

Abstract
Chidamide is a newly designed and synthesized histone deacetylase (HDAC) inhibitor that selectively inhibits HDAC 1, 2, 3, and 10. Our previous study demonstrated that chidamide induces G0/G1 arrest and apoptosis in myelodysplastic syndromes (MDS). Low-dose chemotherapy is effective in treating higher risk MDS patients, and here, we sought to determine whether the combination of chidamide with cytarabine at lowdoses would increase the cytotoxicity to MDS cells. In this study, the combination of chidamide (50 nM) with cytarabine (50 nM) showed synergistic inhibition on cell growth.The mean combination index values were 0.068, 0.158, and 0.226 in SKM-1, MUTZ-1, and KG-1 MDS cell lines, respectively. The combination increased the acetylation levels of histone H3 and decreased HDAC activity in MDS cells.A low concentration (25 and 50 nM) of chidamide combined with low-dose cytarabine (50 nM) inhibited cell proliferation and arrested the cell cycle in the G0/G1 phasevia down-regulating CDK2 and up-regulating p21. Furthermore, the combined treatment induced cell apoptosis via down-regulating Bcl-2 and up-regulating cleaved caspase-3 protein. These results demonstrate the potential utility of combining chidamide and cytarabine in the treatment of MDS.
Keywords: Chidamide, cytarabine, HDAC inhibitor, myelodysplastic syndrome, cell cycle, apoptosis
Introduction
Chidamide is a novel benzamide chemical class of HDAC inhibitor (HDACi) that selectively inhibits HDAC 1, 2, 3, and 10. To date, it has been approved in China for the oral treatment of recurrent or refractory peripheral T-cell lymphoma (PTCL) [1-3]. It is well accepted that HDACs act as transcription repressors that control cell survival, proliferation, angiogenesis, andimmunity. Importantly, multiple human cancers over express HDACs, especially class I HDACs [4,5].
Myelodysplastic syndromes (MDS) are a group of clonal hematopoietic stem cell disorders. The most obvious characteristic of these syndromes is dysplasia of the hematopoietic cell lineages, which increases the risk of developing acute myeloid leukemia (AML) [6]. Low doses of cytarabine are considered one of the most promising strategies for treating high-risk MDS patients [7], because of its safety and efficacy. The combination of cytarabine with the DNA methyl transferase inhibitor decitabine improves the outcomes in MDS patients [8]. Like decitabine, the HDACi, chidamide, acts as a type of epigenetic therapy for cancer. Our previous study has shown that chidamide induces apoptosis and growth inhibition in MDS cell lines [9], and the same results were obtained in other solid cancers such as lung cancer [10] and colon cancer [1]. Therefore, the combination of HDAC is with chemotherapy drugs at a low concentration is considered as one of the most promising strategies to increase the efficacy of cancer treatments. In this study, we sought to determine whether the combination of chidamideand cytarabine at low doses can synergistically enhance apoptosis in MDS cells, and to provide support for the clinical application of this regimen.
Materials and methods
Reagents
Chidamide was custom-synthesized by Shenzhen Chipscreen Biosciences Ltd. (Shenzhen, China) and dissolved in dimethyl sulfoxide (DMSO) at the concentration of 25, 50, 125, 250, and 500 nM. Cytarabinewas a gift from Pfizer, Ltd. (China) and dissolved in DMSO at the concentration of 25, 50, 125, 250, and 500 nM. All the above stock solutions were stored at -20°C until use.
Cell culture
Two MDS cell lines (SKM-1 and MUTZ-1) and the AML cell line KG-1 were used in our study.SKM-1 was obtained from the Health Science Research Resources Bank in Japan [11]. MUTZ-1, a cell line established from Childhood Myelodysplastic Syndrome with del (5q), was obtained from the German Brauschweig cell center [12]. KG-1 was obtained from the American Type Culture Collection (Rockville, Maryland, USA). All cell lines were cultured in RPMI 1640 medium (Boehringer, Ingelheim, Germany) containing 10% heat-inactivated fetal calf serum (Boehringer), 100 μg/mL penicillin (Gibco), and 100 U/mL streptomycin (Gibco) in a humidified atmosphere (37.5°C and 5% CO2).
