

Abstract
Cerebral ischemic injury has been the leading cause of death and long term disability in the world because of the lack of successful therapies to it, leading to neurological and behavioral deficits. The present study aims to investigate the effects of combined preconditioning (PC) with hypoxia and GYKI-52466 (GYKI) on cerebral ischemic injury and to explore the mechanism. The results showed that combined preconditioning with hypoxia and GYKI-52466 increased the survival rate of cerebral ischemia rats, alleviated the neurological deficit, increased the object recognition and social recognition memory of rats and suppressed the inflammatory reaction induced by cerebral ischemia. Further experiments found that preconditioning with hypoxia and GYKI-52466 significantly increased the HIF-1α and eNOS expression as well as eNOS activity, while inhibitors of HIF-1α and eNOS abolished the protective effects of hypoxia+GYKI PC on neurological deficit. Taken together, these results indicate that combined preconditioning with hypoxia and GYKI-52466 is effective to prevent cerebral ischemia injury, while HIF-1α and eNOS may be involved in the mechanism.
Keywords: Cerebral ischemic injury, preconditioning, hypoxia, GYKI-52466, HIF-1α, eNOS
Introduction
Ischemic stroke has become the third leading cause of death and the most causative factor of long term disability worldwide [1], leading to serious neuron damage and loss of neuronal function. As brain needs large amount of oxygen because of its high intrinsic oxygen consumption rate, minutes of cerebral blood supply stoppage can cause oxygen deficiency and energy metabolism disturbance to compromised areas [2]. Cerebral ischemic injury often causes deficits in spatial learning and memory and seriously reduces the life quality of patients. Unfortunately, although decades of efforts have been done to seek possible treatment for ischemic stroke induced neuron damage and deficits in spatial learning and memory, effective approach for prevention and treatment of them remains unavailable [3].
Hypoxia preconditioning (PC) refers to a brief period of sub-lethal hypoxia that provides protection against subsequent lethal hypoxia or ischemia insults [4]. Many researchers have found that it can trigger multiple endogenous protective mechanisms to restore O2 homeostasis at the cellular, tissue and organism levels [5,6]. During hypoxia exposure, hypoxia-inducible factor 1-alpha (HIF-1α) is an important protein activated and can provide protection against subsequent lethal insults by regulating the expression of many target genes involved in cell metabolism. However, it still requires more investigation to understand the underlying mechanism of hypoxia PC and to optimize the effect of hypoxia PC in experimental and clinical settings. GYKI-52466 (a 2,3-bezodiazepine derivative) is widely regarded as a selective non-competitive α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor negative allosteric modulator [7,8]. Some previous studies have showed that GYKI-52466 has both anticonvulsant ability and neuroprotection against ischemia stroke [9,10]. However, the therapeutic utility of normal dose of GYKI-52466 is limited by its adverse side effects, such as sedation, ataxia, and confusion [11,12]. Some recent studies showed that low-dose GYKI-52466 PC significantly protects against seizures and cerebral ischemia without producing side effects, making the low-dose GYKI-52466 PC a new promising approach to treat ischemia stroke [13,14]. To optimize the protective effect at the lowest dosage, we hypothesized that a combined PC with hypoxia and GYKI-52466 may produce better protection against cerebral ischemic injury. Therefore, to testify this hypothesis, we examined the protective effects of combined PC with hypoxia and GYKI-52466 in an animal model of cerebral ischemia by examining the survival rate, neurological deficit, changes in spatial learning and memory abilities and proimflammatory factors, then explored the involvement and interaction of HIF-1α and eNOSas the underlying mechanism.
Methods
Animals and chemicals
Adult male Sprague-Dawley rats (220-250 g) were purchased from Shanghai Animal Center and used in this study. The study was approved by the institutional animal care and use committee in the Shuguang Hospital, Shanghai University of Traditional Chinese Medicine. All rats were housed in clean, pathogen-free polycarbonate cages in the animal care facility on a 12 h light/dark cycle with free access to food and water. The room temperature was kept at 22±4°C. All experimental procedures are in accordance with the Guide for the Care and Use of Laboratory Animals, 8th Edition (National Academies Press, Washington, DC, 2010).
Experimental protocol
The experimental protocol was summarized in Figure 1. Rats were randomized into five groups (N=10 per group): Sham; Ischemia; hypoxia PC; GYKI PC; hypoxia+GYKI PC. Rats in Sham group received a sham surgery similar to middle cerebral artery occlusion (MCAO), except that the cerebral artery was not occluded; Rats in Ischemia group received a MCAO surgery; Rats in hypoxia PC group received preconditioning with hypoxia for four constitutive days before MCAO surgery; Rats in GYKI PC group received preconditioning with GYKI-52466 for four constitutive days before MCAO surgery; Rats in hypoxia+GYKI PC group received combined preconditioning with hypoxia and GYKI-52466 for four constitutive days before MCAO surgery. For GYKI PC, GYKI-52466 hydrochloride (Sigma-Aldrich, Australia) was repeatedly s.c.Injected (0.5 mg/kg weight) for four times before MCAO surgery, once a day.
Figure 1.
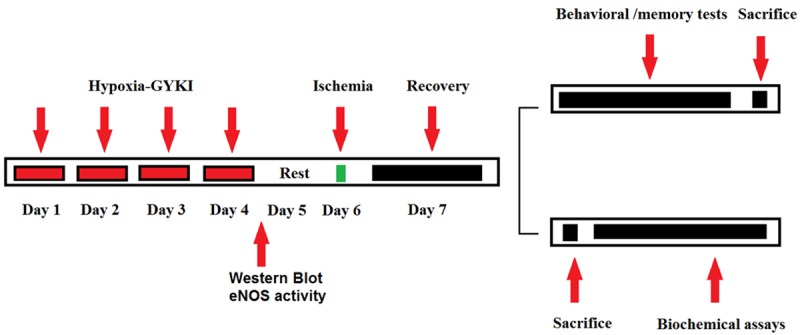
Experimental design. Rats received preconditioning with hypoxia or GYKI-52466 or hypoxia+GYKI-52466 for four constitutive days before MCAO surgery, then rested for one day. Next, rats received MCAO or sham surgery, then recovered for another day. Some rat then received behavioral or memory tests and sacrifice; some rats were sacrificed for biochemical assays.
