

Abstract
Objective: RAGE pathway plays crucial effects in causing acute lung injury (ALI). Dexmedetomidine (DEX) is showed to mitigate sepsis-stimulated ALI. However, its mechanisms have not been verified. The study was to evaluate whether the RAGE pathway participated in the actions of DEX on sepsis-stimulated ALI in rats. Methods: Male rats were administrated with intravenously DEX 30 min after sepsis. At 24 h of sepsis, lung myeloperoxidase (MPO) and macrophages in the bronchoalveolarlavage fluid (BALF) were observed. The actions of DEX on pro-inflammatory molecules and related mechanisms were determined by immunological methods. Results: It was indicated that DEX markedly attenuated CLP-stimulated augment of lung inflammatory cells infiltration, along with significantly mitigated MPO activity. Besides, DEX obviously reduced lung wet/dry weight ratio and the levels of HMGB1 and RAGE in BALF and lung tissue. Moreover, DEX post-treatment apparently attenuated the histopathological lung injury compared with CLP model group. Furthermore, western blot analysis revealed that DEX efficiently restrained the activation of IκB-α, NF-κB p65, and MAPK. Conclusion: Our studies demonstrated that DEX attenuates the aggravation of sepsis-stimulated ALI via down regulation of RAGE pathway, which has a potential value in the clinical therapy.
Keywords: Dexmedetomidine, CLP, HMGB1, RAGE, ALI
Introduction
Acute lung injury (ALI) is characterized by a clinical disorders that derives from complicated reactions of the lungs to numerous direct and consequential injury [1]. ALI represents a strong inflammatory response to the interstitial edema, neutrophil infiltration, the destruction of the integrity of the epithelium, and lung parenchyma lesion [2]. The mechanism of ALI includes inflammation/anti-inflammation maladjustment, up-regulation of inflammatory mediators, and the increased production of adhesion molecules [3,4]. Neutrophil activation is conducive to lung inflammation via producing reactive oxygen species, proteolytic enzymes, and pro-inflammatory molecules which contribute to the development of ALI. Inflammatory mediators, for example, HMGB1, IL-1β, and IL-6, exert a crucial effect in involving and enlarging ALI via triggering neutrophils infiltration and chemotaxis. There is no particular drug to relieve ALI in clinic so far. Therefore, exploring for the original treatment and searching the novel reagent to remedy ALI may stand for a latent target for treatment.
The receptor for advanced glycation end-products (RAGE) results in continuing downstream signaling pathway activation, mediates hyper-inflammatory responses, and contributes to high mortality during both endotoxin shock and severe sepsis [5-7]. Su et al. have reported that RAGE is a direct index of lung inflammation in experimental lung inflammation models [8]. RAGE regulates the expression of pro-inflammatory cytokines by promoting the activation of MAPK and NF-kappaB (NF-κB) [9]. These pro-inflammatory molecules are regarded as vital in the development of ALI. Consequently, it has been shown that control of RAGE activity may be serviceable as anti-inflammation drugs.
Dexmedetomidine (DEX) has been reported to guard creatures against harm via its immunoregulation and anti-inflammation roles [10-12]. DEX can inhibite the pro-inflammatory molecules reactions in rats with sepsis [13], and likewise has underlying worth in dealing with several other disorders creature models, consisting of transient focal cerebral ischemia/reperfusion [14] and ALI [15]. Nevertheless, whether DEX attenuates RAGE expression and endow with its inhibition of ALI remain ill-defined. In the current research, we applied sepsis-stimulated ALI animal model to determine the role of DEX on the inflammation reaction, especially RAGE expression, and to further investigate its underlying pathological process.
Materials and methods
Animals
Six-week-old male RAGE deficient (RAGE-/-) rats (180-200 g) were obtained from the Shanghai Experimental Animal Center. All rats were arranged for a chamber (25±2°C, 50-75% humidity) and free choice feeding. The animal scheme was authorized by the Institutional Animal Ethics Committee of Zhejiang Provincial Hospital of TCM.
Experimental design and grouping
For the roles of DEX on CLP-stimulated ALI, WT rats were assigned to six groups: Sham, DEX, CLP, CLP+DEX (2.5), CLP+DEX (5), and CLP+DEX (10) groups (n=10). The details of various treatment of each group are summarized in Table 1. The rats were anesthetized by the intraperitoneal administration of sodium pentobarbital (50 mg/kg, Sigma-Aldrich, USA) before surgery. CLP animal model was performed to mimic polymicrobial sepsis as mentioned earlier [16]. Fluid resuscitation was performed to all animals by injecting 10 mL normal saline (NS) promptly after surgical procedures. DEX (2.5, 5 or 10 μg·kg-1·h-1) or NS were infused continuously through the femoral vein cannula 30 min after CLP. The dosages of these reagents were in accordance with Takumi Taniguchi’s former study and our preliminary experiments [17]. Lung samples and BALF were harvested at 24 h following sepsis for the following experiments.
Table 1.
Experimental treatment and grouping
| Treatment | Group | |
|---|---|---|
| Effects of dexmedetomidine (DEX) on CLP-induced ALI in WT rats (n=10 in each group) | ||
| Sham | Sham-operation plus normal saline (NS) | |
| DEX | Sham-operation plus DEX (10 μg·kg-1·h-1) | |
| CLP | CLP plus NS | |
| CLP+DEX (2.5) | CLP plus DEX (2.5 μg·kg-1·h-1) | |
| CLP+DEX (5) | CLP plus DEX (5 μg·kg-1·h-1) | |
| CLP+DEX (10) | CLP plus DEX (10 μg·kg-1·h-1) | |
| Effects of Anti-RAGE Ab on CLP-induced lung inflammatory cytokines production in the BALF from WT rats (n=10 in each group) | ||
| Sham | Sham-operation plus NS | |
| Anti-RAGE Ab | Sham-operation plus Anti-RAGE Ab (10 mg/kg) | |
| DEX | Sham-operation plus DEX (10 μg·kg-1·h-1) | |
| CLP | CLP plus NS | |
| CLP+Anti-RAGE Ab | CLP plus Anti-RAGE Ab (10 mg/kg) | |
| CLP+DEX (10) | CLP plus DEX (10 μg·kg-1·h-1) | |
| CLP+Anti-RAGE Ab+DEX (10) | CLP plus Anti-RAGE Ab (10 mg/kg) plus DEX (10 μg·kg-1·h-1) | |
| Effects of DEX on CLP-induced lung NF-κB activation and phosphorylation of MAPK in WT rats (n=10 in each group) | ||
| Sham | Sham-operation plus NS | |
| CLP | CLP plus NS | |
| CLP+DEX (2.5) | CLP plus DEX (2.5 μg·kg-1·h-1) | |
| CLP+DEX (5) | CLP plus DEX (5 μg·kg-1·h-1) | |
| CLP+DEX (10) | CLP plus DEX (10 μg·kg-1·h-1) | |
| Effects of DEX on CLP-induced ALI in WT and RAGE-/- rats (n=10 in each group) | ||
| WT | Sham | Sham-operation plus NS |
| CLP | CLP plus NS | |
| CLP+DEX (10) | CLP plus DEX (10 μg·kg-1·h-1) | |
| RAGE-/- | Sham | Sham-operation plus NS |
| CLP | CLP plus NS | |
| CLP+DEX (10) | CLP plus DEX (10 μg·kg-1·h-1) | |
For the roles of Anti-RAGE Ab on CLP-induced lung inflammatory mediators (such as, IL-1β, IL-6, TNF-α, and HMGB1) production in BALF, WT rats were allocated to one of the seven groups: Sham, Anti-RAGE Ab, DEX, CLP, CLP+Anti-RAGE Ab, CLP+DEX (10), and CLP+Anti-RAGE Ab+DEX (10) groups (n=10). The details of various treatment of each group are summarized in Table 1. WT rats were intraperitoneal administrated with Anti-RAGE Ab (10 mg/kg) 1 h prior to sepsis, and subsequently were administrated with DEX (10 μg·kg-1·h-1, i.v.) or a normal saline (NS) following sepsis; BALF was harvested at 24 h of sepsis.
