbc.M701465200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004;18:1272–1282. doi: 10.1101/gad.1199904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Davis K, Banerjee S, Friggeri A, Bell C, Abraham E, Zerfaoui M. Poly (ADP-ribosyl) ation of high mobility group box 1 (HMGB1) protein enhances inhibition of efferocytosis. Mol Med. 2012;18:359–69. doi: 10.2119/molmed.2011.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang H, Nace GW, McDonald KA, Tai S, Klune JR, Rosborough BR, Ding Q, Loughran P, Zhu X, Beer-Stolz D, Chang EB, Billiar T, Tsung A. Hepatocyte specific HMGB1 deletion worsens the injury in liver ischemia/reperfusion: a role for intracellular HMGB1 in cellular protection. Hepatology (Baltimore, Md.) 2014;59:1984–97. doi: 10.1002/hep.26976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bustin M, Lehn DA, Landsman D. Structural features of the HMG chromosomal proteins and their genes. Biochim Biophys Acta. 1990;1049:231–243. doi: 10.1016/0167-4781(90)90092-g. [DOI] [PubMed] [Google Scholar]
- 63.Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89–95. doi: 10.1067/mcp.2001.113989. [DOI] [PubMed] [Google Scholar]
- 64.Fukami A, Adachi H, Yamagishi S, Matsui T, Ueda S, Nakamura K, Enomoto M, Otsuka M, Kumagae S, Nanjo Y, Kumagai E, Esaki E, Murayama K, Hirai Y, Imaizumi T. Factors associated with serum high mobility group box 1 (HMGB1) levels in a general population. Metabolism. 2009;58:1688–1693. doi: 10.1016/j.metabol.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 65.Urbonaviciute V, Fürnrohr BG, Weber C, Haslbeck M, Wilhelm S, Herrmann M, Voll RE. Factors masking HMGB1 in human serum and plasma. J Leukoc Biol. 2007;81:67–74. doi: 10.1189/jlb.0306196. [DOI] [PubMed] [Google Scholar]
- 66.Barnay-Verdier S, Gaillard C, Messmer M, Borde C, Gibot S, Maréchal V. PCA-ELISA: a sensitive method to quantify free and masked forms of HMGB1. Cytokine. 2011;55:4–7. doi: 10.1016/j.cyto.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 67.Gaillard C, Borde C, Gozlan J, Maréchal V, Strauss F. A high-sensitivity method for detection and measurement of HMGB1 protein concentration by high-affinity binding to DNA hemicatenanes. PLoS One. 2008;3:e2855. doi: 10.1371/journal.pone.0002855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kornblit B, Munthe-Fog L, Petersen S, Madsen H, Vindeløv L, Garred P. The genetic variation of the human HMGB1 gene. Tissue Antigens. 2007;70:151–156. doi: 10.1111/j.1399-0039.2007.00854.x. [DOI] [PubMed] [Google Scholar]
- 69.Kornblit B, Munthe-Fog L, Madsen HO, Strøm J, Vindeløv L, Garred P. Association of HMGB1 polymorphisms with outcome in patients with systemic inflammatory response syndrome. Crit Care. 2008;12:R83. doi: 10.1186/cc6935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deng CQ, Deng GH, Wang YM. HMGB1 gene polymorphisms in patients with chronic hepatitis B virus infection. World J Gastroenterol. 2013;19:5144–5149. doi: 10.3748/wjg.v19.i31.5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zeng L, Zhang AQ, Gu W, Chen KH, Jiang DP, Zhang LY, Du DY, Hu P, Huang SN, Wang HY, Jiang JX. Clinical relevance of single nucleotide polymorphisms of the high mobility group box 1 protein gene in patients with major trauma in southwest China. Surgery. 2012;151:427–436. doi: 10.1016/j.surg.2011.07.075. [DOI] [PubMed] [Google Scholar]