MCAO surgery
The MCAO surgery was similar to the study of Ren et al. [15]. First of all, rats were anaesthetized by chloral hydrate (350 mg/kg, intraperitoneally). After the depth of anesthesia was confirmed, a midline ventral incision was made in the neck to expose the right common carotid artery and the right external carotid artery. A 4-0 nylon suture with a blunted tip was inserted into the middle cerebral artery after the external carotid artery was coagulated. After 90 min (in survival experiment) or 60 min (in the rest experiments) of occlusion, the suture was withdrawn to restore the blood supply. The body temperature was maintained at 37±0.5°C with a thermostatically controlled infrared lamp during the procedure. Buprenorphine hydrochloride was injected (0.65 mg/kg, intramuscularly) to reduce postoperative pain.
Evaluation of neurological deficit scores
The neurological deficit scores were evaluated 24 h after MCAO with the modified Neurological Severity Score by a professional investigator who is blinded to the experimental groups, as previously described [16]. The parameters include motor, sensory, reflex and balance tests.
Control behavioral tests
The control behavioral tests were performed after rats have recovered from ischemia surgery for one day using an open field test similarly to Martins et al. [17]. Each rat was placed in the left quadrant of a 50 × 50 × 39 cm open field arena made with wood, and with a frontal glass wall. Black lines were drawn to divide the floor into 12 quadrants. Crossing and rearing were considered as measures for locomotor and exploratory activities, respectively, over 5 min [18].
Object recognition and social recognition memory tests
The object recognition and social recognition memory tests were carried out similarly to Martins et al. [17]. The object recognition task was performed in an open-field arena (50 × 50 × 50 cm) in a transparent acrylic. Rats were allowed to freely explore objects in the arena for 5 min. The object recognition index=(Tnovel - Tfamiliar)/(Tnovel + Tfamiliar) × 100%). The social recognition task was performed with the same apparatus. Social recognition training was performed with inclusion of one unfamiliar rat in the arena for 1 h of free exploration. After 24 h, testing was performed when the same rat of training (now a familiar rat) and a new rat were placed for 5-min exploration. The social recognition index= (Tnovel - Tfamiliar)/(Tnovel + Tfamiliar) × 100%).
Morris water maze study
The water maze study was performed similarly to Yang et al. [19]. The water maze was a black circular pool filled with water (21°C-23°C) and divided into four equal imaginary quadrants. A black circular platform was hidden 2 cm below the water surface. When rats were placed in the water, the swimming time and path were recorded with a video camera. Training was done for five constitutive days with the hidden platform kept at the same position. The time used to find the platform (escape latency) and the travel length before they arrived at the platform was measured for five constitutive days, once a day. The average escape latency and travel length was obtained and presented in a line graph.
Measurement of proinflammatory cytokines
After rats recovered from ischemia for one day, they were scarified by quick decapitation. The injured brain section was collected and 20 mg of tissue was homogenized in 2 ml PBS on ice. After they were centrifuged at 10,000 g for 15 min, the proinflammatory cytokines (IL-6, IL-1β, TNF-α) in the supernatant were measured using sandwich enzyme-linked immunosorbent assay (ELISA) kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer’s instructions.
Western blot analysis
The brains of rats were removed by quick decapitation after the preconditioning was finished for the western blot analysis of HIF-1α and eNOS. The brain tissue was homogenized at 12,000 g for 20 min before the protein was extracted and determined using a BCA Protein Assay Reagent Kit (Beyotime Biotech, Haimen, China). An equivalent amount of total protein (40 μg) was separated by SDS-PAGE on 10% polyacrylamide gels and then transferred to polyvinylidine membranes. Next, the membranes were blocked with 5% dry milk in PBS/0.1% Tween 20 (PBST) and then incubated with primary antibodies (anti-HIF-1α, anti-eNOS and anti-β-actin) diluted in 2% BSA in PBST overnight at 4°C. The next day, membranes were washed thrice with PBST, and then incubated with horseradish peroxidase-conjugated secondary antibody at room temperature for 30 min, and then washed thrice with PBST again. Finally, the immunoreactive bands were visualized by enhanced chemiluminescence (ECL, Amersham Biosciences, USA). The blots were exposed to X-ray film for radiographic detection and the protein bands were quantified by densitometry using Quantity One (Bio-Rad Laboratories, CA, USA). Antibodies were all bought from Santa Cruz Biotechnologies (Santa Cruz, USA).
Measurement of eNOS activity
The method was previously described by Mookerjee et al. [20]. Briefly, the brains of rats were removed by quick decapitation after the preconditioning was finished. Tissues were then homogenized with ice-cold HEPES buffer and centrifuged at 1000 g for 10 minutes at 4°C. 5 μl of the supernatant were incubated with reaction buffer [Tris-HCL buffer (30 mmol/L, ph7.4); NADPH (1.25 mmol/L); 3H-arginine (10 μCi/ml); norvaline (5 mmol/L); and cacl2 (400 μmol/L)] at 30°C for 30 minutes. After the reaction was terminated with ice-cold citrate buffer, the scintillation activity was measured with a commercial kit (Ultima Gold scintillant, MA, USA) and a liquid scintillation analyzer (Tri-Carb 2100TR, Packard Biosciences, UK) to obtain the eNOSactivity, which was expressed as Unit (micromoles citrulline/mg protein/h)/mg protein.
Statistical analysis
Data were expressed as the Mean ± SEM and statistically analyzed with one-way Analysis of Variance (ANOVA) followed by Tukey’s post hoc test using SPSS 17.0. P < 0.05 was considered significant.
Results
Effect of hypoxia and/or GYKI preconditioning on rat survival rate after 90-min cerebral ischemia
Table 1 shows the effect of hypoxia and/or GYKI preconditioning on rat survival rate after 90-min cerebral ischemia. In the Ischemic group, the survival rate at 24 h, 48 h and 72 h after cerebral ischemia was all significantly lower than in Sham group. Only 33% of rats survived at 72 h after cerebral ischemia. In the hypoxia PC or GYKI PC groups, the survival rate was not significantly different from that in the Ischemic group. In the hypoxia+GYKI PC group, however, the survival rate was significantly increased compared to that in the Ischemic group (p<0.05) at every time point. 78% of rats survived at 72 h after cerebral ischemia.
