

Abstract
TRPC6 plays a critical role in proteinuric kidney diseases, and TRPC3 is involved in tubulointerstitial damage and renal fibrosis in obstructed kidneys. Podocyte loss is a characteristic event in diabetic nephropathy (DN). The aim of this study was to examine whether deletion of the closely related diacylglycerol (DAG)-responsive TRPCs in mice (TRPC3/6/7-/-) affects diabetes-induced renal dysfunction and podocyte loss. We compared urine volume, kidney hypertrophy, glomerular enlargement, albuminuria and podocyte loss between wild type (WT) and TRPC3/6/7-/- diabetic mice. Finally, we examined whether the TGFβ1 signaling pathway is changed in diabetic WT and TRPC3/6/7-/- mice. TRPC6 protein in the renal cortex was increased in WT diabetic mice. High glucose (HG) treatment increased TRPC6 expression in human podocytes. TRPC3 protein, however, was not altered in either diabetic mice or HG-treated human podocytes. Although diabetic WT and TRPC3/6/7-/- mice had similar levels of hyperglycemia, the TRPC3/6/7-/- diabetic mice showed less polyuria, kidney hypertrophy, glomerular enlargement, albuminuria, and had lost less podocytes compared with WT diabetic mice. In addition, we observed decreased expression of anti-apoptotic Bcl2 and increased expression of pro-apoptotic cleaved caspase 3 in WT diabetic mice, but such changes were not significant in TRPC3/6/7-/- diabetic mice. Western blot and immunohistochemistry revealed that TGFβ1, p-Smad2/3, and fibronectin were upregulated in WT diabetic mice; however, expression of these signaling molecules was not changed in TRPC3/6/7-/- diabetic mice. In conclusion, deletion of DAG-responsive TRPCs attenuates diabetic renal injury via inhibiting the upregulation of TGFβ1 signaling in diabetic kidneys.
Keywords: Diabetic nephropathy, diacylglycerol, TRPC3/6/7, podocyte, albuminuria, TGFβ1
Introduction
Diabetic nephropathy (DN) is a major microvascular complication of diabetes mellitus and the leading cause worldwide of end-stage renal disease [1]. The characteristic pathological changes of DN include severe albuminuria, renal hyperfiltration, glomerular basement membrane thickening, and glomerulosclerosis [2]. Podocytes are crucial components of the glomerular capillary filter. Damage to the glomerular capillary filter, particularly at the level of the podocyte and the slit diaphragm, is of crucial importance in the pathogenesis of DN, which eventually leads from microalbuminuria to overt proteinuria with renal function decline.
The canonical transient receptor potential (TRPC) channels are a family of Ca2+-permeable non-selective cation channels with seven members, i.e., TRPC1 to TRPC7. This family can be further divided into two major subfamilies based on their functional similarities and sequence alignment: TRPC1/4/5 and TRPC3/6/7 (TRPC2 is a pseudogene in humans) [3]. In addition to sequence similarity, TRPC3/6/7 share the property of being activated by diacylglycerols (DAG). TRPC6 has been identified as a slit diaphragm associated protein in podocytes [4,5]. Gain of function mutants of TRPC6 were identified in patients with proteinuria due to inherited forms of focal segmental glomerulosclerosis (FSGS) in their kidney glomeruli [4,5]. Transient in vivo gene delivery of TRPC6 into mice leads to the expression of TRPC6 protein at the slit diaphragm that causes proteinuria [6]. Moreover, TRPC6-deficient (TRPC6-/-) mice show significantly less angiotensin II-induced proteinuria compared with wild type mice [7], indicating that TRPC6 activation is also involved in the pathology of acquired proteinuric kidney disease, such as DN. In vitro studies have shown that high glucose levels induce podocyte apoptosis via upregulation of TRPC6 [8,9]. Besides TRPC6, a recent study demonstrated that TRPC3 is involved in renal injury, by showing that TRPC3 knockout in mice ameliorates tubulointerstitial damage and renal fibrosis in obstructed kidneys [10].
It has been established that transforming growth factor β1 (TGFβ1) is involved in the pathogenesis of DN [11]. Gain-of-function mutation of TRPC6 results in extracellular signal-regulated kinases 1/2 (ERK1/2) activation in cultured podocytes [12], followed by activation of the TGFβ-Smad2/3 signaling pathway, downregulation of Bcl2, and upregulation of cleaved caspase 3, which leads to a reduction in the podocyte number per glomerulus and the development of albuminuria [13]. Conversely, TGFβ1 induces podocyte injury through TRPC6 phosphorylation [14], suggesting that there may be a positive feedback between TGFβ1 and TRPC6.
Given that TRPC3 and TRPC6 are deleterious to kidney disease and that they are of the same DAG-activated channel subfamily, we hypothesized that deletion of DAG-responsive class of TRPC genes (TRPC3/6/7) would attenuate diabetic renal injury. In this study, type 1 diabetes mellitus was induced in wild type (WT) and TRPC3/6/7-deleted (TRPC3/6/7-/-) mice on a mixed C57BL/6J-129SvEv background using streptozotocin (STZ). Compared with their diabetic WT counterparts, diabetic TRPC3/6/7-/- mice displayed less polyuria, kidney hypertrophy, glomerular enlargement, albuminuria, and had reduced the loss of podocytes. The protective effect of TRPC3/6/7 deletion may be attributed to the inhibition of up-regulated TGFβ1 signaling in diabetic kidneys.
Materials and methods
Reagents
The primary antibodies used for western blot or immunohistochemistry were as follows: rabbit polyclonal anti-TRPC3 antibody (Cat#: ACC-016, Alomone Labs, Israel); rabbit polyclonal anti-TRPC6 antibody (Cat#: ACC-017, Alomone Labs, Israel); rabbit polyclonal anti-Bcl2 antibody (Cat#: 12789-1-AP, Proteintech, USA); rabbit polyclonal anti-Cleaved caspase 3 antibody (Cat#: 9661, Cell Signaling Technology, USA); rabbit polyclonal anti-WT-1 antibody (Cat#: sc-192, Santa Cruz, USA); rabbit polyclonal anti-TGFβ1 antibody (Cat#: 18978-1-AP, Proteintech, USA); rabbit polyclonal anti-p-Smad2/3 antibody (Cat#: 8828, Cell Signaling Technology, USA); mouse monoclonal anti-Fibronectin antibody (Cat#: F7387, Sigma-aldrich, USA); mouse monoclonal anti-β-actin antibody (Cat#: TA-09, Zhongshan jinqiao, China). STZ, D-glucose and mannitol were purchased from Sigma (Louis, MO).