- 72.Kornblit B, Masmas T, Petersen SL, Madsen HO, Heilmann C, Schejbel L, Sengeløv H, Müller K, Garred P, Vindeløv L. Association of HMGB1 polymorphisms with outcome after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2010;16:239–252. doi: 10.1016/j.bbmt.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 73.Urbonaviciute V, Voll RE. High-mobility group box 1 represents a potential marker of disease activity and novel therapeutic target in systemic lupus erythematosus. J Intern Med. 2011;270:309–318. doi: 10.1111/j.1365-2796.2011.02432.x. [DOI] [PubMed] [Google Scholar]
- 74.Popovic K, Ek M, Espinosa A, Padyukov L, Harris HE, Wahren-Herlenius M, Nyberg F. Increased expression of the novel proinflammatory cytokine high mobility group box chromosomal protein 1 in skin lesions of patients with lupus erythematosus. Arthritis Rheum. 2005;52:3639–3645. doi: 10.1002/art.21398. [DOI] [PubMed] [Google Scholar]
- 75.Kokkola R, Sundberg E, Ulfgren AK, Palmblad K, Li J, Wang H, Ulloa L, Yang H, Yan XJ, Furie R, Chiorazzi N, Tracey KJ, Andersson U, Harris HE. High mobility group box chromosomal protein 1: a novel proinflammatory mediator in synovitis. Arthritis Rheum. 2002;46:2598–2603. doi: 10.1002/art.10540. [DOI] [PubMed] [Google Scholar]
- 76.Andersson U, Erlandsson-Harris H. HMGB1 is a potent trigger of arthritis. J Intern Med. 2004;255:344–350. doi: 10.1111/j.1365-2796.2003.01303.x. [DOI] [PubMed] [Google Scholar]
- 77.Ek M, Popovic K, Erlandsson Harris H, Söderberg Nauclér C, Wahren-Herlenius M. Increased extracellular levels of the novel proinflammatory cytokine high mobility group box chromosomal protein 1 in minor salivary glands of patients with Sjögren’s syndrome. Arthritis Rheum. 2006;54:2289–2294. doi: 10.1002/art.21969. [DOI] [PubMed] [Google Scholar]
- 78.Kim JB, Choi JS, Yu YM, Nam K, Piao CS, Kim SW, Lee MH, Han PL, Park Js, Lee JK. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci. 2006;26:6413–6421. doi: 10.1523/JNEUROSCI.3815-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jiang W, Pisetsky DS. Mechanisms of disease: the role of high-mobility group protein 1 in the pathogenesis of inflammatory arthritis. Nat Clin Pract Rheumatol. 2007;3:52–58. doi: 10.1038/ncprheum0379. [DOI] [PubMed] [Google Scholar]
- 80.Båve U, Magnusson M, Eloranta ML, Perers A, Alm GV, Rönnblom L. FcγRIIa is expressed on natural IFN-α-producing cells (plasmacytoid dendritic cells) and is required for the IFN-α production induced by apoptotic cells combined with lupus IgG. J Immunol. 2003;171:3296–3302. doi: 10.4049/jimmunol.171.6.3296. [DOI] [PubMed] [Google Scholar]
- 81.Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest. 2005;115:407–417. doi: 10.1172/JCI23025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, Hua J, An LL, Audoly L, La Rosa G, Bierhaus A, Naworth P, Marshak-Rothstein A, Crow MK, Fitzgerald KA, Latz E, Kiener PA, Coyle AJ. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8:487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 83.Czura CJ, Yang H, Tracey KJ. High mobility group box-1 as a therapeutic target downstream of tumor necrosis factor. J Infect Dis. 2003;187(Suppl 2):S391–S396. doi: 10.1086/374753. [DOI] [PubMed] [Google Scholar]