Proliferation assay (CCK-8)
Cell viability was measured by using the tetrazolium salt-based CCK-8 assay (Dojindo Molecular Technologies Inc.). Cells were seeded into 96-well plates at 1.5×105 cells/mL in 200 µL complete medium. Plates were put at 37°C in 5% CO2, and then 20 µL of CCK-8 reagent was added to the wells and incubated for 1.5 h. The OD was then read at 450 nm within 15 min. This experiment was repeated three times and each sample had three replicates. Cell proliferation was calculated using the following equation:
Proliferation (%)=(OD450 of isogarcinol group/OD450 of control group)×100%.
HDAC activity analysis
Chidamide (25 and 50 nM) and 50 nM cytarabine were added to the MDS cells and incubated at 37°C for 72 h. HDAC activity was detected as described in the Colorimetric HDAC Activity Assay kit (BioVision). Each reaction (100 μL)contained the nuclear protein (50 μg) extract from SKM-1, MUTZ-1, or KG-1 cells. The HDAC activities were measured with a microplate reader (SpectramMaxM5) at 405 nm. The positive control (i.e., only the nuclear extract and vehicle) was set as 100% and double-distilled water containing 10 μM Trichostatin A, a strong HDACi, wasused as a negative control and set as 0%.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from cells using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA). One microgram of purified total RNA was used for the RT-PCR analysis with the SuperScript First-Strand Synthesis System (Invitrogen). The qRT-PCR was performed using SYBR Premix Ex Taq (Takara Biomedical, Siga, Japan) and the Thermal Cycler Dice Real Time system (Takara Biomedical) in a 96-well plate according to the manufacturer’s instructions. Amplification proceeded through 40 cycles of 94°C for 10 s and 56.7°C for 30 s with anextension at 72°C for 30 s. The primers used for RT-PCR were as follows: human HDAC1 F: 5’-ACCGGGCAACGTTACGAAT-3’, R: 5’-CTATCAAAGGACACGCCAAGTG-3’; HDAC2F: 5’-TCATTGGAAAATTGACAGCATAGT-3’, R: 5’-CATGGTGATGGTGTTGAAGAAG-3’; HDAC3 F: 5’-TTGAGTTCTGCTCGCGTTACA-3’ R: 5’-CCCAGTTAATGGCAATATCACAGAT-3’. Human β-actin was used as a housekeeping gene forquantity normalization, F: 5’-CTGGAACGGTGAAGGTGACA-3’, R: 5’-AAGGGACTTCCTGTAACAATGCA-3’. The levels were calculated using the 2-ΔΔCt method [(Ct, target gene Ct, β-actin) sample-(Ct, target gene Ct, β-actin) control] after normalizing the data according to the β-actin mRNA expression [13].
Cell cycle analysis by flow cytometry
Cell cycle analysis was performed with anAccuri C6 (BD Bioscience, Franklin Lakes, USA).Cells were fixed in 70% ethanol for at least 4 h at 4°C, and stained with 20 μg/mL propidium iodide (PI) containing 10 μg/mL RNase A for 30 min at room temperature. The resulting DNA distributions were analyzed by Modifit (Verify Software House Inc., Topsham, ME, USA) for the proportions of cells in the phases of the cell cycle.
Cell apoptosis analysis by flow cytometry
Cell apoptosis [14] was detected by flow cytometry and cells were stained with fluorescein-conjugated annexinV and PI. After washing with phosphate-buffered saline (PBS) two times, cells were resuspended in binding buffer (10 mM N-2-hydroxyl piperazine-N0-2-ehane sulfonicacid/NaOH, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2). One hundred microliters of cell suspension were incubated with 10 µL annexin V-FITC and 10 µL PI for 20 min at room temperature in the dark. Cells were then detected by flow cytometry within 30 min, and the percentage of apoptotic cell sin the total number of cells in each group was compared.