Experimental animals and protocol
TRPC3/6/7-/- mice on a mixed C57BL/6J-129SvEv background and WT C57BL/6J/129SvEv mice were reconstituted from cryopreserved embryos. TRPC3/6/7 triple knockout (KO) parents were generated by crossing TRPC3 KO [15], TRPC6 KO [16], and TRPC7 KO [17] mice to the desired homozygosity at the Comparative Medicine Branch (CMB) of the National Institute of Environmental Health Sciences, North Carolina. The animals were housed at 22°C, 50% humidity with a 12 h-light/12 h-dark cycle with unlimited access to water and food.
Male TRPC3/6/7-/- and WT mice at 6 weeks of age were injected with three consecutive intraperitoneal injections of STZ 100 mg/kg body weight (Sigma, St. Louis, Mo, USA) or vehicle (Veh), as described by the Animal Models of Diabetic Complications Consortium. Unrestricted food and water were provided throughout the duration of the experiment. Diabetes was defined as a non-fasting blood glucose level above 17 mmol/l. All mice were sacrificed 12 weeks after the first injection for tissue collection. This study was carried out in strict accordance with the recommendations in the Guide for Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Tongji Medical College, Huazhong University of Science and Technology.
Cell culture
The human podocyte cell line was a gift from Dr. Chun Zhang (Union Hospital, Tongji Medical college) [18], which was established and cultured as described previously [19]. In brief, cells were cultured in RPMI-1640 medium (Hyclone, USA) supplemented with 10% fetal bovine serum (Hyclone, USA) at 33°C (‘permissive’ conditions). To induce differentiation, the cells were transferred to 37°C (‘non-permissive’ conditions) and kept in culture for 10 days. After 16 h of serum starvation, cells were treated for 6 days with high glucose (HG, 40 mM, D-glucose), normal glucose (NG, 5.6 mM), and normal glucose with added 34.4 mM mannitol (NG+M, 40 mM).
RT-PCR
Total RNA from the WT mouse brain and renal cortex and from the TRPC3/6/7-/- mouse brain was isolated using the Trizol reagent (Invitrogen, USA). First strand cDNA was synthesized using SuperScript II Reverse Transcriptase (Invitrogen, USA) and Oligo (dT)12-18 oligonucleotide primers in accordance with the manufacturer’s instructions. The primer sequences for TRPC1-TRPC7 are listed in Table 1.
Table 1.
Primer sequences for TRPC1-TRPC7
| Gene | Forward | Reverse |
|---|---|---|
| TRPC1 | 5’-CTCAAAGTGGTGGCTCACAACAAG-3’ | 5’-CATAGAGCTGAGTCAGTCCAATCG-3’ |
| TRPC2 | 5’-GGCTGTCAACTACAACCAGAAACAG-3’ | 5’-AGCTGGCAGAATATATGCCAGTCG-3’ |
| TRPC3 | 5’-GGACTGTCTGAAGTGACTTCTGTTG-3’ | 5’-CTCGAGTGAGACTGTGTGAAGAGG-3’ |
| TRPC4 | 5’-GTGGAGTGGATGATATTACCGTGG-3’ | 5’-TCAGGATGTCCAGAAGCATCCTTC-3’ |
| TRPC5 | 5’-TCCATGATTGGTGGAACCTGATGG-3’ | 5’-AGCATATTCAGCAGCACTACCAGG-3’ |
| TRPC6 | 5’-CCTACTGATCCTCAGATCATCTCTG-3’ | 5’-CTCTGCATCTTCTTGGAAGCCTTG-3’ |
| TRPC7 | 5’-GACGGAGATGCTCATCATGAAGTG-3’ | 5’-CGGTAGTAGGAGTACAGGTTGAAC-3’ |
Blood glucose measurement
Body weights and blood glucose levels were recorded every 4 weeks. Blood glucose levels were measured between 8:30 AM and 10:30 AM using Accu-chek Performa (Roche, Mannheim, Germany). Approximately 3 μl of blood were collected in conscious mice via tail vein puncture.
24 h albuminuria assessment
24 h albuminuria was assessed as described [20]. In brief, 24 h urine collections were obtained from mice before sacrifice by housing them in individual mouse metabolic cages with free access to water and rodent mash, and adjusted to the same volume using pure water. Albumin standards were prepared by serial dilution of BSA (Sigma, St. Louis, Mo, USA). Samples of standards or urine were mixed with 5× SDS-sample buffer, boiled for 5 min, and analyzed by 10% SDS-polyacrylamide gel electrophoresis followed by Coomassie blue staining. The BSA bands (~65 kDa) were scanned and quantified using NIH ImageJ software.
Glomerular volume calculation
Before harvesting kidney tissues, mice were anaesthetized and perfused with PBS via left ventricle puncture. Kidneys were removed, and the kidney capsule was stripped. The kidney weight was then recorded. Kidneys were fixed in 4% paraformaldehyde for 16-18 h and embedded in paraffin, cut to 3 μm thickness, and stained with periodic acid-Schiff (PAS) reagent. Mean glomerular volumes (VG) were calculated from mean cross-sectional areas (AG) by Aperio ImageScope software (Aperio Technologies Inc., Vista, CA, USA) using the equation VG = β/k × AG 3/2 where the shape coefficient for sphere β = 1.38 and the size distribution coefficient k = 1.1 [21]. All glomerular profiles from each PAS section were used for quantification of glomerular volume, with an average of approximately 100 glomerular profiles for each animal.