Table 1.
Effect of hypoxia and/or GYKI preconditioning on rat survival rate after 90-min cerebral ischemia
| Survival | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| 24 h | 48 h | 72 h | ||||
|
| ||||||
| N | % | N | % | N | % | |
| Sham | 10/10 | 100% | 10/10 | 100% | 10/10 | 100% |
| Ischemia | 10/18& | 56% | 8/18& | 44% | 6/18& | 33% |
| Hypoxia PC | 11/18 | 61% | 9/18 | 50% | 8/18 | 44% |
| GYKI PC | 10/18 | 56% | 8/18 | 44% | 8/18 | 44% |
| Hypoxia+GYKI PC | 15/18* | 83% | 14/18* | 78% | 14/18* | 78% |
The survival rate at 24 h, 48 h and 72 h after cerebral ischemia was all significantly lower than in Sham group. The hypoxia PC or GYKI PC alone did alter the survival rate. The hypoxia+GYKI PC group significantly increased the survival rate at every time point. N: number of survivors/number of rats in the group. PC: preconditioning; GYKI: GYKI-52466.
p<0.05 compared to Ischemia group;
p<0.05 compared to Ischemia group.
N=10 or 18 per group.
Changes in neurological deficit score
The changes in neurological deficit score were demonstrated in Figure 2. There was significant increase in neurological deficit score in Ischemia rats compared to Sham rats (p<0.05). Compared to Ischemia rats, the neurological deficit scores in the hypoxia PC or GYKI PC group were significantly decreased (p<0.05). When rats received combined PC with hypoxia and GYKI-52466, the neurological deficit score was further decreased (p<0.05 compared to hypoxia PC or GYKI PC group).
Figure 2.
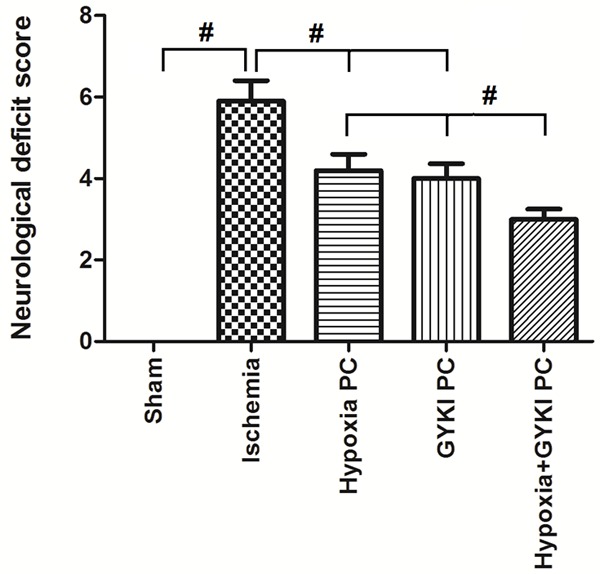
Effect of hypoxia and/or GYKI preconditioning on rat neurological deficit after 90-min cerebral ischemia. Rats received preconditioning with hypoxia or GYKI-52466 or hypoxia+GYKI-52466 for four constitutive days and 90-min cerebral ischemia surgery, then tested for neurological deficit. Ischemia significantly increased the neurological deficit score compared to Sham rats (p<0.05). Hypoxia PC or GYKI PC significantly decreased the neurological deficit scores (p<0.05). Combined PC with hypoxia and GYKI-52466 further decreased the neurological deficit score. PC: preconditioning; GYKI: GYKI-52466. #: p<0.05. N=10 per group.
Influence of the procedures on exploratory and locomotor activities
Table 2 shows the influence of the procedures on exploratory and locomotor activities. There is no significant difference in the exploratory and locomotor activities between the Sham group, Ischemia group, hypoxia PC group, GYKI PC group, or hypoxia+GYKI PC group, indicating that the exploratory and locomotor activities were not influenced by the procedures.
Table 2.
Results of control behavioral tasks
| Sham | Ischemia | Hypoxia PC | GYKI PC | Hypoxia+GYKI PC | |
|---|---|---|---|---|---|
| Total exploration time in OR training (s) | 36.6±6.8 | 40.1±5.7 | 36.4±8.1 | 33.5±6.4 | 39.7±7.1 |
| Total exploration time in OR test (s) | 39.3±5.2 | 42.5±6.2 | 39.6±6.8 | 41.7±8.9 | 39.9±6.7 |
| Total exploration time in SR test (s) | 71.4±9.6 | 72.9±9.8 | 69.8±8.4 | 75.5±7.9 | 68.4±8.1 |
| Open field Crossing (n) | 40.5±10.2 | 45.6±13.2 | 51.5±10.4 | 47.9±9.8 | 45.7±8.7 |
| Open field Rearing (n) | 16.9±3.5 | 19.8±4.5 | 17.2±3.2 | 19.1±3.7 | 14.2±4.2 |
Data are expressed as mean ± SEM of the total exploration time in OR and SR. No significant difference in the exploratory and locomotor activities was shown between the Sham group, Ischemia group, hypoxia PC group, GYKI PC group, orhypoxia+GYKI PC group, indicating that the exploratory and locomotor activities were not influenced by the procedures. OR: object recognition; SR: social recognition; PC: preconditioning; GYKI: GYKI-52466. N=12 pre group.
Changes in object recognition and social recognition memory
Figure 3 shows the changes in object recognition memory and social recognition memory. Both object recognition index and social recognition index were significantly decreased in Ischemia group compared to Sham group (p<0.05). Either hypoxia PC or GYKI PC increased the object recognition index and social recognition index compared to the Ischemia group, while the hypoxia+GYKI PC increased them more dramatically (p<0.05 compared to hypoxia PC or GYKI PC group).
Figure 3.