For the roles of DEX on CLP-stimulated the activation of NF-κB and phosphorylation of MAPK, WT rats were assigned to five groups: Sham, CLP, CLP+DEX (2.5), CLP+DEX (5), and CLP+DEX (10) groups (n=10). The details of various treatment of each group are summarized in Table 1. Lung samples were harvested at 24 h of sepsis.
For the roles of DEX on sepsis-stimulated ALI, WT and RAGE-/- rats were allocated to one of the six groups: Sham in WT rats, CLP in WT rats, CLP+DEX (10) in WT rats, Sham in RAGE-/- rats, CLP in RAGE-/- rats, and CLP+DEX (10) in RAGE-/- rats groups (n=10). The details of various treatment of each group are summarized in Table 1. Lung samples and BALF were harvested at 24 h of sepsis.
Lung wet-to-dry ratio and MPO assay
The wet weight was recorded as the removed lung weight while the rats were euthanized. Dry weight was obtained from oven dried lung tissues (80°C for 48 h). Lung edema evaluation was performed to lung wet to dry ratio. Homogenates (5%) was obtained from homogenized lung samples (100 mg) by fluidizing in extraction buffer. MPO measurement was performed by the centrifuged homogenates (14,000 g for 30 min). MPO levels were determined by assay kits obtained from Beyotime Institute of Biotechnology (Shanghai, China) in accordance with the manuals.
Protein content assay and inflammatory cell counts of BALF
BCA test kit (Pierce, Rockford, Illinois) was used to determine the BALF total protein level in accordance with the manufacturer’s manuals. The cell pellets were collected from the centrifuged samples (800 g for 15 min at 4°C) and resuspended in PBS (1 mL). A hemocytometer was applied to measure the total cell counts. The Wright-Giemsa staining was used to differential cell counts in cytospins.
Assessment of inflammatory mediators
Production of inflammatory mediators, including IL-1β, IL-6, TNF-α, and HMGB1, in lung samples and BALF, was determined via ELISA assay kits on the basis of the manufacturer’s instructions.
Histopathological examination of the lungs
Rats that were not performed to collect BALF were subjected to lung histopathological evaluation. The lung sections were obtained from the process of excising, paraformaldehyde (4%) fixing, imbedding in paraffin, and sliced. Pathogenic manifestations of lung samples were stained by H&E and evaluated under an optical microscope.
Western blot analysis
Liquid nitrogen was used for freezing and collecting lung samples for storage until homogenization at 24 h after CLP. Cell Lysis Buffer on ice was used to homogenize lung samples. The protein was quantified by BCA determination kit (Beyotime Institute of Biotechnology, Shanghai, China). A PVDF membrane (Millipore, MA, USA) was used to electro-transferee proteins (50 μg) separated by SDS-PAGE. Buffer (3% BSA in TBST) was used to block the membrane at room temperature for 1 h. Shortly afterwards, the primary antibody was used to incubate the membrane overnight at 4°C by diluting in blocking buffer. An appropriate HRP-conjugated secondary antibody was used to incubate the membrane by diluting in blocking buffer after washing three times in TBST. The enhanced chemiluminescence system (Santa Cruz) was used to visualize and capture the images.
Real-time PCR
Trizol (Invitrogen Corporation, Carlsbad, CA) was used to extract total RNA from lung in accordance with the manufacturer’s manuals. The LightCycler 2.0 System (Roche Applied Science, Indianapolis, IN) was used to accomplish Real-time PCR. Specific primers for inflammatory cytokine’s gene expression were used to amplify the complementary DNA, and β-actin gene expression was used to normalize the findings. The 2-ΔΔCt method was used to determine the relative mean fold change of inflammatory cytokine’s gene expression [18].
Statistical analysis
Data were expressed as means ± standard deviation (S.D.). Statistically significant differences between groups were measured by one-way analysis of variance (ANOVA) followed by post hoc Bonferroni test. P<0.05 was considered statistically significant.
Results
Role of DEX on CLP-stimulated vascular edema
CLP administration generated a marked enhancement in capillary leakage, as illustrated by the BALF total protein level and lung W/D ratio. The BALF total protein level and lung W/D ratio were investigated at 24 h after CLP. As shown in Figure 1A and 1C, CLP administration markedly enhanced BALF total protein level and lung W/D ratio. Nevertheless, post-treatment with DEX markedly reduced BALF total protein level and lung W/D ratio compared to CLP group. These findings showed that DEX could attenuate lung edema in CLP-stimulated ALI rats.
Figure 1.