Western blot
Mouse renal cortex or human podocytes were lysed in RIPA buffer (150 mM NaCl, 50 mM Tris-HCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, pH 7.4) containing 1% protease inhibitor cocktail and 1 mmol/l PMSF on ice. Equal amounts of protein lysate (50 μg/well) were separated by electrophoresis in 12% or 15% SDS-PAGE gels, transferred to polyvinylidene difluoride membranes, and blocked for 1 h at room temperature with Tris-buffered saline with 0.1% Tween 20 (TBST) and 5% nonfat dry milk. After blocking, membranes were incubated with the corresponding primary antibodies (anti-TRPC6 1:500; anti-TRPC3 1:500; anti-Wilms tumor protein (WT-1) 1:1000; anti-Bcl2 1:1000; anti-Cleaved caspase 3 1:500; anti-TGFβ1 1:500; anti-p-Smad2/3 1:500; anti-β-actin 1:500) in TBST overnight at 4°C. After being washed in TBST, membranes were exposed to horseradish peroxidase (HRP)-conjugated goat anti-mouse or goat anti-rabbit IgGs secondary antibody (1:20000) and washed once again. The immunoreactive bands were visualized by chemiluminescence. Quantification was performed by measuring the intensity of the bands with the aid of NIH ImageJ software. The expression level of each protein was normalized to β-actin on the same blot.
Immunohistochemistry
Dewaxed tissue sections were boiled with antigen-retrieval solution (0.01 M Citrate Buffer, pH 6.0) for 15-20 min in a microwave oven. Sections were incubated with the corresponding primary antibodies (anti-TGFβ1 1:100, anti-Fibronectin 1:100) overnight at 4°C, followed by incubation with the secondary antibody for 1 h, revealed with DAB (Vector Laboratories), and counterstained with hematoxylin.
Statistical analysis
Data were expressed as means ± SEM. GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA) was used for statistical analysis. For multiple group comparisons, one-way ANOVA with Bonferroni’s post hoc test was performed and for two group comparisons, unpaired t-test was performed. p<0.05 was considered statistically significant.
Results
Hyperglycemia or high glucose increases TRPC6 expression
The expression of TRPC mRNAs in the renal cortex was examined by RT-PCR (Figure 1B). Figure 1A demonstrates the presence of mRNA for TRPC1-TRPC7 in the mouse brain (positive controls). As shown in Figure 1B, only TRPC1, TRPC2, TRPC3, TRPC4 and TRPC6 were expressed in the mouse renal cortex, TRPC5 and TRPC7 were not detectable.
Figure 1.
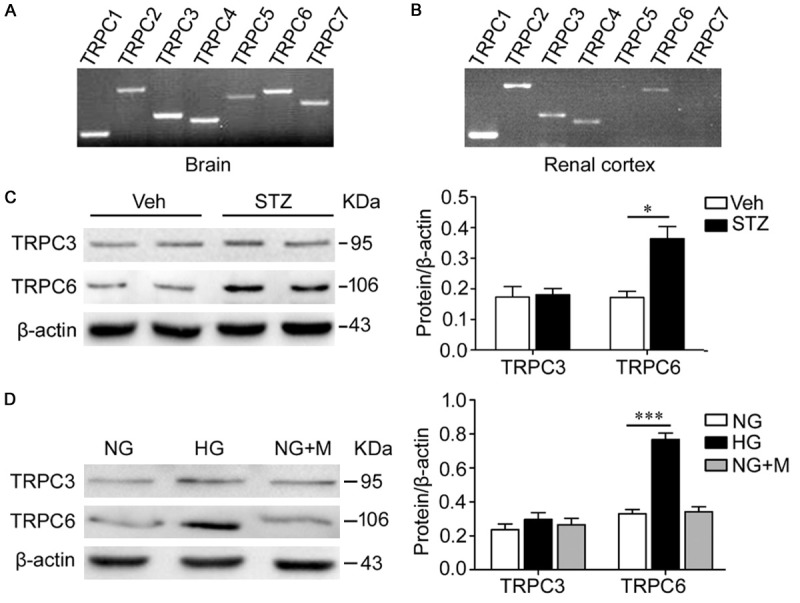
Hyperglycemia (high glucose) increases TRPC6. A, B. RT-PCR showed that TRPC1, TRPC2, TRPC3, TRPC4 and TRPC6 were expressed in the mouse renal cortex, while TRPC5 and TRPC7 were not detectable. C. Western blot analysis demonstrated that the TRPC6 protein level of kidney cortical lysates was significantly increased in diabetic mice compared with nondiabetic mice (*p<0.05), while TRPC3 was unaltered. n=5 mice/group. D. Differentiated human podocytes were treated with high glucose for 6 days. Western blot analysis showed that high glucose increased TRPC6 protein level obviously (***p<0.0001), while TRPC3 was not affected. The administration of mannitol was performed to exclude osmotic effects. NG, normal glucose (5.6 mmol/l); HG, high glucose (40 mmol/l); NG+M, normal glucose with added 34.4 mmol/l mannitol. Data are representative of three separate experiments.
To assess the potential role of the DAG-responsive TRPC3 and TRPC6 in DN, the expression of TRPC3/6 in the renal cortex of the WT diabetic and nondiabetic mice were examined by western blot. TRPC6 protein expression of kidney cortical lysates was significantly upregulated in diabetic WT mice compared with nondiabetic WT mice (Figure 1C). The protein expression of TRPC3 in the two groups was not significantly different (Figure 1C). Western blot analysis also demonstrated the increased expression of TRPC6 protein in human podocytes treated with HG for 6 days compared to NG; no change in TRPC3 protein expression was observed between HG-treated and NG-treated podocytes (Figure 1D). To exclude osmotic effects, the administration of NG+M did not significantly affect the protein levels of TRPC3 and TRPC6 (Figure 1D).
TRPC3/6/7 deletion reduces the extent of diabetes-induced polyuria, kidney hypertrophy and glomerular enlargement
RT-PCR analysis of the TRPC3/6/7-/- mouse brain showed that TRPC3, TRPC6 and TRPC7 mRNA are not detectable in tissues from TRPC3/6/7-/- mice (Figure 2A). Moreover, TRPC3 and TRPC6 proteins were undetectable in the renal cortex of TRPC3/6/7-/- mice by western blot (Figure 2B), indicating that the renal cortex of TRPC3/6/7-/- mice are indeed deficient in TRPC3 and TRPC6.
Figure 2.