Western blotting
Western blot was used to evaluate the content of acetyl-histone H3, p21, CDK2, cleaved caspase-3, Bcl-2, and Bax in cell extracts after using chidamide (0, 25, and 50 nM), cytarabine (50 nM), and the combination of chidamide with cytarabine (50 nM for both substances) for 72 h. Cells were lysed with a lysis buffer (20 mM Tris-HCl, pH8.0, 150 mM NaCl, 2 mM EDTA, 100 mM NaF, 1% NP-40, 1 μg/mL leupeptin, 1 μg/mL anti-pain, and 1 mM phenylmethylsulfonyl fluoride), and the protein concentrations were determined with the BCA protein assay kit (Pierce, Rockford, IL, USA). Proteins (30 μg) were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). After electrophoresis on an 8% SDS gel, the proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad Laboratories, Hercules, CA, USA). PVDF membranes were blocked with a solution containing 5% skim milk and then incubated overnight at 4°C with the following antibodies: anti-acetyl-histoneH3 (#9649), anti-p21 (#2947), anti-CDK2 (#2546), anti-cleaved caspase-3 (#9661), anti-Bcl-2 (#15071), anti-Bax (#5023), anti-GAPDH antibody (#5174) (Cell Signaling Technology, Beverly, MA, USA) antibody. After washing with Tris-buffered saline with Tween-20 (TBST), the membranes were incubated for 1 h at room temperature with anti-rabbit IgG sheep antibody or anti-mouse IgG sheep antibody coupled to horseradish peroxidase (Amer-sham). Reactive proteins were visualized using a chemiluminescence kit (Millipore, Bedford, MA, USA).
Statistical analysis
All results are expressed as means and S.D. of several independent experiments. Pairwise comparison of CCK-8 results was done by the Student’s t-test. Multiple comparisons of the PCR, HDAC activity, apoptosis,and cell cycle data were madeu sing ANOVA test to determine statistical significance of the detected differences. A value of P < 0.05 was considered statistically significant.
Results
Chidamide synergistically enhanced the histone H3 acetylation level and inhibited HDAC activity with cytarabine
To explore whether the combination of chidamide and cytarabine might influence histone H3 acetylation in MDS cells, we investigated the histone H3 acetylation level when cells were exposed to just chidamide or cytarabine alone, and when they were given together. Chidamide and cytarabine together showed ahigher acetylation level of histone H3 as compared to when chidamide was given alone (Figure 1A). The HDAC activity results indicate that the combination of chidamide with cytarabine caused greater HDAC inhibition as compared with chidamide alone (Figure 1B). Furthermore, we measured the expression of HDAC1, HDAC2, and HDAC3, since chidamide mainly suppresses the activity of class I HDACs. Our results showeda decrease in HDAC expression when chidamide with cytarabine were given together as compared to when chidamide was given alone (Figure 1C). Thus, the combination of chidamide (50 nM) and cytarabine (50 nM) lead to higher levels of histone H3 acetylation and an inhibition of HDAC activity as compared to when MDS cells were exposed to chidamide alone.