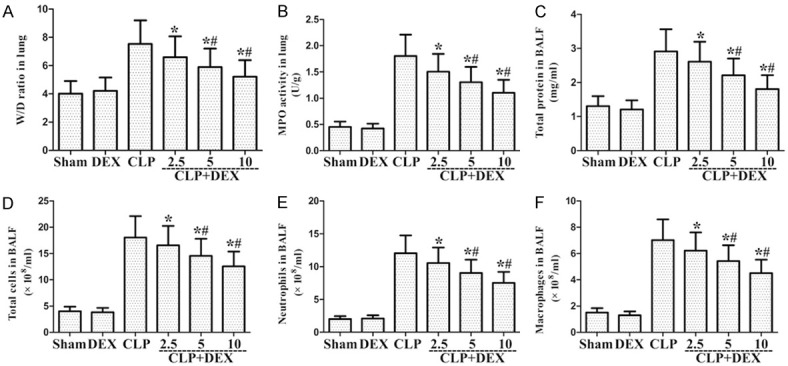
Role of DEX on lung MPO activity and W/D ratio in lung and the total protein levels and the number of total cells, neutrophils, and macrophages in the BALF of CLP-stimulated ALI in WT rats. Rats were infused continuously with DEX or NS through the femoral vein cannula 30 min after CLP. Lung W/D ratio (A) and MPO activity (B) were evaluated in lung samples at 24 h after CLP. BALF was harvested at 24 h after CLP to determine the total protein levels (C) and the count of total cells (D), neutrophils (E), and macrophage (F). *P<0.05, #P<0.01 CLP vs. CLP+DEX. DEX, dexmedetomidine.
Role of DEX on MPO content and cell content in CLP-Stimulated ALI WT rats
24 h following CLP treatment, the number of total cells, macrophages, and neutrophils in the BALF were markedly enhanced relative to the sham group. As shown in Figure 1D-F, post-treatment with DEX markedly reduced the count of total cells, neutrophils, and macrophages compared to CLP-stimulated ALI rats. MPO content was evaluated to farther explore the role of DEX on the infiltration of lung neutrophils in CLP-stimulated ALI rats, the activity of. As illustrated in Figure 1B, CLP administration markedly enhanced MPO activity compared to the sham group. In addition, this enhancement was obviously attenuated via post-treatment with DEX (Figure 1B). These findings demonstrated that DEX reduced lung hypernomic neutrophils infiltration in CLP-stimulated ALI rats.
Role of DEX on inflammatory molecule levels in lung tissue and BALF of CLP-Stimulated ALI WT rats
To determine the inflammatory molecules production, including IL-1β, IL-6, and TNF-α, in lung tissue and BALF, lung tissues and BALF were collected at 24 h following CLP treatment. As shown in Figure 2, CLP treatment markedly enhanced the production of IL-1β, IL-6, and TNF-α in lung tissues and BALF compared to the sham group. DEX markedly reduced IL-1β, IL-6, and TNF-α level compared to the CLP-stimulated ALI group (Figure 2).
Figure 2.
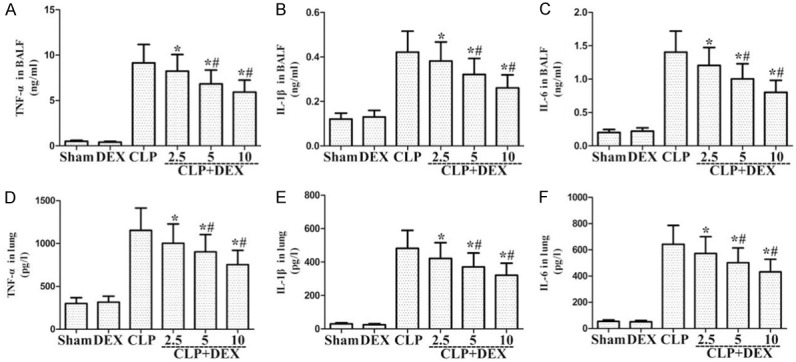
Role of DEX on the levels of inflammatory molecules in lung sample and BALF from WT rats with CLP-stimulated ALI. Rats were infused continuously with DEX or NS through the femoral vein cannula 30 min after CLP. Lung sample and BALF were harvested at 24 h after CLP to determine inflammatory molecules TNF-α (A, D), IL-1β (B, E), and IL-6 (C, F). *P<0.05, #P<0.01 CLP vs. CLP+DEX. DEX, dexmedetomidine.
Role of DEX on CLP-stimulated lung histopathological changes in WT rats
To determine the role of DEX on CLP-stimulated ALI in rats, lung manifestations were evaluated under an optical microscope. As illustrated in Figure 3C, lung samples collected from the CLP group indicated markedly pathological lesion, such as thickened alveolar wall, pulmonary hemorrhage, and infiltration of inflammatory cell. In addition, these manifestations were ameliorated via post-treatment with DEX (Figure 3D-F).
Figure 3.
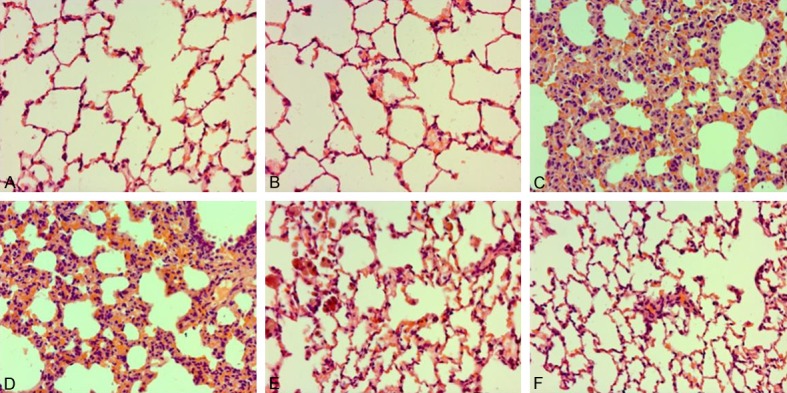
Role of DEX on pathological manifestations in lung samples in CLP-stimulated ALI. Rats were infused continuously with DEX or NS through the femoral vein cannula 30 min after CLP. Lung samples (n=10) were subjected to pathological assessment at 24 h following sepsis. Representative pathological manifestations of lung sample collected from different groups. (A) Control group, (B) DEX (10) group, (C) CLP group, (D) CLP+DEX (2.5) group, (E) CLP+DEX (5) group group, (F) CLP+DEX (10) group (H&E staining, magnification 200×). DEX, dexmedetomidine.
Role of DEX on RAGE and HMGB1 levels in sepsis-stimulated ALI
ELISA indicated that HMGB1 releases in the CLP group were markedly higher than in the sham group. DEX administration has been shown to obviously attenuate HMGB1 production compared with the CLP group at 24 h (Figure 4A). Furthermore, ELISA indicated that BALF sRAGE release in the CLP group was obviously higher than in the sham group. DEX administration failed to markedly attenuate sRAGE expression compared with the CLP group at 24 h (Figure 4B). Western blot and Real-time PCR indicated that lung RAGE and HMGB1 levels in the CLP group were obviously higher than in the sham group (Figure 4C-F). DEX administration markedly downregulated RAGE and HMGB1 levels compared with the CLP group at 24 h (Figure 4C-F).