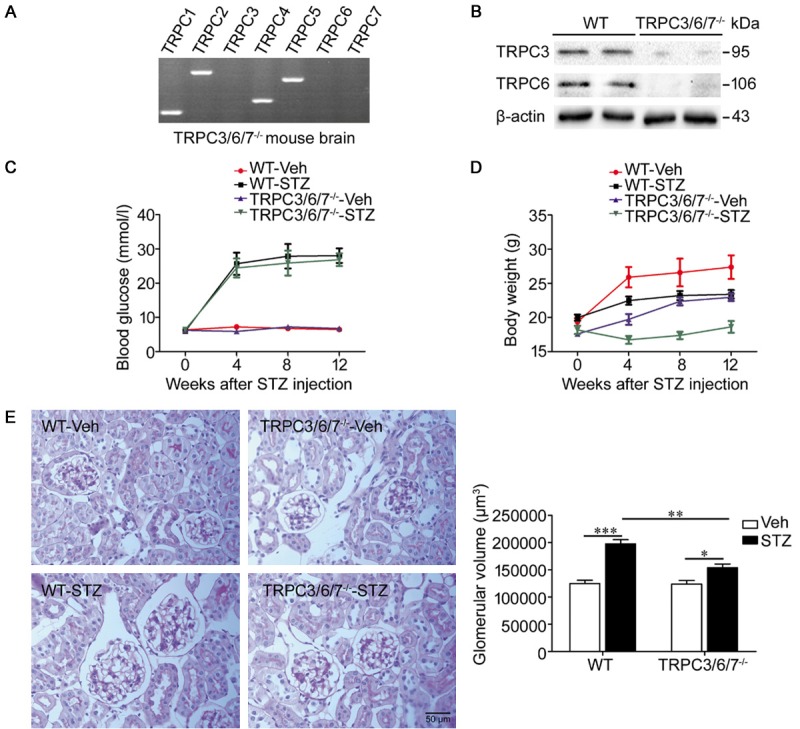
TRPC3/6/7 deletion reduces the extent of diabetes-induced glomerular enlargement. A, B. RT-PCR showed that TRPC3, TRPC6, and TRPC7 were undetectable in the TRPC3/6/7-/- mouse brain, and western blot analysis showed that TRPC3 and TRPC6 were indeed deficient in the TRPC3/6/7-/- mouse renal cortex. C, D. Changes in blood glucose and body weight. WT-STZ and TRPC3/6/7-/--STZ mice developed similar degrees of hyperglycemia. The extent of body weight increase among both diabetic groups was less compared with corresponding nondiabetic mice. n=5-11 mice/group. E. PAS staining and its quantitative analysis of glomerular volume in kidney sections, magnification ×400. The result showed that glomerular enlargement occurred in both STZ groups (***p<0.001 and *p<0.05), while the severity of glomerular enlargement in WT-STZ group was greater (**p<0.01). n=5 mice/group.
After 2 weeks of STZ administration, WT-STZ and TRPC3/6/7-/--STZ mice became stably diabetic with comparable rises in blood glucose levels (Figure 2C, Table 2). The four groups of experimental mice exhibited an increase in body weight during the whole experiment, except for the body weight of mice in the TRPC3/6/7-/--STZ group, which slightly decreased during the first 4 weeks after STZ injection. However, the extent of body weight increase of the two diabetic groups was less compared with the nondiabetic groups (Figure 2D). In addition, the body weights of mice in the TRPC3/6/7-/--Veh and TRPC3/6/7-/--STZ groups were lower compared with the WT-Veh and WT-STZ groups, respectively (Table 2).
Table 2.
Characteristics of WT and TRPC3/6/7-/- mice 12 weeks after vehicle or STZ injection
| Variable | WT | TRPC3/6/7-/- | ||
|---|---|---|---|---|
|
| ||||
| Veh | STZ | Veh | STZ | |
| n | 5 | 9 | 5 | 11 |
| Blood glucose (mmol/l) | 6.46±0.27 | 28.06±2.13aa | 6.74±0.39 | 26.86±1.84bbb |
| Body weight (g) | 27.37±1.73 | 23.40±0.62a | 22.95±0.55a | 18.62±0.87bb,cc |
| Urine volume (ml/24 h) | 0.58±0.26 | 28.93±9.44aa | 0.41±0.13 | 12.97±3.87bbb,c |
| Kidney/body weight ratio (%) | 1.50±0.04 | 2.42±0.09aa | 1.37±0.05 | 1.68±0.01b,c |
Results are shown as means ± SEM.
p<0.05 vs. WT-Veh;
p<0.001 vs. WT-Veh;
p<0.05 vs. TRPC3/6/7-/--Veh.
p<0.01 vs. TRPC3/6/7-/--Veh.
p<0.001 vs. TRPC3/6/7-/--Veh.
p<0.01, vs. WT-STZ.
p<0.001, vs. WT-STZ.
Urine volumes were increased in both diabetic groups, but polyuria was alleviated in TRPC3/6/7-/--STZ mice compared with WT-STZ mice (Table 2). Kidney hypertrophy occurred in both diabetic groups as characterized by increased kidney/body weight ratios, though the extent of kidney hypertrophy was significantly attenuated in diabetic TRPC3/6/7-/- mice compared with diabetic WT mice (Table 2). Glomerular enlargement was assessed by glomerular volume in PAS-stained kidney sections of mice in four groups (Figure 2E). Mice in the diabetic groups presented glomerular enlargement, which was attenuated in TRPC3/6/7-/- mice (glomerular volume: WT-STZ versus TRPC3/6/7-/--STZ: 197651±7643 μm3 versus 153922±6539 μm3, p<0.01, n=5 mice/group) (Figure 2E).
TRPC3/6/7 deletion attenuates albumin excretion and podocyte loss induced by hyperglycemia
24 h urine was collected, and albumin levels were measured by SDS-polyacrylamide electrophoresis followed by Coomassie blue staining. As shown in Figure 3A, both STZ groups developed albuminuria; however, the diabetic TRPC3/6/7-/- mice showed significantly less albuminuria than the diabetic WT mice (24 h urinary albumin: WT versus TRPC3/6/7-/-: 278.40±53.84 μg versus 117.6±13.51 μg, p<0.05).
Figure 3.