Figure 1.
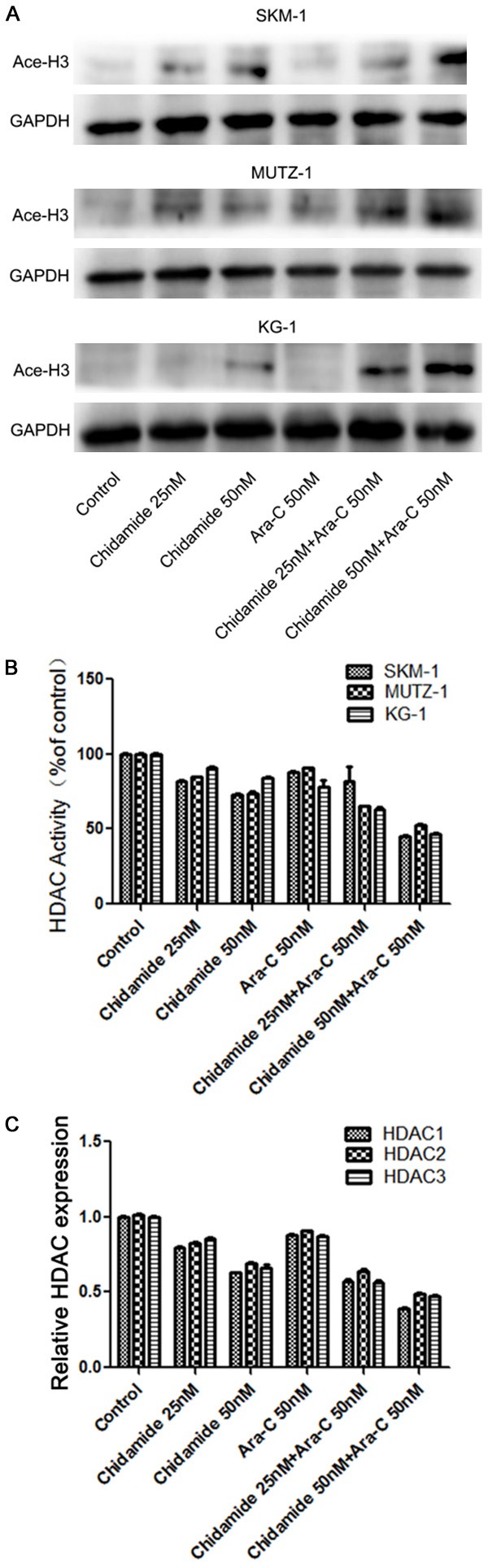
The combination of chidamide with cytarabine enhanced histone H3 acetylation and inhibited HDAC activity in MDS cell linesat 72 h. (A) The combination of chidamide with cytarabine promotedan increase in the level of histone H3 acetylation in human SKM-1, MUTZ-1, and KG-1 cells. (B) The combination of chidamide and cytarabine inhibitedHDAC activity in SKM-1, MUTZ-1, and KG-1 cell lines.(C) The combination of chidamide and cytarabine decreased the expression of HDACs in SKM-1, MUTZ-1, and KG-1 cell lines.
Chidamide synergistically enhanced the inhibitory effects in MDS cells with cytarabine
The concentration of chidamide in human plasma is 25-250 nM [15], and a low dose of cytarabine (50-500 nM) is widely accepted as a therapy for clinical MDS patients [16]. Considering that the combination of chidamide (50 nM) and cytarabine (50 nM) increased histone H3 acetylation, we next tested their effects in MDS cells. To this end, the IC50 of chidamide and cytarabine in SKM-1, MUTZ-1, and KG-1 cell lines was measured. The IC50 for chidamide varied from 301.29-802.81 nM and the IC50 for cytarabine varied from 71.80-300.49 nM (Table 1). To evaluate the effects of chidamide and cytarabineon MDS cell proliferation, we used afixed ratio (1:1) of chidamide (25, 125, 250, and 500 nM) and cytarabine (25, 125, 250, and 500 nM). The combination of chidamide and cytarabine showed significant anti-proliferative/anti-survival effects on MDS cells as compared with cytarabineor chidamide alone at all four doses (Figure 2B). The combination index (CI) values were calculated according to the Chou-Talalay equation [17]. The mean CIs of the combined chidamide and cytarabine group were 0.068, 0.158, and 0.226 in the SKM-1, MUTZ-1, and KG-1 cell lines, respectively (Table 2); all of the CIs were lower than 1, which suggests good synergy with both drugs (Figure 2A). Thus, chidamide synergistically enhanced cell toxicity with cytarabinein MDS cells.
Table 1.
Measurement of IC50 of chidamide and cytarabine in MDS cell lines
| Cell lines | Chidamide (nM) | Ara-C (nM) | ||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| IC50 | m | r | IC50 | m | r | |
| SKM-1 | 802.812 | 0.475±0.028 | 0.983 | 300.491 | 0.458±0.021 | 0.989 |
| MUZT-1 | 345.892 | 0.582±0.021 | 0.994 | 161.158 | 0.631±0.029 | 0.991 |
| KG-1 | 301.291 | 0.731±0.025 | 0.994 | 71.803 | 0.698±0.045 | 0.979 |
m indicates the slope of the median-effect plot. r indicates the linear correlation coefficient of the median-effect plot. r = 1.0 indicates perfect conformity of the dose-effect data with respect to the median-effect principle of the mass-action law. r < 1.0 indicates less than a perfect correlation.
Figure 2.