Figure 4.

Role of DEX on the HMGB1 and RAGE levels in lung sample and BALF from WT rats with CLP-stimulated ALI. Rats were infused continuously with DEX or NS through the femoral vein cannula 30 min after CLP. A, B. BALF HMGB1 and sRAGE protein levels were measured by ELISA; C, D. Lung HMGB1 and RAGE protein levels were measured by Western Blot; E, F. Lung HMGB1 and RAGE mRNA expression were measured by Real-Time PCR. *P<0.05, #P<0.01 CLP vs. CLP+DEX. DEX, dexmedetomidine.
Role of anti-RAGE Ab on inflammatory molecule levels from sepsis-stimulated ALI
We demonstrated that sepsis-stimulated inflammatory molecule production was markedly attenuated by both DEX administration and Anti-RAGE Ab administration individually (Figure 5); the CLP+DEX (10) group has a stronger inhibitory effects on CLP-induced releases of inflammatory cytokines than the CLP+Anti-RAGE Ab group (P<0.05, Figure 5); furthermore, the CLP+Anti-RAGE Ab+DEX (10) group has a stronger inhibitory effects on CLP-induced release of inflammatory cytokines than the CLP+Anti-RAGE Ab group or the CLP+DEX (10) group (Figure 5). These findings suggested that RAGE is associated with DEX-involved amelioration of sepsis-stimulated inflammatory responses.
Figure 5.
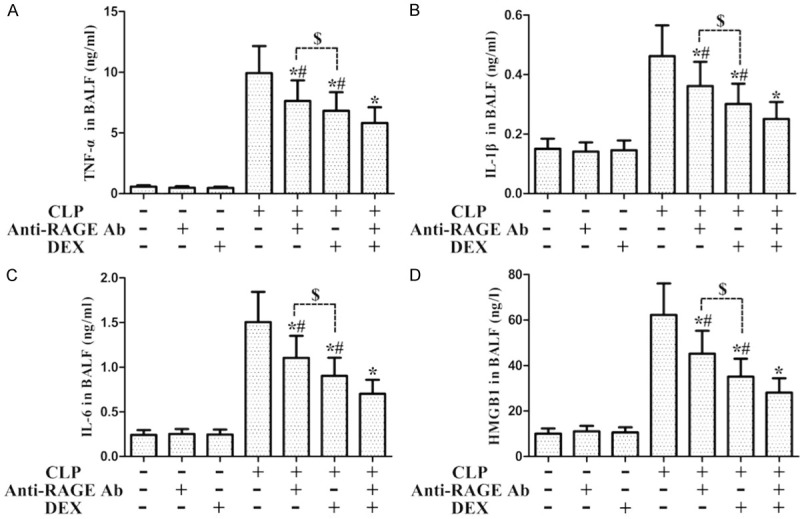
Role of Anti-RAGE Ab on the levels of inflammatory molecules from WT rats with CLP-stimulated ALI. Rats were intraperitoneal administration with Anti-RAGE Ab (10 mg/kg) 1 h before CLP, and then the rats were administration with DEX (10 μg·kg-1· h-1, i.v.) or normal saline after CLP. BALF was harvested to measure the inflammatory molecules TNF-α (A), IL-1β (B), IL-6 (C), and HMGB1 (D). *P<0.05 CLP vs. CLP+DEX, #P<0.05 CLP+Anti-RAGE Ab/DEX vs. CLP+Anti-RAGE Ab+DEX, $P<0.05 CLP+Anti-RAGE Ab vs. CLP+DEX. DEX, dexmedetomidine.
Role of DEX on the phosphorylation of NF-κB in CLP-stimulated ALI WT rats
The activation of NF-κB p65 and degradation of IκB-α were markedly enhanced after CLP administration (Figure 6). Post-treatment with DEX significantly attenuated the activation of NF-κB p65 and degradation of IκB-α (Figure 6). These findings demonstrated that DEX attenuated the phosphorylation of NF-κB in CLP-stimulated ALI rats.
Figure 6.
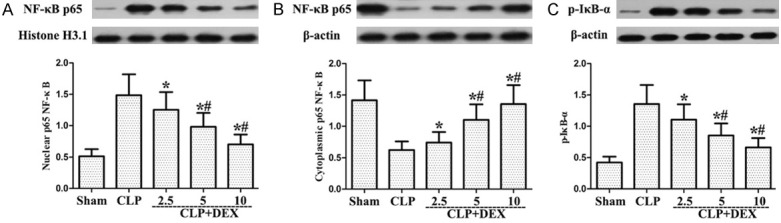
Roles of DEX on the phosphorylation of NF-κB from WT rats with CLP-stimulated ALI. Rats were infused continuously with DEX (2.5, 5 or 10 μg·kg-1·h-1) or NS through the femoral vein cannula 30 min after CLP. NF-κB (p65) in the nucleus (A) and the cytosol (B) and p-IκB-α (C) protein levels were individually determined by Western blot. *P<0.05, #P<0.01 CLP vs. CLP+DEX. DEX, dexmedetomidine.
Role of DEX on activation of MAPK in sepsis-stimulated ALI
As shown in Figure 7, post-treatment with DEX markedly reduced MAPK activation, including ERK, JNK, and p38 compared to the CLP group. Our findings showed that DEX suppressed MAPK activation in CLP-stimulated ALI.
Figure 7.
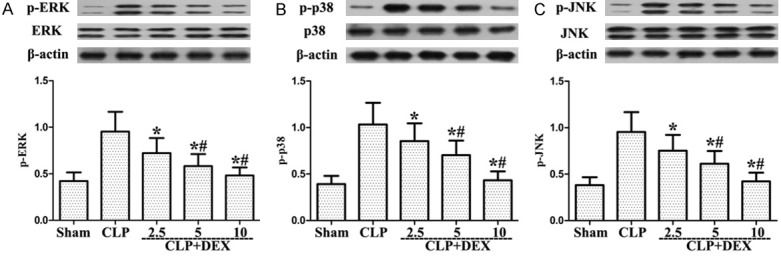
Role of DEX on the activation of MAPK from WT rats with CLP-stimulated ALI. Rats were infused continuously with DEX or NS through the femoral vein cannula 30 min after CLP. Western blotting was used to measure the ERK, p38 MAPK, and JNK levels later. Activated proteins levels were also evaluated. *P<0.05, #P<0.01 CLP vs. CLP+DEX. DEX, dexmedetomidine.