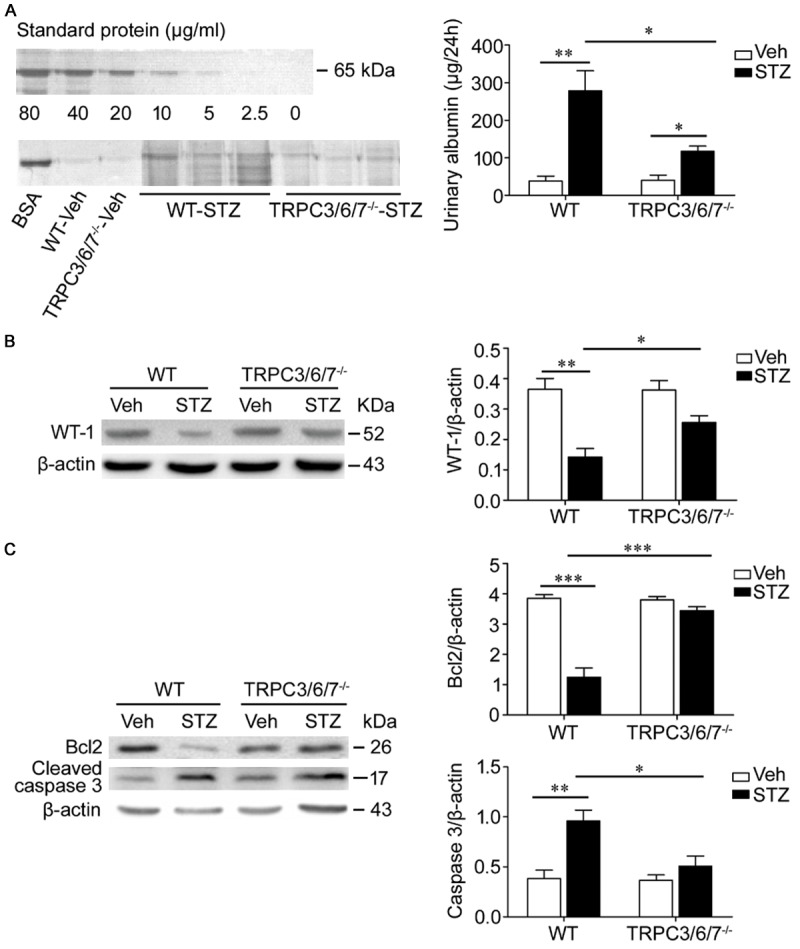
TRPC3/6/7 deletion attenuates albumin excretion and the changes of WT-1, Bcl2, and Cleaved caspase 3 induced by hyperglycemia. A. 24 h albumin excretion was assessed using SDS electrophoresis and staining of gels with Coomassie blue. Both STZ groups developed albuminuria (**p<0.01 and *p<0.05), but albuminuria of TRPC3/6/7-/--STZ mice was significantly less than that of WT-STZ mice (*p<0.05). BSA: bovine serum albumin. n=5-11 mice/group. B. Western blot showed that WT-1 protein of the renal cortex was decreased obviously in WT-STZ mice, compared with WT-Veh mice (**p<0.01); however, the decrease were prevented in TRPC3/6/7-/- mice (*p<0.05). n=3-5 mice/group. C. Western blot analysis demonstrated that the downregulation of Bcl2 and upregulation of Cleaved caspase 3 in WT-STZ mice (***p<0.001 and **p<0.01) was prevented in TRPC3/6/7-/--STZ mice (***p<0.001 and *p<0.05). n=3-5 mice/group.
Podocyte loss was assessed by the amount of WT-1 protein in the renal cortex. As shown in Figure 3B, the WT-1 protein levels of WT diabetic mice was reduced significantly. This alteration was not obviously observed in TRPC3/6/7-/- diabetic mice, though the WT-1 protein levels of TRPC3/6/7-/- diabetic mice showed a declining trend, indicating that podocyte loss was significantly attenuated in TRPC3/6/7-/- diabetic mice compared with WT diabetic mice.
The expression levels of Bcl2, an anti-apoptotic protein, and cleaved caspase 3, a pro-apoptotic protein, of kidney cortical lysates were analyzed by western blot. The expression of Bcl2 was downregulated, and the amount of cleaved caspase 3 was upregulated in the WT diabetic renal cortex, although these alterations were not significant in TRPC3/6/7-/- mice (Figure 3C).
TRPC3/6/7 deletion inhibits activation of TGFβ1 signaling and fibronectin deposition in diabetic kidney
Most subtypes of glomerular diseases, including DN, are associated with increased TGFβ1 signaling activity [22]. To determine whether the protective effect of the TRPC3/6/7 deletion on renal dysfunction and podocyte loss involves decreased activation of TGFβ1 signaling, the expression of TGFβ1-related proteins was studied by western blot and immunohistochemistry. Western blot analysis showed that the levels of TGFβ1 and p-Smad2/3 in the renal cortex in diabetic WT mice were significantly increased (Figure 4A). Moreover, immunohistochemistry showed that TGFβ1 and fibronectin in renal glomeruli and tubules were clearly increased in WT diabetic mice (Figure 4B). These effects, however, were not found in TRPC3/6/7-/- diabetic mice (Figure 4).
Figure 4.
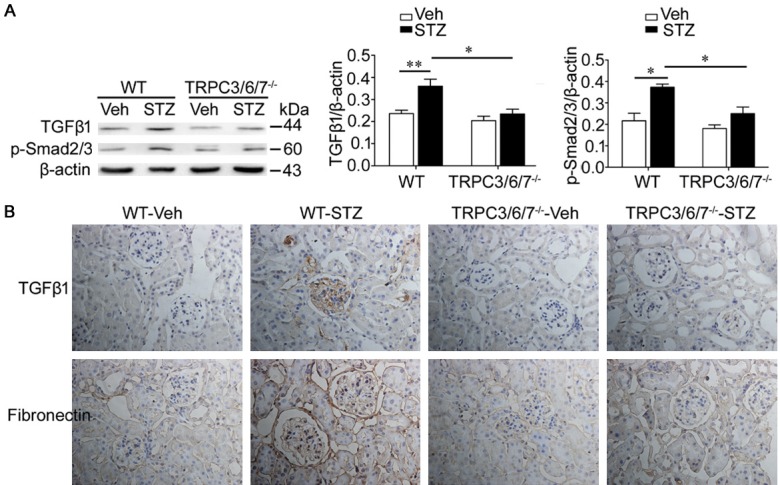
TRPC3/6/7 deletion inhibits the activation of TGFβ1 signaling and fibronectin deposition in diabetic kidney. A. Western blot analysis showed that TGFβ1 and p-Smad2/3 of renal cortex of WT-STZ mice were increased compared with WT-Veh mice (**p<0.01 and *p<0.05); however, the increase was inhibited in TRPC3/6/7-/--STZ mice (both *p<0.05). n=3-5 mice/group. B. Immunohistochemistry showed that the deposition of TGFβ1 and Fibronectin in renal cortex of WT-STZ mice were not obvious in TRPC3/6/7-/--mice. n=3-5 mice/group.