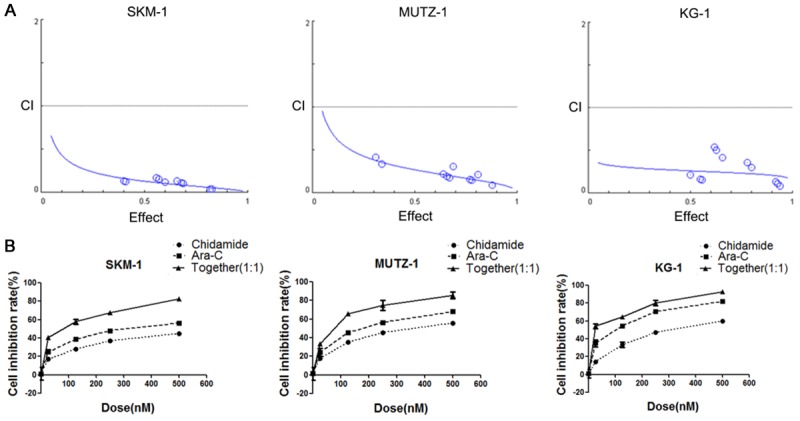
Chidamide and cytarabine synergistically inhibited cell proliferation in MDS cellsat 72 h. (A) Combination index (CI) values for chidamide combined with cytarabine in MDS cells were lower than 1. The effects of these combinations were estimated using the CompuSyn software, which was developed based on the CIequation of Chou-Talalay. CI < 1 indicates synergism; CI =1 indicates additive effect in the absence of synergism or antagonism; CI > 1 indicates antagonism. (B) The combination of chidamide and cytarabine inhibited cell proliferation. Cell inhibition rate (%)=(1-OD450 of isogarcinol group/OD450 of control group)×100%.
Table 2.
Combination index value of the combination of chidamide and cytarabine in MDS cell lines
| Cell lines | Combination index | ||||
|---|---|---|---|---|---|
|
| |||||
| ED50 | ED75 | ED90 | ED95 | Mean | |
| SKM-1 | 0.131 | 0.072 | 0.041 | 0.027 | 0.068 |
| MUZT-1 | 0.271 | 0.171 | 0.109 | 0.081 | 0.158 |
| KG-1 | 0.262 | 0.234 | 0.211 | 0.195 | 0.226 |
Combination index values < 1.0 are consistent with synergism, and the lower the value, the greater the synergism.
Combination of chidamide with cytarabine resulted in cell cycle arrestin the G0/G1 phase
Flow cytometry analysis was used to investigate the cell cycle. The results showed the percentage of cells in the G0/G1 phase increased significantly in the combination of chidamide (25 and 50 nM) and cytarabine (50 nM) group as compared to the chidamide alone (25 and 50 nM) group at 72 h (P < 0.05; Figure 3A and 3B). Western blot analysis indicated that CDK2 expression was down-regulated and p21 was up-regulated after treatment with both drugs, which was consistent with the cell-cycle analysis (Figure 3C). The decreased level of CDK2 and increased level of p21 suggests a possible mechanism by which cell-cycle arrest is occurring in the G0/G1 phase after treatment with both chidamide and cytarabine.
Figure 3.

The combination of chidamide and cytarabine arrested the cell cyclein the G0/G1 phase. (A, B) The combination of chidamide and cytarabine induced cell cycle arrest at the G0/G1 phase. (C) Western blot analysis showed the expression of cell-cycle protein p21 was increased and CDK2 was down-regulated.* indicates a significant difference as compared with 25 nM chidamide. & indicates a significant difference as compared with 50 nM chidamide.
Combination of chidamide and cytarabine induces apoptosis in MDS cells
To further investigate the mechanism by which cell proliferation is arrested in MDS cells treated with chidamide with cytarabine, cell apoptosiswas tested by flow cytometry. Our results indicated that the number of apoptotic cells significantly increased when cells were treated with acombination of chidamide (25 and 50 nM) with cytarabine (50 nM) as compared to chidamide (25 and 50 nM) or cytarabine (50 nM) alone for 72 h (P < 0.05; Figure 4A and 4B). Furthermore, the expression of Bcl-2 was down-regulated, and cleaved caspase-3 and Bax were up-regulated when cells were exposed to both compounds (Figure 3C).