Role of DEX on the manifestations of sepsis-stimulated ALI
As shown in Figure 8, CLP administration markedly enhanced the lung MPO activity and W/D ratio in WT and RAGE-/- rats. Nevertheless, post-administration with DEX (10 μg·kg-1·h-1) significantly decreased lung MPO activity and W/D ratio compared to CLP group (Figure 8A and 8B). Amelioration role of DEX in RAGE-/- rats had higher MPO activity and W/D ratio in lung samples at 24 hours after sepsis compared to those in WT rats (Figure 8A and 8B). As illustrated in Figure 8, CLP treatment markedly increased the count of total cells, macrophages, and neutrophils and the total protein levels in the BALF from WT and RAGE-/- rats. Post-administration with DEX (10 μg·kg-1·h-1) markedly decreased the count of total cells, macrophages, and neutrophils and the total protein levels in the BALF compared with CLP group in rats (Figure 8C-F). Amelioration roles of DEX in RAGE-/- rats had larger count of total cells, macrophages, and neutrophils and the total protein concentration in the BALF at 24 hours after sepsis when relative to with those in WT rats (Figure 8C-F).
Figure 8.
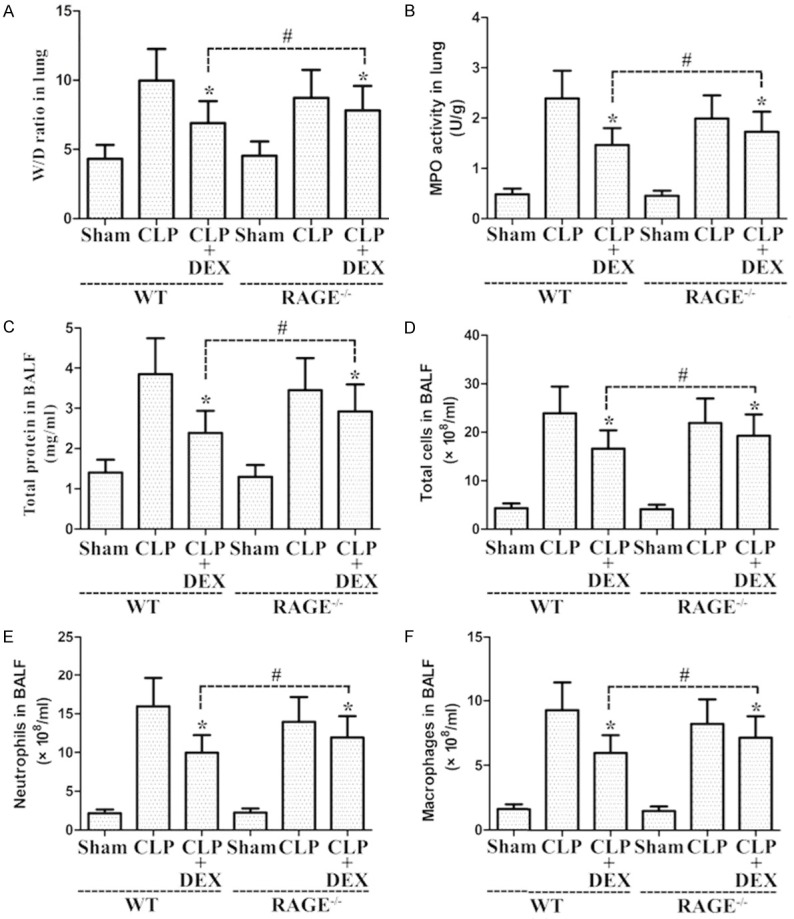
Roles of DEX on the pathological manifestations of CLP-stimulated ALI in WT and RAGE-/- rats. Rats were administration continuously with DEX (10 μg·kg-1·h-1) or NS through the femoral vein cannula 30 min after CLP. The lung W/D ratio (A) and MPO activity (B) were evaluated in lung samples obtained at 24 h after CLP. BALF was harvested to evaluate the total protein levels (C) and the count of total cells (D), neutrophils (E), and macrophage (F). *P<0.05 vs. CLP in WT or RAGE-/- rats, #P<0.05 vs. CLP+DEX in WT rats. DEX, dexmedetomidine.
Role of DEX on inflammatory cytokines release in lung samples and BALF from sepsis-stimulated ALI
As shown in Figure 9, CLP administration markedly enhanced the production of inflammatory cytokines in lung samples and BALF from rats compared with the sham group. DEX (10 μg·kg-1·h-1) significantly inhibited inflammatory cytokines release relative to the CLP-stimulated ALI in WT and RAGE-/- rats (Figure 9). Amelioration roles of DEX in RAGE-/- rats had higher levels of inflammatory molecule production in lung samples and BALF at 24 hours after sepsis when compared to those in WT rats (Figure 9).
Figure 9.
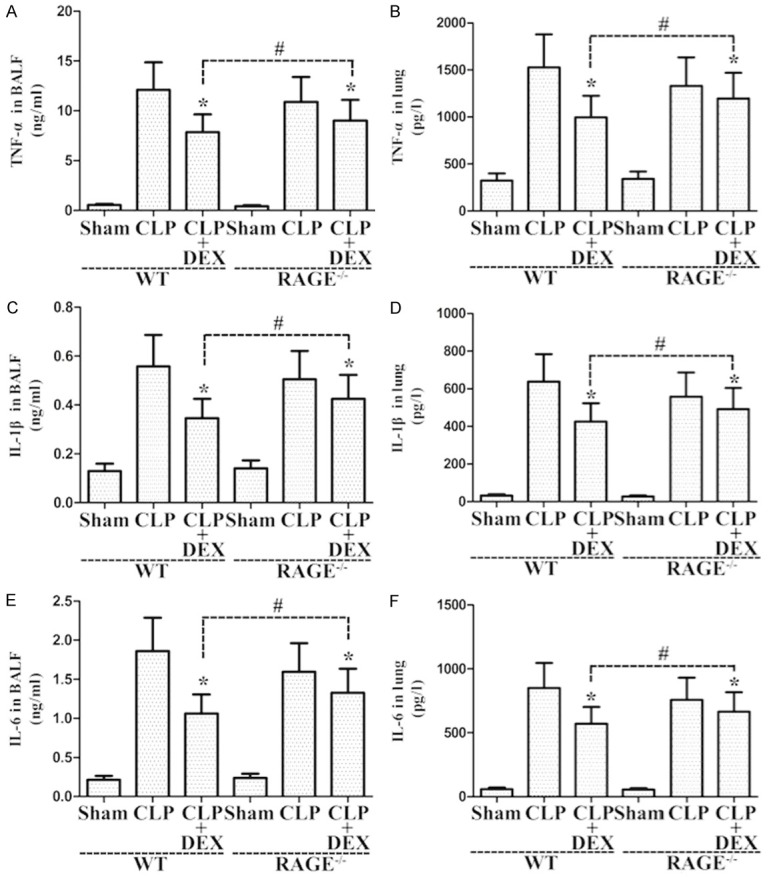
Roles of DEX on the levels of inflammatory molecules in lung samples and BALF from WT and RAGE-/- rats. Rats were infused continuously with DEX (10 μg·kg-1·h-1) or NS through the femoral vein cannula 30 min after CLP. Lung samples and BALF were harvested at 24 h after CLP to determine inflammatory molecules TNF-α (A, B), IL-1β (C, D), and IL-6 (E, F). *P<0.05 vs. CLP in WT or RAGE-/- rats, #P<0.05 vs. CLP+DEX in WT rats. DEX, dexmedetomidine.