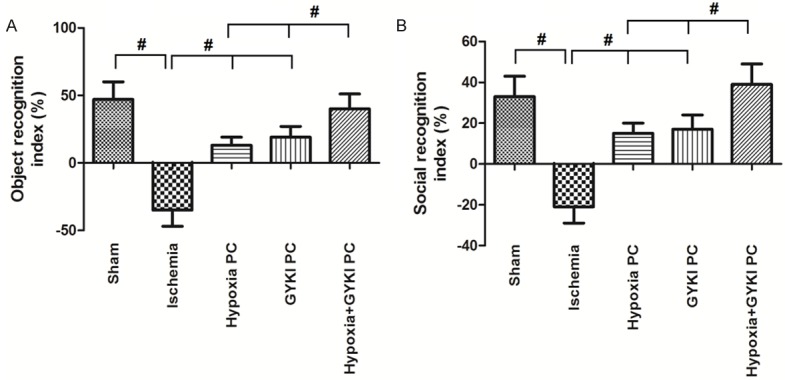
Changes in object recognition and social recognition memory. Object recognition and social recognition memory capacity was indicated by object recognition index and social recognition index, respectively. Ischemia significantly decreased object recognition index and social recognition index. Hypoxia PC or GYKI PC increased the object recognition index and social recognition index; hypoxia+GYKI PC increased them more dramatically. PC: preconditioning; GYKI: GYKI-52466. #: p<0.05. N=10 per group.
Changes in escape latency and travel length in Morris water maze
The escape latency and travel length in Morris water maze were shown in Figure 4. Figure 4A shows the representive traces of rats traveled in different groups. Figure 4B shows the trends of escape latency in Morris water maze study and Figure 4D demonstrates the area under the curve (AUC) calculated from Figure 4B. Escape latency was significantly increased in Ischemia group compared to Sham group (p<0.05). Bothhypoxia PC and GYKI PC decreased the escape latency, while the hypoxia+GYKI PC decreased it to a significantly lower level (p<0.05 compared to hypoxia PC or GYKI PC group). Figure 4C shows the trends of travel length and Figure 4E demonstrates the AUC calculated from Figure 4C. Similarly to escape latency, the travel length was significantly increased by Ischemia (p<0.05) but decreased by both hypoxia PC and GYKI PC. Hypoxia+GYKI PC decreased it to a significantly lower level compared to hypoxia PC or GYKI PC group (p<0.05).
Figure 4.
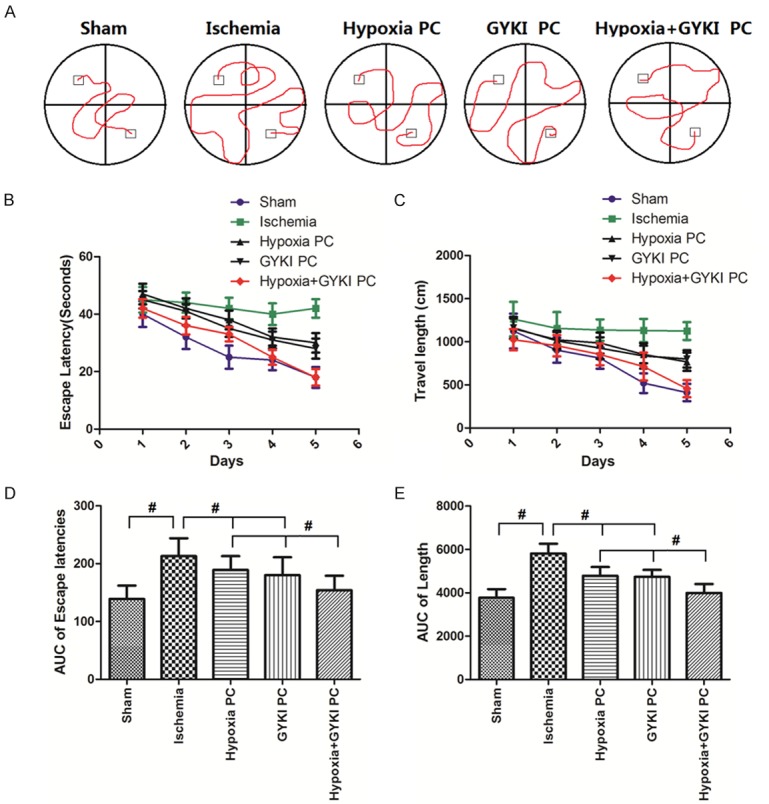
Changes in escape latency and travel length in Morris water maze. (A) Shows the representive traces of rats traveled in different groups. (B) Shows the trends of escape latency in Morris water maze study and (D) demonstrates the area under the curve (AUC) calculated from (B). (C) Shows the trends of travel length and (E) demonstrates the AUC calculated from (C). Escape latency andtravel length was significantly increased in Ischemia group, but decreased by hypoxia PC or GYKI PC. Hypoxia+GYKI PC decreased it to a significantly lower level. PC: preconditioning; GYKI: GYKI-52466. #: p<0.05. N=10 per group.
Changes in the proinflammatory factors (IL-6, IL-1β and TNF-α)
Figure 5 shows the changes in the proinflammatory factors (IL-6, IL-1β and TNF-α). Cerebral ischemia caused significant increase in cerebral proinflammatory factors (IL-6, IL-1β and TNF-α), which was reversed by treatment of hypoxia PC, GYKI PC or hypoxia+GYKI PC (P<0.05). The levels of proinflammatory factors (IL-6, IL-1β and TNF-α) in the hypoxia+GYKI PC group was significantly different from either hypoxia PC or GYKI PC group (P<0.05).
Figure 5.
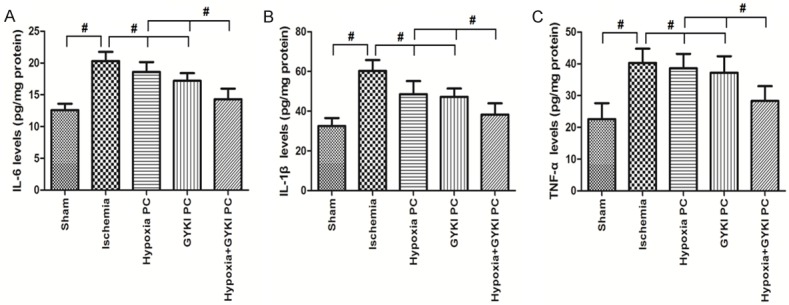
Changes in the proinflammtory factors (IL-6, IL-1β and TNF-α) in the brain. Rats received preconditioning with hypoxia or GYKI-52466 or hypoxia+GYKI-52466 for four constitutive days before MCAO surgery, then rested for one day before they were sacrificed for proinflammtory factor assays in the brain. Cerebral ischemia caused significant increase in cerebral proinflammatory factors, which was reversed by treatment of hypoxia PC, GYKI PC or hypoxia+GYKI PC. The levels of proinflammatory factors in the hypoxia+GYKI PC group was significantly lower thanhypoxia PC or GYKI PC group. PC: preconditioning; GYKI: GYKI-52466. #: p<0.05. N=10 per group.