Discussion
This study provides evidence for the first time that deletion of the DAG-responsive class of TRPC genes TRPC3/6/7 in mice attenuates DN. Although WT and TRPC3/6/7-/- mice developed similar degrees of hyperglycemia during the study, the TRPC3/6/7-/- mice exhibited lower body weights, less polyuria, lower kidney/body weight ratios, less glomerular enlargement, less albuminuria, and lowered loss of podocytes in the diabetic kidney. To determine the underlying mechanism, we found that the TRPC3/6/7 deletion blunted the alteration of pro-apoptotic mediators in the renal cortex, and inhibited activation of TGFβ1 signaling in the diabetic kidney.
Because previous studies had shown that a gain-of-function mutation in TRPC6 is associated with the onset of focal segmental glomerulosclerosis (FSGS) [4,5], efforts of numerous research groups have been focused on the role of TRPC6 in proteinuric kidney disease. Recently, the role of TRPC6 in DN has attracted the considerable attention. However, there are controversies regarding the role of TRPC6 in DN. At the beginning of our experiment, we observed that the total TRPC6 protein in the renal cortex from STZ-induced diabetic mice is increased compared with that of nondiabetic mice. Moreover, we observed that HG-treated cultured human podocytes for 6 days cause upregulation of TRPC6 protein. Interestingly, Graham et al. demonstrated that TRPC6 protein in glomeruli isolated from STZ-induced diabetic rats was decreased compared with control rats [23]. The discrepancy with the findings reported here might be attributed to the different stage of DN, since Graham et al examined the expression of TRPC6 protein in glomeruli 2 weeks after STZ injection. A recent study in TRPC3 knockout mice proposed that TRPC3 channels might constitute important therapeutic targets for improving renal remodeling in kidney disease [10]. However, we did not observe significant increases in TRPC3 protein in either the renal cortex of STZ-induced diabetic mice or in HG-treated podocytes. The cause is not clear, and it may be that the model used in their research [10] is different than ours or that the DN model in our study has not developed to the degree of renal fibrosis. These data suggest that increased DAG-responsive TRPCs may aggravate diabetes-induced renal injury.
Among the TRPC channels, TRPC3, 6 and 7 are approximately 75% identical in their amino acid sequence. These proteins are believed to associate as heterotetramers and form functional ion channels on the cell surface [24]. We and others have performed various experiments in the combined TRPC3/6/7 knockout mouse model [25-27], and it has been shown that there is an interdependent functional profile of TRPC channels in smooth muscle cells of TRPC6-deficient mice [16]. Since our data and other findings support the role of TRPC3 and TRPC6 in kidney disease, in order to more fully characterize the impact of deleting closely related DAG-responsive TRPC genes (TRPC3/6/7) on the diabetic kidney, we used the available TRPC3/6/7 triple gene knockout mouse strain for our study. We found that the body weight of TRPC3/6/7-/- mice was lower than that of age-matched WT mice. TRP channels are present in most mammalian tissues, and can influence diverse physiological processes, including adipocyte function, energy intake and energy expenditure [28], which may be the cause of the small body size. However, the mechanism of how TRPC channels influence body weight is unclear. It was reported that dwarf mice are protected from DN [29]. The body weight may thus be related to the development of DN. With the exception of the effect of TRPC3/6/7 deletion on body weight, no effect on normal glomerular structure or function was observed in TRPC3/6/7 knockout mice.
The early stage of diabetic mellitus is characterized by renal hyperfiltration, kidney hypertrophy, and glomerular enlargement, which promotes the eventual development of DN. Both WT and TRPC3/6/7-/- diabetic mice developed polyuria and an increase in kidney/body weight ratio and glomerular volume. These features were attenuated in TRPC3/6/7-/- diabetic mice, suggesting that the TRPC3/6/7 deletion plays a protective role against renal disorder in STZ-induced diabetic mice. In contrast, Luan et al. [30] reported that tempol improves renal hyperfiltration in STZ-induced diabetic rats via increased expression of TRPC6 protein. The reasons for these conflicting results are not known.
Recent biopsy studies in humans have provided evidence that podocytes are functionally and structurally injured very early in the natural history of DN [31]. Podocyte injury or loss will lead to proteinuria and is associated with glomerulosclerosis [32]. Here, we found that podocyte loss was significantly attenuated in TRPC3/6/7-/- diabetic mice compared with WT diabetic mice and that there was a lower albuminuria. Similarly, the downregulation of the anti-apoptotic protein, Bcl2 and upregulation of pro-apoptotic protein, cleaved caspase 3 observed in WT diabetic mice were not obvious in TRPC3/6/7-/- diabetic mice, which is consistent with the finding of podocyte loss. Moreover, in vitro studies have shown attenuation of HG-induced podocyte apoptosis by TRPC6 gene knockdown [8]. These results suggest that the TRPC3/6/7 deletion attenuates diabetes-induced podocyte loss and albuminuria.
TGFβ1 is a key mediator in chronic kidney disease associated with progressive renal fibrosis [33]. The renal expression of TGFβ1 mRNA as well TGFβ1 protein is increased in patients with diabetes mellitus [34]. Considerable evidence showed that TGFβ1 increases urinary excretion of water, electrolytes and glucose by suppressing tubular reabsorption in both normal and diabetic conditions [35-37]. In addition, a number of previous studies have demonstrated that TGFβ1 promotes the development of diabetic albuminuria. Transgenic animal models overexpressing TGFβ1 exhibit increased urinary excretion of albumin [35]. Furthermore, it was reported that TGFβ1 induces podocyte apoptosis via activation of execution caspase 3 [14,38]. To determine whether reducing the upregulation of TGFβ1 signaling in TRPC3/6/7-/- diabetic mice contributes to the renal protective effect of TRPC3/6/7 deletion, we evaluated TGFβ1 signaling-related proteins in kidneys of diabetic WT and KO mice. Results showed that upregulation of TGFβ1, p-Smad2/3 and fibronectin in the renal cortex of WT diabetic mice were not found in TRPC3/6/7-/- diabetic mice, which may be the molecular basis for the presence of decreased renal dysfunction and podocyte loss in TRPC3/6/7-/- diabetic mice.