Role of DEX on CLP-stimulated lung injury in WT and RAGE-/- rats
To investigate the effect of DEX on sepsis-stimulated ALI in WT and RAGE-/- rats, lung histopathological injury was observed under an optical microscope. As illustrated in Figure 10, lung samples collected from the CLP in WT and RAGE-/- rats exhibited markedly pathological lesion, such as thickened alveolar wall, pulmonary hemorrhage, and infiltration of inflammatory cell. Nevertheless, these manifestations were ameliorated via post-treatment with DEX (Figure 10). Furthermore, amelioration roles of DEX in RAGE-/- rats had severer manifestations in lung samples at 24 hours after sepsis when relative to those in WT rats (Figure 10).
Figure 10.
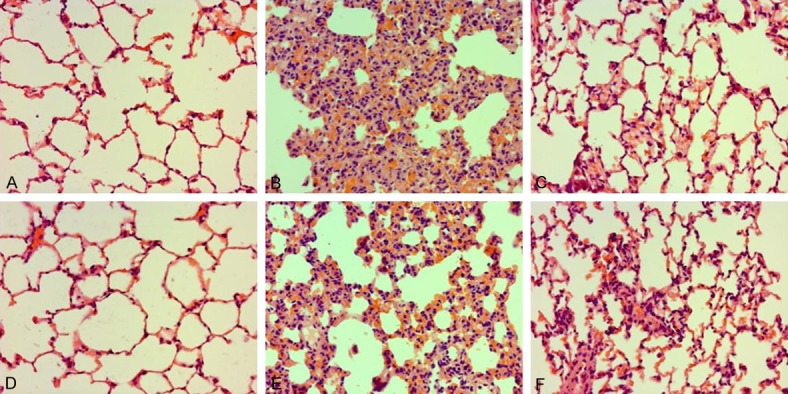
Roles of DEX on pathological manifestation in lung samples in WT and RAGE-/- rats. Rats were infused continuously with DEX (10 μg·kg-1·h-1) or NS through the femoral vein cannula 30 min after CLP. Lung samples (n=10) were subjected to histological measurements at 24 h after CLP. Representative pathological manifestation of lung samples collected from WT and RAGE-/- rats. (A) Sham group in WT rats, (D) Sham group in RAGE-/- rats, (B) CLP group in WT rats, (E) CLP group in RAGE-/- rats, (C) CLP+DEX (10) group in WT rats, (F) CLP+DEX (10) group in RAGE-/- rats (H&E staining, magnification 200×). DEX, dexmedetomidine.
Discussion
CLP triggered systemic inflammatory response represents an enhancement in systemic pro-inflammatory molecules and stimulates lung inflammatory responses that can result in ALI [19]. Since the manifestations of ALI in humans cannot be completely reproduced, CLP rat model was deemed as the best imitation model of the pathogenesis of CLP-stimulated ALI [20]. Widespread damage and leucocyte activation in ALI induces the increased permeability of the alveolar-capillary barrier [21]. Although some recent progress has been made, there is no useful therapy for ALI. Therefore, it is exceedingly significant to investigate a useful reagent and to explore the pathogenesis which can be applied to CLP-stimulated ALI. In the current research, we showed that DEX has a strong anti-inflammatory capacity which prevents the infiltration of leucocyte and protein leakage.
As one of the most abundant forms of human white blood cells, neutrophil is a key player of the lung inflammatory reaction [22]. Patients with the deterioration of pulmonary function showed that neutrophil is an important factor in lung inflammation [23]. Furthermore, MPO is considered to be a symbol of the presence and activity of neutrophil in the circulation of blood [24]. Our results indicated that the post-treatment of DEX inhibited lung MPO activity. This data indicated that DEX displays a protective effect against CLP-stimulated ALI by attenuating MPO activity.
DEX has been regarded as a highly selective α2-adrenoceptor agonist, which is mostly applied to different clinical settings for sedative or analgesic requirements [25]. Clinical evidence suggested that DEX will suppress the inflammatory response mediated by TNF-α, IL-1β, IL-6, and so on, in septic patients [26]. In addition, MDA and MPO levels were obviously lower in the CLP+DEX group compared to those in the CLP group. DEX post-treatment provides protection roles and may be involved in its obvious biological activities in CLP-stimulated lung inflammation. Our results suggest that DEX administration may supply protection and may be related to pro-inflammatory mediators’ changes. Therefore, DEX may act as a remedial drug for multiple medical conditions relating to sepsis-associated lung inflammation.
Sepsis is involved in the production of HMGB1 and S100A12 [27]. Inhibition of RAGE by different ligands leads to the activation of NF-κB [27]. Besides, RAGE is considered to be an endothelial cell adhesion receptor and contributes to the recruitment of leukocytes [27]. Engagement of RAGE pathway inhibits the host defense in sepsis and S. pneumonia animal models [27]. HMGB1and RAGE have been associated with the molecular mechanisms of ALI and adjusted with NF-κB and MAPK [28]. These experimental researches provide evidence to further understand the effect of RAGE signaling in the inflammatory response during medically major infectious diseases, which ultimately promotes better remedies resisting in pathogenic bacteria. Therefore, we suggested that DEX attenuated CLP-stimulated ALI by inhibiting HMGB1 and RAGE activation.