Changes in the expression of HIF-1α and eNOSand eNOS activity
Figure 6 shows the expression of HIF-1α and eNOS and eNOS activity. As demonstrated in Figure 6A, the expression of HIF-1α was significantly increased by hypoxia PC and hypoxia+GYKI PC, but not changed by ischemia or GYKI PC. Figure 6B shows the expression of eNOS (See “Supplementary Figure 1” for original bands). Either ischemia, hypoxia PC or GYKI PC did not change the expression of eNOS, but the hypoxia+GYKI PC significantly increased it. Similarly to the expression of eNOS, the eNOS activity was also only increased by hypoxia+GYKI PC, as shown in Figure 6C.
Figure 6.
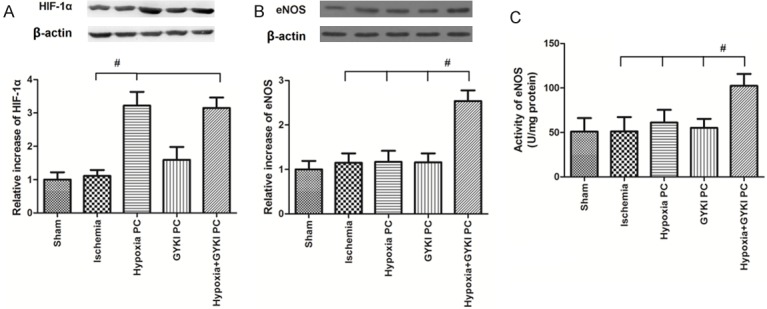
Changes in the expression of HIF-1α and eNOS and eNOS activity in the brain. Rats received preconditioning with hypoxia or GYKI-52466 or hypoxia+GYKI-52466 for four constitutive days before MCAO surgery, then rested for one day before they were sacrificed for Western blot and eNOS activity assays in the brain. The expression of HIF-1α was significantly increased by hypoxia PC and hypoxia+GYKI PC, but not changed by ischemia or GYKI PC. The expression of eNOS and eNOS activity was only increased by hypoxia+GYKI PC. PC: preconditioning; GYKI: GYKI-52466. #: p<0.05. N=10 per group.
Changes in neurological deficit scores by inhibitors of HIF-1α and eNOS
We treated with rats with hypoxia+GYKI PC and HIF-1α inhibitor PX-478 or eNOS inhibitor L-NIO, then re-measured the neurological deficit score (Figure 7). The results showed that both HIF-1α inhibitor PX-478 and eNOS inhibitor L-NIO abolished the effects of hypoxia+GYKI PC on neurological deficit score (P<0.05 compared to hypoxia+GYKI PC). Treatment of PX-478 or L-NIO alone had no impact on neurological deficit score.
Figure 7.
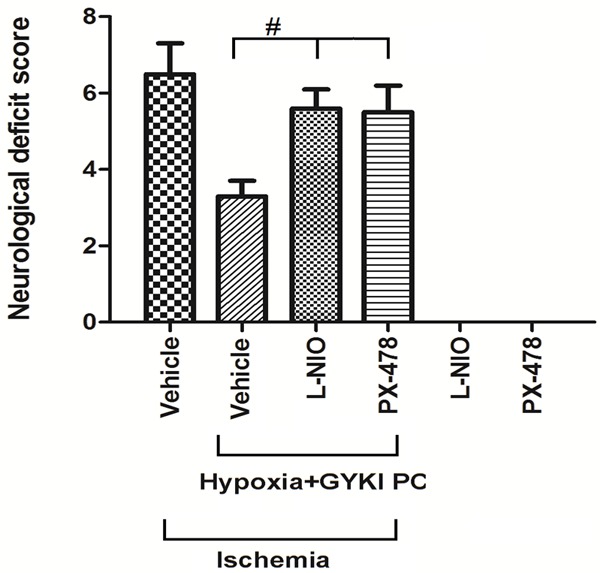
Changes in neurological deficit scores by inhibitors of HIF-1α and eNOS. We treated rats with preconditioning with hypoxia+GYKI PC for four constitutive days and HIF-1α inhibitor PX-478 or eNOS inhibitor L-NIO before MCAO surgery, then re-measured the neurological deficit score. Both HIF-1α inhibitor PX-478 and eNOS inhibitor L-NIO abolished the effects of hypoxia+GYKI PC on neurological deficit score, while treatment of PX-478 or L-NIO alone had no impact on neurological deficit score. PC: preconditioning; GYKI: GYKI-52466. #: p<0.05. N=10 per group.
Discussion
In clinical level, cerebral ischemic injury has been the leading cause of death and long term disability, which targets approximately 20 million people globally per year [21]. Prolonged circulatory disturbance in cerebral blood flow leads to broad range of neurological and behavioral deficits, which has been the mostly characterized behavioral abnormality induced by global cerebral ischemic injury. Due to lack of successful therapies to cerebral ischemic injury, it has become a major cause of mortality and adult physical disability all over the world [22]. The present study, for the first time, found out that combined preconditioning with hypoxia and GYKI-52466 increased the survival rate of cerebral ischemia rats, alleviated the neurological deficit, increased the object recognition and social recognition memory of rats and suppressed the inflammatory reaction induced by cerebral ischemia. Further studies found that preconditioning with hypoxia and GYKI-52466 significantly increased the HIF-1α and eNOS expression as well as eNOS activity, while inhibitors of HIF-1α and eNOS abolished the protective effects of hypoxia+GYKI PC on neurological deficit. These results indicate that HIF-1α and eNOS may be involved in the mechanism of neuroprotection of hypoxia+GYKI PC.