In conclusion, the results obtained here provide in vivo evidence that knockout of DAG-responsive class of TRPC genes TRPC3/6/7 resulted in decreased susceptibility to the development of renal dysfunction and podocyte loss in DN in mice. Additionally, this effect is closely associated with inhibiting the activation of the TGFβ1 signaling pathway, but the individual contributions of TRPC3, TRPC6 and TRPC7 to this phenotype are not known. Our data suggest that global inhibition of DAG-responsive TRPCs may have a therapeutic benefit in the prevention and treatment of DN.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (Grant 31171087 and Grant 30970662 to Y. L.) and NIH Intramural Research Program Project (Z01-ES-101684 to L. B.).
Disclosure of conflict of interest
None.
References
- 1.Kato M, Natarajan R. Diabetic nephropathyemerging epigenetic mechanisms. Nat Rev Nephrol. 2014;10:517–530. doi: 10.1038/nrneph.2014.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tervaert TW, Mooyaart AL, Amann K, Cohen AH, Cook HT, Drachenberg CB, Ferrario F, Fogo AB, Haas M, de Heer E, Joh K, Noël LH, Radhakrishnan J, Seshan SV, Bajema IM, Bruijn JA. Pathologic classification of diabetic nephropathy. J Am Soc Nephrol. 2010;21:556–563. doi: 10.1681/ASN.2010010010. [DOI] [PubMed] [Google Scholar]
- 3.Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- 4.Reiser J, Polu KR, Möller CC, Kenlan P, Altintas MM, Wei C, Faul C, Herbert S, Villegas I, Avila-Casado C, McGee M, Sugimoto H, Brown D, Kalluri R, Mundel P, Smith PL, Clapham DE, Pollak MR. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet. 2005;37:739–744. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB. A Mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 6.Möller CC, Wei C, Altintas MM, Li J, Greka A, Ohse T, Pippin JW, Rastaldi MP, Wawersik S, Schiavi S, Henger A, Kretzler M, Shankland SJ, Reiser J. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J Am Soc Nephrol. 2007;18:29–36. doi: 10.1681/ASN.2006091010. [DOI] [PubMed] [Google Scholar]
- 7.Eckel J, Lavin PJ, Finch EA, Mukerji N, Burch J, Gbadegesin R, Wu G, Bowling B, Byrd A, Hall G, Sparks M, Zhang ZS, Homstad A, Barisoni L, Birbaumer L, Rosenberg P, Winn MP. TRPC6 enhances angiotensin II-induced albuminuria. J Am Soc Nephrol. 2011;22:526–535. doi: 10.1681/ASN.2010050522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu BC, Song X, Lu XY, Li DT, Eaton DC, Shen BZ, Li XQ, Ma HP. High glucose induces podocyte apoptosis by stimulating TRPC6 via elevation of reactive oxygen species. Biochim Biophys Acta. 2013;1833:1434–1442. doi: 10.1016/j.bbamcr.2013.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sonneveld R, van der Vlag J, Baltissen MP, Verkaart SA, Wetzels JF, Berden JH, Hoenderop JG, Nijenhuis T. Glucose specifically regulates TRPC6 expression in the podocyte in an AngIIdependent manner. Am J Pathol. 2014;184:1715–1726. doi: 10.1016/j.ajpath.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 10.Saliba Y, Karam R, Smayra V, Aftimos G, Abramowitz J, Birnbaumer L, Farès N. Evidence of a Role for Fibroblast Transient Receptor Potential Canonical 3 Ca2+ Channel in Renal Fibrosis. J Am Soc Nephrol. 2015;26:1855–1876. doi: 10.1681/ASN.2014010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang AS, Hathaway CK, Smithies O, Kakoki M. Transforming growth factor beta1 and diabetic nephropathy. Am J Physiol Renal Physiol. 2016;310:F689–F696. doi: 10.1152/ajprenal.00502.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiluiza D, Krishna S, Schumacher VA, Schlöndorff J. Gain-of-function mutations in transient receptor potential C6 (TRPC6) activate extracellular signal-regulated kinases 1/2 (ERK1/2) J Biol Chem. 2013;288:18407–18420. doi: 10.1074/jbc.M113.463059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen J, Chen JK, Harris RC. EGF receptor deletion in podocytes attenuates diabetic nephropathy. J Am Soc Nephrol. 2015;26:1115–1125. doi: 10.1681/ASN.2014020192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu L, Lin Q, Liao H, Feng J, Dong X, Ye J. TGF-β1 induces podocyte injury through Smad3-ERKNFkB pathway and Fyn-dependent TRPC6 phosphorylation. Cell Physiol Biochem. 2010;26:869–878. doi: 10.1159/000323996. [DOI] [PubMed] [Google Scholar]
- 15.Hartmann J, Dragicevic E, Adelsberger H, Henning HA, Sumser M, Abramowitz J, Blum R, Dietrich A, Freichel M, Flockerzi V, Birnbaumer L, Konnerth A. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron. 2008;59:392–398. doi: 10.1016/j.neuron.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dietrich A, Mederos Y Schnitzler M, Gollasch M, Gross V, Storch U, Dubrovska G, Obst M, Yildirim E, Salanova B, Kalwa H, Essin K, Pinkenburg O, Luft FC, Gudermann T, Birnbaumer L. Increased vascular smooth muscle contractility in TRPC6-/- mice. Mol Cell Biol. 2005;25:6980–6989. doi: 10.1128/MCB.25.16.6980-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perez-Leighton CE, Schmidt TM, Abramowitz J, Birnbaumer L, Kofuji P. Intrinsic phototransduction persists in melanopsin-expressing ganglion cells lacking diacylglycerol-sensitive TRPC subunits. Eur J Neurosci. 2011;33:856–867. doi: 10.1111/j.1460-9568.2010.07583.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Su H, Wan Q, Tian XJ, He FF, Gao P, Tang H, Ye C, Fan D, Chen S, Wang YM, Meng XF, Zhang C. MAD2B contributes to podocyte injury of diabetic nephropathy via inducing cyclin B1 and Skp2 accumulation. Am J Physiol Renal Physiol. 2015;308:F728–F736. doi: 10.1152/ajprenal.00409.2014. [DOI] [PubMed] [Google Scholar]