Figure 4.

The combination of chidamide and cytarabine induced cell apoptosis in MDS cells at 72 h. (A, B) The combination of chidamide and cytarabine induced cell apoptosis. (C) Western blot analysis showed the expression of the cell apoptosis protein cleaved caspase-3 was up-regulated, but Bcl-2 and Bax were down-regulated following treatment with both agents. * indicates a significant difference as compared with 25 nM chidamide. & indicates a significant difference as compared with 50 nM chidamide. # indicates a significant difference as compared with 50 nM cytarabine.
Discussion
Our previous study found that chidamide induces cell cycle arrest and apoptosis in MDS and another study demonstrated that chidamide inhibits the viability of MDS and AML cells [9,18]. Indeed, chidamide is a potential therapeutic agent in MDS patients. The deoxycytidine analogue1-h-D-arabinofuranosylcy-tosine (cytarabine, ara-C) is the most commonly used drug in thetreatment of AML [19]. Moreover, use of lowdose of cytarabineis beneficial for elderly patients with high-risk MDS [16]. Ara-C exerts its cytotoxicity by inhibiting DNA polymerase or by terminating chain elongation upon being incorporated into newly synthesized DNA [20,21]. During a demethylation therapy,chidamide increases the acetylation level of histone H3 and inhibits HDAC activity. Both chidamide and ara-C are effective in treating myeloidleukemia through different mechanisms, thus triggering interest in combining them as a therapeutic treatment for MDS. However, there is little known regarding the effects of their combination on MDS cells.
Our current study showed that chidamide and cytarabine synergistically enhanced histone H3 acetylation and inhibited HDAC activityin MDS cells. The effect was significant in the group that received 50 nM of both chidamide and cytarabine. Therefore, our results indicate that the fixed ratio (1:1) of chidamide tocytarabineis more efficient in killing MDS cells. Moreover, our CCK-8 results further support our hypothesis regarding the synergistic effect of both compounds on MDS cell proliferation. These results provide some clues as to the potential application of the combination of chidamide and cytarabine in MDS patients.However,the mechanism by which these chemicals inhibit MDS cell proliferation and survival needs to be clarified.
HDACis induce cell cycle arrest in both normal cells and cancer cells at low concentration [22,23]. Chidamide induces G1 arrest and ROS-dependent apoptosis in human leukemia cells [24].
Our results showed that the expression of CDK2, which is a positive regulator of the cell cycle, [25] was down-regulated and the expression of p21, which is a negative regulator of the cell cycle, was up-regulated inthe chidamide/cytarabine combination group. All of these modulations resulted in cell growth arresting at the G0/G1 phase. Furthermore, several critical apoptotic regulators were modulated by the administration of chidamide and cytarabine.Specifically, we observed a down-regulation Bcl-2 (a negatively regulator of cell apoptosis) [26,27], and an up-regulation of cleaved caspase-3 and Bax (a positively regulator of cell apoptosis) [28,29] expression with the combination of chidamide and cytarabine treatment in MDS cell lines. All of these modulations resulted in cell apoptosis.
In summary, we provide compelling evidence that a non-toxic dose of chidamide and cytarabine synergistically induced cell cycle arrest and apoptosis in MDS cells. This is an interesting clinical therapy strategy for MDS, whichcombines epigenetic and traditional chemical drugs at safe concentrations. Our data suggest that chidamide in combination with cytarabine at low concentration may be a promising treatment for the patients with MDS. Future studies will likely need to focus on the in vivo efficacy of such a treatment regimen.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant nos. 81570106, 81400088, 81400085), the Tianjin Municipal Natural Science Foundation (Grant nos. 14JCYBJC25400, 15JCYBJC24300, 16ZXMJSY00180, 20140118), The youth incubation fund of Tianjin Medical University General Hospital (ZYYFY 2016006) and Tianjin institution of lung cancer for their support in the lab.
Disclosure of conflict of interest
None.