According to previous studies, NF-κB is a main transcription factor and exerts a critical regulatory action in ALI to numerous stimuli [29-31]. NF-κB exists in an inactive form and binds to its inhibitor IκB in the cytosol. Activated NF-κB is associated with immune regulation, inflammatory response, and anti-apoptosis via phosphorylating IκB and shifting into the nucleus [32,33]. Notably, previous researches indicated that the suppression of NF-κB pathway was recommended as a potential useful remedial tactics for ALI [34,35]. Therefore, we explored whether DEX could regulate NF-κB signaling pathway. Our results confirmed that DEX inhibited the activation of NF-κB via reducing the phosphorylation of p65 and I-κBα. MAPK signaling pathway, including ERK, JNK, and p38 MAPK, is important for the administration of pro-inflammatory mediator genes and inflammatory responses in ALI [15,28]. Thus, we investigated whether DEX exerts its anti-inflammatory effect through the MAPK signaling. Our data showed that DEX suppressed the activation of MAPK in CLP-stimulated ALI. The present study demonstrated that CLP resulted in the activation of NF-κB and MAPK, and DEX post-treatment attenuated these features. These findings showed that the control of CLP-stimulated ALI though DEX might be through the repression of NF-κB and MAPK activation.
Certain study limitations do exist. First, pulmonary gas exchange was not assessed even though a histologic change in the lung correlates with clinical lung injury. Secondly, the question of whether short term reduction in cytokine activation will translate to a sustained outcome of reduced inflammation once the DEX is metabolized in eventual clinical utility remains unanswered. Thirdly, our study only focuses on the activation of NF-κB and MAPK signal. Thirdly, the question of whether other pathways, e.g. PI3K/Akt and HO-1, participate in mediating the lung protective effects of DEX remains unanswered. Fourthly, this study did not use the antagonist of the α2-adrenergic receptors. The roles of the α2-adrenergic receptors on mediating the beneficial roles of DEX against sepsis-stimulated ALI remain to be determined.
In summary, our results indicated the alternatives of applying DEX as an impactful reagent with preventing CLP-stimulated ALI. The molecular mechanism of DEX is via downregulating HMGB1 and RAGE expression and reducing the activation of NF-κB and MAPK. Further researches have contributed to explore whether DEX is a potential agents to attenuate the occurrence of CLP-stimulated ALI.
Acknowledgements
This work was supported by the Zhejiang Province Traditional Chinese Medicine Research Fund (NO. 2012ZA048).
Disclosure of conflict of interest
None.
References
- 1.Caser EB, Zandonade E, Pereira E, Gama AM, Barbas CS. Impact of distinct definitions of acute lung injury on its incidence and outcomes in Brazilian ICUs: prospective evaluation of 7,133 patients*. Crit Care Med. 2014;42:574–582. doi: 10.1097/01.ccm.0000435676.68435.56. [DOI] [PubMed] [Google Scholar]
- 2.van Soeren MH, Diehl-Jones WL, Maykut RJ, Haddara WM. Pathophysiology and implications for treatment of acute respiratory distress syndrome. AACN Clin Issues. 2000;11:179–197. doi: 10.1097/00044067-200005000-00004. [DOI] [PubMed] [Google Scholar]
- 3.Bhatia M, Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol. 2004;202:145–156. doi: 10.1002/path.1491. [DOI] [PubMed] [Google Scholar]
- 4.Jiang S, Park DW, Tadie JM, Gregoire M, Deshane J, Pittet JF, Abraham E, Zmijewski JW. Human resistin promotes neutrophil proinflammatory activation and neutrophil extracellular trap formation and increases severity of acute lung injury. J Immunol. 2014;192:4795–4803. doi: 10.4049/jimmunol.1302764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Zoelen MA, van der Poll T. Targeting RAGE in sepsis. Crit Care. 2008;12:103. doi: 10.1186/cc6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lutterloh EC, Opal SM, Pittman DD, Keith JC Jr, Tan XY, Clancy BM, Palmer H, Milarski K, Sun Y, Palardy JE, Parejo NA, Kessimian N. Inhibition of the RAGE products increases survival in experimental models of severe sepsis and systemic infection. Crit Care. 2007;11:R122. doi: 10.1186/cc6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christaki E, Opal SM, Keith JC Jr, Kessimian N, Palardy JE, Parejo NA, Tan XY, Piche-Nicholas N, Tchistiakova L, Vlasuk GP, Shields KM, Feldman JL, Lavallie ER, Arai M, Mounts W, Pittman DD. A monoclonal antibody against RAGE alters gene expression and is protective in experimental models of sepsis and pneumococcal pneumonia. Shock. 2011;35:492–498. doi: 10.1097/SHK.0b013e31820b2e1c. [DOI] [PubMed] [Google Scholar]
- 8.Su X, Looney MR, Gupta N, Matthay MA. Receptor for advanced glycation end-products (RAGE) is an indicator of direct lung injury in models of experimental lung injury. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1–5. doi: 10.1152/ajplung.90546.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qin YH, Dai SM, Tang GS, Zhang J, Ren D, Wang ZW, Shen Q. HMGB1 enhances the proinflammatory activity of lipopolysaccharide by promoting the phosphorylation of MAPK p38 through receptor for advanced glycation end products. J Immunol. 2009;183:6244–6250. doi: 10.4049/jimmunol.0900390. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y, Miao L, Yao Y, Wu W, Wu X, Gong C, Qiu L, Chen J. Dexmedetomidine ameliorate CLP-induced rat intestinal injury via inhibition of inflammation. Mediators Inflamm. 2015;2015:918361. doi: 10.1155/2015/918361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J, Wang Z, Wang Y, Zhou G, Li H. The effect of dexmedetomidine on inflammatory response of septic rats. BMC Anesthesiol. 2015;15:68. doi: 10.1186/s12871-015-0042-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu Y, Liu Y, Huang H, Zhu Y, Zhang Y, Lu F, Zhou C, Huang L, Li X. Dexmedetomidine inhibits inflammatory reaction in lung tissues of septic rats by suppressing TLR4/NF-kappaB pathway. Mediators Inflamm. 2013;2013:562154. doi: 10.1155/2013/562154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu L, Bao H, Si Y, Wang X. Effects of dexmedetomidine on early and late cytokines during polymicrobial sepsis in mice. Inflamm Res. 2013;62:507–514. doi: 10.1007/s00011-013-0604-5. [DOI] [PubMed] [Google Scholar]