GYKI-52466 hydrochloride is a non-competitive AMPA and kainate receptor antagonist, which blocks gluk 3 homomeric receptors and gluk 2b (R)/gluk 3 hetero receptors. Due to its antagonist ability for non-competitive AMPA receptor, GYKI 52466 can prevent the excitotoxicity induced by high extracellular glutamate levels and protect animals against convulsions evoked by AMPA or kainate, electroshock and audiogenic stimulation [23]. Moreover, recent studies demonstrated that GYKI-52466 has neuroprotective actions in experimental models of global and focal cerebral ischemia. It is shown that the acute administration of GYKI-52466 at doses greater than 10 mg/kg rescues CA1 neurons in animal models of global ischemia [24]. However, protection against cerebral injury following ischemia with preconditioning with low doses of GYKI-52466 has not been investigated. As the middle cerebral artery is the most commonly occluded artery in embolic and thrombotic stroke, it makes the transient MCAO model a widely accepted experimental model to study cerebral ischemic injury [25]. As death is the most severe consequence of cerebral ischemic injury, we treated rats with a prolonged duration of ischemia (90 min), then examined the impacts of treatments on survival rate. In the Ischemic group, the survival rate at 24 h, 48 h and 72 h after cerebral ischemia was all significantly decreased. Only 33% of rats survived at 72 h after cerebral ischemia. Either hypoxia PC or GYKI PC did not alter the survival rate, but hypoxia+GYKI PC significantly increased the survival rate at every time point. 78% of rats survived at 72 h after cerebral ischemia, indicating the effectiveness of hypoxia+GYKI PC on survival protection.
The damage induced by middle cerebral artery occlusion to neurons in hippocampus and cortex region causes memory impairments and motor dysfunction [26]. The results in our study revealed that ischemia rats had significant neurological deficit, which was attenuated by hypoxia PC or GYKI PC, and most reduced by combined PC with hypoxia and GYKI-52466. Similarly, both object recognition index and social recognition index were significantly decreased by ischemia group, but increased by hypoxia PC and GYKI PC. Hypoxia+GYKI PC increased them more dramatically compared to hypoxia PC or GYKI PC. The escape latency and travel length in Morris water maze showed a similar trend, which were decreased by both hypoxia PC and GYKI PC, and decreased to a significantly lower level by hypoxia+GYKI PC. These results indicate that combined PC with hypoxia and GYKI-52466 had a better effect on neurological functions than hypoxia PC or GYKI PC alone.
Inflammation is critically involved in the molecular and cellular mechanisms of cerebral ischemia and results in cell death eventually [27]. It was reported that excessive inflammatory response functions prominently in the pathogenesis of cerebral ischemia [28]. Cerebral ischemia induces a complex series of inflammatory events, including activation of microglia and astrocytes and influx of hematogenous cells across the blood vessel wall. These inflammatory events eventually cause an increase in behavioral deficits and secondary brain damage. Therefore, targeting inflammation may be an important therapeutic strategy in the prevention and treatment of cerebral ischemia injury. As shown in Figure 5, cerebral ischemia caused significant increase in cerebral proinflammtory factors (IL-6, IL-1β and TNF-α), which was reversed by treatment of hypoxia PC, GYKI PC or hypoxia+GYKI PC. The levels of proinflammtory factors in the hypoxia+GYKI PC group was significantly different from either hypoxia PC or GYKI PC group, suggesting that hypoxia+GYKI PC had a significant higher ability to repress the inflammatory reaction in cerebral ischemia.
HIF-1α is a key protein involved in hypoxic preconditioning. Exposure to hypoxia prevents HIF-1α degradation and allows it to dimerize with HIF-1β protein. The HIF-1 heterodimer binds to a specific hypoxia-response element in the promoter and enhancer regions of many hypoxia-inducible genes (such as vascular endothelial growth factor and erythropoietin), which increases ischemic tolerance through upregulation of glucose uptake and glycolysis [29]. Bernaudin et al. Demonstrated that hypoxia PC 24 hours before ischemia could increase HIF-1α expression and reduce infarct volume by 22% to 36% in mice [29]. Prass et al. Showed that hypoxic PC markedly activated HIF-1 DNA-binding activity and reduced infarct volume in subsequent cerebral ischemia [30]. It has been widely accepted that eNOS can regulate cerebral blood flow and inhibit neutrophil adhesion to the vascular endothelium [31,32]. NOinducing chemicals are able to promote endothelial cell proliferation and migration in vivo and in vitro [33]. To study the role of HIF-1α and eNOSin the mechanism of hypoxia+GYKI PC, we measured the expression of HIF-1α and eNOS and eNOS activity after preconditioning. The expression of HIF-1α was significantly increased by hypoxia PC and hypoxia+GYKI PC, but not changed by ischemia or GYKI PC. Interestingly, either ischemia, hypoxia PC or GYKI PC did not change the expression of eNOS, but the hypoxia+GYKI PC significantly increased it. Similarly to the expression of eNOS, the eNOS activity was also only increased by hypoxia+GYKI PC. These results revealed that hypoxia PC or GYKI PC alone could not change the expression of eNOS, but the combined preconditioning of hypoxia and GYKI-52466 could induce the expression of eNOS and increase the eNOS activity, which eventually protects rats from cerebral ischemia injury. The direct regulation of GYKI-52466 on eNOS, has not been investigate priviouly, but some studies has suggested the potential relationship between GYKI-52466, AMPA/kainate receptor and NOS [34,35]. The present first observed the effects of combined preconditioning with hypoxia and GYKI-52466 on NOS expression, suggesting a new mechanism and target to prevent cerebral ischemia injury. To further confirm the involvement of HIF-1α and eNOS, we treated rats with hypoxia+GYKI PC and HIF-1α inhibitor PX-478 or eNOS inhibitor L-NIO, then re-measured the neurological deficit score. The results showed that both HIF-1α inhibitor PX-478 and eNOS inhibitor L-NIO abolished the effects of hypoxia+GYKI PC on neurological deficit score, indicating the involvement of HIF-1α and eNOS in the neuroprotection of hypoxia+GYKI PC.