- 19.Saleem MA, O’Hare MJ, Reiser J, Coward RJ, Inward CD, Farren T, Xing CY, Ni L, Mathieson PW, Mundel P. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol. 2002;13:630–638. doi: 10.1681/ASN.V133630. [DOI] [PubMed] [Google Scholar]
- 20.Canaud G, Bienaimé F, Viau A, Treins C, Baron W, Nguyen C, Burtin M, Berissi S, Giannakakis K, Muda AO, Zschiedrich S, Huber TB, Friedlander G, Legendre C, Pontoglio M, Pende M, Terzi F. AKT2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat Med. 2013;19:1288–1296. doi: 10.1038/nm.3313. [DOI] [PubMed] [Google Scholar]
- 21.Hirose K, Osterby R, Nozawa M, Gundersen HJ. Development of glomerular lesions in experimental long-term diabetes in the rat. Kidney Int. 1982;21:689–695. doi: 10.1038/ki.1982.82. [DOI] [PubMed] [Google Scholar]
- 22.Herman-Edelstein M, Weinstein T, Gafter U. TGFβ1-dependent podocyte dysfunction. Curr Opin Nephrol Hypertens. 2013;22:93–99. doi: 10.1097/MNH.0b013e32835b4870. [DOI] [PubMed] [Google Scholar]
- 23.Graham S, Gorin Y, Abboud HE, Ding M, Lee DY, Shi H, Ding Y, Ma R. Abundance of TRPC6 protein in glomerular mesangial cells is decreased by ROS and PKC in diabetes. Am J physiol Cell Physiol. 2011;301:C304–C315. doi: 10.1152/ajpcell.00014.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dietrich A, Kalwa H, Rost BR, Gudermann T. The diacylgylcerol-sensitive TRPC3/6/7 subfamily of cation channels: functional characterization and physiological relevance. Pflugers Arch. 2005;451:72–80. doi: 10.1007/s00424-005-1460-0. [DOI] [PubMed] [Google Scholar]
- 25.Chen X, Lu M, He X, Ma L, Birnbaumer L, Liao Y. TRPC3/6/7 Knockdown Protects the Brain from Cerebral Ischemia Injury via Astrocyte Apoptosis Inhibition and Effects on NF-κB Translocation. Mol Neurobiol. 2017;54:7555–7566. doi: 10.1007/s12035-016-0227-2. [DOI] [PubMed] [Google Scholar]
- 26.He X, Li S, Liu B, Susperreguy S, Formoso K, Yao J, Kang J, Shi A, Birnbaumer L, Liao Y. Major contribution of the 3/6/7 class of TRPC channels to myocardial ischemia/reperfusion and cellular hypoxia/reoxygenation injuries. Proc Natl Acad Sci U S A. 2017;114:E4582–E4591. doi: 10.1073/pnas.1621384114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seo K, Rainer PP, Shalkey Hahn V, Lee DI, Jo SH, Andersen A, Liu T, Xu X, Willette RN, Lepore JJ, Marino JP Jr, Birnbaumer L, Schnackenberg CG, Kass DA. Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc Natl Acad Sci U S A. 2014;111:1551–1556. doi: 10.1073/pnas.1308963111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahern GP. Transient receptor potential channels and energy homeostasis. Trends Endocrinol Metab. 2013;24:554–560. doi: 10.1016/j.tem.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen NY, Chen WY, Bellush L, Yang CW, Striker LJ, Striker GE, Kopchick JJ. Effects of streptozotocin treatment in growth hormone (GH) and GH antagonist transgenic mice. Endocrinology. 1995;136:660–667. doi: 10.1210/endo.136.2.7835300. [DOI] [PubMed] [Google Scholar]
- 30.Luan J, Li W, Han J, Zhang W, Gong H, Ma R. Renal protection of in vivo administration of tempol in streptozotocin-induced diabetic rats. J Pharmacol Sci. 2012;119:167–176. doi: 10.1254/jphs.12002fp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolf G, Chen S, Ziyadeh FN. From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes. 2005;54:1626–1634. doi: 10.2337/diabetes.54.6.1626. [DOI] [PubMed] [Google Scholar]
- 32.Pavenstädt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- 33.Lin J, Shi Y, Peng H, Shen X, Thomas S, Wang Y, Truong LD, Dryer SE, Hu Z, Xu J. Loss of PTEN promotes podocyte cytoskeletal rearrangement, aggravating diabetic nephropathy. J Pathol. 2015;236:30–40. doi: 10.1002/path.4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamoto T, Nakamura T, Noble NA, Ruoslahti E, Border WA. Expression of transforming growth factor beta is elevated in human and experimental diabetic nephropathy. Proc Natl Acad Sci U S A. 1993;90:1814–1818. doi: 10.1073/pnas.90.5.1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hathaway CK, Gasim AM, Grant R, Chang AS, Kim HS, Madden VJ, Bagnell CR Jr, Jennette JC, Smithies O, Kakoki M. Low TGFβ1 expression prevents and high expression exacerbates diabetic nephropathy in mice. Proc Natl Acad Sci U S A. 2015;112:5815–5820. doi: 10.1073/pnas.1504777112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kakoki M, Pochynyuk OM, Hathaway CM, Tomita H, Hagaman JR, Kim HS, Zaika OL, Mamenko M, Kayashima Y, Matsuki K, Hiller S, Li F, Xu L, Grant R, Bertorello AM, Smithies O. Primary aldosteronism and impaired natriuresis in mice underexpressing TGFβ1. Proc Natl Acad Sci U S A. 2013;110:5600–5605. doi: 10.1073/pnas.1302641110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fujimoto M, Maezawa Y, Yokote K, Joh K, Kobayashi K, Kawamura H, Nishimura M, Roberts AB, Saito Y, Mori S. Mice lacking Smad3 are protected against streptozotocin-induced diabetic glomerulopathy. Biochem Biophys Res Commun. 2003;305:1002–1007. doi: 10.1016/s0006-291x(03)00885-4. [DOI] [PubMed] [Google Scholar]
- 38.Schiffer M, Bitzer M, Roberts IS, Kopp JB, ten Dijke P, Mundel P, Böttinger EP. Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin Invest. 2001;108:807–816. doi: 10.1172/JCI12367. [DOI] [PMC free article] [PubMed] [Google Scholar]