- 14.Zhu YM, Wang CC, Chen L, Qian LB, Ma LL, Yu J, Zhu MH, Wen CY, Yu LN, Yan M. Both PI3K/Akt and ERK1/2 pathways participate in the protection by dexmedetomidine against transient focal cerebral ischemia/reperfusion injury in rats. Brain Res. 2013;1494:1–8. doi: 10.1016/j.brainres.2012.11.047. [DOI] [PubMed] [Google Scholar]
- 15.Xu Y, Zhang R, Li C, Yin X, Lv C, Wang Y, Zhao W, Zhang X. Dexmedetomidine attenuates acute lung injury induced by lipopolysaccharide in mouse through inhibition of MAPK pathway. Fundam Clin Pharmacol. 2015;29:462–471. doi: 10.1111/fcp.12138. [DOI] [PubMed] [Google Scholar]
- 16.Yu M, Shao D, Liu J, Zhu J, Zhang Z, Xu J. Effects of ketamine on levels of cytokines, NFkappaB and TLRs in rat intestine during CLPinduced sepsis. Int Immunopharmacol. 2007;7:1076–1082. doi: 10.1016/j.intimp.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 17.Taniguchi T, Kurita A, Kobayashi K, Yamamoto K, Inaba H. Dose- and time-related effects of dexmedetomidine on mortality and inflammatory responses to endotoxin-induced shock in rats. J Anesth. 2008;22:221–228. doi: 10.1007/s00540-008-0611-9. [DOI] [PubMed] [Google Scholar]
- 18.Schmittgen TD, Livak KJ. Analyzing realtime PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 19.Lingaraju MC, Pathak NN, Begum J, Balaganur V, Bhat RA, Ramachandra HD, Ayanur A, Ram M, Singh V, Kumar D, Tandan SK. Betulinic acid attenuates lung injury by modulation of inflammatory cytokine response in experimentally-induced polymicrobial sepsis in mice. Cytokine. 2015;71:101–108. doi: 10.1016/j.cyto.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 20.Wu R, Dong W, Zhou M, Zhang F, Marini CP, Ravikumar TS, Wang P. Ghrelin attenuates sepsis-induced acute lung injury and mortality in rats. Am J Respir Crit Care Med. 2007;176:805–813. doi: 10.1164/rccm.200604-511OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meng F, Meliton A, Moldobaeva N, Mutlu G, Kawasaki Y, Akiyama T, Birukova AA. Asef mediates HGF protective effects against LPSinduced lung injury and endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2015;308:L452–463. doi: 10.1152/ajplung.00170.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mondrinos MJ, Zhang T, Sun S, Kennedy PA, King DJ, Wolfson MR, Knight LC, Scalia R, Kilpatrick LE. Pulmonary endothelial protein kinase C-delta (PKCdelta) regulates neutrophil migration in acute lung inflammation. Am J Pathol. 2014;184:200–213. doi: 10.1016/j.ajpath.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Le NP, Channabasappa S, Hossain M, Liu L, Singh B. Leukocyte-specific protein 1 regulates neutrophil recruitment in acute lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2015;309:L995–1008. doi: 10.1152/ajplung.00068.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirano Y, Aziz M, Yang WL, Wang Z, Zhou M, Ochani M, Khader A, Wang P. Neutralization of osteopontin attenuates neutrophil migration in sepsis-induced acute lung injury. Crit Care. 2015;19:53. doi: 10.1186/s13054-015-0782-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reardon DP, Anger KE, Adams CD, Szumita PM. Role of dexmedetomidine in adults in the intensive care unit: an update. Am J Health Syst Pharm. 2013;70:767–777. doi: 10.2146/ajhp120211. [DOI] [PubMed] [Google Scholar]
- 26.Memis D, Hekimoglu S, Vatan I, Yandim T, Yuksel M, Sut N. Effects of midazolam and dexmedetomidine on inflammatory responses and gastric intramucosal pH to sepsis, in critically ill patients. Br J Anaesth. 2007;98:550–552. doi: 10.1093/bja/aem017. [DOI] [PubMed] [Google Scholar]
- 27.van Zoelen MA, Achouiti A, van der Poll T. RAGE during infectious diseases. Front Biosci (Schol Ed) 2011;3:1119–1132. doi: 10.2741/215. [DOI] [PubMed] [Google Scholar]
- 28.Li K, Yang J, Han X. Ketamine attenuates sepsis-induced acute lung injury via regulation of HMGB1-RAGE pathways. Int Immunopharmacol. 2016;34:114–128. doi: 10.1016/j.intimp.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 29.Peng S, Hang N, Liu W, Guo W, Jiang C, Yang X, Xu Q, Sun Y. Andrographolide sulfonate ameliorates lipopolysaccharide-induced acute lung injury in mice by down-regulating MAPK and NF-kappaB pathways. Acta Pharm Sin B. 2016;6:205–211. doi: 10.1016/j.apsb.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu M, Cao FL, Zhang YF, Shan L, Jiang XL, An XJ, Xu W, Liu XZ, Wang XY. Tanshinone IIA therapeutically reduces LPS-induced acute lung injury by inhibiting inflammation and apoptosis in mice. Acta Pharmacol Sin. 2015;36:179–187. doi: 10.1038/aps.2014.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu ZN, Zhao M, Zheng Q, Zhao HY, Hou WJ, Bai SL. Inhibitory effects of rosiglitazone on paraquat-induced acute lung injury in rats. Acta Pharmacol Sin. 2013;34:1317–1324. doi: 10.1038/aps.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fukumoto J, Fukumoto I, Parthasarathy PT, Cox R, Huynh B, Ramanathan GK, Venugopal RB, Allen-Gipson DS, Lockey RF, Kolliputi N. NLRP3 deletion protects from hyperoxia-induced acute lung injury. Am J Physiol Cell Physiol. 2013;305:C182–189. doi: 10.1152/ajpcell.00086.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J, Qin Y, Mi X. The protective effects of bone marrow-derived mesenchymal stem cell (BMSC) on LPS-induced acute lung injury via TLR3-mediated IFNs, MAPK and NF-kappaB signaling pathways. Biomed Pharmacother. 2016;79:176–187. doi: 10.1016/j.biopha.2016.02.037. [DOI] [PubMed] [Google Scholar]
- 34.Jiang W, Luo F, Lu Q, Liu J, Li P, Wang X, Fu Y, Hao K, Yan T, Ding X. The protective effect of Trillin LPS-induced acute lung injury by the regulations of inflammation and oxidative state. Chem Biol Interact. 2016;243:127–134. doi: 10.1016/j.cbi.2015.09.010. [DOI] [PubMed] [Google Scholar]
- 35.Wang NA, Su Y, Che XM, Zheng H, Shi ZG. Penehyclidine ameliorates acute lung injury by inhibiting Toll-like receptor 2/4 expression and nuclear factor-kappaB activation. Exp Ther Med. 2016;11:1827–1832. doi: 10.3892/etm.2016.3154. [DOI] [PMC free article] [PubMed] [Google Scholar]