In conclusion, our study demonstrated that hypoxia+GYKI PC ameliorated the cerebral ischemia injury, alleviated the neurological deficit, increased the recognition and memory function of rats and suppressed the inflammatory reaction. Preconditioning with hypoxia and GYKI-52466 significantly increased the HIF-1α and eNOS expression as well as eNOS activity, while inhibitors of HIF-1α and eNOS abolished the protective effects of hypoxia+GYKI PC on neurological deficit. Although the underlying mechanism needs further exploration, these results indicate that HIF-1α and eNOS may be involved in the mechanism of neuroprotection of hypoxia+GYKI PC and provides a new target for cerebral stroke prevention.
Acknowledgements
This work was supported by a grant from the National Natural Science Foundation of China, (No. 81573782) and a grant from the clinical science and technology innovation project of Shanghai Shen-Kang Hospital Development Center (No.SHDC22015028).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Siniscalchi A, Gallelli L, Malferrari G, Pirritano D, Serra R, Santangelo E, De Sarro G. Cerebral stroke injury: the role of cytokines and brain inflammation. J Basic Clin Physiol Pharmacol. 2014;25:131–137. doi: 10.1515/jbcpp-2013-0121. [DOI] [PubMed] [Google Scholar]
- 2.An H, Liu Q, Chen Y, Vo KD, Ford AL, Lee JM, Lin W. Oxygen metabolism in ischemic stroke using magnetic resonance imaging. Transl Stroke Res. 2012;3:65–75. doi: 10.1007/s12975-011-0141-x. [DOI] [PubMed] [Google Scholar]
- 3.Lagali PS, Corcoran CP, Picketts DJ. Hippocampus development and function: role of epigenetic factors and implications for cognitive disease. Clin Genet. 2010;78:321–333. doi: 10.1111/j.1399-0004.2010.01503.x. [DOI] [PubMed] [Google Scholar]
- 4.Stetler RA, Leak RK, Gan Y, Li P, Zhang F, Hu X, Jing Z, Chen J, Zigmond MJ, Gao Y. Preconditioning provides neuroprotection in models of CNS disease: paradigms and clinical significance. Prog Neurobiol. 2014;114:58–83. doi: 10.1016/j.pneurobio.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bader AM, Klose K, Bieback K, Korinth D, Schneider M, Seifert M, Choi YH, Kurtz A, Falk V, Stamm C. Hypoxic preconditioning increases survival and pro-angiogenic capacity of human cord blood mesenchymal stromal cells in vitro. PLoS One. 2015;10:e0138477. doi: 10.1371/journal.pone.0138477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang JA, Chen TL, Jiang J, Shi H, Gui C, Luo RH, Xie XJ, Xiang MX, Zhang X. Hypoxic preconditioning attenuates hypoxia/reoxygenation-induced apoptosis in mesenchymal stem cells. Acta Pharmacol Sin. 2008;29:74–82. doi: 10.1111/j.1745-7254.2008.00716.x. [DOI] [PubMed] [Google Scholar]
- 7.Donevan SD, Rogawski MA. GYKI 52466, a 2,3-benzodiazepine, is a highly selective, noncompetitive antagonist of AMPA/kainate receptor responses. Neuron. 1993;10:51–59. doi: 10.1016/0896-6273(93)90241-i. [DOI] [PubMed] [Google Scholar]
- 8.De Sarro G, Ferreri G, Gareri P, Russo E, De Sarro A, Gitto R, Chimirri A. Comparative anticonvulsant activity of some 2,3-benzodiazepine derivatives in rodents. Pharmacol Biochem Behav. 2003;74:595–602. doi: 10.1016/s0091-3057(02)01040-7. [DOI] [PubMed] [Google Scholar]
- 9.Gigler G, Móricz K, Agoston M, Simó A, Albert M, Benedek A, Kapus G, Kertész S, Vegh M, Barkóczy J, Markó B, Szabó G, Matucz E, Gacsályi I, Lévay G, Hársing LG Jr, Szénási G. Neuroprotective and anticonvulsant effects of EGIS-8332, a non-competitive AMPA receptor antagonist, in a range of animal models. Br J Pharmacol. 2007;152:151–160. doi: 10.1038/sj.bjp.0707362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erdo F, Berzsenyi P, Andrasi F. The AMPAantagonist talampanel is neuroprotective in rodent models of focal cerebral ischemia. Brain Res Bull. 2005;66:43–49. doi: 10.1016/j.brainresbull.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 11.Borowicz KK, Duda AM, Kleinrok Z, Czuczwar SJ. Interaction of GYKI 52466, a selective non-competitive antagonist of AMPA/kainate receptors, with conventional antiepileptic drugs in amygdala-kindled seizures in rats. Pol J Pharmacol. 2001;53:101–108. [PubMed] [Google Scholar]
- 12.Czuczwar SJ, Gasior M, Kaminski R, Kleinrok Z, Kozicka M, Ossowska G, Pietrasiewicz T. GYKI 52466 [1-(4-aminophenyl)-4-methoxy-7,8-methylenedioxy-5H-2,3-benzodiazepine hydrochloride. And the anticonvulsive activity of conventional antiepileptics against pentetrazol in mice. Mol Chem Neuropathol. 1998;33:149–162. doi: 10.1007/BF02815178. [DOI] [PubMed] [Google Scholar]
- 13.Goulton CS, Patten AR, Kerr JR, Kerr DS. Pharmacological Preconditioning with GYKI 52466: a prophylactic approach to neuroprotection. Front Neurosci. 2010:4. doi: 10.3389/fnins.2010.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nayak PK, Kerr DS. Low-dose GYKI-52466: prophylactic preconditioning confers long-term neuroprotection and functional recovery following hypoxic-ischaemic brain injury. Neuroscience. 2013;232:128–138. doi: 10.1016/j.neuroscience.2012.11.063. [DOI] [PubMed] [Google Scholar]