- 84.Andersson U, Tracey KJ. HMGB1 as a mediator of necrosis-induced inflammation and a therapeutic target in arthritis. Rheum Dis Clin North Am. 2004;30:627–637. xi. doi: 10.1016/j.rdc.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 85.Sama AE, D’Amore J, Ward MF, Chen G, Wang H. Bench to bedside: HMGB1-a novel proinflammatory cytokine and potential therapeutic target for septic patients in the emergency department. Acad Emerg Med. 2004;11:867–873. doi: 10.1197/j.aem.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 86.Kokkola R, Andersson Å, Mullins G, Östberg T, Treutiger CJ, Arnold B, Nawroth P, Andersson U, Harris RA, Harris HE. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol. 2005;61:1–9. doi: 10.1111/j.0300-9475.2005.01534.x. [DOI] [PubMed] [Google Scholar]
- 87.Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 88.Kokkola R, Li J, Sundberg E, Aveberger AC, Palmblad K, Yang H, Tracey K, Andersson U, Harris HE. Successful treatment of collagen-induced arthritis in mice and rats by targeting extracellular high mobility group box chromosomal protein 1 activity. Arthritis Rheum. 2003;48:2052–2058. doi: 10.1002/art.11161. [DOI] [PubMed] [Google Scholar]
- 89.Zetterström CK, Jiang W, Wähämaa H, Östberg T, Aveberger AC, Schierbeck H, Lotze MT, Andersson U, Pisetsky DS, Harris HE. Pivotal advance: inhibition of HMGB1 nuclear translocation as a mechanism for the antirheumatic effects of gold sodium thiomalate. J Leukoc Biol. 2008;83:31–38. doi: 10.1189/jlb.0507323. [DOI] [PubMed] [Google Scholar]
- 90.Östberg T, Wähämaa H, Palmblad K, Ito N, Stridh P, Shoshan M, Lotze MT, Harris HE, Andersson U. Oxaliplatin retains HMGB1 intranuclearly and ameliorates collagen type IIinduced arthritis. Arthritis Res Ther. 2008;10:R1. doi: 10.1186/ar2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ellerman JE, Brown CK, de Vera M, Zeh HJ, Billiar T, Rubartelli A, Lotze MT. Masquerader: high mobility group box-1 and cancer. Clin Cancer Res. 2007;13:2836–2848. doi: 10.1158/1078-0432.CCR-06-1953. [DOI] [PubMed] [Google Scholar]
- 92.Lotze MT, DeMarco RA. Dealing with death: HMGB1 as a novel target for cancer therapy. Curr Opin Investig Drugs (London, England: 2000) 2003;4:1405–1409. [PubMed] [Google Scholar]
- 93.Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2009;28:367–388. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- 94.Tang D, Kang R, Zeh HJ 3rd, Lotze MT. High-mobility group box 1 and cancer. Biochim Biophys Acta. 2010;1799:131–140. doi: 10.1016/j.bbagrm.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li JH, CL , Richard SA, Liu YH. Giant solitary primary intracranial lymphoma masquerading as meningioma: a case and review of literature. Pan African Medical Journal. 2017:28. doi: 10.11604/pamj.2017.28.196.13996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kang R, Zhang Q, Zeh HJ 3rd, Lotze MT, Tang D. HMGB1 in cancer: good, bad, or both? Clin Cancer Res. 2013;19:4046–4057. doi: 10.1158/1078-0432.CCR-13-0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 98.Jube S, Rivera ZS, Bianchi ME, Powers A, Wang E, Pagano I, Pass HI, Gaudino G, Carbone M, Yang H. Cancer cell secretion of the DAMP protein HMGB1 supports progression in malignant mesothelioma. Cancer Res. 2012;72:3290–3301. doi: 10.1158/0008-5472.CAN-11-3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tafani M, Schito L, Pellegrini L, Villanova L, Marfe G, Anwar T, Rosa R, Indelicato M, Fini M, Pucci B, Russo MA. Hypoxia-increased RAGE and P2X7R expression regulates tumor cell invasion through phosphorylation of Erk1/2 and Akt and nuclear translocation of NF-kB. Carcinogenesis. 2011;32:1167–75. doi: 10.1093/carcin/bgr101. [DOI] [PubMed] [Google Scholar]