References
- 1.Liu L, Chen B, Qin S, Li S, He X, Qiu S, Zhao W, Zhao H. A novel histone deacetylase inhibitor Chidamide induces apoptosis of human colon cancer cells. Biochem Biophys Res Commun. 2010;392:190–195. doi: 10.1016/j.bbrc.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 2.Shi Y, Dong M, Hong X, Zhang W, Feng J, Zhu J, Yu L, Ke X, Huang H, Shen Z, Fan Y, Li W, Zhao X, Qi J, Huang H, Zhou D, Ning Z, Lu X. Results from a multicenter, open-label, pivotal phase II study of chidamide in relapsed or refractory peripheral T-cell lymphoma. Ann Oncol. 2015;26:1766–1771. doi: 10.1093/annonc/mdv237. [DOI] [PubMed] [Google Scholar]
- 3.Xie A, Liao C, Li Z, Ning Z, Hu W, Lu X, Shi L, Zhou J. Quantitative structure-activity relationship study of histone deacetylase inhibitors. Curr Med Chem Anticancer Agents. 2004;4:273–299. doi: 10.2174/1568011043352948. [DOI] [PubMed] [Google Scholar]
- 4.Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: what are the cancer relevant targets. Cancer Lett. 2009;277:8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 5.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi J, Shao ZH, Liu H, Bai J, Cao YR, He GS, Tu MF, Wang XL, Hao YS, Yang TY, Yang CL. Transformation of myelodysplastic syndromes into acute myeloid leukemias. Chin Med J (Engl) 2004;117:963–967. [PubMed] [Google Scholar]
- 7.Wang FX, Zhang WG, He AL, Cao XM, Chen YX, Zhao WH, Yang Y, Wang JL, Zhang PY, Gu LF. Effect of granulocyte colony-stimulating factor priming combined with low-dose cytarabine and homoharringtonine in higher risk myelodysplastic syndrome patients. Leuk Res. 2016;48:57–61. doi: 10.1016/j.leukres.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 8.Ye L, Ren Y, Zhou X, Mei C, Ma L, Ye X, Wei J, Xu W, Meng H, Qian W, Mai W, Lou Y, Xu G, Qian J, Lou Y, Luo Y, Xie L, Lin P, Hu C, Jin J, Tong H. Decitabine priming prior to low-dose chemotherapy improves patient outcomes in myelodysplastic syndromes-RAEB: a retrospective analysis vs. chemotherapy alone. J Cancer Res Clin Oncol. 2017;143:873–882. doi: 10.1007/s00432-016-2331-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Z, Ding K, Li L, Liu H, Wang Y, Liu C, Fu R. A novel histone deacetylase inhibitor Chidamide induces G0/G1 arrest and apoptosis in myelodysplastic syndromes. Biomed Pharmacother. 2016;83:1032–1037. doi: 10.1016/j.biopha.2016.08.023. [DOI] [PubMed] [Google Scholar]
- 10.Zhou Y, Pan DS, Shan S, Zhu JZ, Zhang K, Yue XP, Nie LP, Wan J, Lu XP, Zhang W, Ning ZQ. Non-toxic dose chidamide synergistically enhances platinum-induced DNA damage responses and apoptosis in Non-Small-Cell lung cancer cells. Biomed Pharmacother. 2014;68:483–491. doi: 10.1016/j.biopha.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 11.Nakagawa T, Matozaki S, Murayama T, Nishimura R, Tsutsumi M, Kawaguchi R, Yokoyama Y, Hikiji K, Isobe T, Chihara K. Establishment of a leukaemic cell line from a patient with acquisition of chromosomal abnormalities during disease progression in myelodysplastic syndrome. Br J Haematol. 1993;85:469–476. doi: 10.1111/j.1365-2141.1993.tb03334.x. [DOI] [PubMed] [Google Scholar]
- 12.Steube KG, Gignac SM, Hu ZB, Teepe D, Harms D, Kabisch H, Gaedicke G, Hansen-Hagge T, Macleod RA, Quentmeier H, Drexler HG. In vitro culture studies of childhood myelodysplastic syndrome: establishment of the cell line MUTZ-1. Leuk Lymphoma. 1997;25:345–363. doi: 10.3109/10428199709114174. [DOI] [PubMed] [Google Scholar]
- 13.Zhao B, He T. Chidamide, a histone deacetylase inhibitor, functions as a tumor inhibitor by modulating the ratio of Bax/Bcl-2 and P21 in pancreatic cancer. Oncol Rep. 2015;33:304–310. doi: 10.3892/or.2014.3595. [DOI] [PubMed] [Google Scholar]