- 15.Ren C, Li N, Wang B, Yang Y, Gao J, Li S, Ding Y, Jin K, Ji X. Limb ischemic perconditioning attenuates blood-brain barrier disruption by inhibiting activity of MMP-9 and occludin degradation after focal cerebral ischemia. Aging Dis. 2015;6:406–417. doi: 10.14336/AD.2015.0812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen J, Li Y, Wang L, Zhang Z, Lu D, Lu M, Chopp M. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke. 2001;32:1005–1011. doi: 10.1161/01.str.32.4.1005. [DOI] [PubMed] [Google Scholar]
- 17.Martins A, Schimidt HL, Garcia A, Colletta Altermann CD, Santos FW, Carpes FP, da Silva WC, Mello-Carpes PB. Supplementation with different teas from Camellia sinensis prevents memory deficits and hippocampus oxidative stress in ischemia-reperfusion. Neurochem Int. 2017;29:30026–30028. doi: 10.1016/j.neuint.2017.04.019. [DOI] [PubMed] [Google Scholar]
- 18.Bonini JS, Bevilaqua LR, Zinn CG, Kerr DS, Medina JH, Izquierdo I, Cammarota M. Angiotensin II disrupts inhibitory avoidance memory retrieval. Horm Behav. 2006;50:308–313. doi: 10.1016/j.yhbeh.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 19.Yang J, Pan Y, Li X, Wang X. Atorvastatin attenuates cognitive deficits through Akt1/caspase-3 signaling pathway in ischemic stroke. Brain Res. 2015;10:231–239. doi: 10.1016/j.brainres.2015.10.032. [DOI] [PubMed] [Google Scholar]
- 20.Mookerjee RP, Wiesenthal A, Icking A, Hodges SJ, Davies NA, Schilling K, Sen S, Williams R, Novelli M, Muller-Esterl W, Jalan R. Increased gene and protein expression of the novel eNOSregulatory protein NOSTRIN and a variant in alcoholic hepatitis. Gastroenterology. 2007;132:2533–2541. doi: 10.1053/j.gastro.2006.12.035. [DOI] [PubMed] [Google Scholar]
- 21.Rodrigo R, Fernandez-Gajardo R, Gutierrez R, Matamala JM, Carrasco R, Miranda-Merchak A, Feuerhake W. Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets. 2013;12:698–714. doi: 10.2174/1871527311312050015. [DOI] [PubMed] [Google Scholar]
- 22.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, mcguire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB. Executive Summary: Heart Disease and stroke statistics--2016 update: a report from the American heart association. Circulation. 2016;133:447–454. doi: 10.1161/CIR.0000000000000366. [DOI] [PubMed] [Google Scholar]
- 23.Szabados T, Gigler G, Gacsalyi I, Gyertyan I, Levay G. Comparison of anticonvulsive and acute neuroprotective activity of three 2,3-benzodiazepine compounds, GYKI 52466, GYKI 53405, and GYKI 53655. Brain Res Bull. 2001;55:387–391. doi: 10.1016/s0361-9230(01)00516-0. [DOI] [PubMed] [Google Scholar]
- 24.Block F, Schmitt W, Schwarz M. Pretreatment but not posttreatment with GYKI 52466 reduces functional deficits and neuronal damage after global ischemia in rats. J Neurol Sci. 1996;139:167–172. [PubMed] [Google Scholar]
- 25.Tabassum R, Vaibhav K, Shrivastava P, Khan A, Ejaz Ahmed M, Javed H, Islam F, Ahmad S, Saeed Siddiqui M, Safhi MM. Centella asiatica attenuates the neurobehavioral, neurochemical and histological changes in transient focal middle cerebral artery occlusion rats. Neurol Sci. 2013;34:925–933. doi: 10.1007/s10072-012-1163-1. [DOI] [PubMed] [Google Scholar]
- 26.Vaibhav K, Shrivastava P, Khan A, Javed H, Tabassum R, Ahmed ME, Khan MB, Moshahid Khan M, Islam F, Ahmad S, Siddiqui MS, Safhi MM. Azadirachta indica mitigates behavioral impairments, oxidative damage, histological alterations and apoptosis in focal cerebral ischemia-reperfusion model of rats. Neurol Sci. 2013;34:1321–1330. doi: 10.1007/s10072-012-1238-z. [DOI] [PubMed] [Google Scholar]
- 27.Hou ST, macmanus JP. Molecular mechanisms of cerebral ischemia-induced neuronal death. Int Rev Cytol. 2002;221:93–148. doi: 10.1016/s0074-7696(02)21011-6. [DOI] [PubMed] [Google Scholar]
- 28.Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010;87:779–789. doi: 10.1189/jlb.1109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bernaudin M, Nedelec AS, Divoux D, mackenzie ET, Petit E, Schumann-Bard P. Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and VEGF, in the adult mouse brain. J Cereb Blood Flow Metab. 2002;22:393–403. doi: 10.1097/00004647-200204000-00003. [DOI] [PubMed] [Google Scholar]
- 30.Prass K, Scharff A, Ruscher K, Lowl D, Muselmann C, Victorov I, Kapinya K, Dirnagl U, Meisel A. Hypoxia-induced stroke tolerance in the mouse is mediated by erythropoietin. Stroke. 2003;34:1981–1986. doi: 10.1161/01.STR.0000080381.76409.B2. [DOI] [PubMed] [Google Scholar]
- 31.Dawson VL, Dawson TM, Bartley DA, Uhl GR, Snyder SH. Mechanisms of nitric oxidemediated neurotoxicity in primary brain cultures. J Neurosci. 1993;13:2651–2661. doi: 10.1523/JNEUROSCI.13-06-02651.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci U S A. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma FX, Han ZC. Statins, nitric oxide and neovascularization. Cardiovasc Drug Rev. 2005;23:281–292. doi: 10.1111/j.1527-3466.2005.tb00173.x. [DOI] [PubMed] [Google Scholar]
- 34.Lonart G, Johnson KM. Characterization of nitric oxide generator-induced hippocampal [3H] norepinephrine release. I. The role of glutamate. J Pharmacol Exp Ther. 1995;275:7–13. [PubMed] [Google Scholar]
- 35.Monteith TS, Goadsby PJ. Acute migraine therapy: new drugs and new approaches. Curr Treat Options Neurol. 2011;13:1–14. doi: 10.1007/s11940-010-0105-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.