- 100.Van Beijnum J, Nowak-Sliwinska P, Van den Boezem E, Hautvast P, Buurman W, Griffioen A. Tumor angiogenesis is enforced by autocrine regulation of high-mobility group box 1. Oncogene. 2013;32:363–374. doi: 10.1038/onc.2012.49. [DOI] [PubMed] [Google Scholar]
- 101.Yang EJ, Lee W, Ku SK, Song KS, Bae JS. Anti-inflammatory activities of oleanolic acid on HMGB1 activated HUVECs. Food Chem Toxicol. 2012;50:1288–1294. doi: 10.1016/j.fct.2012.02.026. [DOI] [PubMed] [Google Scholar]
- 102.Bald T, Quast T, Landsberg J, Rogava M, Glodde N, Lopez-Ramos D, Kohlmeyer J, Riesenberg S, van den Boorn-Konijnenberg D, Hömig-Hölzel C. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature. 2014;507:109–113. doi: 10.1038/nature13111. [DOI] [PubMed] [Google Scholar]
- 103.Gebhardt C, Riehl A, Durchdewald M, Németh J, Fürstenberger G, Müller-Decker K, Enk A, Arnold B, Bierhaus A, Nawroth PP, Hess J, Angel P. RAGE signaling sustains inflammation and promotes tumor development. J Exp Med. 2008;205:275–285. doi: 10.1084/jem.20070679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mittal D, Saccheri F, Vénéreau E, Pusterla T, Bianchi ME, Rescigno M. TLR4-mediated skin carcinogenesis is dependent on immune and radioresistant cells. EMBO J. 2010;29:2242–2252. doi: 10.1038/emboj.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tang D, Kang R, Livesey KM, Kroemer G, Billiar TR, Van Houten B, Zeh HJ 3rd, Lotze MT. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab. 2011;13:701–711. doi: 10.1016/j.cmet.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sasahira T, Akama Y, Fujii K, Kuniyasu H. Expression of receptor for advanced glycation end products and HMGB1/amphoterin in colorectal adenomas. Virchows Arch. 2005;446:411–415. doi: 10.1007/s00428-005-1210-x. [DOI] [PubMed] [Google Scholar]
- 107.He Y, Zha J, Wang Y, Liu W, Yang X, Yu P. Tissue damage-associated “danger signals” influence T-cell responses that promote the progression of preneoplasia to cancer. Cancer Res. 2013;73:629–639. doi: 10.1158/0008-5472.CAN-12-2704. [DOI] [PubMed] [Google Scholar]
- 108.Sasahira T, Kirita T, Bhawal UK, Ikeda M, Nagasawa A, Yamamoto K, Kuniyasu H. The expression of receptor for advanced glycation end products is associated with angiogenesis in human oral squamous cell carcinoma. Virchows Arch. 2007;450:287–295. doi: 10.1007/s00428-006-0359-2. [DOI] [PubMed] [Google Scholar]
- 109.Jiao Y, Wang HC, Fan SJ. Growth suppression and radiosensitivity increase by HMGB1 in breast cancer1. Acta Pharmacol Sin. 2007;28:1957–1967. doi: 10.1111/j.1745-7254.2007.00669.x. [DOI] [PubMed] [Google Scholar]
- 110.Polanská E, Dobšáková Z, Dvořáčková M, Fajkus J, Štros M. HMGB1 gene knockout in mouse embryonic fibroblasts results in reduced telomerase activity and telomere dysfunction. Chromosoma. 2012;121:419–431. doi: 10.1007/s00412-012-0373-x. [DOI] [PubMed] [Google Scholar]
- 111.Zhao Z, Oh S, Li D, Ni D, Pirooz SD, Lee JH, Yang S, Lee JY, Ghozalli I, Costanzo V, Stark JM, Liang C. A dual role for UVRAG in maintaining chromosomal stability independent of autophagy. Dev Cell. 2012;22:1001–1016. doi: 10.1016/j.devcel.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hou W, Zhang Q, Yan Z, Chen R, Zeh Iii H, Kang R, Lotze M, Tang D. Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell Death Dis. 2013;4:e966. doi: 10.1038/cddis.2013.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–875. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 114.Suzuki Y, Mimura K, Yoshimoto Y, Watanabe M, Ohkubo Y, Izawa S, Murata K, Fujii H, Nakano T, Kono K. Immunogenic tumor cell death induced by chemoradiotherapy in patients with esophageal squamous cell carcinoma. Cancer Res. 2012;72:3967–3976. doi: 10.1158/0008-5472.CAN-12-0851. [DOI] [PubMed] [Google Scholar]