- 14.Aubry JP, Blaecke A, Lecoanet-Henchoz S, Jeannin P, Herbault N, Caron G, Moine V, Bonnefoy JY. Annexin V used for measuring apoptosis in the early events of cellular cytotoxicity. Cytometry. 1999;37:197–204. doi: 10.1002/(sici)1097-0320(19991101)37:3<197::aid-cyto6>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 15.Gu R, Liu T, Zhu X, Gan H, Wu Z, Li J, Zheng Y, Dou G, Meng Z. Development and validation of a sensitive HPLC-MS/MS method for determination of chidamide (epidaza), a new benzamide class of selective histone deacetylase inhibitor, in human plasma and its clinical application. J Chromatogr B Analyt Technol Biomed Life Sci. 2015;1000:181–186. doi: 10.1016/j.jchromb.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 16.Wu L, Li X, Su J, He Q, Zhang X, Chang C, Pu Q. Efficacy and safety of CHG regimen (low-dose cytarabine, homoharringtonine with G-CSF priming) as induction chemotherapy for elderly patients with high-risk MDS or AML transformed from MDS. J Cancer Res Clin Oncol. 2011;137:1563–1569. doi: 10.1007/s00432-011-1020-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 18.Zhao S, Guo J, Zhao Y, Fei C, Zheng Q, Li X, Chang C. Chidamide, a novel histone deacetylase inhibitor, inhibits the viability of MDS and AML cells by suppressing JAK2/STAT3 signaling. Am J Transl Res. 2016;8:3169–3178. [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson SA. Nucleoside analogues in the treatment of haematological malignancies. Expert Opin Pharmacother. 2001;2:929–943. doi: 10.1517/14656566.2.6.929. [DOI] [PubMed] [Google Scholar]
- 20.Sampath D, Rao VA, Plunkett W. Mechanisms of apoptosis induction by nucleoside analogs. Oncogene. 2003;22:9063–9074. doi: 10.1038/sj.onc.1207229. [DOI] [PubMed] [Google Scholar]
- 21.Hileman EO, Liu J, Albitar M, Keating MJ, Huang P. Intrinsic oxidative stress in cancer cells: a biochemical basis for therapeutic selectivity. Cancer Chemother Pharmacol. 2004;53:209–219. doi: 10.1007/s00280-003-0726-5. [DOI] [PubMed] [Google Scholar]
- 22.Xu W, Ngo L, Perez G, Dokmanovic M, Marks PA. Intrinsic apoptotic and thioredoxin pathways in human prostate cancer cell response to histone deacetylase inhibitor. Proc Natl Acad Sci U S A. 2006;103:15540–15545. doi: 10.1073/pnas.0607518103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ungerstedt JS, Sowa Y, Xu WS, Shao Y, Dokmanovic M, Perez G, Ngo L, Holmgren A, Jiang X, Marks PA. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005;102:673–678. doi: 10.1073/pnas.0408732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gong K, Xie J, Yi H, Li W. CS055 (Chidamide/HBI-8000), a novel histone deacetylase inhibitor, induces G1 arrest, ROS-dependent apoptosis and differentiation in human leukaemia cells. Biochem J. 2012;443:735–746. doi: 10.1042/BJ20111685. [DOI] [PubMed] [Google Scholar]
- 25.Harper JW, Elledge SJ, Keyomarsi K, Dynlacht B, Tsai LH, Zhang P, Dobrowolski S, Bai C, Connell-Crowley L, Swindell E. Inhibition of cyclindependent kinases by p21. Mol Biol Cell. 1995;6:387–400. doi: 10.1091/mbc.6.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H, Guo Y, Fu M, Liang X, Zhang X, Wang R, Lin C, Qian H. Antitumor activity of Chidamide in hepatocellular carcinoma cell lines. Mol Med Rep. 2012;5:1503–1508. doi: 10.3892/mmr.2012.858. [DOI] [PubMed] [Google Scholar]
- 27.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 28.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 29.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]