- 115.Yamazaki T, Hannani D, Poirier-Colame V, Ladoire S, Locher C, Sistigu A, Prada N, Adjemian S, Catani JP, Freudenberg M, Galanos C, André F, Kroemer G, Zitvogel L. Defective immunogenic cell death of HMGB1-deficient tumors: compensatory therapy with TLR4 agonists. Cell Death Differ. 2014;21:69–78. doi: 10.1038/cdd.2013.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Su Z, Ni P, She P, Liu Y, Richard SA, Xu W, Zhu H, Wang J. Bio-HMGB1 from breast cancer contributes to M-MDSC differentiation from bone marrow progenitor cells and facilitates conversion of monocytes into MDSC-like cells. Cancer Immunol Immunother. 2017;66:391–401. doi: 10.1007/s00262-016-1942-2. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 117.Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K, Edwards MR, Michelsen KS, Kroeger KM, Liu C. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009;6:e1000010. doi: 10.1371/journal.pmed.1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, Fujioka Y, Ohba Y, Gorman JV, Colgan JD. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. 2012;13:832–842. doi: 10.1038/ni.2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Luo Y, Chihara Y, Fujimoto K, Sasahira T, Kuwada M, Fujiwara R, Fujii K, Ohmori H, Kuniyasu H. High mobility group box 1 released from necrotic cells enhances regrowth and metastasis of cancer cells that have survived chemotherapy. Eur J Cancer. 2013;49:741–751. doi: 10.1016/j.ejca.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 120.Kang R, Tang D, Schapiro NE, Livesey KM, Farkas A, Loughran P, Bierhaus A, Lotze MT, Zeh HJ. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 2010;17:666–676. doi: 10.1038/cdd.2009.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rabik CA, Dolan ME. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat Rev. 2007;33:9–23. doi: 10.1016/j.ctrv.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Huang J, Ni J, Liu K, Yu Y, Xie M, Kang R, Vernon P, Cao L, Tang D. HMGB1 promotes drug resistance in osteosarcoma. Cancer Res. 2012;72:230–238. doi: 10.1158/0008-5472.CAN-11-2001. [DOI] [PubMed] [Google Scholar]
- 123.Livesey KM, Kang R, Vernon P, Buchser W, Loughran P, Watkins SC, Zhang L, Manfredi JJ, Zeh HJ 3rd, Li L, Lotze MT, Tang D. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 2012;72:1996–2005. doi: 10.1158/0008-5472.CAN-11-2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liu K, Huang J, Xie M, Yu Y, Zhu S, Kang R, Cao L, Tang D, Duan X. MIR34A regulates autophagy and apoptosis by targeting HMGB1 in the retinoblastoma cell. Autophagy. 2014;10:442–452. doi: 10.4161/auto.27418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ni Z, Dai X, Wang B, Ding W, Cheng P, Xu L, Lian J, He F. Natural Bcl-2 inhibitor (-)-gossypol induces protective autophagy via reactive oxygen species-high mobility group box 1 pathway in Burkitt lymphoma. Leuk Lymphoma. 2013;54:2263–2268. doi: 10.3109/10428194.2013.775437. [DOI] [PubMed] [Google Scholar]
- 126.Krynetskaia N, Xie H, Vucetic S, Obradovic Z, Krynetskiy E. High mobility group protein B1 is an activator of apoptotic response to antimetabolite drugs. Mol Pharmacol. 2008;73:260–269. doi: 10.1124/mol.107.041764. [DOI] [PMC free article] [PubMed] [Google Scholar]